

Abstract
The accelerated development of atherosclerosis with increased risk of cardiovascular disease in systemic lupus erythematosus (SLE) patients is not well understood. An appropriate mouse model would greatly help to understand the mechanisms of this association. We have therefore combined the ApoE-/- model of atherosclerosis with three different murine models of SLE. We found that induction of cGVH in B6.ApoE-/- mice, breeding a Fas null gene onto these B6/lpr.ApoE-/- mice, and breeding the ApoE-/- defect onto MRL/lpr mice all caused a modest increase of atherosclerosis at 24 weeks of age compared to B6.ApoE-/- controls. B cells in B6.ApoE-/- mice had certain phenotypic differences compared to congenic C57BL/6 mice, as indicated by high expression of MHC II, Fas, CD86, and by increased number of cells bearing marginal-zone phenotype. Furthermore, B6ApoE-/- mice had significant titers of anti-oxLDL and anti-cardiolipin autoantibodies compared to their B6 counterparts. Our studies also indicate that, following induction of cGVH, marginal zone B cells in B6.ApoE-/- are depleted, and there is considerable increase in anti-oxLDL and anti-cardiolipin abs along with secretion of lupus-specific autoantibodies, such as anti-dsDNA and anti-chromatin abs. Histological sections showed that cGVH and/or Fas deficiency could exacerbate atherosclerosis. The production of anti-oxLDL and anti-cardiolipin in ApoE-/- mice was also increased. These observations define a connection between induction of lupus-like symptoms and development of severe atherosclerosis in apoE deficient lupus mouse models.
Keywords: Autoimmunity, Atherosclerosis, Apolipoprotein E, Graft Versus Host Disease, lpr, Systemic Lupus Erythematosus
Introduction
Systemic lupus erythematosus (SLE) is characterized by the production of autoantibodies against chromatin, Sm, DNA, and other nuclear antigens, as well as the deposition of Ig in various organs, including the kidneys and skin. These immunologic events are accompanied by the development of glomerulonephritis. However, the underlying mechanisms are not fully understood [1]. There are reports suggesting that deposition of immune complexes exacerbates the local inflammatory and autoimmune processes and thereby, accelerates atherosclerosis in SLE patients [2; 3]. In fact, SLE patients exhibit a higher incidence of atherosclerosis and are at significant risk for premature cardiovascular disease [4]. Elevated levels of antiphospholipid antibodies, which are often found in people with lupus, have traditionally been linked to an increased risk of blood clotting and may increase the risk of clot formation at the plaque site [5]. Accumulating evidence supports an autoimmune mechanism as one of the prime pathogenic processes involved in the development of atherosclerosis [6]. More recent evidence suggests that these antibodies may also facilitate the uptake of oxidized low density lipoprotein into inflammatory cells in the vessel wall, which is a key step in the formation of atherosclerotic plaque [7; 8]. In order to determine whether the general inflammatory process of lupus enhances atherogenesis, we bred several different lupus susceptible mouse strains with the B6ApoE-/- strain (apolipoprotein-E deficient) and tested the hypothesis that the combination of lupus and genetic mutations favored atherosclerosis in mice. We found that each model could modestly accelerate atherosclerosis compared to control B6ApoE-/- mice. Our findings extend the work of others in the gld, lpr, and Sle1.2.3 models [9; 10; 11]. These mouse models may allow us further understanding of the interaction between SLE and atherosclerosis.
Materials and Methods
Mice
C57BL/6J (B6), B6.C-H-2bm12/KhEg (bm12), and C57BL/6J ApoE knockout (B6ApoE-/-) mice were originally obtained from The Jackson Laboratory (Bar Harbor, ME). B6ApoE-/- mice were backcrossed to C57BL/6J-lpr/lpr (B6/lpr) for four generations, and to MRL/MpJ-Faslpr (MRL/lpr) for ten generations. All mice were maintained in our mouse colony at the University of Pennsylvania Medical Center. Recipient and donor mice were sex- and age-matched for induction of cGVH experiments. In the spontaneous SLE models, we did not find any significant difference between female and male mice in atherosclerosis, and so they are pooled in the presentation of data. The ApoE-/- and lpr loci were tracked by PCR of tail DNA [12; 13]. cGVH disease was induced as previously described [1]. Briefly, recipient mice (8 weeks old) were injected (i.p.) with single cell suspensions of 1 ×108 donor splenocytes, prepared by pressing donor spleens through a wire mesh screen in HBSS. Mice were sacrificed 16 weeks after the induction of cGVH (24-weeks of age). Blood samples, heart and aorta were collected for cholesterol, antibodies, and histology analysis. B6/lprApoE-/- and MRL/ lprApoE-/- were sacrificed at 24 weeks of age.
En face quantification of atherosclerotic lesions
At sacrifice, the aorta was perfused with PBS via the left ventricle and then removed. The heart and ascending and descending aorta were dissected free as far as the iliac bifurcation. Adventitial and adipose tissues were removed, and the outer curvature of the arch was cut longitudinally. The aortic tree was briefly rinsed with 70% ethanol, stained with Sudan IV for 5 minutes, and then destained for 5 minutes. The average lesion areas were calculated with Image 5.0 software systems (MediaCybernetics, Inc., Silver Spring, MD). Results were expressed as the percentage of intimal surface involved [14].
Histochemistry
The aortic arch was transected at the level of the aortic sinuses, snap-frozen in embedding compound (OTC), and stored at -80°C. Eight μm sections were serially cut from the aortic root starting at the aortic sinus and ending where the aortic sinus disappeared, a distance of approximately 280-300 μm, and were then stained with Oil red O. The average lesion areas were calculated with Image 5.0 software. Results were expressed as square micrometer/section [14].
Plasma lipids
Total cholesterol levels were measured by an enzymatic colorimetric kit (Wako Chemicals USA, Inc.). Briefly, 10 μl of plasma (diluted at different concentrations based on mouse strains) and 10 μl of standard solution (provided by the kit) were put into tubes, and 3.0 ml of color reagent solution was then added. The tubes were mixed well and incubated at 37°C for 5 minutes. The color development was detected at 505 nM with Beckman DU 640 spectrophotometer.
Immunofluorescence staining of spleen cell suspensions
The following conjugated Abs were purchased from BD PharMingen (San Diego, CA): anti-CD19 (clone 1D3), anti-CD21 (clone 7G6), anti-CD23 (clone B3B4), anti-CD24 (clone M1/69), anti-CD80 (clone16-10A1), anti-CD86 (clone GL1), anti-MHC class II (clone AF6-120.1), and anti-Fas (clone Jo2). Cell surface staining was routinely performed with age- and sex-matched controls, as previously described [15; 16]. A total of 1.5 × 106 cells were blocked with 50 μl of 2.4G2 culture supernatant. The cells were then incubated with directly-labeled Abs for 30 min and washed. An additional 20 min incubation with streptavidin-CyChrome was performed to detect biotinylated Abs. Cells were fixed in PBS containing 1% paraformaldehyde and analyzed on a Becton Dickinson Biosciences FACSCalibur. Relative fluorescence intensity was plotted on a logarithmic scale using FlowJo software (Tree Star, Inc., Ashland, OR).
Detection of total IgG, anti-dsDNA, and anti-chromatin
Total IgG, anti-dsDNA, and anti-chromatin were assessed by ELISA, as previously described [17; 18]. For total IgG, goat anti-mouse Fab (Jackson ImmunoResearch Laboratories) at 3 μg/ml was coated onto plates, and mouse IgG (clone HB63) was used as standard. For anti-dsDNA quantitation, DNA was purified from calf thymus (Sigma) and used at 3 μg/ml. For anti-chromatin ELISAs, chromatin was purified from chicken erythrocyte nuclei and used at 5μg/ml. In both cases, the secondary antibody was biotinylated goat anti-mouse pFc’ (Jackson ImmunoResearch Laboratories, West Grove, PA), followed by avidin-alkaline phosphatase. Para-nitrophenyl phosphate substrate (Sigma, St. Louis, MO), 1 mg/ml in 0.01 M diethanolamine, pH 9.8, was added to develop the color. The plates were read at various time points with an automated ELISA reader (Molecular Devices, Sunnyvale, CA). Autoantibody results from individual ELISAs were standardized against a reference serum [19].
Detection of anti-phospholipids
Anti-oxLDL and anti-cardiolipin were assessed by ELISA, as previously described [20; 21]. Cardiolipin was purchased from Avanti Polar Lipids (Alabaster, AL) and LDL from Calbiochem (San Diego, CA). LDL was oxidized as described previously [22] Briefly, polyvinyl chloride microtiter plates (Becton Dickinson, Franklin Lakes, NJ) were coated with 2 μg/ml of ox-LDL and 20 μg/ml of cardiolipin, respectively, and after blocking with PBTN (PBS+1%BSA+0.05%Tween20+0.02%NaN3), diluted serum samples (1:100) or purified monoclonal antibody FB1 as a standard were added to the plates. FB1 is a CL-reactive IgG2b mAb that was obtained from a 5-month-old female (NZW × BXSB) F1 mouse. The plates were incubated for 2 hours at room temperature. After washing, the binding was detected with goat anti-mouse IgG-alkaline phosphatase (Southern Biotechnology, Birmingham, AL), followed by color development with the appropriate substrate [21].
Statistical and data analyses
Statistical analysis was performed using Student’s t test. A value of p < 0.05 was considered to be significant. Results were expressed as mean ± SD.
Results
Lipid profiles in ApoE deficient lupus mice
The ApoE-/- locus was backcrossed onto B6/lpr and MRL/lpr mice. In addition, cGVH was induced in B6.ApoE-/- mice as an experimental model of SLE. Figure 1 shows total plasma cholesterol levels in these mice at 24 weeks of age. As expected, the ApoE-/- locus raised cholesterol levels 4-5 fold in each strain. Surprisingly, total plasma cholesterol was reduced in B6/lprApoE-/- mice compared to B6- ApoE-/-. The MRL/lpr.ApoE-/- mice had increased plasma cholesterol levels compared to B6- ApoE-/- (Figure 1). In mice that were undergoing cGVH, we could detect no significant difference in cholesterol levels compared to non-cGVH controls (B6 and B6- ApoE-/-) at 16 weeks, a time at which autoantibodies were readily detectable (see Figure 3).
Figure 1. Total plasma cholesterol in SLE mice.
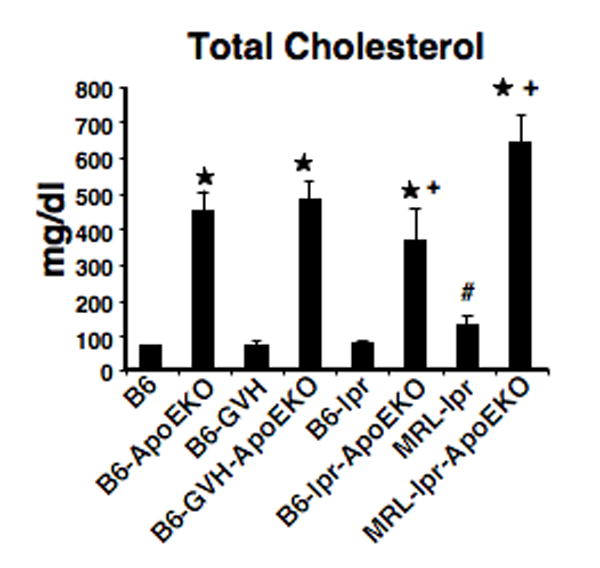
Plasma total cholesterol levels were significantly increased in all ApoEKO mice compared to B6 control. *, p<0.05, comparing lupus-prone-ApoEKO and corresponding controls. #, p<0.05, comparing MRL/lpr and B6 control. +, p<0.05, comparing B6/lpr.ApoE-/- and B6.ApoE-/-; and comparing MRL/lpr.ApoE-/- and B6.ApoE-/-, respectively. For each strain, data are pooled from 5 females and 4 males.
Figure 3. Autoantibodies in ApoE-/- SLE mice.
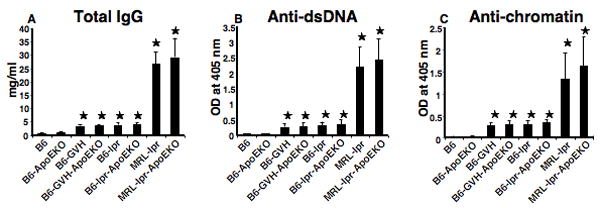
ELISA results are shown in for total IgG (A); anti-dsDNA (B); and anti-chromatin (C). Serum total IgG, anti-dsDNA, and anti-chromatin were increased both in lupus-prone mice and in lupus-prone-ApoE deficient mouse. However, no significant differences were found between SLE mice for each model and its comparable ApoE-/- strain. *, p<0.05, comparing lupus-prone, lupus-prone-ApoEKO with B6 control. Data are pooled from 5 females and 4 males in each strain.
Further Acceleration of atherosclerosis in ApoE lupus mice
To test for an interaction of SLE and atherosclerosis in ApoE deficient mice, we quantified lipid deposition in the aorta by both en face examinations and cross-sectional aortic root measurements. Figure 2A shows a typical set of en face specimens, while Figure 2C shows a comparable set of aortic root sections. Compared to B6.ApoE-/- mice, each of the three SLE models also lacking ApoE showed modest increases in the total area of lipid deposits by both methods. These differences were consistent in the two experimental comparisons done with each model, and were statistically significant (Figure 2B and 2D).
Figure 2. Accelerated atherosclerosis in lupus ApoE deficient mouse.
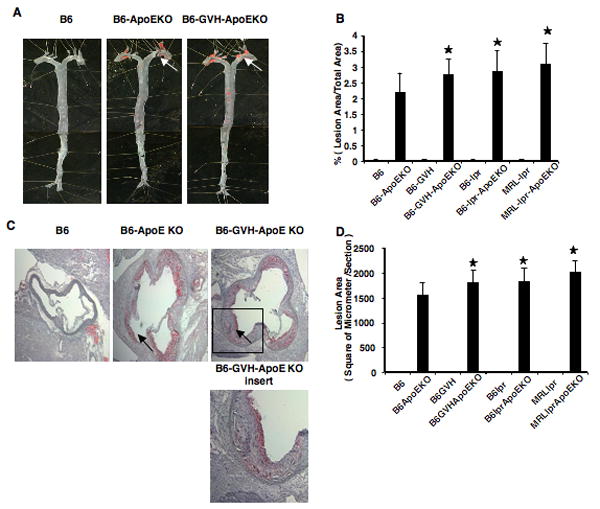
The aortic tree was stained with Sudan IV for en face analysis (A). Data pooled from 7-9 mice are shown in B. Aorta root lesions were stained in cross section with Oil red O (C). Lesion quantification for 9 mice (5 females and 4 males) is shown in D. *, p<0.05, comparing lupus-prone-ApoEKO and B6.ApoEKO. The arrows indicate atherosclerotic plaques.
Lupus ApoE deficient mice show a selective increase in anti-phospholipid autoantibodies
SLE is associated with a variety of anti-nuclear antibodies, and atherosclerosis is characterized by autoantibodies that react with certain lipid moieties. To determine if ApoE deficiency could affect the development of autoantibodies in lupus-prone mice, we assayed total IgG, anti-dsDNA, and anti-chromatin levels. Each of these serological markers was greatly increased in the SLE mice compared to B6 or B6.ApoE-/- mice (Figure 3). Despite a slight trend towards increased autoantibodies in the presence of the ApoE-/- locus, no significant differences were seen between SLE mice with or without ApoE. Conversely, we also tested how the anti-phospholipid antibodies potentially associated with atherosclerosis might be influenced by ApoE deficiency and SLE. As shown in Figure 4, both IgG and IgM anti-oxidized LDL and anti-cardiolipin antibodies were increased in SLE mice compared to non-SLE controls. In addition, ApoE-/- mice showed increased antibodies compared to non-knockout controls. For each of the three SLE models, the addition of the ApoE-/- locus caused a further increase in antibody titers.
Figure 4. Anti-phospholipid antibodies in SLE mice.
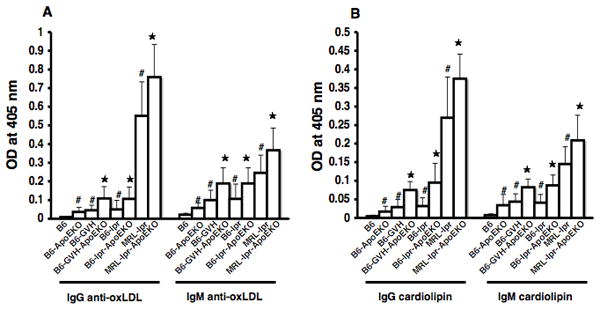
Anti-oxLDL IgG and IgM are shown in (A). Anti-cardiolipin IgG and IgM are shown in (B). *, p<0.05, comparing lupus-prone-ApoEKO and corresponding lupus-prone controls. #, p<0.05, comparing B6.ApoEKO, lupus-prone-ApoEKO with B6 control. Data are pooled from 5 females and 4 males in each strain.
B-cell phenotypic changes in B6ApoE deficient mice
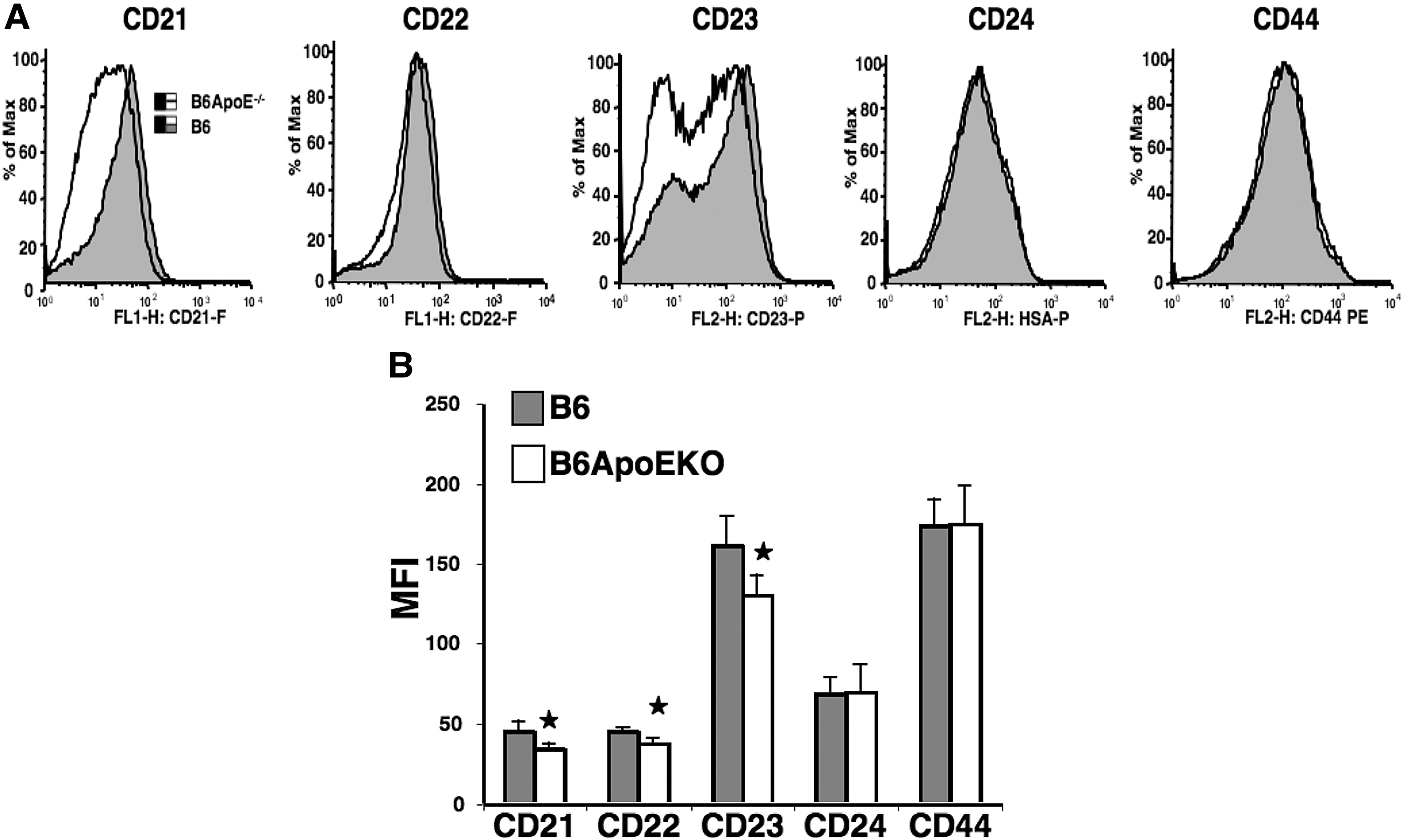
It was possible that the interaction of atherogenesis and SLE might be reflected in changes in lymphocytes subsets. We therefore performed phenotypic analysis of spleen cells from B6 and B6.ApoE-/- mice, both with and without cGVH-induced SLE. Strikingly, the ApoE-/- locus caused an apparent polyclonal B-cell activation, as evidenced by increased expression of MHC II, Fas, and CD86 (Figure 5). We also found lower expression of CD21, CD22, and CD23 on the B cells in B6.ApoE-/- compared to B6 (Supplemental Figure 1). The marginal zone B-cell subset in the spleen has been implicated in both autoimmunity and interaction with the innate immune system, and some lupus-prone mouse strains show expansion of MZ B cells. It is therefore of note that the marginal zone B cells were increased nearly two-fold in B6.ApoE-/- compared to B6 mice, as were the newly formed B cells, while the follicular B cells were decreased (Figure 6). The induction of cGVH in both of these strains reduced the number of MZ B cells. In the B6.ApoE-/- strain, cGVH altered the pattern of B-cell subsets so that they resembled those in the B6-cGVH mice.
Figure 5. B cell activation in B6.ApoE-/- mice.
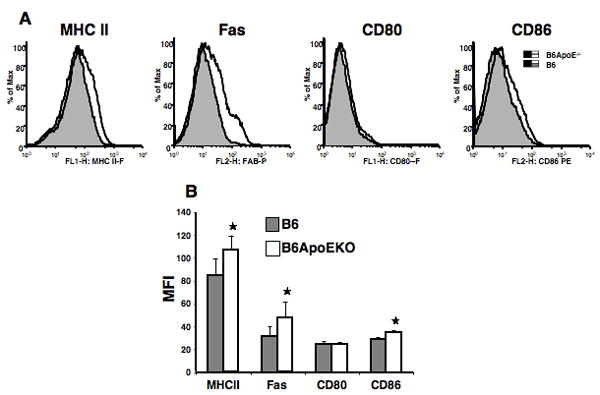
Splenic lymphocytes were analyzed at thirteen weeks post induction of cGVH. Cells were gated by size and CD19. (A) histograms from one representative mouse in each group; (B) means and SD. *, p<0.05, comparing B6.ApoEKO (N = 5 females) and B6 control (N = 3 females). MFI=Mean Fluorescence Intensity.
Figure 6. B cell subsets in B6.ApoE-/- mice.
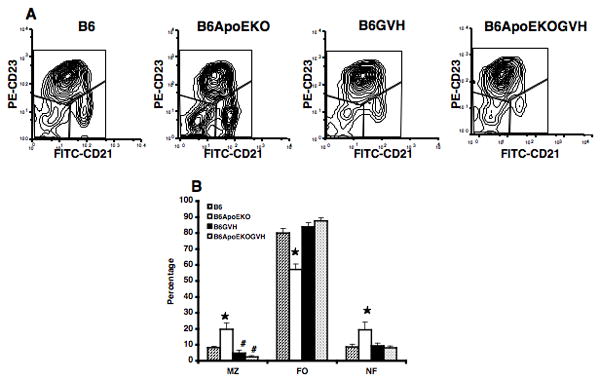
Splenic lymphocytes were analyzed at thirteen weeks post induction of cGVH. Cells were gated by size and CD19, and stained for CD21 and CD23. Marginal zone (MZ), follicular (FO) and newly formed (NF) subsets were gated as shown in A. (A) scatter plots of data from one representative mouse in each group; (B) means and SD for all mice examined. *, p<0.05, comparing cGVH and control recipients. #, p<0.05, comparing B6-GVH (N = 5 females), B6.ApoE-GVH (N = 5 females), with corresponding controls (N = 3 females).
Discussion
The accelerated development of atherosclerosis and increased risk of cardiovascular disease in young women with systemic lupus erythematosus (SLE) is a disturbing feature of the disease that is not well understood. Although the general risk factors for atherosclerosis appear to play some role in this population, additional mechanisms related to the disease or its treatment must also be involved. Perhaps this is not surprising, since atherosclerosis is an inflammatory disease and is accompanied by an autoimmune response [23]. In order to develop a system in which potential mechanisms of synergy between SLE and atherosclerosis might be explored, we have combined different mouse models of SLE with a model of atherosclerosis. Mice can be induced to develop atherosclerosis in their aorta through several genetic mechanisms, either alone or in combination with a “Western” (high lipid) diet. We chose the ApoE-/- locus as one of the best models. We did not subject the mice to an atherogenic diet, since we wanted to be able to distinguish exacerbations resulting from SLE. We tested three different SLE models. The cGVH had the advantage of providing an excellent control group, as did the B6/lpr model. The MRL/lpr model gets the most severe SLE-like disease, but lacks a fully appropriate comparison strain (MRL/+ also gets SLE, albeit milder and delayed).
Not surprisingly, the ApoE-/- locus caused a marked increase in plasma cholesterol levels in all strains we studied. The SLE models had little effect on this parameter, except that the cholesterol levels in the MRL/lpr strain were somewhat higher than in B6, and the MRL/lpr.ApoE-/- had higher levels than the B6. ApoE-/-, although the latter did not reach statistical significance. By serological analysis, we did not see any effect of ApoE deficiency on anti-DNA or anti-chromatin autoantibodies. However, two autoantibody species directed at phospholipids, anti-cardiolipin and anti-oxidized LDL were increased both in the SLE models and in the ApoE deficient mice, while the combination of these factors led to further increased titers. SLE autoantibodies were slightly, but not significantly higher in our ApoE-/- groups (Figure 3). The anti-DNA results differ from those reported by Aprahamian et al [10] and Feng et al [9], while the increased anti-oxidized LDL antibodies are similar to results from Stanic et al [11] and Feng et al [9]. Two of these previous studies each used SLE models different from the three we explored, while the Feng study also cross ApoE-/- with B6/lpr.
By both en face staining of the dissected aorta, and histological analysis of aortic root cross sections, all three SLE models showed a modest, but significant, increase in lipid deposition. We detected little lipid in the coronary arteries in all cases (data not shown). The autoimmune MRL/lpr mouse strain exhibited an unusual plasma lipoprotein profile and was shown to spontaneously develop hyperlipidemia, suggesting a possible interaction of autoimmune disease and lipoprotein metabolism [24; 25].
Interestingly, ApoE deficiency caused several changes in the B-cell populations. Compared to B6 mice, B cells in B6.ApoE-/- mice had increased expression of the activation markers MHC II, Fas and CD86, and decreased expression of CD21, CD22, and CD23. In addition, the numbers of splenic marginal zone B cells were increased in B6.ApoE-/-. Both in this strain and in B6, the numbers of MZ B cells were decreased after the induction of cGVH. Marginal zone (MZ) B cells in the spleen have the ability to respond vigorously to antigen and polyclonal activators, and this makes them key players in the early response to blood-borne pathogens. The specialized functions of these lymphocytes bridge the gap between the early innate immune response and the slower adaptive antibody response, which depends mainly on the more prolific follicular B cells. MZ B cells, like B1 cells, are important not only to combat infections but also in the maintenance of host homeostasis [26]. Some lupus-prone mouse strains showed expansion of MZ B cells [27]. The MZ B cell subset has been implicated in both autoimmunity and interaction with the innate immune system [28; 29]. However, it is not clear how our findings of marginal zone B-cell increase in ApoE-/- mice or their depletion fit in with the generation of atherosclerosis or of autoimmunity in cGVH.
Accumulation of T cells and macrophages in atherosclerotic plaques and the formation of antibodies directed against plaque proteins suggests that adaptive immunity contributes to the development of atherosclerosis [30]. Zhou and Hansson demonstrated the presence of CD22 positive B cells and IgM in the aortic root lesions of ApoE-/- mice [31]. However, what role these B cells play in the development of atherosclerosis is unclear. The frequent occurrence of antibodies to oxidized LDL in SLE and anti-phospholipid syndrome may be associated with enhanced oxidative stress in these autoimmune conditions [5]. Autoantibodies could play a role in the disease by forming immune complexes and aggravating the inflammatory process [2]. In contrast, these antibodies may protect against the disease, and may act as scavengers and clear oxLDL from the circulation or prevent their uptake by macrophages [32; 33]. In fact, in vitro these antibodies enhance the accumulation of LDL into macrophages, while in vivo immunization with oxidized LDL has been shown to protect from atherosclerosis in ApoE deficient mice [34]. Recent studies have suggested that antiphospholipid antibodies contribute to the development of atherothrombosis by enhancing atherogenesis and/or by interfering with blood coagulation. In addition, the uptake of immune complexes consisting of oxidized LDL bound to anti-LDL antibodies by the macrophages via their Fc receptors can lead to foam cell formation in a manner similar to the scavenger receptor-mediated uptake of oxidized LDL [35; 36]. Overall, the role of these anti-phospholipid antibodies in atherosclerosis remains controversial, and whether they are agonistic, antagonistic or neutral may depend on particular attributes of each antibody clone, such as specificity or isotype [37; 38]. The enhanced atherosclerosis in SLE mice that were ApoE deficient correlated with increased levels of these antibodies, but the modest degree of the effect may in part be due to the opposing mechanistic roles of different autoantibodies.
It is possible that Fas deficiency may also play a more direct effect in exacerbating atherosclerosis. We found increased atherosclerosis in the B6/lpr.ApoE-/- strain compared to B6.ApoE-/-, although the magnitude of the synergy was considerably less than was reported in B6/gld.ApoE-/- mice [10] and somewhat less than the results of Feng et al [9]. The MRL/lpr.ApoE-/- strain also had enhanced atherosclerosis, but we did not compare it to a Fas sufficient congenic. Others have reported greatly enhanced atherogenesis in MRL/lpr mice put on a high fat diet [39], but we have not been able to reproduce that finding (data not shown). We did not test the role of a high-lipid diet in conjunction with the ApoE-/- locus. Both Aprahamian et al and Stanic et al found an additional contribution of Western diet to both atherosclerosis and autoimmunity in their models [10; 11]. Recently, it has been found that failure to degrade chromatin or properly clear apoptotic cells contributes to the development of autoimmunity. Impaired apoptotic debris clearance leads to the development of an autoimmune phenotype in mouse models [10; 40; 41]. ApoE deficiency may result in impaired clearance of apoptotic cell remnants and a functionally relevant systemic proinflammatory condition in mice, independent of its role in lipoprotein metabolism [42].
In part, the differences between our work and the previous publications may be due to the distinct models investigated. Stanic et al combined the B6.Sle 1.2.3 lupus model with the LDLr-/- + Western diet atherosclerosis model [11]. Aprahamian et al utilized the B6.gld model with the ApoE-/- transgene [10], which is similar to the B6.lpr.ApoE-/- group in our study. In general, lpr and gld mice show identical phenotypes, except in cell transfer studies [43; 44]. Feng et al also looked at B6.lpr.ApoE-/-, although they derived their strain independently of ours[9]. In fact, these latter results are parallel to ours where they measured similar parameters.
In conclusion, we found a modest increased degree of lipid deposits in the aortas of ApoE deficient mice that also developed systemic autoimmunity on a genetic or experimental basis. This was associated with cellular changes in B cells and increased levels of autoantibodies to phospholipids. The synergy that we found between the three models of SLE tested and the ApoE-/- locus was less that that seen in several recent publications [9; 10; 11]. Nevertheless, these systems may permit further mechanistic understanding of the interaction between SLE and atherosclerosis, and thus allow more effective interventions in patients with this disorder.
Supplementary Material
{kind=link}
Acknowledgments
We appreciate the excellent technical assistance of Dr. Daniel Rader’s laboratory, and of Dr. Lei Zhao, Shuqin Wang, Magda Cuevas, Jing Jiao, and Vivian Ji.
Footnotes
Supported by the Lupus Research Institute, the Arthritis Foundation and the Alliance for Lupus Research, the Lupus Foundation of South New Jersey, the Department of Veterans’ Affairs, and the NIH (R01-AR-34156; U19-AI-46358; R01-AI063626-22; and RO1-DK53088).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Morris SC, Cheek RL, Cohen PL, Eisenberg RA. Autoantibodies in chronic graft versus host result from cognate T-B interactions. Journal of Experimental Medicine. 1990;171:503–17. doi: 10.1084/jem.171.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Palinski W, Witztum JL. Immune responses to oxidative neoepitopes on LDL and phospholipids modulate the development of atherosclerosis. J Intern Med. 2000;247:371–80. doi: 10.1046/j.1365-2796.2000.00656.x. [DOI] [PubMed] [Google Scholar]
- 3.Lopez LR, Salazar-Paramo M, Palafox-Sanchez C, Hurley BL, Matsuura E, Garcia-De La Torre I. Oxidized low-density lipoprotein and beta2-glycoprotein I in patients with systemic lupus erythematosus and increased carotid intima-media thickness: implications in autoimmune-mediated atherosclerosis. Lupus. 2006;15:80–6. doi: 10.1191/0961203306lu2267oa. [DOI] [PubMed] [Google Scholar]
- 4.Bruce IN. Coronary heart disease (CHD) in lupus: round up the usual suspects? Lupus. 2004;13:557–60. doi: 10.1191/0961203304lu1066ed. [DOI] [PubMed] [Google Scholar]
- 5.Vaarala O. Autoantibodies to modified LDLs and other phospholipid-protein complexes as markers of cardiovascular diseases. J Intern Med. 2000;247:381–4. doi: 10.1046/j.1365-2796.2000.00657.x. [DOI] [PubMed] [Google Scholar]
- 6.Mandal K, Jahangiri M, Xu Q. Autoimmune mechanisms of atherosclerosis. Handb Exp Pharmacol. 2005:723–43. doi: 10.1007/3-540-27661-0_27. [DOI] [PubMed] [Google Scholar]
- 7.Matsuura E, Kobayashi K, Tabuchi M, Lopez LR. Oxidative modification of low-density lipoprotein and immune regulation of atherosclerosis. Prog Lipid Res. 2006;45:466–86. doi: 10.1016/j.plipres.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 8.Matsuura E, Kobayashi K, Inoue K, Lopez LR, Shoenfeld Y. Oxidized LDL/beta2-glycoprotein I complexes: new aspects in atherosclerosis. Lupus. 2005;14:736–41. doi: 10.1191/0961203305lu2211oa. [DOI] [PubMed] [Google Scholar]
- 9.Feng X, Li H, Rumbin AA, Wang X, La Cava A, Brechtelsbauer K, Castellani LW, Witztum JL, Lusis AJ, Tsao BP. ApoE-/-Fas-/- C57BL/6 mice: a novel murine model simultaneously exhibits lupus nephritis, atherosclerosis, and osteopenia. J Lipid Res. 2007;48:794–805. doi: 10.1194/jlr.M600512-JLR200. [DOI] [PubMed] [Google Scholar]
- 10.Aprahamian T, Rifkin I, Bonegio R, Hugel B, Freyssinet JM, Sato K, Castellot JJ, Jr, Walsh K. Impaired clearance of apoptotic cells promotes synergy between atherogenesis and autoimmune disease. J Exp Med. 2004;199:1121–31. doi: 10.1084/jem.20031557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stanic AK, Stein CM, Morgan AC, Fazio S, Linton MF, Wakeland EK, Olsen NJ, Major AS. Immune dysregulation accelerates atherosclerosis and modulates plaque composition in systemic lupus erythematosus. Proc Natl Acad Sci U S A. 2006;103:7018–23. doi: 10.1073/pnas.0602311103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Piedrahita JA, Zhang SH, Hagaman JR, Oliver PM, Maeda N. Generation of mice carrying a mutant apolipoprotein E gene inactivated by gene targeting in embryonic stem cells. Proc Natl acad Sci USA. 1992;89:4471–4475. doi: 10.1073/pnas.89.10.4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Weintraub JP, Godfrey V, Wolthusen PA, Cheek RL, Eisenberg RA, Cohen PL. Immunological and pathological consequences of mutations in both Fas and Fas ligand. Cellular Immunology. 1998;186:8–17. doi: 10.1006/cimm.1998.1290. [DOI] [PubMed] [Google Scholar]
- 14.Kozarsky KF, Donahee MH, Glick JM, Krieger M, Rader DJ. Gene transfer and hepatic overexpression of the HDL receptor SR-BI reduces atherosclerosis in the cholesterol-fed LDL receptor-deficient mouse. Arterioscler Thromb Vasc Biol. 2000;20:721–7. doi: 10.1161/01.atv.20.3.721. [DOI] [PubMed] [Google Scholar]
- 15.Sekiguchi DR, Jainandunsing SM, Fields ML, Maldonado MA, Madaio MP, Erikson J, Weigert M, Eisenberg RA. Chronic graft-versus-host in Ig knockin transgenic mice abrogates B cell tolerance in anti-double-stranded DNA B cells. J Immunol. 2002;168:4142–53. doi: 10.4049/jimmunol.168.8.4142. [DOI] [PubMed] [Google Scholar]
- 16.Reap EA, Felix NJ, Wolthusen PA, Kotzin BL, Cohen PL, Eisenberg RA. bcl-2 transgenic Lpr mice show profound enhancement of lymphadenopathy. Journal of Immunology. 1995;155:5455–62. [PubMed] [Google Scholar]
- 17.Bradley DS, Jennette JC, Cohen PL, Eisenberg RA. Chronic graft versus host disease-associated autoimmune manifestations are independently regulated by different MHC class II loci. Journal of Immunology. 1994;152:1960–9. [PubMed] [Google Scholar]
- 18.Chen F, Maldonado MA, Madaio M, Eisenberg RA. The role of host (endogenous) T cells in chronic graft-versus-host autoimmune disease. J Immunol. 1998;161:5880–5. [PubMed] [Google Scholar]
- 19.Choudhury A, Maldonado MA, Cohen PL, Eisenberg RA. The Role of Host CD4 T Cells in the Pathogenesis of the Chronic Graft-versus-Host Model of Systemic Lupus Erythematosus. J Immunol. 2005;174:7600–9. doi: 10.4049/jimmunol.174.12.7600. [DOI] [PubMed] [Google Scholar]
- 20.Monestier M, Kandiah DA, Kouts S, Novick KE, Ong GL, Radic MZ, Krilis SA. Monoclonal antibodies from NZW x BXSB F1 mice to beta2 glycoprotein I and cardiolipin. Species specificity and charge-dependent binding. J Immunol. 1996;156:2631–41. [PubMed] [Google Scholar]
- 21.Nicolo D, Goldman BI, Monestier M. Reduction of atherosclerosis in low-density lipoprotein receptor-deficient mice by passive administration of antiphospholipid antibody. Arthritis Rheum. 2003;48:2974–8. doi: 10.1002/art.11255. [DOI] [PubMed] [Google Scholar]
- 22.Varadhachary AS, Monestier M, Salgame P. Reciprocal induction of IL-10 and IL-12 from macrophages by low-density lipoprotein and its oxidized forms. Cell Immunol. 2001;213:45–51. doi: 10.1006/cimm.2001.1855. [DOI] [PubMed] [Google Scholar]
- 23.George J, Afek A, Gilburd B, Harats D, Shoenfeld Y. Autoimmunity in atherosclerosis: lessons from experimental models. Lupus. 2000;9:223–7. doi: 10.1191/096120300678828190. [DOI] [PubMed] [Google Scholar]
- 24.Gu L, Johnson MW, Lusis AJ. Quantitative trait locus analysis of plasma lipoprotein levels in an autoimmune mouse model : interactions between lipoprotein metabolism, autoimmune disease, and atherogenesis. Arterioscler Thromb Vasc Biol. 1999;19:442–53. doi: 10.1161/01.atv.19.2.442. [DOI] [PubMed] [Google Scholar]
- 25.Yamaguchi Y, Kitagawa S, Imaizumi N, Kunitomo M, Fujiwara M. Effects of cholesterol loading on autoimmune MRL-lpr/lpr mice: susceptibility to hypercholesterolemia and aortic cholesterol deposition. Jpn J Pharmacol. 1993;61:291–8. doi: 10.1254/jjp.61.291. [DOI] [PubMed] [Google Scholar]
- 26.Lopes-Carvalho T, Kearney JF. Development and selection of marginal zone B cells. Immunol Rev. 2004;197:192–205. doi: 10.1111/j.0105-2896.2004.0112.x. [DOI] [PubMed] [Google Scholar]
- 27.Wither JE, Paterson AD, Vukusic B. Genetic dissection of B cell traits in New Zealand black mice. The expanded population of B cells expressing up-regulated costimulatory molecules shows linkage to Nba2. Eur J Immunol. 2000;30:356–65. doi: 10.1002/1521-4141(200002)30:2<356::AID-IMMU356>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 28.Viau M, Zouali M. B-lymphocytes, innate immunity, and autoimmunity. Clin Immunol. 2005;114:17–26. doi: 10.1016/j.clim.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 29.Li Y, Li H, Ni D, Weigert M. Anti-DNA B cells in MRL/lpr mice show altered differentiation and editing pattern. J Exp Med. 2002;196:1543–52. doi: 10.1084/jem.20021560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Davenport P, Tipping PG. The role of interleukin-4 and interleukin-12 in the progression of atherosclerosis in apolipoprotein E-deficient mice. Am J Pathol. 2003;163:1117–25. doi: 10.1016/S0002-9440(10)63471-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhou X, Hansson GK. Detection of B cells and proinflammatory cytokines in atherosclerotic plaques of hypercholesterolaemic apolipoprotein E knockout mice. Scand J Immunol. 1999;50:25–30. doi: 10.1046/j.1365-3083.1999.00559.x. [DOI] [PubMed] [Google Scholar]
- 32.Horkko S, Bird DA, Miller E, Itabe H, Leitinger N, Subbanagounder G, Berliner JA, Friedman P, Dennis EA, Curtiss LK, Palinski W, Witztum JL. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid-protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. J Clin Invest. 1999;103:117–28. doi: 10.1172/JCI4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shaw PX, Horkko S, Tsimikas S, Chang MK, Palinski W, Silverman GJ, Chen PP, Witztum JL. Human-derived anti-oxidized LDL autoantibody blocks uptake of oxidized LDL by macrophages and localizes to atherosclerotic lesions in vivo. Arterioscler Thromb Vasc Biol. 2001;21:1333–9. doi: 10.1161/hq0801.093587. [DOI] [PubMed] [Google Scholar]
- 34.George J, Afek A, Gilburd B, Levkovitz H, Shaish A, Goldberg I, Kopolovic Y, Wick G, Shoenfeld Y, Harats D. Hyperimmunization of apo-E-deficient mice with homologous malondialdehyde low-density lipoprotein suppresses early atherogenesis. Atherosclerosis. 1998;138:147–52. doi: 10.1016/s0021-9150(98)00015-x. [DOI] [PubMed] [Google Scholar]
- 35.Griffith RL, Virella GT, Stevenson HC, Lopes-Virella MF. Low density lipoprotein metabolism by human macrophages activated with low density lipoprotein immune complexes. A possible mechanism of foam cell formation. J Exp Med. 1988;168:1041–59. doi: 10.1084/jem.168.3.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsuura E, Kobayashi K, Koike T, Shoenfeld Y. Autoantibody-mediated atherosclerosis. Autoimmun Rev. 2002;1:348–53. doi: 10.1016/s1568-9972(02)00084-8. [DOI] [PubMed] [Google Scholar]
- 37.Nicolo D, Monestier M. Antiphospholipid antibodies and atherosclerosis. Clin Immunol. 2004;112:183–9. doi: 10.1016/j.clim.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 38.Nicolo D, Varadhachary AS, Monestier M. Atherosclerosis, antiphospholipid syndrome, and antiphospholipid antibodies. Front Biosci. 2007;12:2171–82. doi: 10.2741/2220. [DOI] [PubMed] [Google Scholar]
- 39.Qiao JH, Castellani LW, Fishbein MC, Lusis AJ. Immune-complex-mediated vasculitis increases coronary artery lipid accumulation in autoimmune-prone MRL mice. Arteriosclerosis & Thrombosis. 1993;13:932–943. doi: 10.1161/01.atv.13.6.932. [DOI] [PubMed] [Google Scholar]
- 40.Potter PK, Cortes-Hernandez J, Quartier P, Botto M, Walport MJ. Lupus-prone mice have an abnormal response to thioglycolate and an impaired clearance of apoptotic cells. J Immunol. 2003;170:3223–32. doi: 10.4049/jimmunol.170.6.3223. [DOI] [PubMed] [Google Scholar]
- 41.Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–75. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 42.Grainger DJ, Reckless J, McKilligin E. Apolipoprotein E modulates clearance of apoptotic bodies in vitro and in vivo, resulting in a systemic proinflammatory state in apolipoprotein E-deficient mice. J Immunol. 2004;173:6366–75. doi: 10.4049/jimmunol.173.10.6366. [DOI] [PubMed] [Google Scholar]
- 43.Cohen PL, Eisenberg RA. The lpr and gld genes in systemic autoimmunity: life and death in the Fas lane. Immunology Today. 1992;13:427–8. doi: 10.1016/0167-5699(92)90066-G. published erratum appears in Immunol Today 1993 Feb;14(2):97. [DOI] [PubMed] [Google Scholar]
- 44.Sobel ES, Kakkanaiah VN, Kakkanaiah M, Cohen PL, Eisenberg RA. Co-infusion of normal bone marrow partially corrects the gld T-cell defect. Evidence for an intrinsic and extrinsic role for Fas ligand. Journal of Immunology. 1995;154:459–64. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.