

Abstract
Cleavage of the intracellular carboxyl terminus of the N-methyl-D-aspartate (NMDA) receptor 2 subunit (NR2) by calpain regulates NMDA receptor function and localization. Here, we show that Fyn-mediated phosphorylation of NR2B controls calpain-mediated NR2B cleavage. In cultured neurons, calpain-mediated NR2B cleavage is significantly attenuated by blocking NR2B phosphorylation of Tyr-1336, but not Tyr-1472, via inhibition of Src family kinase activity or decreasing Fyn levels by small interfering RNA. In HEK cells, mutation of Tyr-1336 eliminates the potentiating effect of Fyn on calpain-mediated NR2B cleavage. The potentiation of NR2B cleavage by Fyn is limited to cell surface receptors and is associated with calpain translocation to plasma membranes during NMDA receptor activation. Finally, reducing full-length NR2B by calpain does not decrease extrasynaptic NMDA receptor function, and truncated NR1/2B receptors similar to those generated by calpain have electrophysiological properties matching those of wild-type receptors. Thus, the Fyn-controlled regulation of NMDA receptor cleavage by calpain may play critical roles in controlling NMDA receptor properties during synaptic plasticity and excitotoxicity.
The N-methyl-D-aspartate (NMDA)2 receptor is a calcium-permeable ionotropic glutamate receptor requiring glutamate, glycine, and membrane depolarization for activation. These receptors play crucial roles in brain development, excitatory neurotransmission, and synaptic plasticity but also may contribute to the pathogenesis of neurological disorders, including ischemia, epilepsy, and Huntington’s disease (1–4). NMDA receptors are made from heteromeric assemblies of different subunits called NR1 and NR2 (NR2A to -D) (5–8). The NR2B subunit is essential for both neonatal and mature NMDA receptors and is highly expressed in the entire embryonic brain and the adult forebrain (9). In the hippocampus, the major neonatal receptor is believed to contain two NR1 and two NR2B subunits, whereas the major receptor in more mature synapses probably contains two NR1 subunits, an NR2A subunit, and an NR2B subunit. Analysis of NR2B-null mice has suggested the physiological importance of NR2B, since the absence of NR2B-containing NMDA receptors in NR2B−/−mice leads to perinatal death, whereas mice lacking NR2A, NR2C, and NR2D subtypes are viable (10–12).
NR2B subunits have long C-terminal tails that link NR2B to intracellular pathways. Like NR2B-deficient mice, mice expressing NR2B subunits with truncated C-terminal domains die shortly after birth (13), showing the physiological importance of the intracellular region of NR2B. Therefore, processes that modify the C-terminal region of NR2B may alter the physiological events mediated by NR2B-containing receptors. Calpain and tyrosine kinases regulate NMDA receptors through the C-terminal domain. Calpain, a Ca2+-dependent neutral cysteine protease, cleaves the C-terminal domain of NR2B in HEK cells, neuronal cultures, and animal models of ischemia and seizures (14–17). In acutely isolated and cultured cortical neurons, prolonged stimulation of NMDA receptors reduces NMDA receptor-mediated currents in a manner requiring calpain activation (18). In cultures of hippocampal neurons, calpain-mediated cleavage of NR2B produces truncated receptors that remain on the cell surface but potentially have novel properties (16, 19). These studies indicate that calpain-mediated truncation of NR2B may regulate NMDA receptor function and localization.
The NR2B subunit is tyrosine-phosphorylated in neurons (20–22). Fyn, a Src family tyrosine kinase, phosphorylates Tyr-1252, Tyr-1336, and Tyr-1472 in the C-terminal domain of NR2B subunit in vitro, with Tyr-1336 and Tyr-1472 being the most readily phosphorylated sites in cell culture systems (23). A previous study limited to in vitro experiments suggested that Fyn and Src might regulate cleavage of NR2 subunits in opposing manners. Here, using in situ approaches in transfected cells and neurons, we have used molecular biological, physiological, and receptor trafficking approaches to show that phosphorylation of tyrosine residue Tyr-1336 by Fyn controls calpain-mediated proteolysis of NR2B without revealing any evidence for involvement of Src in situ. In addition, the effects of Fyn are modulated by NMDA receptor activation. This provides a novel mechanism through which Fyn regulates the NMDA receptor as part of a feedback loop.
EXPRIMENTAL PROCEDURES
Materials
Glutamate and glycine were obtained from Sigma; dizocilpine (MK-801) was obtained from Research Biochemicals International (Natick, MA). Benzyloxycar-bonyl-Val-Phe-aldehyde (MDL 28170), calpain inhibitor III (CalI3), calpain I, and 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2) were purchased from Calbiochem. Anti-NR2B antibody (made to the N-terminal 251 amino acids) was obtained from Zymed Laboratories Inc. (San Francisco, CA). AB38, which recognizes calpain-cleaved spectrin, was produced as described previously (16). Anti-NR1 and anti-PSD-95 monoclonal antibodies were obtained from BD Transduction Laboratories (Lexington, KY). Anti-actin antibody and antibody to NR2B phospho-Tyr-1472 were obtained from Sigma. Anti-calpain I antibody was obtained from Chemicon (Temecula, CA). Anti-Fyn antibody, anti-Src antibody, and antibody to NR2B phospho-Tyr-1336, as well as pKD-Fyn-v6 and pKD-NegCon-v1, were obtained from Upstate Biotechnology, Inc. (Lake Placid, NY).
The following full-length cDNA constructs were gifts: Fyn Y531F from Dr. Tohru Tezuka (University of Tokyo, Tokyo, Japan), synapse-associated protein 102 (SAP-102) from Dr. Richard L. Huganir (Johns Hopkins University, Baltimore, MD), PSD-95 from Dr. David S. Bredt (University of California, San Francisco, CA), and SrcY527F from Dr. Michael W. Salter (University of Toronto, Toronto, Ontario, Canada).
Site-directed Mutagenesis of NR2B
A PCR-based site-directed mutagenesis method was employed to generate the single amino acid mutants NR2BY1336F and NR2BY1472F. Wild-type NR2B in pRK7 was used as a template, and PCR was carried out for each mutant with a set of sense and antisense mutagenic primers (using the Stratagene QuikChange kit). PCR products were treated with DpnI restriction enzyme and purified by agarose gel electrophoresis. Purified DNA was used to transform Escherichia coli (DH5α), and transformants harboring mutant constructs were identified. Mutations were confirmed by DNA sequencing in the Molecular Biology Core of the Mental Retardation Research Center of the Children’s Hospital of Philadelphia. Truncated NR2B (designated NR2Bdel1036) was constructed using PCR by replacing the CelII fragment (bases 3102–4116) with a modified corresponding fragment in which the 5′ end contained two stop codons at base 3109 (amino acid 1037). NR2B in pRK7 was primed with 5′-AACACAGCCAGCTCAGCTAGTAGTACGGCAAGTTCTCTTTC-3′ and 5′-GTCAGGGTAGAGGGACTTGCTGAGCATGTAGCCGCTGCC-3′ to create this construct.
Small Interfering RNA (siRNA) Preparation and Transfection
An siRNA plasmid, pKD-Fyn-v6, specific for Fyn protein silencing, and a negative control plasmid for the siRNA, pKD-NegCon-v1, were amplified in DH5α cells and purified. HEK293 cells were transfected using calcium phosphate, and neurons were transfected via siIMPORTER™ transfection reagent (Upstate Biotechnology) according to the manufacturer’s instruction. We transfected 10 or 11 DIV neurons in 12-well plates with 500 ng of corresponding siRNA plasmid (pKD-Fyn-v6 or pKD-NegCon-v1), and Fyn protein expression was assessed 96 h after transfection (15–16 DIV) by immunoblotting. The species of the target for pKD-Fyn-V6 is human, but pKD-Fyn-V6 has a ~95% sequence homology to Rat Fyn and has no homology to other known rodent genes (based on BLAST searches).
In Vitro Phosphorylation and Proteolysis of NR2B by Purified Calpain I
Twenty-four hours after transfection, HEK293 cells were rinsed with phosphate-buffered saline and then scraped into phosphorylation buffer (200 mM Tris-HCl, 10 mM MgCl2, 5 mM MnCl2, 0.4 μM Na2VO4, 500 μM ATP, 0.8 mM EGTA). Samples were briefly sonicated on ice, incubated at 56 °C for 30 min to inactivate endogenous kinases and phosphatases, and then incubated with recombinant active Fyn protein (100 ng/ml) for 40 min at 30 °C. After phosphorylation, samples were incubated with 2 units/ml calpain I (Calbiochem) for 2 min at 30 °C in a buffer containing final concentrations of 1 mM CaCl2, 5 mM dithiothreitol, and 40 mM HEPES, pH 7.2. The reaction was stopped by an addition of SDS sample buffer, and the reaction mixtures were then loaded on an 8% SDS-polyacrylamide gel.
Whole-cell Recording
Experiments were performed with standard whole-cell voltage clamp techniques using transfected HEK293 cells 12–24 h after transfection and rat embryonic hippocampal neurons maintained in culture for 17–21 days DIV. NMDA-evoked currents were recorded at room temperature using NMDA (100 μM) along with glycine (10 μM) in a Mg2+-free extracellular recording medium containing 500 nM tetrodotoxin, 155 mM NaCl, 3 mM KCl, 3 mM CaCl2, and 10 mM HEPES, pH 7.35. To examine NMDA receptor function, cells were voltage-clamped at −50 mV using an intrapipette solution containing 100 mM Trizma phosphate (dibasic), 28 mM Trizma base, 11 mM EGTA, 2 mM MgCl2, 0.5 mM CaCl2, and 10 mM Mg-ATP, pH 7.35, 290 mosM/kg H2O. Each application was 2 s, and the interval between applications was more than 1 min to prevent current desensitization. Solution switching was carried out using an SF-77B fast step solution delivery device (Warner Instrument Co., Hamden, CT). Recording signals were amplified using an Axopatch-1D amplifier, filtered at 5 kHz, and then saved using an IBM PC running pCLAMP 8.01 software (Axon Instruments, Inc. Foster City, CA) for off-line analysis. NMDA, glutamate, and CalI3 were dissolved in extracellular solution. Some aliquots of CalI3 were solubilized in Me2SO; final Me2SO concentration was 0.001% (v/v). This concentration had no effect on whole-cell NMDA currents (data not shown). In order to observe the extrasynaptic NMDA receptor-mediated effects, synaptic NMDA receptors in hippocampal culture neurons were blocked with 50 μM bicuculline and 10 μM MK801 for 10 min. In addition, ifenprodil (10 μM) was used to confirm that NMDA receptor-mediated current in bicuculline/MK801-pre-treated neurons was primarily from extrasynaptic NMDA receptor (since they contain primarily NR1/2B). To examine the pharmacological effect of ifenprodil on NMDA receptors, ifenprodil was preadministered to the recorded neurons alone for more than 30 s before neurons were co-applied with NMDA/glycine and ifenprodil. Since the effect of ifenprodil could not be washed away within 30 min (data not shown), only one neuron per culture dish was recorded during these experiments.
Neuronal Cultures
Primary cultures of hippocampal and cortical neurons from embryonic day 17–19 Sprague-Dawley rats (Charles River Laboratories, Wilmington, MA) were prepared as described previously (16, 24). Briefly, the hippocampi were dissected, gently minced, trypsinized (0.027%, 37 °C; 7% CO2 for 20 min), and then washed with 1× HBSS. Neurons were seeded to a density of 6 × 105 viable cells/35-mm culture dish or similar relative density onto 12-well plates or 60-mm culture dishes. All plates and culture dishes were coated with poly-D-lysine (100 μg/ml) prior to seeding neurons. Cultures were maintained at 37 °C with 5% CO2, fed with neurobasal medium with B27 supplement, and the cultures were used at 17–21 DIV.
Western Blotting Analysis
Western blotting was performed as described previously (16, 24). Protein content was estimated using BCA Protein Assay (Pierce). Equal amounts of total protein (15–20 μg/lane) were resolved on denaturing 8–10% SDS-polyacrylamide gels, and transferred to nitrocellulose membranes. Membranes were blocked with 3% nonfat milk and incubated with primary antibody for 2 h at room temperature or overnight at 4 °C. Blots were then incubated with appropriate horseradish peroxidase-conjugated secondary antibodies (Sigma) for 2 h at room temperature and then washed; bands were visualized using enhanced chemiluminescence (Pierce). Reaction product levels were quantified by scanning densitometry using NIH Image 1.62 (available on the World Wide Web).
Transfection of HEK293 Cells
HEK293 cells were grown on tissue culture dishes in minimum Eagle’s medium containing 5% horse serum, 5% fetal bovine serum supplemented with 2 mM glutamine, 100 units/ml penicillin/streptomycin and placed in a 5% CO2 incubator at 37 °C. Transfection was performed using calcium phosphate as previously described (25). Treatments were performed 18–24 h following transfection. Ketamine (500 μM) was included in the media during transfection to prevent NMDA receptor activation as previously described (25).
Calcium Imaging
HEK293 cells were cotransfected with NR1, SAP-102, and GFP with NR2B wild type, NR2BY1336F, NR2BY1472F, or NR2Bdel1036 (NR2B truncated at amino acid 1036) each with or without FynY531F. Eighteen to twenty hours later, transfected cells were identified via green fluorescence protein fluorescence and selected for measurement of intracellular calcium responses. After loading with 5 μM fura-2-acetoxymethyl ester (Molecular Probes) for 30 min, cells were washed with Hanks’ balanced salt solution twice. Intracellular calcium was measured by a dual excitation wavelength method (340/380 nm) using a Nikon microscope in conjunction with Metafluor imaging software (Universal Imaging, West Chester, PA) (26).
Biotinylation Assays
Surface biotinylation was used to assess receptor expression on the cell surface. HEK293 cells, grown in 60-mm dishes, were transfected with various cDNA combinations. Twenty-four hours after transfection, cell surface proteins were labeled with biotin and isolated (16). Cells were washed twice with cold rinsing solution (phosphate-buffered saline with Ca/Mg (1 mM MgCl2 and 0.1 mM CaCl2, pH 7.35)) (27) and incubated in rinsing solution containing 1 mg/ml EZ-Link Sulfo-NHS-Biotin (Pierce) for 30 min at 4 °C. Cells were washed twice with quenching solution (rinsing solution with 100 mM glycine added), incubated in quenching solution for 20 min, and solubilized at 4 °C in radioimmune precipitation buffer ((150 mM NaCl, 1 mM EDTA, and 100 mM Tris-HCl, pH 7.4), 1% (v/v) Triton X-100, 1% (w/v) deoxycholate, and 0.1% (w/v) SDS containing protease inhibitors (1 μg/ml leupeptin, 250 μM phenylmethylsulfonyl fluoride, 1 μg/ml aprotinin, 1 mg/ml trypsin inhibitor, and 1 mM iodoacetamide). Samples were cleared by centrifugation at 16,300 × g at 4 °C for 20 min. A small sample of cleared lysates was saved for analysis (designated the lysate fraction); the remaining portion of lysates was incubated with avidin beads (Pierce) at 4 °C overnight. After overnight incubation, samples were centrifuged at 16,300 × g for 15 min, and the supernatant was saved as the intracellular fraction. The beads were washed once in radio-immune precipitation buffer, twice in cold high salt buffer (0.1% Triton X-100, 500 mM NaCl, 5 mM EDTA, 50 mM Tris, pH 7.5), and once in low salt buffer (50 mM NaCl, pH 7.5), and biotinylated proteins were eluted with SDS sample buffer (62.5 mM Tris-Cl, pH 6.8, 2% SDS, and 100 mM mercaptoethanol) at 37 °C for 30 min. The remainder of the procedure followed standard Western blotting protocols. To confirm that biotin only labeled surface proteins, the integrity of the cell membrane during biotinylation was tested by immunoblotting with an anti-actin antibody. Actin immunoreactivity in the biotinylated fraction was 9 ± 2% (n = 24) of that in the whole cell lysate.
In selected experiments, biotinylations were performed under nondenaturing conditions by solubilizing cell membranes in radioimmune precipitation buffer (lysis buffer) lacking SDS. Without SDS in the radioimmune precipitation buffer, integral membrane proteins exposed to the extracellular space and thus labeled by biotin are not dissociated from membrane-associated proteins bound to such integral membrane proteins by noncovalent interactions. This allows us to possibly detect the changes in association of nonintegral membrane proteins (such as calpain) with biotinylated, cell surface-exposed proteins in different experimental conditions.
Statistical Analysis
Data are shown as the mean ± S.E. Experiments were analyzed using Student’s t test (paired) to compare two conditions or ANOVA followed by planned comparisons of multiple conditions (28). For most comparisons, nonnormalized OD values were used. Significance was set at p < 0.05. All electrophysiology data, such as current amplitude, current desensitization, and time constant (fitted with a single exponential component function using the Levenberg-Marquardt nonlinear least squares algorithm), were calculated using Clampfit software (pCLAMP 8.01; Axon Instruments, Inc., Foster City, CA). We have designated “n” to refer to independent experiments in biochemical studies and independent single cells in electrophysiological studies.
RESULTS
Reduction of Calpain-mediated Proteolysis of NR2B by Inhibition of Src Family Kinase (SFK) Activity in Neurons
SFK-mediated tyrosine phosphorylation and calpain-mediated proteolysis occur in the NR2B C terminus (23, 29–31). To test the possibility that phosphorylation of NR2B by SFK changes its sensitivity to calpain, we investigated the effect of PP2, a specific inhibitor of SFK (32, 33), on calpain-mediated cleavage of NR2B. In primary hippocampal cultures (Fig. 1A), neurons pre-treated with or without 10 μM PP2 for 30 min had similar levels of full-length NR2B (180 kDa, detected by an N-terminal antibody), showing that inhibition of SFK activity did not acutely change NR2B protein expression. Exposure of cultured neurons to 100 μM glutamate and 100 μM glycine for 30 min in the absence of PP2 preapplication decreased full-length NR2B levels and generated a 115–120-kDa N-terminal breakdown product (previously shown to be generated by calpain) (16) (Fig. 1, A and B). In parallel cultures, the application of 10 μM PP2 30 min before glutamate and glycine stimulation decreased the loss of full-length NR2B and production of the breakdown product (Fig. 1, A–C; full-length NR2B, without PP2, 48 ± 10%; with PP2, 82 ± 4%; p = 0.0069; NR2B breakdown product, without PP2, 53 ± 6%; with PP2, 30 ± 4%; p = 0.0068; n = 6). To control for the possibility that PP2 application globally decreased calpain activity, we examined calpain-mediated spectrin breakdown. Glutamate treatment produced similar levels of a 150-kDa calpain-generated spectrin breakdown product in neurons with or without PP2, showing that PP2 application did not decrease calpain activity toward all substrates (Fig. 1, A and D). Taken together, these results show that blocking SFK activity slows calpain-mediated cleavage of NR2B specifically and suggest that SFK activity controls NR2B subunit levels in hippocampal neurons in response to glutamatergic stimulation.
FIGURE 1. Prevention of calpain-mediated NR2B cleavage by inhibition of SFK activity in cultures of hippocampal neurons.

A, cultures of hippocampal neurons (15–21 DIV) either untreated (0 min) or treated with 100 μM glutamate and 100 μM glycine for 30 min in the presence of PP2 (10 μM) or Me2SO (PP2−) (control). Neurons were harvested 30 min after the addition of agonists, and samples were subjected to SDS-PAGE and Western blot analysis with an N-terminal NR2B antibody (top) and AB38 (recognizing a calpain-generated spectrin breakdown product) (bottom). The lower band in the top is a calpain-mediated NR2B breakdown product (16). B and C, bar graphs demonstrate the levels of full-length NR2B and NR2B-derived breakdown after the indicated treatments. The expression level of each control was normalized to 100% (based on full-length NR2B), and the levels of each band after various treatments were calculated as the percentages of corresponding control values. D, bar graph shows elevated levels of the calpain-generated spectrin breakdown after the indicated treatments. Data are means ± S.E. from six experiments. *, p < 0.05; **, p < 0.001, ANOVA.
NR2B Is Phosphorylated at Tyrosine 1336 during NMDA Receptor Activation in Neurons
NR2B can be phosphorylated at Tyr-1336 and Tyr-1472 by Fyn, an SFK directly implicated in tyrosine phosphorylation of NMDA receptors in the brain (23, 34). Since Fyn may change NR2 cleavage by calpain in vitro (35), we first tested whether Fyn mediated phosphorylation of NR2B at Tyr-1336, Tyr-1472, or both sites during NMDA receptor activation. Cultures were treated with glutamate and glycine, and the time course of Tyr-1336 and Tyr-1472 phosphorylation was determined using NR2B phosphospecific anti-Tyr-1336 or anti-Tyr-1472 antibodies (Fig. 2, A and B). Levels of phosphorylation of Tyr-1472 were similar over the period of 30 min of glutamate stimulation (Fig. 2A). However, increased phosphorylation of Tyr-1336 was noted rapidly (by 7.5 min after NMDA receptor activation) and increased 2.2-fold by 30 min (0 min, 100 ± 2%; 7.5 min, 151 ± 22%; 30 min, 219 ± 14%, p = 0.0095 at 7.5 min and p = 0.0004 at 30 min both versus 0 min, n = 5 each) (Fig. 2B). The progressive phosphorylation of NR2B at Tyr-1336 was associated with a gradual reduction of full-length NR2B and generation of the NR2B breakdown product (Fig. 2, C and D). Thus, whereas the level of full-length NR2B declined with NMDA receptor activation, the immunoreactivity of phospho-Tyr-1336 NR2B increased. In addition, phosphorylation of Tyr-1336 in this condition is mediated by SFK, since PP2 (10 μM) eliminated phospho-Tyr-1336 immunoreactivity at all of the indicated time points (Fig. 2, A and B). Furthermore, PP2 slowed both the reduction of full-length NR2B and the generation of NR2B-derived breakdown products over a time course that generally resembled that for phosphorylation of NR2B-Tyr-1336 (Fig. 2, A, C, and D). As in our previous work (16, 19), inclusion of a calpain inhibitor decreased NR2B breakdown (data not shown) but increased the amount of Tyr-1336 phosphorylation (Glu, 190 ± 7%; CalI3 +Glu, 330 ± 18%; n = 6; p = 0.0016 versus Glu) (Fig. 2E), suggesting that NR2B-Tyr-1336 is a substrate for calpain. MK-801 also blocked Tyr-1336 phosphorylation (control, 100 ± 6%; MK801 + Glu, 94 ± 8%; n = 6, p = 0.57 versus control; Fig. 2E). These results indicate that SFK-mediated phosphorylation and calpain-mediated proteolysis, which both regulate NR2B through the C terminus, are facilitated by NMDA receptor activation in neurons. Since SFK inhibition decreases calpain-mediated NR2B cleavage, this also suggests that SFK-mediated phosphorylation of NR2B directly accelerates the calpain-generated NR2B breakdown.
FIGURE 2. NR2B is phosphorylated at Tyr1336, and inhibition of Tyr1336 phosphorylation prevents calpain-mediated NR2B cleavage in cultures of hippocampal neurons.

A, cultures of hippocampal neurons (15–21 DIV) were treated with 100 μM glutamate and 100 μM glycine for various amounts of time in the presence of 10 μM PP2 (PP2 (+)) or Me2SO control (PP2 (−)). Neurons were harvested at each indicated time after the addition of agonist, and samples were subjected to SDS-PAGE and Western blot analysis with an NR2B phosphospecific anti-Tyr-1336 antibody (top), an N-terminal NR2B antibody (middle), or an antibody to phosphorylated NR2B Tyr-1472. B–D, quantification of immunoreactivity of Tyr-1336 phosphorylation, full-length NR2B, and the 115-kDa NR2B-derived breakdown product for the indicated length of time in the presence or absence of PP2 during agonist stimulation. E, NR2B-Tyr-1336 was a calpain substrate, and phosphorylation of NR2B-Tyr-1336 was blocked by MK-801 in cultured neurons. Neurons were treated with or without glutamate (100 μM) and glycine (100 μM) for 30 min in the presence or absence of CalI3 (10 μM) or MK-801 (10 μM), and equal amounts of protein were then analyzed by Western blot using an antibody to NR2B Tyr-1336. Data are means ± S.E. from six experiments. *, p < 0.05; **, p < 0.001, ANOVA.
Fyn Accelerates Calpain-mediated NR2B Cleavage in Neurons
To assess whether calpain-mediated NR2B cleavage during NMDA receptor activation in neurons is controlled by Fyn, we decreased endogenous Fyn protein expression using an siRNA against Fyn coded in a DNA-based vector (36). The Fyn siRNA, which has no homology to other known rodent genes (based on BLAST searches of the sequences), decreased endogenous Fyn protein in HEK293 cells and in cultured hippocampal neurons to 30–35% of levels detected in mock expression 96 h after transfection (Fig. 3, A and B). As noticed by protein levels of actin (data not shown), spectrin breakdown (frequently an early marker of cell death), and the NR1 NMDA receptor subunit (Fig. 3D) from untransfected and transfected neurons, siRNA transfection did not produce any significant neuronal toxicity in our culture paradigm. The levels of Src protein were not affected (data not shown). At this time point, Fyn knockdown substantially diminished phosphorylation of NR2B Tyr-1336 (Fig. 3C) and protected NR2B from calpain-mediated proteolysis after NMDA receptor activation (Fig. 3D). After a 30-min treatment of 100 μM glutamate and 100 μM glycine, full-length NR2B decreased 20% in neurons transfected with Fyn siRNA, 45% in untransfected neurons, and 44% in neurons transfected with a mock construct (Fig. 3, D and E; for untransfected neurons, control, 100 ± 6%; Glu, 55 ± 0.3%; for siRNA, control, 100 ± 11%, Glu, 81.0 ± 1%; for mock, control, 100 ± 0.4%, Glu, 56 ± 12%; n = 5 each). Fyn siRNA-transfected neurons also generated lower levels of the NR2B-derived breakdown product (Fig. 3, D and F; untransfected neurons, control, 11 ± 1%; Glu, 73 ± 5%; siRNA, control, 12 ± 1%, Glu, 33 ± 4%; mock, control, 10 ± 0.3%, Glu, 69 ± 5%; n = 5 each). Transfection of the control siRNA construct (mock) did not alter NR2B breakdown. In each transfection, calpain-mediated cleavage of spectrin was similar after agonist treatment for 30 min (untransfected neuron, control, 100 ± 6%; Glu, 560 ± 80%; siRNA, control, 100 ± 6%; Glu, 570 ± 60%; for mock, control, 100 ± 13%; Glu, 540 ± %; n = 5 each; data not shown). Thus, decreasing Fyn protein expression by siRNA decreases calpain-mediated proteolysis of NR2B in neurons but does not alter calpain-mediated cleavage of other substrates.
FIGURE 3. Prevention of calpain-mediated NR2B cleavage by targeting Fyn with siRNA in neurons.

A, knockdown of endogenous Fyn protein by siRNA in HEK cells. Lysates from mock (mock)- and Fyn RNA interference-transfected cells (siRNA) were analyzed by Western blotting 96 h after transfection. Mock, an siRNA construct designed to have no effect on Fyn expression. B and C, Western blot analysis of Fyn protein expression and NR2B Tyr-1336 phosphorylation in rat hippocampal neurons transfected with mock or siRNA. Neurons were transfected at 10–11 DIV, and knockdown of Fyn protein expression was assessed 96 h after siRNA nucleofection (15–16 DIV). Phosphorylation of NR2B Tyr-1336 (C) was compared between mock- and siRNA-transfected neurons with or without agonist stimulation (100 μM glutamate and 100 μM glycine for 30 min). D, representative immunoblots indicate protein levels of NR2B, spectrin breakdown, and NR1 during NMDA receptor activation in nontransfected, Fyn siRNA-transfected and mock-transfected hippocampal neurons. E and F, quantification of relative expression full-length NR2B and calpain-generated NR2B-derived breakdown product from D. Data are mean ± S.E. from five or more experiments. *, p < 0.05; **, p < 0.001, ANOVA.
Facilitation of Calpain-mediated NR2B Cleavage by Fyn-mediated Phosphorylation of NR2B Tyr1336 in HEK293 Cells
To determine whether calpain-mediated NR2B cleavage is controlled selectively by Fyn-mediated phosphorylation, we investigated the effects of Fyn on cleavage of NR2B in HEK cells. We transfected HEK cells with an expression plasmid encoding FynY531F, a constitutively active form of Fyn, along with cDNAs for NR1, NR2A, and NR2B. NR2A expression is needed for calpain activation in HEK cells (24). The membrane-associated guanylate kinase (MAGUK) protein PSD-95 promotes Fyn-mediated phosphorylation of NR2B in vitro (23) but also protects NR2B from cleavage by calpain (24). We expressed NMDA receptor subunits with PSD-95 or with SAP-102 (synapse-associated protein 102), a MAGUK protein similar to PSD-95 that binds to NR2 subunits but does not alter cleavage of NR2B by calpain (24). In agreement with others (23), recombinant NR2B could be phosphorylated by FynY531F at Tyr-1336 when NR2B was cotransfected with PSD-95 (Fig. 4A, last lane). Phospho-Tyr-1336 was also detected in cells transfected with NR1/2A/2B/FynY531F and SAP-102 (Fig. 4A, middle lane). Replacing PSD-95 or SAP-102 with an empty vector control led to no detectable phosphorylation of NR2B-Tyr-1336 (Fig. 4A, first lane), suggesting that PSD-95 or SAP-102 is needed for Fyn-mediated phosphorylation of NR2B-Tyr-1336 (Fig. 4A). We next studied whether Fyn-mediated phosphorylation of NR2B at Tyr-1336 altered calpain-mediated cleavage of NR2B in this system. Following 100 μM glutamate and 100 μM glycine application, full-length NR2B immunoreactivity decreased by 29% in cells transfected with NR1/2A/2B/SAP-102/vector compared with those cells without agonist stimulation (control, 100 ± 5%; Glu, 71 ± 1%; p = 0.0042 versus control; n = 6 each) (Fig. 4, B and C), whereas full-length NR2B immunoreactivity in cells expressing NR1/2A/2B/SAP-102/FynY531F decreased by 62% (control, 100 ± 3%; Glu, 38 ± 2%; p = 0.0001 versus control; n = 6 each) (Fig. 4, B and C). The decreases in NR2B immunoreactivity in both transfected conditions were completely blocked by 10 μM calpain inhibitor III (CalI3 + Glu, 103 ± 4% in cells without FynY531F (p = 0.65 versus control); 95 ± 2% in cells with FynY531F (p = 0.24 versus control); n = 6 each) (Fig. 4, B and C). The facilitation of calpain-mediated NR2B cleavage by FynY531F was selective for NR2B, since actin (Fig. 4B) and NR1 (data not shown) were unaltered by FynY531F, and calpain-generated spectrin breakdown increased to similar levels in cells with and without FynY531F application (increase of 650 ± 20% in cells without FynY531F; increase of 670 ± 35% in cells with FynY531F; n = 6) (Fig. 4, B and E). Thus, in this heterologous system, FynY531F does not globally increase calpain activity but selectively facilitates calpain-mediated cleavage of NR2B. In addition, as observed in neuronal cultures, we detected increased levels of Tyr-1336 phosphorylation in cells cotransfected with FynY531F (immunoreactivity, control, 9500 ± 470; Glu, 11,500 ± 480; CalI3 + Glu, 36,100 ± 2070; n = 6; p = 0.04 versus control) (Fig. 4, B and D), further demonstrating that the increase in the immunoreactivity of NR2B phospho-Tyr-1336 and the reduction of the level of NR2B both occurred after NMDA receptor activation. Preapplication of calpain inhibitor III substantially augmented the level of NR2B phosphorylation at Tyr-1336 at 30 min after glutamate and glycine application (p = 0.0003 versus Glu) (Fig. 4, B and D) but had no effect without agonist treatment (data not shown). This further suggests that calpain preferentially cleaves Fyn-phosphorylated NR2B subunits.
FIGURE 4. Facilitation of calpain-mediated NR2B cleavage by Fyn Y531F, which phosphorylates NR2B Tyr1336 in HEK293 cells.
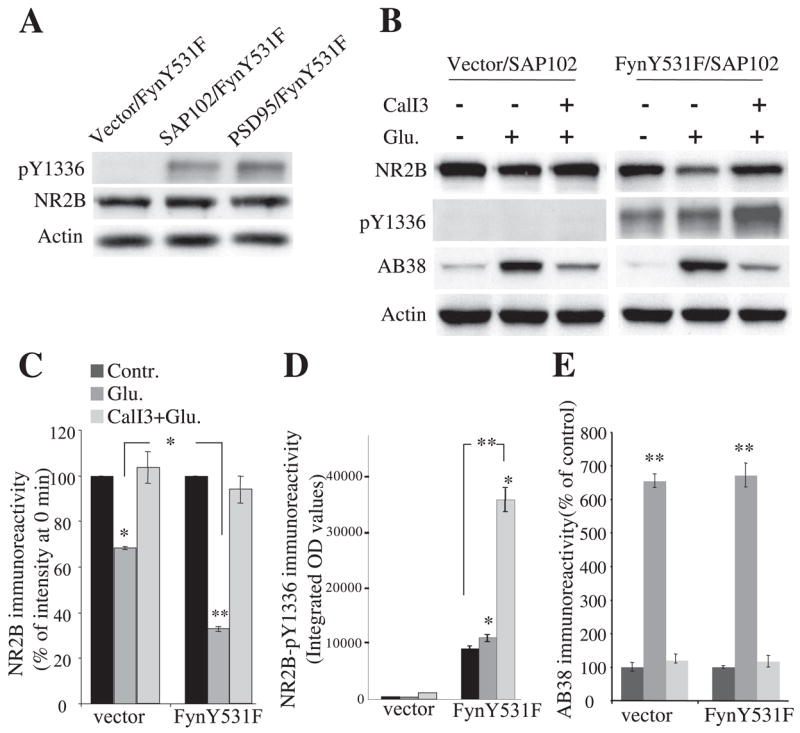
A, coexpression of either PSD-95 or SAP-102 with FynY531F and NR2B promoted phosphorylation of NR2B Tyr-1336 (top) compared with vector control. PSD-95 and SAP-102 cotransfection with FynY531F did not increase levels of NR2B (middle) and actin (bottom). B, representative immunoblots after designated treatments (sham, Glu/Gly, CalI3) detected by NR2B antibody, phospho-Tyr-1336-specific antibody, AB38, and actin antibody. C–E, quantification of relative levels of full-length NR2B, Tyr-1336 phosphorylation, and spectrin breakdown from results of B. A greater decrease in full-length NR2B was observed 30 min after glutamate and glycine treatment in cells coexpressing NR1/2A/2B/SAP-102 with FynY531F compared with that from cells not transfected with FynY531F. In both types of transfection, calpain-generated spectrin products increased to a similar extent. No change in actin signal was detected. Data are mean ± S.E. from five or more experiments. D, data are expressed as absolute values of signal intensity determined by NIH Image. *, p < 0.05; **, p < 0.001, ANOVA.
The Effect of PSD-95 on Fyn-mediated Facilitation of NR2B Cleavage in HEK Cells
We previously found that PSD-95 protects NR2B from calpain-mediated proteolysis during NMDA receptor activation (24). To examine whether Fyn-mediated phosphorylation of NR2B alters the protection of NR2B from calpain-mediated cleavage by PSD-95, HEK cells cotransfected with NR1/2A/2B/FynY531F and either PSD-95 or vector control were incubated with NMDA receptor agonists and analyzed by Western blots using anti-N-terminal NR2B, anti-phospho-NR2B-Tyr-1336, and AB38. Immunoreactivity of phospho-Tyr-1336 of NR2B was detected at base line and increased with the application of NMDA receptor agonists for 30 min (control, 100 ± 4%; Glu, 196 ± 17%; n = 5; p = 0.016 versus control) (Fig. 5A). In contrast to results from SAP-102-cotransfected cells (Fig. 4, B and D), 100 μM glutamate and 100 μM glycine applications did not significantly decrease the immunoreactivity of full-length NR2B (control, 100 ± 1%; Glu, 85 ± 8%; n = 5; p = 0.10 versus control) (Fig. 5B), whereas increased levels of spectrin breakdown by calpain were detected 30 min after agonist application and were inhibited by preapplication of calpain inhibitor III (control, 100 ± 12%; Glu, 370 ± 16%; p = 0.0035 versus control; CalI3 + Glu, 130 ± 8%; p = 0.14 versus control; n = 5 each) (Fig. 5C). Preapplication of calpain inhibitor III did not further increase the phosphorylation of NR2B Tyr-1336 (CalI3 + Glu, 205 ± 9%; p = 0.0046 versus control; p = 0.57 versus Glu; n = 5 each) (Fig. 5A), suggesting that even Tyr-1336-phosphorylated NR2B is not readily degraded by calpain in this paradigm. These results indicate that PSD-95 protects NR2B from calpain-mediated cleavage in NMDA receptor-transfected HEK cells, consistent with our previous finding (24), and phosphorylation of NR2B at Tyr-1336 by FynY531F did not remove the ability of PSD-95 to block calpain-mediated cleavage of NR2B.
FIGURE 5. The effect of PSD95 on Fyn-mediated facilitation of NR2B cleavage and the role of Src in calpain-mediated cleavage of NR2B.
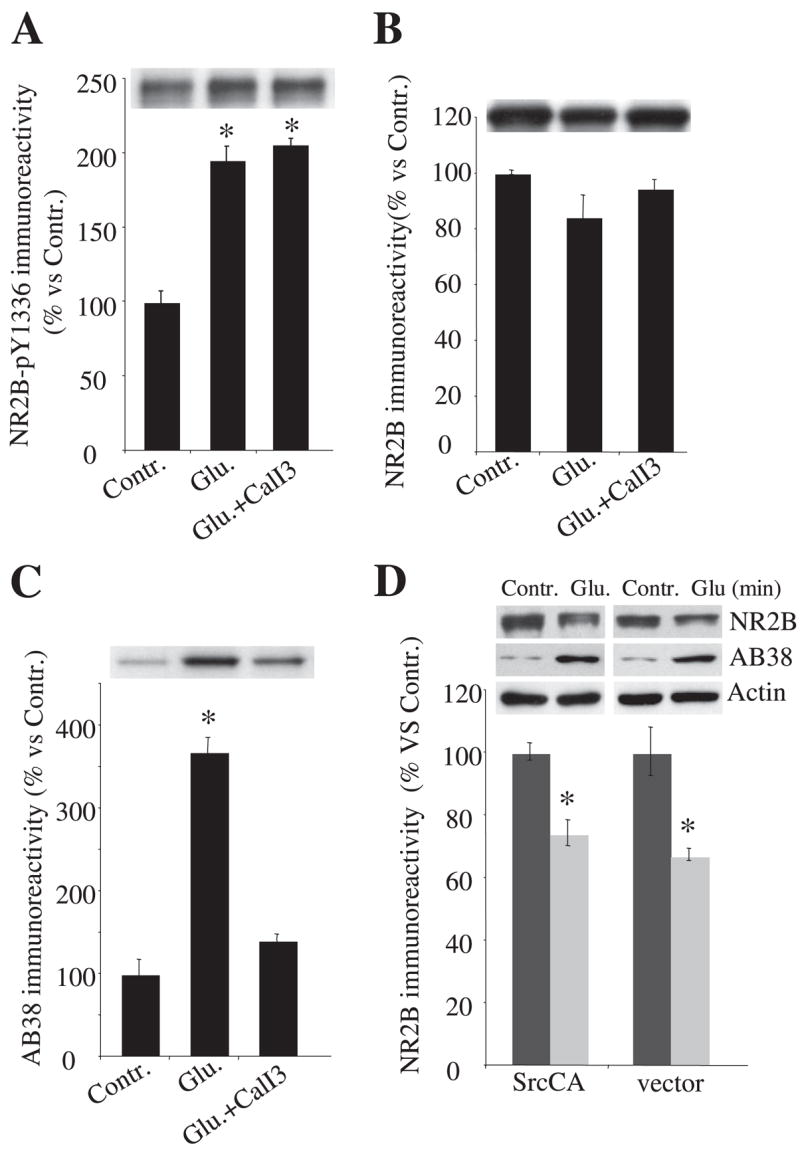
A–C, HEK cells were transfected with NR1/2A/2B/PSD-95 and Fyn and treated with 100 μM glutamate and 100 μM glycine in the presence or absence of calpain inhibitor III. Cells were harvested 30 min after agonist application, and samples were subjected to 8% SDS-PAGE and Western blot analysis with an NR2B phosphospecific anti-Tyr-1336 antibody (A), an N-terminal NR2B antibody (B), and AB38 antibody (C). Even with FynY531F phosphorylation of NR2B-Tyr-1336 after NMDA receptor activation, PSD-95 coexpression prevented NR2B cleavage by calpain. D, representative blots of NR2B, spectrin breakdown, and actin as well as quantitative levels of NR2B from HEK cells expressing constitutively active Src or a control vector with NR1/2A/2B/SAP-102. SrcCA neither prevented nor facilitated calpain-mediated cleavage of NR2B at 30 min after NMDA receptor activation in HEK cells. Data are representative of four independent experiments and are shown as mean ± S.E. wt, wild type.
Because another SFK, Src, changes calpain-mediated cleavage of NR2A in vitro (35), we examined whether Src kinase can alter calpain-mediated cleavage of NR2B and phosphorylation of NR2B-Tyr-1336. Constitutively active Src (SrcCA) (SrcY527F) or a vector control was expressed in HEK cells with NR1/2A/NR2B/SAP-102 (Fig. 5D). Compared with the corresponding 0 min control, 30-min application of glutamate and glycine reduced the NR2B immunoreactivity to 70% of control levels (n = 5; p = 0.011) in cells transfected with SrcCA and to 72% (n = 5; p = 0.019) in cells transfected with a vector control. Calpain-generated spectrin breakdown increased to the same extent at 30 min after agonist application in both transfected conditions (Fig. 5D). Both the reduction of NR2B levels and the increase in spectrin breakdown were completely blocked by pretreatment with calpain inhibitor III (data not shown). The immunoreactivity of actin did not change before or after agonist application in any transfected condition (Fig. 5D). In addition, immunoreactivity of phosphorylated NR2B Tyr-1336 was undetectable before or after agonist treatment in cells either with or without SrcCA coexpression, showing that NR2B Tyr-1336 was not a substrate for Src kinase in this paradigm (data not shown). Taken together, these results indicate that in HEK cells, SrcCA does not alter calpain-mediated cleavage of NR2B in this transfected cell paradigm; nor does it readily mediate phosphorylation of Tyr-1336 after NMDA receptor activation induced by glutamate and glycine. Thus in this system, Fyn (but not all SFKs) accelerates calpain-mediated NR2B cleavage.
Mutation of NR2B Tyr1336 Blocks Fyn-mediated Facilitation of NR2B Cleavage
We examined the specific role of NR2B Tyr-1336 using the mutants NR2BY1336F and NR2BY1472F. Before agonist treatment, full-length NR2B immunoreactivity in cells transfected with NR2BY1336F or NR2BY1472F along with NR1/2A/SAP-102/FynY531F was comparable with that of NR2B wild-type (Fig. 6A). Following a 30-min agonist treatment, the immunoreactivity of the NR2BY1336F mutant decreased by 32%, a decrease similar to that noted with wild-type NR2B without FynY531F cotransfection (Fig. 6, A and B; NR2Bwt/vector, 71 ± 8%; NR2BY1336F/FynY531F, 68 ± 1%; p = 0.59; n = 6 each), whereas the immunoreactivity of wild-type NR2B with FynY531F declined by 62% (Fig. 6, A and B, NR2Bwt/FynY531F, 38 ± 3%; p = 0.023 versus NR2Bwt/vector and p = 0.036 versus NR2BY1336F/FynY531F; n = 6 each). In contrast, with FynY531F cotransfection, the immunoreactivity of NR2BY1472F decreased by 43%, a similar level to that of NR2B wild type with FynY531F cotransfection (Fig. 6, A and B, NR2BY1472F/FynY531F, 57 ± 2%; p = 0.45 versus NR2Bwt/FynY531F, p = 0.037 versus NR2Bwt/vector, and p = 0.0022 versus NR2BY1336F/FynY531F; n = 6 each). Receptors made by expression of NR1/NR2A/NR2BY1336F, NR2BY1472F, or NR2B wild type with FynY531F had similar NMDA receptor-mediated intracellular calcium responses (Fig. 6D), indicating that the differences in the effect of FynY531F did not result from differential calcium responses to glutamatergic stimulation. Calpain-generated spectrin breakdown was similar in each condition (Fig. 6, A and C). Thus, in HEK cells, prevention of Fyn-mediated phosphorylation of NR2B-Tyr-1336 through mutation eliminates the facilitating effect of FynY531F on calpain-mediated NR2B cleavage.
FIGURE 6. Blockade of FynY531F-induced facilitation of NR2B cleavage by mutation of NR2B Tyr1336 but not Tyr1472.
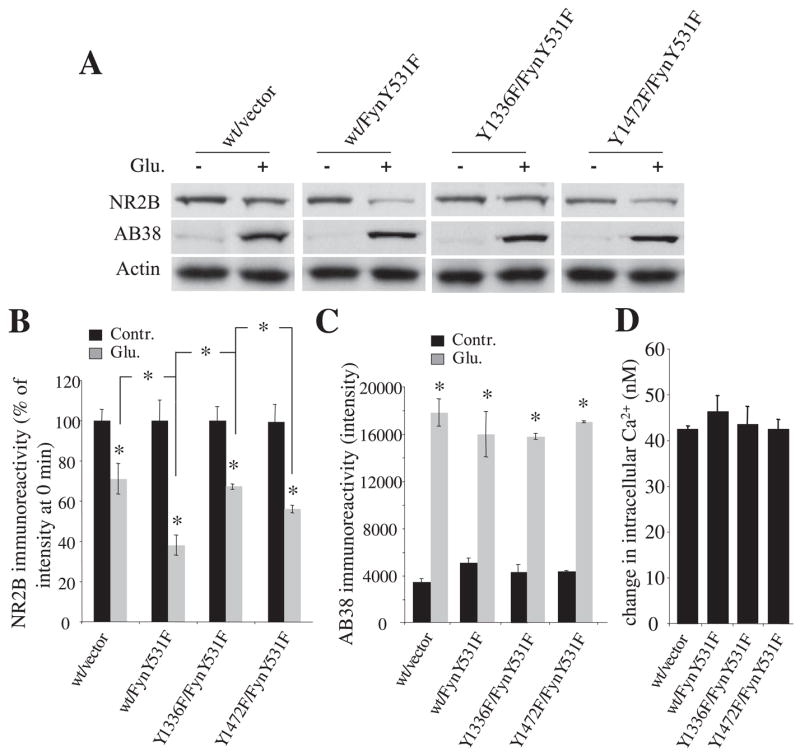
A, representative immunoblots with NR2B antibody, AB38, and anti-actin antibody. HEK293 cells were transfected with combinations of expression plasmids for NR2B wild type or NR2B mutants (Y1336F and Y1472F), and NR1/2A/SAP-102 with or without FynY531F. Twenty-four hours after transfection, cells were treated with glutamate and glycine for 30 min, and lysates were subjected to immunoblotting with the anti-NR2B, AB38, and actin antibodies. B and C, quantification of the relative levels of full-length NR2B and calpain-generated spectrin product. Absolute integrated OD values are displayed. D, NMDA-mediated intracellular calcium responses in transfected HEK293 cells containing wild-type or mutant NR2B. The cells were loaded with 5 μM fura-2 and then stimulated with 100 μM glutamate and 100 μM glycine. Peak calcium concentrations (nM) were typically observed within 30 s. Data from 20–30 individual cells were collected per experiment, and ensemble averages were calculated. n = 10. Data are means ± S.E. wt, wild type.
Cellular Location of Fyn Facilitation of Calpain-mediated NR2B Cleavage in HEK Cells
Since activation of NMDA receptors leads to calpain-mediated NR2B cleavage in HEK cells in both the plasma membrane and intracellular compartments (16, 24), we next investigated whether Fyn-mediated facilitation of NR2B cleavage occurred in a selective cellular compartment in HEK cells. Cells expressing NR1/NR2A/NR2B with or without FynY531F were treated with glutamate and glycine. Surface receptors were biotinylated, separated using avidin beads, and detected by Western blotting using anti-N-terminal NR2B antibody. In each transfected condition (Fig. 7, B and C), before agonist application, NR2B was found in both the cell surface fraction and the intracellular fraction. Actin was detected almost exclusively in the intracellular fraction, confirming that biotinylation occurred only at the cell surface (Fig. 7, B and C). Before agonist treatment, in both the surface and the intracellular fractions, the levels of immunoreactivity of NR2B in cells transfected with NR1/2B with or without FynY531F were similar (Fig. 7, B and C). This suggests that basal Fyn-mediated phosphorylation of NR2B Tyr-1336 does not change the NR2B levels in each compartment in this system, including the subunit delivery to the cell surface. Following 30-min agonist treatment, the immunoreactivity of NR2B in the overall lysate declined compared with the corresponding control (0 min). (Fig. 7A, 35% decrease without FynY531F; 63% decrease in wild type with FynY531F; n = 6 each). In the cell surface fraction, agonist application decreased NR2B immunoreactivity by 33% in cells transfected with NR2B wild type without FynY531F and by 62% in cells transfected with NR2B wild type with FynY531F, suggesting that NR2B cleavage is enhanced by FynY531F in the surface fraction (Fig. 7C; NR1/2B without FynY531F, 67 ± 2%; NR1/2B with FynY531F, 38 ± 3%; p = 0.0063; n = 6 each). In the intracellular fraction, the decrease in levels of NR2B by agonist application was similar among wild-type NR2B with or without FynY531F (Fig. 7B; NR1/2B without FynY531F, control, 100 ± 3%; Glu, 41 ± 4%; NR1/2B with FynY531F, control, 100 ± 7%; Glu, 40 ± 2%; n = 6 each). Overall, the level of NR2B cleavage in the intracellular fraction in the absence of FynY531F was similar to the level cleavage with FynY531F in the surface fraction. This suggests that Fyn may relieve a tonic inhibition of cleavage of NR2B in the surface fraction. Overall, these results show that FynY531F mediated facilitation of NR2B cleavage in the cell surface fraction without significant effects on cleavage of intracellular receptors.
FIGURE 7. Cellular location of the effect of Fyn on calpain-mediated NR2B cleavage in HEK cells.
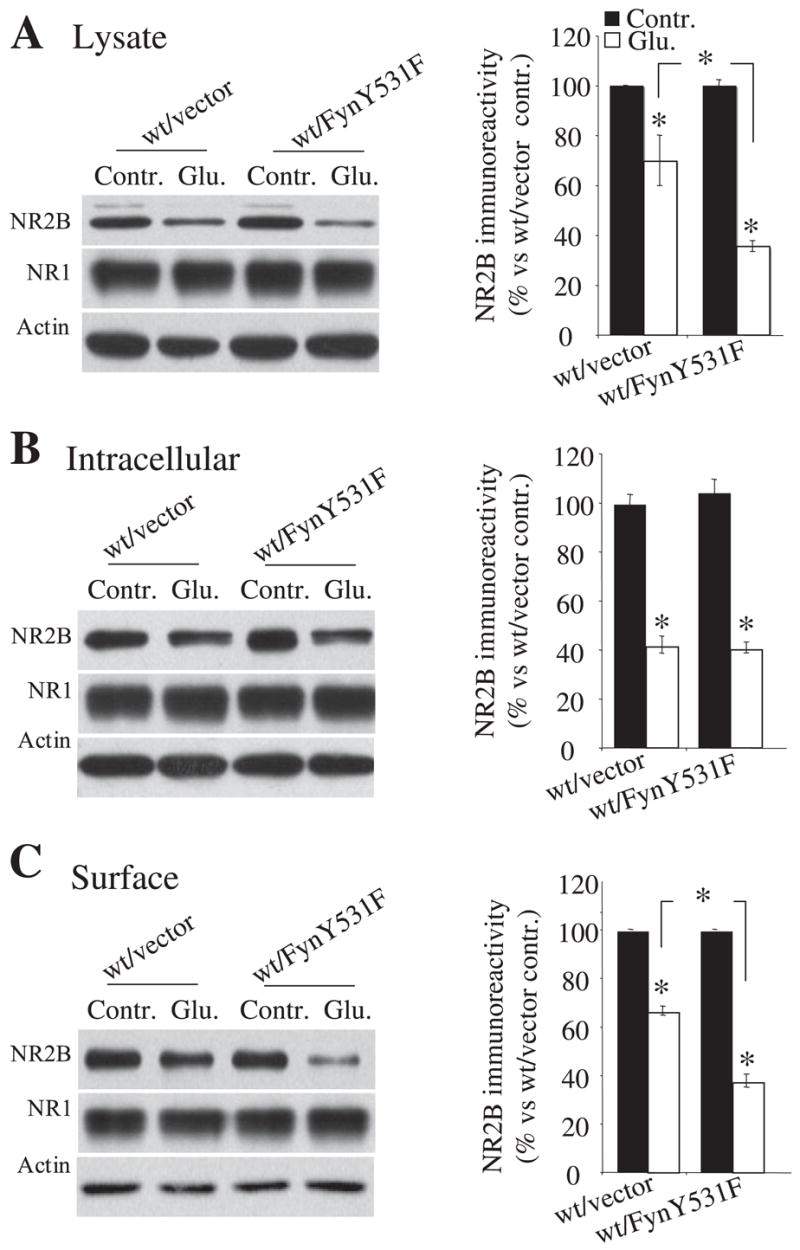
A–C, representative blots and histogram showing NR2B levels in different cellular compartments in cells transfected with NR1/2B with or without FynY531F before (control) and 30 min after agonist application. Quantification of the NR2B levels by biotinylation showed that NR2B levels decreased to a greater degree in the lysate (A) and the surface fraction (C) in cells coexpressing FynY531F than in those expressing a vector control. In the intracellular fraction (B), quantification showed that the levels of NR2B with or without FynY531F decreased to a similar extent after agonist application. Biotinylation data were obtained from six independent experiments performed in triplicate. Error bars, S.E. wt, wild type.
The Mechanism of Fyn-mediated Facilitation of NR2B Cleavage
Since cleavage of intracellular receptors was not enhanced by Fyn, we sought to better define the mechanism by which Fyn enhances calpain-generated cleavage of NR2B. In vitro, Fyn enhanced the speed of NR2B cleavage (Fig. 8, A and B). This increase in the rate of cleavage was not noted in receptors formed from NR2BY1336F, demonstrating that the mechanism of the effects of Fyn in vitro is consistent with that observed in cell culture. Consequently, the effect of Fyn probably does not require processes observed only in situ (such as trafficking of NMDA receptors between cellular compartments). Additionally, FynY531F expression did not increase levels of NR2B on the cell surface either before or 30 min after agonist application in which calpain activity was blocked by its inhibitor, indicating that Fyn does not facilitate NR2B delivery to the cell membrane in this system (Fig. 8C; wt/vector, control, 100 ± 0.2%; Glu, 94% ± 9; p = 0.27 versus control; wt/FynY531F, control, 93 ± 11%; p = 0.44 versus control of wt/vector; Glu, 93 ± 9%; p = 0.24 versus control of wt/vector; n = 6 each). In addition, NR2B Tyr-1336 was phosphorylated to a similar degree in both cell surface and intracellular fractions (Fig. 8D; surface, control, 100 ± 6%; Glu, 248 ± 24%; intracellular, control, 100 ± 8%; Glu, 210 ± 18%; n = 6 each). Thus, the selective enhancement of surface receptor cleavage by Fyn cannot simply reflect the distribution of phosphorylation. However, since calcium entry facilitates association of calpain with cell membranes, including the plasma membrane (37, 38), we investigated whether NMDA receptor activation translocated calpain I from the cytosol to the membrane area using biotinylation assays under nondenaturing conditions before and after agonist stimulation. This protocol should leave associations of calpain with membrane components intact and lead to retention of cell surface-associated calpain in the biotinylated fraction. In transfected HEK cells, 100 μM glutamate treatment increased the calpain I immunoreactivity in the cell surface fraction 1.9-fold relative to levels before agonist stimulation, with no significant changes of calpain I levels in the intracellular fraction or the lysate (Fig. 8E; surface, CalI3, 3300 ± 100; CalI3 + Glu, 6220 ± 380; intracellular, CalI3, 11,600 ± 4600; CalI3 + Glu, 12,400 ± 140; lysate, CalI3, 19,300 ± 360; CalI3 + Glu, 20,800 ± 400). Actin levels in the surface fraction were not detectably altered by 100 μM glutamate application for 30 min. Thus, overall, the facilitation of calpain-mediated cleavage of NR2B appears to reflect an increased sensitivity of NR2B to calpain with phosphorylation at Tyr-1336 and the translocation of activated calpain to the cell membrane area, creating a selective enhancement of cleavage in the cell surface fraction.
FIGURE 8. Mechanistic studies on the facilitation of calpain-mediated cleavage of NR2B by Fyn.
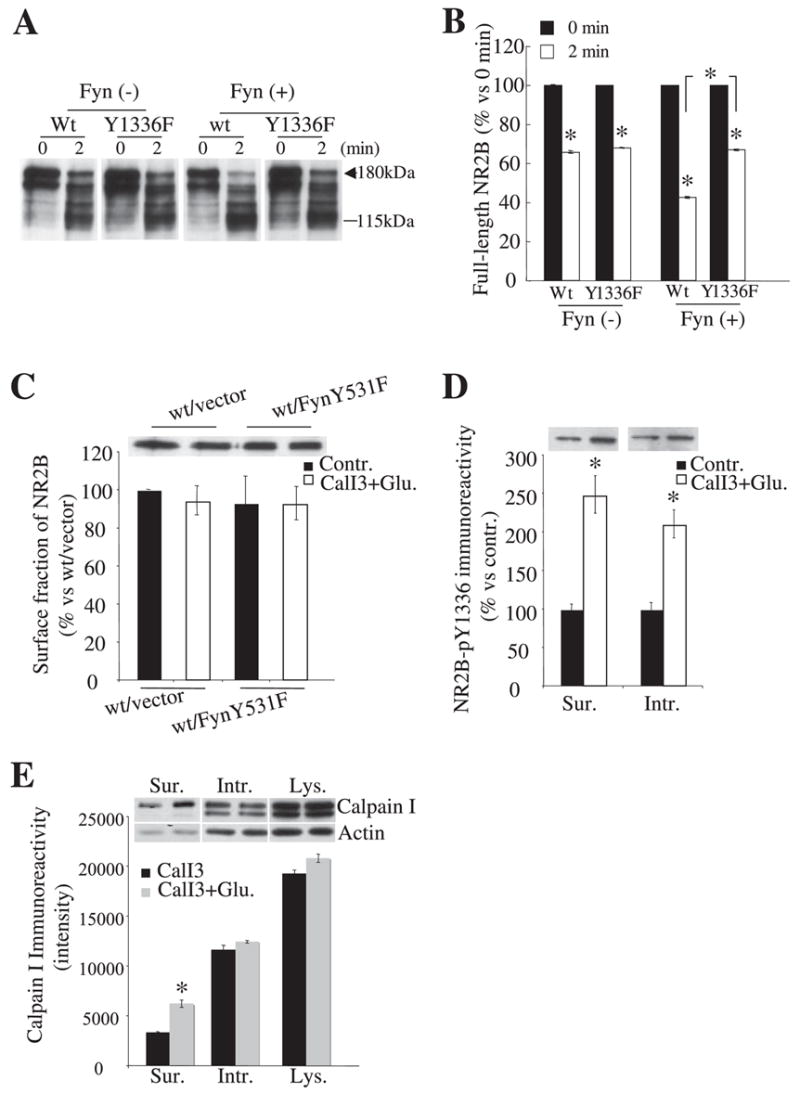
A, representative blots showing cleavage of NR2B in vitro by recombinant calpain I with or without active Fyn protein pretreatment. Homogenates from HEK293 cells transfected with wild-type NR2B or NR2B mutant Y1336F and NR1 were pretreated with or without 100 ng of active Fyn protein for 40 min at 30 °C. After pretreatment, samples were then treated with 2 units/ml recombinant calpain I for 2 min. B, quantitative data obtained from five independent experiments show that Fyn treatment accelerates the cleavage of wild-type NR2B but not NR2BY1336F. C, representative blots and their quantification showed similar levels of NR2B immunoreactivity in the surface fractions of HEK293 cells transfected with NR1/2A/2B with and without FynY531F and with or without agonist stimulation. D, representative blots and quantification of NR2B Tyr(P)1336 immunoreactivity in surface and intracellular fractions from cells transfected with NR1/2A/2B/SAP-102/FynY531F in control conditions and following 30 min of agonist treatment. For C and D, CalI3 was included during agonist treatment to avoid calpain-induced degradation of NR2B. All biotinylated samples were obtained from six independent experiments. Error bars, S.E. E, representative blots and histogram showing levels of calpain I immunoreactivity associated with different cellular compartments (under nondenaturing conditions) in cells transfected with NR1/2A/2B/SAP-102 and FynY531F following agonist stimulation for 30 min. All biotinylated samples were obtained from six independent experiments. Error bars, S.E. Sur, surface; Intr, intracellular; Lys, lysate; wt, wild type.
Cleavage of Full-length NR2B Does Not Decrease Extrasynaptic NMDA Receptor Function
Previously, we demonstrated that calpain does not control the overall size of electrophysiological responses of NMDA receptors in DIV17 hippocampal neurons (19). However, in hippocampal neurons, extrasynaptic NMDA receptors preferentially contain NR2B (39) linked to SAP-102, not PSD-95. This makes extrasynaptic receptors the most likely selective physiologic target of neuronal calpain. We therefore investigated whether calpain-mediated cleavage of the NR2B subunit alters extrasynaptic NMDA receptor function. To monitor pure extrasynaptic NMDA currents in cultures of hippocampal neurons, we selectively blocked synaptic NMDA receptors with an MK801-trapping technique that eliminates the contribution of synaptic receptors from the whole-cell response (39–41). In this paradigm, however, we also wished to examine whether activation of calpain would alter extrasynaptic currents, a goal requiring a control ensuring that the conditions needed to activate calpain did not unblock synaptic receptors. Since extrasynaptic receptors contain largely NR1/2B in hippocampal neurons, one can ensure that response reflects extrasynaptic receptors by their sensitivity to ifenprodil. Consistent with previous reports, blocking synaptic NMDA receptors with 50 μM bicuculline in conjunction with 10 μM MK-801 for 10 min reduced NMDA receptor current density by 69% from the control level of 107 ± 10 pA/pF (n = 12) to 33 ± 5 pA/pF (n = 18) (Fig. 9A). The residual currents were almost completely abolished (5 ± 1 pA/pF, n = 5) by ifenprodil (10 μM), a selective antagonist of NR1/2B receptors, indicating that the whole-cell NMDA-evoked responses after bicuculline/MK801 pre-blocking reflected extrasynaptic currents consisting mainly of NR1/NR2B heteromers (Fig. 9A). With MK801/bicuculline pretreatment, neurons showed no apparent increase in calpain activity based on spectrin cleavage and cleavage of NR2B (Fig. 9B). However, if 100 μM glutamate was then applied to these neurons for 30 min, there was loss of full length NR2B, generation of a 115 kDa NR2B truncation and cleavage of spectrin, showing that isolated NR1/2B extrasynaptic receptors can activate calpain (Fig. 9B). We then compared extrasynaptic NMDA receptor responses before and after calpain-mediated NR2B cleavage in bicuculline/MK801 pre-blocked neurons. As shown in Fig. 9A, after application of 100 μM glutamate for 30 min in bicuculline/MK801 pre-blocked neurons, the NMDA receptor current density was 34 ± 4 pA/pF (n = 21), similar to the corresponding value obtained from bicuculine/MK801 pre-blocked neurons without glutamate treatment (33 ± 5 pA/pF; n = 18, p > 0.8). Inhibition of calpain activation with calpain inhibitor III significantly prevented the glutamate-induced decrease of full-length NR2B (Fig. 9B) but produced similar levels of extra-synaptic NMDA-evoked response (40 ± 5 pA/pF, n = 19, p > 0.3), compared with either glutamate-treated or untreated neurons (Fig. 9A). Current evoked by NMDA after glutamate application for 30 min with or without calpain inhibitor III in bicuculine/MK801 preblocked neurons was completely blocked by ifenprodil (Fig. 9A), indicating no recovery of synaptic NMDA receptors following removal of bicuculline/MK801 during glutamate and calpain inhibitor III treatment (since these receptors typically are ifenprodil-insensitive). This is consistent with previous studies showing that synaptic receptors remain blocked for this period after glutamate application (41). Thus, reducing levels of full-length NR2B without a corresponding change in the current amplitude of response to NMDA indicates that calpain-mediated loss of full-length NR2B does not cause a functional decrease in extrasynaptic NMDA receptor.
FIGURE 9. Decreasing full-length NR2B does not decrease extrasynaptic NMDA receptor function, and calpain-truncated NR2B-containing receptors are functional in HEK cells.

A, extrasynaptic NMDA receptor-mediated currents in hippocampal culture neurons were isolated by blocking synaptic NMDA receptor with 50 μM bicuculline and 10 μM MK801 pretreatment. NMDA receptor current density was reduced. NMDA-evoked currents in glutamate-treated (with or without calpain inhibitor III) in bicuculine/MK801 preblocked neurons were both almost completely blocked by ifenprodil (10 μM; for details, see “Experimental Procedures”), consistent with extrasynaptic receptors being the only population activated by glutamate after bicuculine/MK801 blockade. In addition, ifenprodil did not completely block the NMDA-evoked current in control (no bicuculine/MK801) neurons, indicating the existence of receptors of subunit composition other than NR1/NR2B (presumptively NR1/2A/2B) in control neurons. B, MK801/bicuculine preblocked hippocampal neurons (17–21 DIV) either untreated or treated with 100 μM glutamate and 100 μM glycine for 30 min. Neurons were harvested 30 min after the addition of agonists, and samples were subjected to SDS-PAGE and Western blot analysis with an N-terminal NR2B antibody (upper panel), AB38 (middle panel), and actin (lower panel). C, representative blots from the biotinylation assay, showing that, like wild-type NR2B, NR2Bdel1036 expressed in the cell surface when cotransfected with NR1. D, NMDA receptor current properties were not different between NR1/NR2Bdel1036- and NR1/NR2Bwt-transfected HEK-293 cells. There were no differences in current density (left) or percentage of current desensitization (right) between both groups of transfected cells. **, p = 0.001, ANOVA. wt, wild type.
Calpain-truncated NR2B-containing NMDA Receptors Are Functional in HEK Cells
We then attempted to confirm that receptors containing calpain-truncated NR2B remain active. Based on the expected molecular weight of the truncated form (roughly 1030 amino acids) (16) generated by calpain in neurons, we produced a construct leading to NR2B truncated at amino acid 1036 (NR2Bdel1036) by insertion of two consecutive stop codons and then examined its functional properties. In HEK cells expressing NR1/NR2Bdel1036, Western blots of biotinylated samples demonstrated that the truncated NR2B subunit was expressed on the cell surface (Fig. 9C). In addition, NMDA receptors incorporating NR2Bdel1036 produced intracellular calcium transients similar to wild type receptors when stimulated with agonists (data not shown). Electrophysiologically, brief (2-s) application of 100 μM NMDA activated both wild type- and NR2Bdel1036-containing receptors with no significant difference in current density (current amplitude normalized to cell capacitance; wild-type, 90 ± 15 pA/pF, n = 24; NR1/NR2Bdel1036, 103 ± 13.3 pA/pF, n = 35). In addition, the percentage of current desensitization of NR1/NR2Bdel1036 was statistically indistinguishable from the values found in cells expressing wild-type receptors (Fig. 9D). This result indicates that calpain-truncated NR2B-containing NMDA receptors are functional in HEK cells. These data also demonstrate that Fyn-mediated phosphorylation of NR2B facilitates the generation of truncated receptors that function similarly to full-length receptors but lack the domains necessary for intracellular signal transduction, which may play critical roles in synaptic plasticity or excitotoxicity.
DISCUSSION
In the present study, we have demonstrated that the tyrosine kinase Fyn controls the ability of the NR2B subunit to be cleaved by calpain in neurons and in model expression systems. In neurons, SFK inhibitors and siRNA specific for Fyn slow calpain-mediated cleavage of NR2B but do not alter cleavage of spectrin. In HEK cells, calpain-mediated cleavage is accelerated by constitutively active Fyn but not Src. Calpain-mediated cleavage is enhanced in association with phosphorylation of NR2B Tyr-1336 but not NR2B Tyr-1472, and site-directed mutagenesis demonstrates that removal of Tyr-1336 (but not Tyr-1472) removes the ability of Fyn to potentiate NR2B cleavage by calpain. These data demonstrate that Fyn enhances calpain-mediated cleavage of NR2B in multiple experimental conditions through a mechanism involving direct phosphorylation of NR2B.
The interactions of Fyn, NR2B, and calpain are also controlled by the specific MAGUK protein involved in NR2B binding. Both SAP-102 and PSD-95 enhance phosphorylation of NR2B Tyr-1336 by Fyn. However, as shown previously, PSD-95 blocks the ability of calpain to cleave NR2 subunits (24), and phosphorylation by Fyn was unable to override this effect. This suggests that in neurons, the NR2B cleaved by calpain is most likely bound to SAP-102 and phosphorylated by Fyn. Based on the requirement for SAP-102 (compared with PSD-95) and the preference for cleavage of NR2B rather than NR2A, most calpain-cleaved NMDA receptors are likely to be extrasynaptic NR1/2B receptors. In addition, our data show that such receptors (both extrasynaptic receptors in neurons and truncated NR1/2B in heterologous cells) are predicted to have electrophysiological features similar to full-length receptors. This shows that the primary effect of calpain in our system is not simply degradative but may create receptors with novel properties based on the dissociation of channel activity from downstream messenger systems, such as p38 mitogen-activated protein kinase dephosphorylation and phosphatidylinositol 3-kinase activation. Such receptors could then play crucial roles in excitotoxicity and synaptic modification. The mechanism by which Fyn controls cleavage of NR2B may have several different components. Fyn accelerated the cleavage in vitro of recombinant receptors in a manner that depended on NR2B Tyr-1336, suggesting that Fyn directly enhances the substrate characteristics of the C terminus of NR2B. This has been noted previously for NR2A and NR2B in brain homogenates, and in fusion proteins of NR2B from 1269–1482 (35), suggesting that the effect of Fyn is mediated entirely within these amino acids and matching the dependence on NR2B Tyr-1336 demonstrated here. These data suggest that at least some aspect of the mechanism of the effects of Fyn is a direct biochemical effect on the substrate properties of the C-terminal region of NR2B.
However, in HEK cells, the subcellular localization of the enhancement of calpain-mediated cleavage suggests other aspects to the mechanistic effects of Fyn. Although calpain can cleave NR2B within intracellular compartments and Fyn can phosphorylate NR2B Tyr-1336 in intracellular compartments, Fyn did not enhance calpain-mediated cleavage in these compartments. This is consistent with the localization of NR2B in neurons, where most NR2B is located on the cell surface in hippocampal cultures (16), but suggests that factors beyond biochemical aspects contribute to the effects of Fyn on calpain-mediated cleavage of NR2B. One factor could be increased trafficking of NR2B to the cell surface after activation of Fyn. However, the presence of constitutively active Fyn (with resultant base-line phosphorylation of Tyr-1336) did not increase levels of NR2B on the cell surface; nor did agonist treatment of receptors (with Fyn activation) during calpain blockade alter surface NR2B levels. Thus, increased trafficking of NR2B to the cell surface cannot explain the selective enhancement of cleavage by Fyn in this compartment. Instead, calpain, previously reported to associate with membranes after activation (37, 38), increased in the cell surface fraction under nondenaturing conditions (Fig. 8E), suggesting that targeting of calpain to the plasma membrane mediates the selective enhancement of cleavage in the cell surface fraction. The regulatory subunit of calpain has been associated with synaptic membranes in proteomic experiments (42). These results suggest that the biochemical enhancement of NR2B degradation by Fyn in conjunction with calpain transport to the plasma membrane provides the mechanism for selective enhancement of cleavage by Fyn in the cell surface fraction. However, calpain also can associate with internal membranes after rises in intracellular calcium, suggesting that the translocation of calpain to the plasma membrane may only be a partial explanation for the compartmentalized effect of Fyn. The level of cleavage of NR2B in intracellular compartments was essentially equal to that observed with enhancement by Fyn in the surface fraction, suggesting that unidentified plasma membrane components tonically inhibit cleavage of NR2B. It is conceivable that such components (structural features of the attachment of NR2B to MAGUK proteins or plasma membrane lipids, association with RACK1 (receptor for activated protein kinase C1) or related proteins) selectively inhibit calpain-mediated NR2B cleavage in a manner that is relieved by phosphorylation by Fyn.
Fyn modulates NMDA receptors in several ways (33, 43–46). Although Fyn has not clearly been shown to alter channel function directly, Fyn enhances transport of NR2 subunits to the cell surface and into the synapse under different conditions and blocks internalization of NR2B-containing receptors (47). However, these effects are most likely mediated by Tyr-1472 based on site-directed mutagenesis (23, 45, 46). The control of calpain-mediated cleavage appears to reflect events occurring with phosphorylation of Tyr-1336, a site that is phosphorylated in cellular systems to a slightly lower degree than Tyr-1472. This site also controls activation of phosphatidylinositol 3-kinase and dephosphorylation of p38 mitogen-activated protein kinase, potential protective mechanisms in neurons (22). This suggests that the events occurring with Tyr-1336 phosphorylation provide a set of responses distinct from the role of Fyn in organizing NR2B localization at the synapse through receptor trafficking and internalization.
The effect of Fyn here does not entirely match in vitro results examining calpain-mediated cleavage of NR2 subunits. Whereas Fyn accelerated cleavage of NR2B subunits in vitro, the same effect is seen on NR2A. However, the selective binding of NR2A to PSD-95 in synapses limits its ability to be cleaved in neurons (24). Src blocks calpain-mediated cleavage in vitro (through an unknown mechanism), but constitutively active Src did not lead to phosphorylation at Tyr-1336 in situ; nor did it alter the speed of cleavage of NR2B by calpain. This result agrees with the observation that among MAGUK proteins, only PSD-95 binds Src (48). Our data illustrate that the interaction of MAGUK proteins and SFK regulates the cleavage of NR2 subunits by calpain in living cells and that when calpain-mediated cleavage of NR2B is examined in situ, only Fyn alters this process.
Fyn significantly contributes to tyrosine phosphorylation of NMDA receptors, and Fyn mutant mice show defects in NMDA receptor-dependent LTP (43). However, NMDA receptor activation itself has not been shown to stimulate Fyn activity. In the present study, phosphorylation of NR2B Tyr-1336 increased with NMDA receptor stimulation in cultured hippocampal neurons or HEK cells expressing NR2B-containing NMDA receptors. These effects were blocked with MK801 and suggest that NMDA receptors could directly activate Fyn. However, NMDA receptor stimulation did not enhance phosphorylation of NR2B Tyr-1472, suggesting that NMDA receptor stimulation may instead direct the activity of Fyn to a different site on the NMDA receptor. Selective Fyn-mediated phosphorylation of Tyr-1336 is also seen in neurons treated with ethanol, in which it is controlled by dissociation of RACK1 from NR2B (33). Nevertheless, the present results indicate that the NR2B/Fyn/calpain interactions may act as part of a Fyn kinase cascade, which in turn regulates NMDA receptor features through promoting calpain-mediated cleavage of NR2B subunits. Further experiments are required to investigate the mechanisms by which NMDA receptor activation is coupled to Fyn-mediated phosphorylation of NR2B Tyr-1336 and calpain-mediated cleavage of NR2B in neuronal pathophysiology.
In summary, the present study provides a model for understanding the control and selectivity of neuronal calpain for selected proteins in synaptic and extrasynaptic membranes. Since calpain-cleaved receptors dissociated from intracellular signal transduction elements remain on the cell surface and are active similarly to those of wild-type receptors, the present data demonstrate a new manner in which Fyn may contribute to enhancement of synaptic plasticity or excitotoxicity in neuronal systems.
Acknowledgments
We thank Margie Maronski for hippocampal neuron preparation.
Footnotes
This work was supported by National Institutes of Health Grant NS45986, the Trisomy 21 program and Mental Retardation Research Center of the Children’s Hospital of Philadelphia (to D. R. L.), and American Heart Association Grant 0625390U (to H. Y. W.).
The abbreviations used are: NMDA, N-methyl-D-aspartate; SFK, Src family kinase; MK-801, dizocilpine; HEK, human embryonic kidney; MAGUK, membrane-associated guanylate kinase; CalI3, calpain inhibitor III; MDL 28170, benzyloxycarbonyl-Val-Phe-aldehyde; PP2, 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine; NR2Bdel1036, NR2B truncated at amino acid 1036; siRNA, small interfering RNA; DIV, days in vitro; ANOVA, analysis of variance; pF, picofarads; Glu, treatment with 100 μM glutamate and 100 μM glycine.
References
- 1.Lynch DR, Guttmann RP. J Pharmacol Exp Ther. 2002;300:717–723. doi: 10.1124/jpet.300.3.717. [DOI] [PubMed] [Google Scholar]
- 2.Lynch DR, Guttmann RP. Curr Drug Targets. 2001;2:215–231. doi: 10.2174/1389450013348434. [DOI] [PubMed] [Google Scholar]
- 3.Mayer ML, Westbrook GL. Prog Neurobiol. 1987;28:197–276. doi: 10.1016/0301-0082(87)90011-6. [DOI] [PubMed] [Google Scholar]
- 4.Collingridge GL, Bliss TV. Trends Neurosci. 1995;18:54–56. [PubMed] [Google Scholar]
- 5.Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, Seeburg PH. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- 6.Kutsuwada T, Kashiwabuchi N, Mori H, Sakimura K, Kushiya E, Araki N, Meguro H, Masaki H, Kumanishi T, Arakawa M, Mishina M. Nature. 1992;358:36–41. doi: 10.1038/358036a0. [DOI] [PubMed] [Google Scholar]
- 7.Meguro H, Mori H, Araki K, Kushiya E, Kutsuwada T, Yamazaki M, Kumanishi T, Arakawa M, Sakimura K, Mishima M. Nature. 1992;357:70–74. doi: 10.1038/357070a0. [DOI] [PubMed] [Google Scholar]
- 8.Ishii T, Moriyoshi K, Sugihara H, Sakurada K, Kadotani H, Yokoi M, Akazawa C, Shigemoto R, Mizuno N, Masu M. J Biol Chem. 1993;268:2836–2843. [PubMed] [Google Scholar]
- 9.Watanabe M, Inoue Y, Sakimura K, Mishina M. NeuroReport. 1992;3:1138–1140. doi: 10.1097/00001756-199212000-00027. [DOI] [PubMed] [Google Scholar]
- 10.Kutsuwada T, Sakimura K, Manabe T, Takayama C, Katakura N, Kushiya E, Natsume R, Watanabe M, Inoue Y, Yagi T, Aizawa S, Arakawa M, Takahashi T, Nakamura Y, Mori H, Mishina M. Neuron. 1996;16:333–344. doi: 10.1016/s0896-6273(00)80051-3. [DOI] [PubMed] [Google Scholar]
- 11.Ebralidze AK, Rossi DJ, Tonegawa S, Slater NT. J Neurosci. 1996;16:5014–5025. doi: 10.1523/JNEUROSCI.16-16-05014.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kadotani H, Hirano T, Masugi M, Nakamura K, Nakao K, Katsuki M, Nakanishi S. J Neurosci. 1996;16:7859–7867. doi: 10.1523/JNEUROSCI.16-24-07859.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sprengel R, Suchanek B, Amico C, Brusa R, Burnashev N, Rozov A, Hvalby O, Jensen V, Paulsen O, Andersen P, Kim JJ, Thompson RF, Sun W, Webster LC, Grant SG, Eilers J, Konnerth A, Li J, McNamara JO, Seeburg PH. Cell. 1998;92:279–289. doi: 10.1016/s0092-8674(00)80921-6. [DOI] [PubMed] [Google Scholar]
- 14.Guttmann RP, Baker DL, Seifert KS, Cohen A, Coulter DA, Lynch DR. J Neurochem. 2001;78:1083–1093. doi: 10.1046/j.1471-4159.2001.00493.x. [DOI] [PubMed] [Google Scholar]
- 15.Guttmann RP, Sokol S, Baker DL, Simpkins KL, Dong Y, Lynch DR. J Pharmacol Exp Ther. 2002;302:1023–1030. doi: 10.1124/jpet.102.036962. [DOI] [PubMed] [Google Scholar]
- 16.Simpkins KL, Guttmann RP, Dong Y, Chen Z, Sokol S, Neumar RW, Lynch DR. J Neurosci. 2003;23:11322–11331. doi: 10.1523/JNEUROSCI.23-36-11322.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Araujo I, Xapelli S, Gil J, Mohapel P, Petersen A, Pinheiro PS, Malva JO, Bahr B, Brundin P, Carvalho CM. NeuroReport. 2005;16:393–396. doi: 10.1097/00001756-200503150-00017. [DOI] [PubMed] [Google Scholar]
- 18.Wu HY, Yuen EY, Lu YF, Matsushita M, Matsui H, Yan Z, Tomizawa K. J Biol Chem. 2005;280:21588–21593. doi: 10.1074/jbc.M501603200. [DOI] [PubMed] [Google Scholar]
- 19.Dong Y, Wu HY, Hsu FC, Coulter DA, Lynch DR. J Neurochem. 2006;99:206–217. doi: 10.1111/j.1471-4159.2006.04096.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moon IS, Apperson ML, Kennedy MB. Proc Natl Acad Sci U S A. 1994;91:3954–3958. doi: 10.1073/pnas.91.9.3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gurd JW, Bissoon N. J Neurochem. 1997;69:623–630. doi: 10.1046/j.1471-4159.1997.69020623.x. [DOI] [PubMed] [Google Scholar]
- 22.Waxman EA, Lynch DR. J Biol Chem. 2005;280:29322–29333. doi: 10.1074/jbc.M502080200. [DOI] [PubMed] [Google Scholar]
- 23.Nakazawa T, Komai S, Tezuka T, Hisatsune C, Umemori H, Semba K, Mishina M, Manabe T, Yamamoto T. J Biol Chem. 2001;276:693–699. doi: 10.1074/jbc.M008085200. [DOI] [PubMed] [Google Scholar]
- 24.Dong YN, Waxman EA, Lynch DR. J Neurosci. 2004;24:11035–11045. doi: 10.1523/JNEUROSCI.3722-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grant ER, Basckai BJ, Gallagher MJ, Kendrick SL, Pritchett DB, Kricka L, Pleasure DP, Lynch DR. J Biol Chem. 1997;272:647–656. doi: 10.1074/jbc.272.1.647. [DOI] [PubMed] [Google Scholar]
- 26.Lynch DR, Shim SS, Seifert KM, Kurapathi S, Mutel V, Gallagher MJ, Guttmann RP. Eur J Pharmacol. 2001;416:185–195. doi: 10.1016/s0014-2999(01)00868-8. [DOI] [PubMed] [Google Scholar]
- 27.Sims KD, Straff DJ, Robinson MB. J Biol Chem. 2000;275:5228–5237. doi: 10.1074/jbc.275.7.5228. [DOI] [PubMed] [Google Scholar]
- 28.Anegawa NJ, Lynch DR, Verdoorn TA, Pritchett DB. J Neurochem. 1995;64:2004–2012. doi: 10.1046/j.1471-4159.1995.64052004.x. [DOI] [PubMed] [Google Scholar]
- 29.Yang M, Leonard JP. J Neurochem. 2001;77:580–588. doi: 10.1046/j.1471-4159.2001.00255.x. [DOI] [PubMed] [Google Scholar]
- 30.Cheung HH, Gurd JW. J Neurochem. 2001;78:524–534. doi: 10.1046/j.1471-4159.2001.00433.x. [DOI] [PubMed] [Google Scholar]
- 31.Zheng F, Gingrich MB, Traynelis SF, Conn PJ. Nat Neurosci. 1998;1:185–191. doi: 10.1038/634. [DOI] [PubMed] [Google Scholar]
- 32.Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 33.Yaka R, Phamluong K, Ron D. J Neurosci. 2003;23:3623–3632. doi: 10.1523/JNEUROSCI.23-09-03623.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sala C, Sheng M. Proc Natl Acad Sci U S A. 1999;96:335–337. doi: 10.1073/pnas.96.2.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rong Y, Lu X, Bernard A, Khrestchatisky M, Baudry M. J Neurochem. 2001;79:382–390. doi: 10.1046/j.1471-4159.2001.00565.x. [DOI] [PubMed] [Google Scholar]
- 36.Sui G, Soohoo C, Affare B, Gay F, Shi Y, Forrester WC, Shi Y. Proc Natl Acad Sci U S A. 2002;99:5515–5520. doi: 10.1073/pnas.082117599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pontremoli S, Melloni E, Salamino F, Patrone M, Michetti M, Horecker BL. Biochem Biophys Res Commun. 1989;160:737–743. doi: 10.1016/0006-291x(89)92495-9. [DOI] [PubMed] [Google Scholar]
- 38.Ariyoshi H, Shiba E, Sakon M, Kambayashi J, Yoshida K, Kawashima S, Mori T. Biochem Mol Biol Int. 1993;30:63–72. [PubMed] [Google Scholar]
- 39.Tovar KR, Westbrook GL. J Neurosci. 1999;19:4180–4188. doi: 10.1523/JNEUROSCI.19-10-04180.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tovar KR, Westbrook GL. Neuron. 2002;34:255–264. doi: 10.1016/s0896-6273(02)00658-x. [DOI] [PubMed] [Google Scholar]
- 41.Hardingham GE, Fukunaga Y, Bading H. Nat Neurosci. 2002;5:405–414. doi: 10.1038/nn835. [DOI] [PubMed] [Google Scholar]
- 42.Husi H, Ward MA, Choudhary JS, Blackstock WP, Grant SGN. Nat Neurosci. 2000;3:661–669. doi: 10.1038/76615. [DOI] [PubMed] [Google Scholar]
- 43.Grant SG, O’Dell TJ, Karl KA, Stein PL, Soriano P, Kandel ER. Science. 1992;258:1903–1910. doi: 10.1126/science.1361685. [DOI] [PubMed] [Google Scholar]
- 44.Miyakawa T, Yagi T, Kitazawa H, Yasuda M, Kawai N, Tsuboi K, Niki H. Science. 1997;278:698–701. doi: 10.1126/science.278.5338.698. [DOI] [PubMed] [Google Scholar]
- 45.Lavezzari G, McCallum J, Lee R, Roche KW. Neuropharmacology. 2003;45:729–737. doi: 10.1016/s0028-3908(03)00308-3. [DOI] [PubMed] [Google Scholar]
- 46.Prybylowski K, Chang K, Sans N, Kan LL, Vicini S, Wenthold RJ. Neuron. 2005;47:845–857. doi: 10.1016/j.neuron.2005.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dunah AW, Sirianni AC, Fienberg AA, Bastia E, Schwarzschild MA, Standaert DG. Mol Pharmacol. 2004;65:121–129. doi: 10.1124/mol.65.1.121. [DOI] [PubMed] [Google Scholar]
- 48.Kalia LV, Salter MW. Neuropharmacology. 2003;45:720–728. doi: 10.1016/s0028-3908(03)00313-7. [DOI] [PubMed] [Google Scholar]