

Abstract
Chromatin structure is not fixed. Instead, chromatin is dynamic and is subject to extensive developmental and age-associated remodeling. In some cases, this remodeling appears to counter the aging and age-associated diseases, such as cancer, and extend organismal lifespan. However, stochastic non-deterministic changes in chromatin structure might, over time, also contribute to the break down of nuclear, cell and tissue function, and consequently aging and age-associated diseases.
Introduction
As reviewed in this volume and elsewhere, organismal longevity and aging may be influenced by many complex interacting factors [1]. These include accumulation of nuclear and mitochondrial genome mutations; shortened and dysfunctional chromosome telomeres; oxidative damage of DNA and other macromolecules; systemic hormonal factors, such as insulin/IGF signaling; and senescence, apoptosis and altered differentiation in self-renewing tissues that depend on stem cells. In many cases, these factors are, or are likely to be, overlapping and/or interacting. In this chapter, we discuss another possible determinant of aging, “epigenetics”.
Strictly speaking, “epigenetics” refers to chromatin and DNA modifications that are heritable through cell division, but do not involve changes in the underlying DNA sequence [2]. However, since the aging process is not restricted to proliferating tissues and may, in fact, be influenced by irreversible proliferation arrest, we will use the term more loosely to refer to effects mediated by chromatin structure and DNA methylation, but not DNA sequence.
The dynamic and stochastic properties of chromatin
The basic repeating unit of chromatin structure is the nucleosome, comprised of about 146bp of DNA wrapped around an octamer of histones [3]. Chromatin can be broadly divided into two types, heterochromatin and euchromatin [4]. Euchromatin is decondensed during interphase, relatively transcriptionally active and replicates early in S-phase. Conversely, heterochromatin remains condensed during interphase, is more transcriptionally silent and is late replicating. Heterochromatin is itself further subdivided into two types, constitutive and facultative heterochromatin. Constitutive heterochromatin, for example at repetitive pericentromeric and telomeric DNA, is heterochromatic in all cells of an organism and has typically been considered to be essentially fixed and irreversible through the lifetime of the organism, although this may not be entirely true, as discussed below. In contrast, facultative heterochromatin is usually made as part of regulated cell differentiation processes or other changes in cell phenotype. For example, a single X chromosome is silenced by heterochromatinization in female mammal cells [5], for sex chromosome dosage compensation. Whether chromatin forms euchromatin or heterochromatin is dictated by modifications of the histones and DNA [6]. For example, acetylation of the N-terminal tails of histones promotes formation of euchromatin. Conversely, DNA methylation is characteristic of heterochromatin.
Despite this seemingly clear distinction between heterochromatin and euchromatin, it is increasingly being appreciated that chromatin is a highly dynamic and stochastic entity. The phenomenon of position effect variegation (PEV) first indicated the dynamic and stochastic properties of chromatin [7]. PEV was originally described in flies as a mottled red and white eye phenotype [8]. Due to a chromosomal inversion and change in position of the white gene relative to centromeric heterochromatin, the gene can be silenced by heterochromatin in a subset of cells that give pigment to the eye. The ability of genes subject to PEV to switch between active and inactive states reflects the dynamic nature of chromatin structure. This dynamic nature has been confirmed by more recent cell and molecular studies. First, essential processes, such as gene transcription, DNA replication and repair, all involve disruption and reassembly of chromatin structure [9, 10]. Second, Fluorescence recovery after photobleaching (FRAP) studies have shown that most chromatin-bound proteins are not statically bound, but exhibit relatively high "off" and "on" rates, even in supposedly "closed" heterochromatin [11, 12]. Third, remarkably and paradoxically, formation of heterochromatin actually depends on a degree of transcription, which contributes to heterochromatinization through the RNAi pathway [4]. Consistent with this, recent genome-wide analyses have shown that virtually all DNA nucleotides of the human genome are, to some degree, transcribed into RNA [13–15]. Indeed, telomeric heterochromatin was recently shown to be transcribed [16, 17]. These observations indicate that even heterochromatin is not a static, closed entity. Fourth, heterochromatin can "spread" along chromatin, through cooperative interactions between the enzymes and binding proteins responsible for its formation [18]. The random occurrence of white expression in cells of the fly eye reflects the stochastic nature of heterochromatin's establishment and maintenance. The stochastic nature of chromatin structure and function is reaffirmed by many studies that have demonstrated a stochastic component to the control of gene expression [19].
In sum, an analysis of chromatin structure and its underlying biochemistry indicates that chromatin has dynamic as well as stochastic properties. Chromatin is not fixed, but is continuously remodeled and can redistribute and "breathe", sometimes in a not completely deterministic fashion. As discussed below, in some cases this dynamic property appears to counter the aging process and extend organismal lifespan. However, the stochastic, unpredictable component of chromatin structure might, over time, also contribute to the break down of nuclear, cell and tissue function, and consequently aging and age-associated diseases.
Aging is associated with epigenetic changes, from yeast to humans
The enzymes that add and remove acetyl groups, the histone acetyltransferases (HATs) and histone deacetylases (HDACs) respectively, determine the steady-state level of histone acetylation [10]. In S. cerevisiae, inactivation of the histone deacetylase, Sir2, shortens replicative lifespan [20–22]. Conversely, activation of Sir2 extends lifespan. The anti-aging effects of Sir2 in yeast are, at least in part, due to translocation of a Sir2-containing protein complex from telomeres to ribosomal DNA (rDNA) repeats [23]. These repeats are prone to recombination to form extrachromosomal rDNA circles (ERCs), which curtail yeast lifespan. At the rDNA repeats, Sir2-mediated histone deacetylation, and consequent heterochromatinization by the Sir2-associated proteins (Sir3 and Sir4), prevents recombination and formation of ERCs, thereby extending lifespan [21, 22]. In sum, yeast aging is associated with nuclear redistribution of a histone modifying enzyme, Sir2, and its partner heterochromatin-binding proteins. This epigenetic redistribution counteracts organismal aging.
Orthologs of Sir2 have anti-aging functions in many other species, including nematodes and flies, although the mechanisms do not appear to involve ERCs in these organisms [21, 22]. Significantly, a decreased level of expression of the closest mammalian ortholog of Sir2, SirT1, correlates with apparent premature aging of mice with increased activity of p53 family members [24]. Moreover, in aging wild type mice, SirT1 protein level decreases with age in mitotic tissues [25]. Thus, the ability of Sir2-like proteins to regulate aging appears to be conserved through evolution. However, SirT1 has many non-chromatin substrates [22], so whether any effect on mammalian aging is epigenetically determined remains to be established.
In mammals, there is an age-associated decline in total genomic DNA methylation [26–28]. This occurs mostly at repetitive DNA sequences, and so probably occurs predominantly in domains of constitutive heterochromatin. Since DNA methylation promotes formation of transcriptionally silent heterochromatin [6], this change will facilitate deheterochromatinization of these regions.
However, although genome-wide levels of methylation decrease with age in mammals, at specific sites there is a tendency for DNA methylation to increase [29–37]. This occurs at CpG islands, some of which are in the promoter regions of genes. CpG islands are CG rich sequences that are typically unmethylated, but can become methylated. In addition, the total abundance of histone H4 methylated on lysine 20 (H4K20Me) has also been reported to increase with age in rat liver and kidney [38]. Like DNA methylation, H4K20Me is linked to transcriptional repression [6], supporting the notion that heterochromatin accumulates with tissue aging, at least at some sites.
Histone chaperones are histone binding proteins that mediate assembly of histones into nucleosomes [39]. Consequently, they are key determinants of chromatin structure and function. Significantly, at least one of these, the histone chaperone HIRA, has been shown to increase in expression or undergo some level of regulation in aging baboon skin [40,41]. Although the HIRA histone chaperone may have some functions linked to transcription activation [42–48], this chaperone is known to have an evolutionarily conserved role in heterochromatin formation [49–56].
Together, these observations suggest that mammalian aging is also associated with remodeling of chromatin structure. In particular, analysis of DNA methylation patterns suggests that mammalian aging is associated with an overall decrease in heterochromatin, but an increase at specific sites in the genome. Conceivably, there is a "redistribution" of transcription-silencing heterochromatin from repetitive DNA, that is normally packaged into constitutive heterochromatin, to regions of the genome that are normally transcribed [57–59].
Cellular senescence is associated with redistribution of heterochromatin
Cellular senescence is characterized by an irreversible proliferation arrest [60]. Cellular senescence can be initiated by various triggers, including an excessive number of cell divisions, in part due to shortened chromosome telomeres (so-called replicative senescence). Because of senescence, most primary human cells have a finite proliferative lifespan, and evidence has been presented that senescence contributes to tissue aging in vivo, in part by limiting the self-renewal of tissues [61–63]. Specifically, senescent cells and/or molecular markers of the senescent phenotype have been reported to increase in some aging tissues [40, 41, 64–69], and are linked to some age-associated tissue pathologies, such as osteoarthritis, atherosclerosis and liver cirrhosis [70–72]. Moreover, manipulation of the cell signals that initiate senescence, such as telomere length and expression of the proliferation inhibitor p16INK4a [66–68, 70, 73–80], can modulate some aspects of organismal aging [70–72]. In addition to a likely role in tissue aging, cellular senescence is also a well-established tumor suppression process, by virtue of its ability to arrest proliferation and neoplastic progression of cells harboring oncogenic lesions [81–87].
Many senescent cells, whether triggered by excessive rounds of cell proliferation or activated oncogenes, show dramatic changes in chromatin structure. First, many senescent cells form specialized domains of facultative heterochromatin, called Senescence-Associated Heterochromatin Foci (SAHF) [88]. SAHF are visibly more condensed than interphase chromatin and contain histone modifications and associated proteins characteristic of heterochromatin. These SAHF have been proposed to silence expression of proliferation-promoting genes, and thus contribute to senescence-associated proliferation arrest. Accumulating evidence indicates that SAHF, or similar chromatin changes, are relevant to organismal aging. First, similar changes in chromatin structure have been described in the skin of aging baboons [41]. Second, the histone chaperone HIRA plays a key role in formation of SAHF [89–92]. As noted previously, based on immunostaining assays, this chaperone is upregulated in the dermal fibroblasts of aging baboons [40].
Remarkably, although SAHF appear to result from the condensation of almost entire chromosomes, DNA sequences that are typically contained in constitutive heterochromatin, such as pericentromeres and telomeres, actually appear to be excluded from the bulk of the condensed chromosome [57, 90, 91, 93]. This suggests that these normally constitutively heterochromatic regions are perhaps deheterochromatinized in senescent cells. Consistent with this idea at least for telomeres, Maria Blasco and coworkers have shown that the shortened telomeres in mice lacking telomerase have reduced heterochromatin compared to telomeres from normal cells [94]. In sum, like tissue aging, cellular senescence appears to be accompanied by a redistribution of heterochromatin from constitutive heterochromatin to other normally-euchromatic sites, specifically to specialized domains of facultative heterochromatin, called SAHF.
Progeria cells exhibit accelerated changes in chromatin
Hutchinson-Gilford Progeria Syndrome (HGPS) is a rare syndrome that is characterized by rapid onset of some debilitating phenotypes that are typically indicative of aging [95]. These include severe growth retardation, loss of subcutaneous fat, alopecia, reduced bone density and poor muscle development. The average age of death in HGPS is 12–15 years, usually by myocardial infarction or stroke. However, HGPS patients do not accelerate all aspects of aging. For example, cancer incidence is rare and immune dysfunction is not typically reported. Therefore, whether HGPS is really a mechanistically accelerated aging syndrome or whether it merely phenocopies some aspects of aging through unrelated mechanisms has been debated [96].
Recent studies have demonstrated similarities between the epigenetic defects of HGPS cells and cells from very old normal individuals, supporting the notion that, at this molecular level, HGPS may represent accelerated aging. HGPS is caused by a splicing defect in exon 11 of the Lamin A gene (LMNA), removing 50 amino acids from the carboxy-terminus of the encoded protein. HGPS cells show numerous phenotypes, including dramatic defects in nuclear envelope structure, DNA damage and repair defects, changes in gene expression and a reduced rate of proliferation [95]. Fibroblasts from HGPS cells also exhibit epigenetic defects, including decreased abundance of heterochromatic markers, such as histone H3 lysine 9 methylation (H3K9Me) and HP1 proteins [97–99]. These changes are exacerbated by prolonged passage in culture. Interestingly, after passage in culture, cells from aging normal individuals (>80 years) show similar changes in heterochromatin structure as HGPS individuals [98]. In sum, studies of HGPS cells are consistent with the idea that changes in chromatin structure - specifically loss of heterochromatin - may contribute to the aging phenotype. Whether or not HGPS cells also show increased heterochromatinization at other sites has not been reported.
The consequences of age-associated epigenetic changes
Not surprisingly, the consequence of these age-associated epigenetic changes is best understood in yeast. As discussed above, in this organism redistribution of Sir2 and heterochromatic proteins counteracts the aging process [21, 22]. The consequence of the various modes of age-associated epigenetic regulation in mammals remains somewhat speculative. However, aging is accompanied by various altered phenotypes that might be linked to age-associated epigenetic changes. Studies going back to the 1960's by Richard Doll and coworkers indicated that aging is associated with cell aneuploidy [41]. Since proper chromosome segregation is dependent upon constitutive pericentromeric heterochromatin structure and function [100, 101], decreased DNA methylation and deheterochromatinization of repetitive pericentromeric sequences might contribute to faulty chromosome segregation and age-associated aneuploidy. Recent studies in yeast showed that aneuploidy confers various altered cell phenotypes, including a proliferative impairment, which might contribute to decreased tissue renewal capacity with age [102]. Aneuploidy may also promote cancer [103], a disease for which age is the biggest risk factor.
Aging is also associated with changes in patterns of gene expression. Importantly, linking back to the previous discussion of PEV, some of these changes appear to have a stochastic component. For example, aging is associated with increased variability in gene expression in cardiomyocytes [104]. Aging hematopoietic stem cells also exhibit changes in gene expression. These changes are consistent with a bias towards myeloid differentiation, and so appear to parallel the "lymphoid to myeloid shift" that accompanies aging of the immune system, and contributes to declining adaptive immunity [105, 106]. Whether these age-associated changes in gene expression are due to epigenetic alterations or genetic alterations (i.e. accumulated DNA damage) is not clear, but it is feasible that epigenetics accounts for at least part of the change. Underscoring this possibility, age-associated divergence of DNA methylation and histone acetylation profiles has been found in pairs of monozygous twins that are genetically identical [107] (but see also ref [108]). Remarkably, in the twin study, those genes that were differentially modified were also differentially expressed, suggesting that age-associated epigenetic divergence drives age-associated divergence of gene expression profiles. This age-associated divergence of genetically identical individuals is again consistent with a stochastic component to the change.
Age-associated methylation of CpG islands may have profound functional consequences. Methylation of CpG islands is a well-described mode of silencing of some tumor suppressor genes, such as INK4a and VHL [109]. Remarkably, age-associated CpG methylation of some tumor suppressor genes has been reported in histologically normal tissue, and appears to precede the development of neoplastic changes in tissue organization [32–34, 36, 37]. This suggests that tissue-wide, age-associated CpG methylation may be an early causative event in development of neoplasms. If one consequence of this CpG methylation is to silence tumor suppressor genes, a process that seems disadvantageous to the organism, it is possible that it is linked to other beneficial changes in chromatin structure, but reflects a stochastic, non-determined mis-targeting of that process. For example, since SAHF contribute to the senescence phenotype, they might primarily contribute to senescence-mediated tumor suppression [81–87]. However, stochastic “errors” in the SAHF assembly process might erroneously contribute to methylation of CpG islands of some tumor suppressor genes, accounting for the age-associated silencing of these genes that has been reported. Even if these age-associated stochastic epigenetic errors occur only rarely, like rare genetic mutations, they might confer the first selective advantage on the road to cancer.
Summary, outstanding questions and therapeutic implications
From the preceding discussion, it is apparent that chromatin structure does change with aging, in organisms as diverse as yeast and mammals. However, with the exception of Sir2 in yeast, the extent to which this impacts the aging process has not yet been defined. Although it is early days for this field, we propose that some age-associated epigenetic alterations in mammals, such as formation of SAHF, might extend lifespan by suppressing age-associated diseases, such as cancer. However, as encapsulated within the hypothesis of "antagonistic pleiotropy" [110, 111], SAHF and other events that drive senescence might ultimately promote aging by decreasing the long term renewal capacity of tissues. In addition, we suggest that a stochastic and presumably unregulated component of the age-associated change in chromatin structure, which appears inherent given the underlying biochemistry of chromatin, may lead to gradual deterioration in cell and tissue function with age. By analogy to age-associated genetic alterations (DNA damage) that are thought to contribute to aging, these accumulated epigenetic changes can be considered "chromatin damage".
In sum, the effects of chromatin on aging are likely to be complex and bidirectional. To test and define the impact of specific epigenetic determinants on aging will be a challenging task. Arguably, a pre-requisite to properly understanding the contribution of epigenetics to aging is to better understand the specific cell, tissue and system-wide malfunctions that are responsible for specific aging phenotypes, such as osteoporosis, sarcopenia, declining immune function, alopecia, cancer and many others. Then, it will be feasible to dissect out the contribution to each phenotype of each candidate epigenetic determinant, such as global hypomethylation, CpG island hypermethylation and SAHF.
While these studies are ongoing, we can already consider applying the knowledge gained. For example, we may be able to use age-associated epigenetic alterations as a means of early detection and risk stratification for age-associated diseases. There is already considerable interest in the development of high-throughput methods for early detection of cancer, based on detection of tumor suppressor gene CpG hypermethylation in very small numbers of tumor cells found in blood or other accessible body fluids [112]. However, if CpG hypermethylation is a frequent age-associated, tissue-wide, pre-neoplastic event, it might be possible to extend this technology to cancer risk assessment, based upon age-associated hypermethylation in pre-neoplastic tissue. Likewise, it might also be possible to use age-associated epigenetic changes in cardiac or immune cells for risk assessment or early detection of cardiovascular disease or declining immune function [104–106].
An ultimate goal of aging research is to delay or alleviate some of the most debilitating age-associated diseases, thereby prolonging healthy lifespan. For aspects of aging driven by epigenetics this may be an achievable goal, because in principle, epigenetic alterations are more readily reversible than are genetic alterations. Not only might epigenetic alterations be used in cancer risk assessment and early detection, but reversal of these epigenetic changes might also be a cancer chemoprevention strategy [113]. Chemopreventative strategies may also counter age-associated diseases other than cancer. Remarkably, addition of a SirT1 activator, resveratrol, to the diet of mice fed a high fat diet prolongs the healthy lifespan of those mice [114, 115], apparently mimicking the well-established contribution of dietary caloric restriction to longevity [116]. Although there are questions as to whether resveratrol extends lifespan through SirT1 and whether SirT1 acts through chromatin [117, 118], these studies demonstrate the potential of chemopreventative strategies to combat aging.
Figure 1. A model depicting the proposed impact of heterochromatin on longevity of mammals.
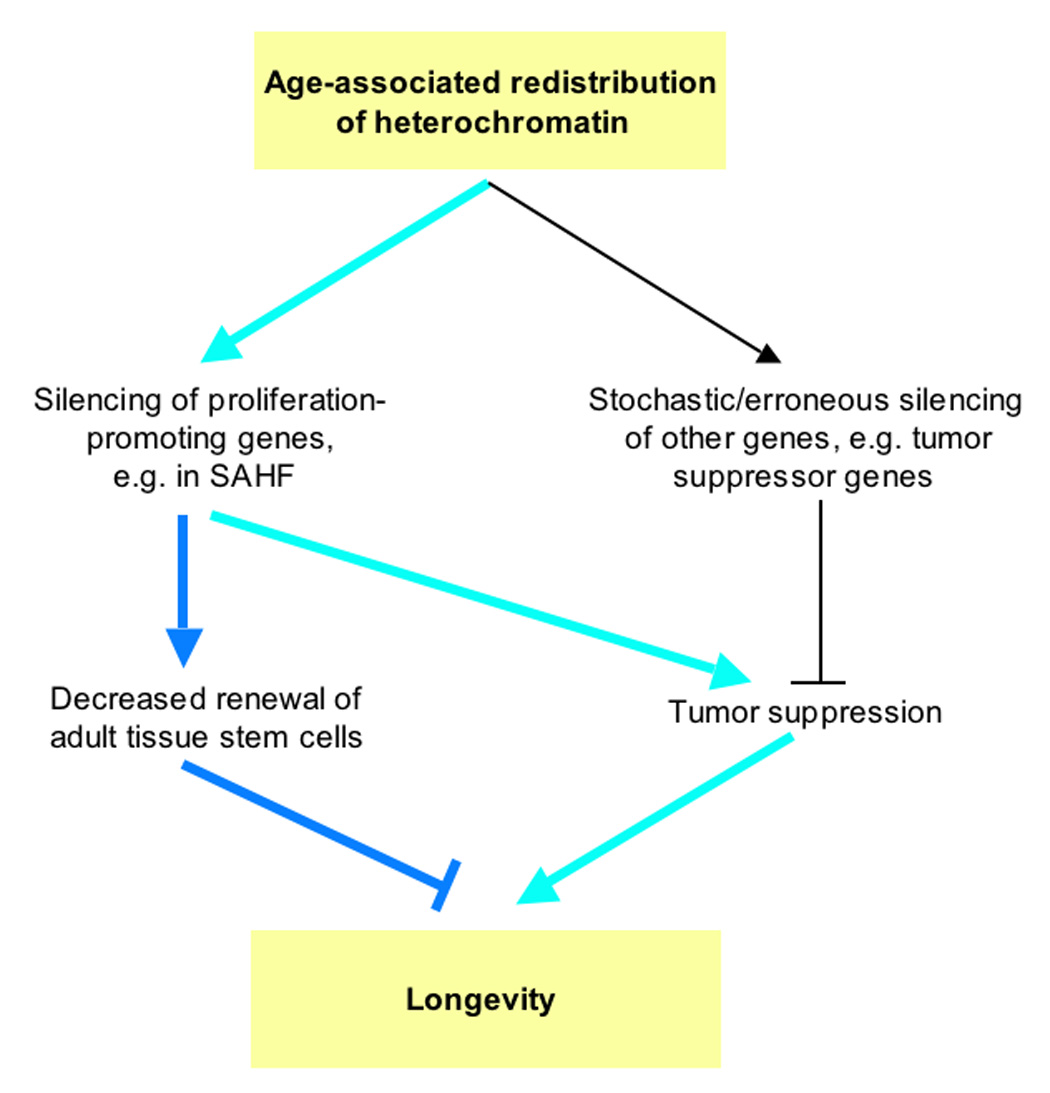
Considerable evidence, discussed in the text, supports the idea that heterochromatin redistributes in cells during cellular and organismal aging. The primary purpose of this redistribution might be to extend longevity by promoting tumor suppression (green arrows). According to the idea of antagonistic pleiotropy, this will also lead to decreased renewal of adult tissue stem cells and renewable tissues, and consequently tissue aging (blue arrows). Stochastic errors in the redistribution of heterochromatin (e.g. excessive spreading of heterochromatin along chromosomes) might lead to erroneous silencing of other genes, e.g. tumor suppressor genes. In this case, redistributed heterochromatin will tend to decrease longevity (thin black lines). Actual organismal longevity will ultimately be determined by the balance between these competing processes.
Acknowledgements
PDA is supported by the NIH (RO1 GM062281, P01 AG031862 and R01 CA129334-01) and is a scholar of the Leukemia and Lymphoma Society. JMS is supported by the NIH (R01 GM041690 and R01 AG016694), and a Senior Scholar Award from the Ellison Medical Foundation. PDA thanks members of the Adams lab as well as Shelley Berger, Ronen Marmorstein and Brad Johnson for discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Kirkwood TB. Understanding the odd science of aging. Cell. 2005;120:437–447. doi: 10.1016/j.cell.2005.01.027. [DOI] [PubMed] [Google Scholar]
- 2.Goldberg AD, Allis CD, Bernstein E. Epigenetics: a landscape takes shape. Cell. 2007;128:635–638. doi: 10.1016/j.cell.2007.02.006. [DOI] [PubMed] [Google Scholar]
- 3.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–260. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
- 4.Grewal SI, Jia S. Heterochromatin revisited. Nat Rev Genet. 2007;8:35–46. doi: 10.1038/nrg2008. [DOI] [PubMed] [Google Scholar]
- 5.Heard E. Delving into the diversity of facultative heterochromatin: the epigenetics of the inactive X chromosome. Curr Opin Genet Dev. 2005;15:482–489. doi: 10.1016/j.gde.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 6.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128:693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 7.Dillon N, Festenstein R. Unravelling heterochromatin: competition between positive and negative factors regulates accessibility. Trends Genet. 2002;18:252–258. doi: 10.1016/s0168-9525(02)02648-3. [DOI] [PubMed] [Google Scholar]
- 8.Muller HJ. Types of visible variations induced by X-rays in Drosophila. J. Genet. 1930;22:299–334. [Google Scholar]
- 9.Groth A, Rocha W, Verreault A, Almouzni G. Chromatin challenges during DNA replication and repair. Cell. 2007;128:721–733. doi: 10.1016/j.cell.2007.01.030. [DOI] [PubMed] [Google Scholar]
- 10.Berger SL. The complex language of chromatin regulation during transcription. Nature. 2007;447:407–412. doi: 10.1038/nature05915. [DOI] [PubMed] [Google Scholar]
- 11.Phair RD, Misteli T. High mobility of proteins in the mammalian cell nucleus. Nature. 2000;404:604–609. doi: 10.1038/35007077. [DOI] [PubMed] [Google Scholar]
- 12.Cheutin T, McNairn AJ, Jenuwein T, Gilbert DM, Singh PB, Misteli T. Maintenance of stable heterochromatin domains by dynamic HP1 binding. Science. 2003;299:721–725. doi: 10.1126/science.1078572. [DOI] [PubMed] [Google Scholar]
- 13.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Mendenhall E, O'Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature. 2007;448:553–560. doi: 10.1038/nature06008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE, Kuehn MS, Taylor CM, Neph S, Koch CM, Asthana S, Malhotra A, Adzhubei I, Greenbaum JA, Andrews RM, Flicek P, Boyle PJ, Cao H, Carter NP, Clelland GK, Davis S, Day N, Dhami P, Dillon SC, Dorschner MO, Fiegler H, Giresi PG, Goldy J, Hawrylycz M, Haydock A, Humbert R, James KD, Johnson BE, Johnson EM, Frum TT, Rosenzweig ER, Karnani N, Lee K, Lefebvre GC, Navas PA, Neri F, Parker SC, Sabo PJ, Sandstrom R, Shafer A, Vetrie D, Weaver M, Wilcox S, Yu M, Collins FS, Dekker J, Lieb JD, Tullius TD, Crawford GE, Sunyaev S, Noble WS, Dunham I, Denoeud F, Reymond A, Kapranov P, Rozowsky J, Zheng D, Castelo R, Frankish A, Harrow J, Ghosh S, Sandelin A, Hofacker IL, Baertsch R, Keefe D, Dike S, Cheng J, Hirsch HA, Sekinger EA, Lagarde J, Abril JF, Shahab A, Flamm C, Fried C, Hackermuller J, Hertel J, Lindemeyer M, Missal K, Tanzer A, Washietl S, Korbel J, Emanuelsson O, Pedersen JS, Holroyd N, Taylor R, Swarbreck D, Matthews N, Dickson MC, Thomas DJ, Weirauch MT, Gilbert J, Drenkow J, Bell I, Zhao X, Srinivasan KG, Sung WK, Ooi HS, Chiu KP, Foissac S, Alioto T, Brent M, Pachter L, Tress ML, Valencia A, Choo SW, Choo CY, Ucla C, Manzano C, Wyss C, Cheung E, Clark TG, Brown JB, Ganesh M, Patel S, Tammana H, Chrast J, Henrichsen CN, Kai C, Kawai J, Nagalakshmi U, Wu J, Lian Z, Lian J, Newburger P, Zhang X, Bickel P, Mattick JS, Carninci P, Hayashizaki Y, Weissman S, Hubbard T, Myers RM, Rogers J, Stadler PF, Lowe TM, Wei CL, Ruan Y, Struhl K, Gerstein M, Antonarakis SE, Fu Y, Green ED, Karaoz U, Siepel A, Taylor J, Liefer LA, Wetterstrand KA, Good PJ, Feingold EA, Guyer MS, Cooper GM, Asimenos G, Dewey CN, Hou M, Nikolaev S, Montoya-Burgos JI, Loytynoja A, Whelan S, Pardi F, Massingham T, Huang H, Zhang NR, Holmes I, Mullikin JC, Ureta-Vidal A, Paten B, Seringhaus M, Church D, Rosenbloom K, Kent WJ, Stone EA, Batzoglou S, Goldman N, Hardison RC, Haussler D, Miller W, Sidow A, Trinklein ND, Zhang ZD, Barrera L, Stuart R, King DC, Ameur A, Enroth S, Bieda MC, Kim J, Bhinge AA, Jiang N, Liu J, Yao F, Vega VB, Lee CW, Ng P, Shahab A, Yang A, Moqtaderi Z, Zhu Z, Xu X, Squazzo S, Oberley MJ, Inman D, Singer MA, Richmond TA, Munn KJ, Rada-Iglesias A, Wallerman O, Komorowski J, Fowler JC, Couttet P, Bruce AW, Dovey OM, Ellis PD, Langford CF, Nix DA, Euskirchen G, Hartman S, Urban AE, Kraus P, Van Calcar S, Heintzman N, Kim TH, Wang K, Qu C, Hon G, Luna R, Glass CK, Rosenfeld MG, Aldred SF, Cooper SJ, Halees A, Lin JM, Shulha HP, Zhang X, Xu M, Haidar JN, Yu Y, Ruan Y, Iyer VR, Green RD, Wadelius C, Farnham PJ, Ren B, Harte RA, Hinrichs AS, Trumbower H, Clawson H, Hillman-Jackson J, Zweig AS, Smith K, Thakkapallayil A, Barber G, Kuhn RM, Karolchik D, Armengol L, Bird CP, de Bakker PI, Kern AD, Lopez-Bigas N, Martin JD, Stranger BE, Woodroffe A, Davydov E, Dimas A, Eyras E, Hallgrimsdottir IB, Huppert J, Zody MC, Abecasis GR, Estivill X, Bouffard GG, Guan X, Hansen NF, Idol JR, Maduro VV, Maskeri B, McDowell JC, Park M, Thomas PJ, Young AC, Blakesley RW, Muzny DM, Sodergren E, Wheeler DA, Worley KC, Jiang H, Weinstock GM, Gibbs RA, Graves T, Fulton R, Mardis ER, Wilson RK, Clamp M, Cuff J, Gnerre S, Jaffe DB, Chang JL, Lindblad-Toh K, Lander ES, Koriabine M, Nefedov M, Osoegawa K, Yoshinaga Y, Zhu B, de Jong PJ. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guenther MG, Levine SS, Boyer LA, Jaenisch R, Young RA. A chromatin landmark and transcription initiation at most promoters in human cells. Cell. 2007;130:77–88. doi: 10.1016/j.cell.2007.05.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schoeftner S, Blasco MA. Developmentally regulated transcription of mammalian telomeres by DNA-dependent RNA polymerase II. Nat Cell Biol. 2008;10:228–236. doi: 10.1038/ncb1685. [DOI] [PubMed] [Google Scholar]
- 17.Azzalin CM, Reichenbach P, Khoriauli L, Giulotto E, Lingner J. Telomeric repeat containing RNA and RNA surveillance factors at mammalian chromosome ends. Science. 2007;318:798–801. doi: 10.1126/science.1147182. [DOI] [PubMed] [Google Scholar]
- 18.Richards EJ, Elgin SC. Epigenetic codes for heterochromatin formation and silencing: rounding up the usual suspects. Cell. 2002;108:489–500. doi: 10.1016/s0092-8674(02)00644-x. [DOI] [PubMed] [Google Scholar]
- 19.Kaern M, Elston TC, Blake WJ, Collins JJ. Stochasticity in gene expression: from theories to phenotypes. Nat Rev Genet. 2005;6:451–464. doi: 10.1038/nrg1615. [DOI] [PubMed] [Google Scholar]
- 20.Kennedy BK, Austriaco NR, Jr, Zhang J, Guarente L. Mutation in the silencing gene SIR4 can delay aging in S. cerevisiae. Cell. 1995;80:485–496. doi: 10.1016/0092-8674(95)90499-9. [DOI] [PubMed] [Google Scholar]
- 21.Haigis MC, Guarente LP. Mammalian sirtuins--emerging roles in physiology, aging, and calorie restriction. Genes Dev. 2006;20:2913–2921. doi: 10.1101/gad.1467506. [DOI] [PubMed] [Google Scholar]
- 22.Longo VD, Kennedy BK. Sirtuins in aging and age-related disease. Cell. 2006;126:257–268. doi: 10.1016/j.cell.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 23.Kennedy BK, Gotta M, Sinclair DA, Mills K, McNabb DS, Murthy M, Pak SM, Laroche T, Gasser SM, Guarente L. Redistribution of silencing proteins from telomeres to the nucleolus is associated with extension of life span in S. cerevisiae. Cell. 1997;89:381–391. doi: 10.1016/s0092-8674(00)80219-6. [DOI] [PubMed] [Google Scholar]
- 24.Sommer M, Poliak N, Upadhyay S, Ratovitski E, Nelkin BD, Donehower LA, Sidransky D. DeltaNp63alpha overexpression induces downregulation of Sirt1 and an accelerated aging phenotype in the mouse. Cell Cycle. 2006;5:2005–2011. doi: 10.4161/cc.5.17.3194. [DOI] [PubMed] [Google Scholar]
- 25.Sasaki T, Maier B, Bartke A, Scrable H. Progressive loss of SIRT1 with cell cycle withdrawal. Aging Cell. 2006;5:413–422. doi: 10.1111/j.1474-9726.2006.00235.x. [DOI] [PubMed] [Google Scholar]
- 26.Wilson VL, Smith RA, Ma S, Cutler RG. Genomic 5-methyldeoxycytidine decreases with age. J Biol Chem. 1987;262:9948–9951. [PubMed] [Google Scholar]
- 27.Singhal RP, Mays-Hoopes LL, Eichhorn GL. DNA methylation in aging of mice. Mech Ageing Dev. 1987;41:199–210. doi: 10.1016/0047-6374(87)90040-6. [DOI] [PubMed] [Google Scholar]
- 28.Romanov GA, Vanyushin BF. Methylation of reiterated sequences in mammalian DNAs. Effects of the tissue type, age, malignancy and hormonal induction. Biochim Biophys Acta. 1981;653:204–218. doi: 10.1016/0005-2787(81)90156-8. [DOI] [PubMed] [Google Scholar]
- 29.Kim JY, Tavare S, Shibata D. Counting human somatic cell replications: methylation mirrors endometrial stem cell divisions. Proc Natl Acad Sci U S A. 2005;102:17739–17744. doi: 10.1073/pnas.0503976102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim JY, Siegmund KD, Tavare S, Shibata D. Age-related human small intestine methylation: evidence for stem cell niches. BMC Med. 2005;3:10. doi: 10.1186/1741-7015-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yatabe Y, Tavare S, Shibata D. Investigating stem cells in human colon by using methylation patterns. Proc Natl Acad Sci U S A. 2001;98:10839–10844. doi: 10.1073/pnas.191225998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ahuja N, Li Q, Mohan AL, Baylin SB, Issa JP. Aging and DNA methylation in colorectal mucosa and cancer. Cancer Res. 1998;58:5489–5494. [PubMed] [Google Scholar]
- 33.Issa JP, Vertino PM, Boehm CD, Newsham IF, Baylin SB. Switch from monoallelic to biallelic human IGF2 promoter methylation during aging and carcinogenesis. Proc Natl Acad Sci U S A. 1996;93:11757–11762. doi: 10.1073/pnas.93.21.11757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Issa JP, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet. 1994;7:536–540. doi: 10.1038/ng0894-536. [DOI] [PubMed] [Google Scholar]
- 35.Issa JP, Ahuja N, Toyota M, Bronner MP, Brentnall TA. Accelerated age-related CpG island methylation in ulcerative colitis. Cancer Res. 2001;61:3573–3577. [PubMed] [Google Scholar]
- 36.So K, Tamura G, Honda T, Homma N, Waki T, Togawa N, Nishizuka S, Motoyama T. Multiple tumor suppressor genes are increasingly methylated with age in non-neoplastic gastric epithelia. Cancer Sci. 2006;97:1155–1158. doi: 10.1111/j.1349-7006.2006.00302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Waki T, Tamura G, Sato M, Motoyama T. Age-related methylation of tumor suppressor and tumor-related genes: an analysis of autopsy samples. Oncogene. 2003;22:4128–4133. doi: 10.1038/sj.onc.1206651. [DOI] [PubMed] [Google Scholar]
- 38.Sarg B, Koutzamani E, Helliger W, Rundquist I, Lindner HH. Postsynthetic trimethylation of histone H4 at lysine 20 in mammalian tissues is associated with aging. J Biol Chem. 2002;277:39195–39201. doi: 10.1074/jbc.M205166200. [DOI] [PubMed] [Google Scholar]
- 39.Polo SE, Almouzni G. Chromatin assembly: a basic recipe with various flavours. Curr Opin Genet Dev. 2006;16:104–111. doi: 10.1016/j.gde.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 40.Jeyapalan JC, Ferreira M, Sedivy JM, Herbig U. Accumulation of senescent cells in mitotic tissue of aging primates. Mech Ageing Dev. 2007;128:36–44. doi: 10.1016/j.mad.2006.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006;311:1257. doi: 10.1126/science.1122446. [DOI] [PubMed] [Google Scholar]
- 42.Henikoff S. Nucleosome destabilization in the epigenetic regulation of gene expression. Nat Rev Genet. 2008;9:15–26. doi: 10.1038/nrg2206. [DOI] [PubMed] [Google Scholar]
- 43.Tagami H, Ray-Gallet D, Almouzni G, Nakatani Y. Histone H3.1 and H3.3 complexes mediate nucleosome assembly pathways dependent or independent of DNA synthesis. Cell. 2004;116:51–61. doi: 10.1016/s0092-8674(03)01064-x. [DOI] [PubMed] [Google Scholar]
- 44.Nakayama T, Nishioka K, Dong YX, Shimojima T, Hirose S. Drosophila GAGA factor directs histone H3.3 replacement that prevents the heterochromatin spreading. Genes Dev. 2007;21:552–561. doi: 10.1101/gad.1503407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nourani A, Robert F, Winston F. Evidence that Spt2/Sin1, an HMG-like factor, plays roles in transcription elongation, chromatin structure, and genome stability in Saccharomyces cerevisiae. Mol Cell Biol. 2006;26:1496–1509. doi: 10.1128/MCB.26.4.1496-1509.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Formosa T, Ruone S, Adams MD, Olsen AE, Eriksson P, Yu Y, Rhoades AR, Kaufman PD, Stillman DJ. Defects in SPT16 or POB3 (yFACT) in Saccharomyces cerevisiae cause dependence on the Hir/Hpc pathway: polymerase passage may degrade chromatin structure. Genetics. 2002;162:1557–1571. doi: 10.1093/genetics/162.4.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Prather D, Krogan NJ, Emili A, Greenblatt JF, Winston F. Identification and characterization of Elf1, a conserved transcription elongation factor in Saccharomyces cerevisiae. Mol Cell Biol. 2005;25:10122–10135. doi: 10.1128/MCB.25.22.10122-10135.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dimova D, Nackerdien Z, Furgeson S, Eguchi S, Osley MA. A role for transcriptional repressors in targeting the yeast Swi/Snf complex. Mol Cell. 1999;4:75–83. doi: 10.1016/s1097-2765(00)80189-6. [DOI] [PubMed] [Google Scholar]
- 49.van der Heijden GW, Derijck AA, Posfai E, Giele M, Pelczar P, Ramos L, Wansink DG, van der Vlag J, Peters AH, de Boer P. Chromosome-wide nucleosome replacement and H3.3 incorporation during mammalian meiotic sex chromosome inactivation. Nat Genet. 2007;39:251–258. doi: 10.1038/ng1949. [DOI] [PubMed] [Google Scholar]
- 50.Kaufman PD, Cohen JL, Osley MA. Hir proteins are required for position-dependent gene silencing in Saccharomyces cerevisiae in the absence of chromatin assembly factor I. Mol Cell Biol. 1998;18:4793–4806. doi: 10.1128/mcb.18.8.4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sharp JA, Franco AA, Osley MA, Kaufman PD, Krawitz DC, Kama T, Fouts ET, Cohen JL. Chromatin assembly factor I and Hir proteins contribute to building functional kinetochores in S. cerevisiae. Genes Dev. 2002;16:85–100. doi: 10.1101/gad.925302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moshkin YM, Armstrong JA, Maeda RK, Tamkun JW, Verrijzer P, Kennison JA, Karch F. Histone chaperone ASF1 cooperates with the Brahma chromatin-remodelling machinery. Genes Dev. 2002;16:2621–2626. doi: 10.1101/gad.231202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Singer MS, Kahana A, Wolf AJ, Meisinger LL, Peterson SE, Goggin C, Mahowald M, Gottschling DE. Identification of high-copy disruptors of telomeric silencing in Saccharomyces cerevisiae. Genetics. 1998;150:613–632. doi: 10.1093/genetics/150.2.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Phelps-Durr TL, Thomas J, Vahab P, Timmermans MC. Maize rough sheath2 and its Arabidopsis orthologue ASYMMETRIC LEAVES1 interact with HIRA, a predicted histone chaperone, to maintain knox gene silencing and determinacy during organogenesis. Plant Cell. 2005;17:2886–2898. doi: 10.1105/tpc.105.035477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sharp JA, Fouts ET, Krawitz DC, Kaufman PD. Yeast histone deposition protein Asf1p requires Hir proteins and PCNA for heterochromatic silencing. Curr Biol. 2001;11:463–473. doi: 10.1016/s0960-9822(01)00140-3. [DOI] [PubMed] [Google Scholar]
- 56.Sherwood PW, Tsang SV, Osley MA. Characterization of HIR1 and HIR2, two genes required for regulation of histone gene transcription in Saccharomyces cerevisiae. Mol Cell Biol. 1993;13:28–38. doi: 10.1128/mcb.13.1.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhang R, Adams PD. Heterochromatin and its relationship to cell senescence and cancer therapy. Cell Cycle. 2007;6:784–789. doi: 10.4161/cc.6.7.4079. [DOI] [PubMed] [Google Scholar]
- 58.Villeponteau B. The heterochromatin loss model of aging. Exp Gerontol. 1997;32:383–394. doi: 10.1016/s0531-5565(96)00155-6. [DOI] [PubMed] [Google Scholar]
- 59.Imai S, Kitano H. Heterochromatin islands and their dynamic reorganization: a hypothesis for three distinctive features of cellular aging. Exp Gerontol. 1998;33:555–570. doi: 10.1016/s0531-5565(98)00037-0. [DOI] [PubMed] [Google Scholar]
- 60.Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007;8:729–740. doi: 10.1038/nrm2233. [DOI] [PubMed] [Google Scholar]
- 61.Serrano M, Blasco MA. Cancer and ageing: convergent and divergent mechanisms. Nat Rev Mol Cell Biol. 2007;8:715–722. doi: 10.1038/nrm2242. [DOI] [PubMed] [Google Scholar]
- 62.Finkel T, Serrano M, Blasco MA. The common biology of cancer and ageing. Nature. 2007;448:767–774. doi: 10.1038/nature05985. [DOI] [PubMed] [Google Scholar]
- 63.Collado M, Blasco MA, Serrano M. Cellular senescence in cancer and aging. Cell. 2007;130:223–233. doi: 10.1016/j.cell.2007.07.003. [DOI] [PubMed] [Google Scholar]
- 64.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, Peacocke M, Campisi J. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, Su L, Sharpless NE. Ink4a/Arf expression is a biomarker of aging. J Clin Invest. 2004;114:1299–1307. doi: 10.1172/JCI22475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Molofsky AV, Slutsky SG, Joseph NM, He S, Pardal R, Krishnamurthy J, Sharpless NE, Morrison SJ. Increasing p16(INK4a) expression decreases forebrain progenitors and neurogenesis during ageing. Nature. 2006;443:448–452. doi: 10.1038/nature05091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Krishnamurthy J, Ramsey MR, Ligon KL, Torrice C, Koh A, Bonner-Weir S, Sharpless NE. p16(INK4a) induces an age-dependent decline in islet regenerative potential. Nature. 2006;443:453–457. doi: 10.1038/nature05092. [DOI] [PubMed] [Google Scholar]
- 68.Janzen V, Forkert R, Fleming HE, Saito Y, Waring MT, Dombkowski DM, Cheng T, Depinho RA, Sharpless NE, Scadden DT. Stem-cell ageing modified by the cyclin-dependent kinase inhibitor p16(INK4a) Nature. 2006;443:421–426. doi: 10.1038/nature05159. [DOI] [PubMed] [Google Scholar]
- 69.Francis MK, Appel S, Meyer C, Balin SJ, Balin AK, Cristofalo VJ. Loss of EPC-1/PEDF expression during skin aging in vivo. J Invest Dermatol. 2004;122:1096–1105. doi: 10.1111/j.0022-202X.2004.22510.x. [DOI] [PubMed] [Google Scholar]
- 70.Wiemann SU, Satyanarayana A, Tsahuridu M, Tillmann HL, Zender L, Klempnauer J, Flemming P, Franco S, Blasco MA, Manns MP, Rudolph KL. Hepatocyte telomere shortening and senescence are general markers of human liver cirrhosis. Faseb J. 2002;16:935–942. doi: 10.1096/fj.01-0977com. [DOI] [PubMed] [Google Scholar]
- 71.Minamino T, Komuro I. Vascular cell senescence: contribution to atherosclerosis. Circ Res. 2007;100:15–26. doi: 10.1161/01.RES.0000256837.40544.4a. [DOI] [PubMed] [Google Scholar]
- 72.Price JS, Waters JG, Darrah C, Pennington C, Edwards DR, Donell ST, Clark IM. The role of chondrocyte senescence in osteoarthritis. Aging Cell. 2002;1:57–65. doi: 10.1046/j.1474-9728.2002.00008.x. [DOI] [PubMed] [Google Scholar]
- 73.Blasco MA, Lee HW, Hande MP, Samper E, Lansdorp PM, DePinho RA, Greider CW. Telomere shortening and tumor formation by mouse cells lacking telomerase RNA. Cell. 1997;91:25–34. doi: 10.1016/s0092-8674(01)80006-4. [DOI] [PubMed] [Google Scholar]
- 74.Lee HW, Blasco MA, Gottlieb GJ, Horner JW, 2nd, Greider CW, DePinho RA. Essential role of mouse telomerase in highly proliferative organs. Nature. 1998;392:569–574. doi: 10.1038/33345. [DOI] [PubMed] [Google Scholar]
- 75.Rudolph KL, Chang S, Lee HW, Blasco M, Gottlieb GJ, Greider C, DePinho RA. Longevity, stress response, and cancer in aging telomerase-deficient mice. Cell. 1999;96:701–712. doi: 10.1016/s0092-8674(00)80580-2. [DOI] [PubMed] [Google Scholar]
- 76.Du X, Shen J, Kugan N, Furth EE, Lombard DB, Cheung C, Pak S, Luo G, Pignolo RJ, DePinho RA, Guarente L, Johnson FB. Telomere shortening exposes functions for the mouse Werner and Bloom syndrome genes. Mol Cell Biol. 2004;24:8437–8446. doi: 10.1128/MCB.24.19.8437-8446.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gonzalez-Suarez E, Samper E, Ramirez A, Flores JM, Martin-Caballero J, Jorcano JL, Blasco MA. Increased epidermal tumors and increased skin wound healing in transgenic mice overexpressing the catalytic subunit of telomerase, mTERT, in basal keratinocytes. Embo J. 2001;20:2619–2630. doi: 10.1093/emboj/20.11.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Herrera E, Samper E, Martin-Caballero J, Flores JM, Lee HW, Blasco MA. Disease states associated with telomerase deficiency appear earlier in mice with short telomeres. Embo J. 1999;18:2950–2960. doi: 10.1093/emboj/18.11.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Garcia-Cao I, Garcia-Cao M, Tomas-Loba A, Martin-Caballero J, Flores JM, Klatt P, Blasco MA, Serrano M. Increased p53 activity does not accelerate telomere-driven ageing. EMBO Rep. 2006;7:546–552. doi: 10.1038/sj.embor.7400667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gonzalez-Suarez E, Geserick C, Flores JM, Blasco MA. Antagonistic effects of telomerase on cancer and aging in K5-mTert transgenic mice. Oncogene. 2005;24:2256–2270. doi: 10.1038/sj.onc.1208413. [DOI] [PubMed] [Google Scholar]
- 81.Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dorken B, Jenuwein T, Schmitt CA. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature. 2005;436:660–665. doi: 10.1038/nature03841. [DOI] [PubMed] [Google Scholar]
- 82.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, Cordon-Cardo C, Pandolfi PP. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–730. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Michaloglou C, Vredeveld LC, Soengas MS, Denoyelle C, Kuilman T, van der Horst CM, Majoor DM, Shay JW, Mooi WJ, Peeper DS. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- 84.Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–665. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 85.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–660. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Feldser DM, Greider CW. Short telomeres limit tumor progression in vivo by inducing senescence. Cancer Cell. 2007;11:461–469. doi: 10.1016/j.ccr.2007.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sun P, Yoshizuka N, New L, Moser BA, Li Y, Liao R, Xie C, Chen J, Deng Q, Yamout M, Dong MQ, Frangou CG, Yates JR, 3rd, Wright PE, Han J. PRAK is essential for ras-induced senescence and tumor suppression. Cell. 2007;128:295–308. doi: 10.1016/j.cell.2006.11.050. [DOI] [PubMed] [Google Scholar]
- 88.Narita M, Nunez S, Heard E, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–716. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- 89.Zhang R, Poustovoitov MV, Ye X, Santos HA, Chen W, Daganzo SM, Erzberger JP, Serebriiskii IG, Canutescu AA, Dunbrack RL, Pehrson JR, Berger JM, Kaufman PD, Adams PD. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell. 2005;8:19–30. doi: 10.1016/j.devcel.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 90.Ye X, Zerlanko B, Zhang R, Somaiah N, Lipinski M, Salomoni P, Adams PD. Definition of pRB- and p53-dependent and -independent steps in HIRA/ASF1a-mediated formation of senescence-associated heterochromatin foci. Mol Cell Biol. 2007;27:2452–2465. doi: 10.1128/MCB.01592-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhang R, Chen W, Adams PD. Molecular Dissection of Formation of Senescent Associated Heterochromatin Foci. Mol. Cell. Biol. 2007;27:2343–2358. doi: 10.1128/MCB.02019-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ye X, Zerlanko B, Kennedy A, Banumathy G, Zhang R, Adams PD. Downregulation of Wnt signaling is a trigger for formation of facultative heterochromatin and onset of cell senescence in primary human cells. Mol Cell. 2007;27:183–196. doi: 10.1016/j.molcel.2007.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Funayama R, Saito M, Tanobe H, Ishikawa F. Loss of linker histone H1 in cellular senescence. J. Cell. Biol. 2006;175:869–880. doi: 10.1083/jcb.200604005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Benetti R, Garcia-Cao M, Blasco MA. Telomere length regulates the epigenetic status of mammalian telomeres and subtelomeres. Nat Genet. 2007;39:243–250. doi: 10.1038/ng1952. [DOI] [PubMed] [Google Scholar]
- 95.Kudlow BA, Kennedy BK, Monnat RJ., Jr Werner and Hutchinson-Gilford progeria syndromes: mechanistic basis of human progeroid diseases. Nat Rev Mol Cell Biol. 2007;8:394–404. doi: 10.1038/nrm2161. [DOI] [PubMed] [Google Scholar]
- 96.Miller RA. 'Accelerated aging': a primrose path to insight? Aging Cell. 2004;3:47–51. doi: 10.1111/j.1474-9728.2004.00081.x. [DOI] [PubMed] [Google Scholar]
- 97.Shumaker DK, Dechat T, Kohlmaier A, Adam SA, Bozovsky MR, Erdos MR, Eriksson M, Goldman AE, Khuon S, Collins FS, Jenuwein T, Goldman RD. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging. Proc Natl Acad Sci U S A. 2006;103:8703–8708. doi: 10.1073/pnas.0602569103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science. 2006;312:1059–1063. doi: 10.1126/science.1127168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med. 2005;11:440–445. doi: 10.1038/nm1204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Obuse C, Iwasaki O, Kiyomitsu T, Goshima G, Toyoda Y, Yanagida M. A conserved Mis12 centromere complex is linked to heterochromatic HP1 and outer kinetochore protein Zwint-1. Nat Cell Biol. 2004;6:1135–1141. doi: 10.1038/ncb1187. [DOI] [PubMed] [Google Scholar]
- 101.Amor DJ, Kalitsis P, Sumer H, Choo KH. Building the centromere: from foundation proteins to 3D organization. Trends Cell Biol. 2004;14:359–368. doi: 10.1016/j.tcb.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 102.Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science. 2007;317:916–924. doi: 10.1126/science.1142210. [DOI] [PubMed] [Google Scholar]
- 103.Weaver BA, Silk AD, Montagna C, Verdier-Pinard P, Cleveland DW. Aneuploidy acts both oncogenically and as a tumor suppressor. Cancer Cell. 2007;11:25–36. doi: 10.1016/j.ccr.2006.12.003. [DOI] [PubMed] [Google Scholar]
- 104.Bahar R, Hartmann CH, Rodriguez KA, Denny AD, Busuttil RA, Dolle ME, Calder RB, Chisholm GB, Pollock BH, Klein CA, Vijg J. Increased cell-to-cell variation in gene expression in ageing mouse heart. Nature. 2006;441:1011–1014. doi: 10.1038/nature04844. [DOI] [PubMed] [Google Scholar]
- 105.Rossi DJ, Bryder D, Weissman IL. Hematopoietic stem cell aging: mechanism and consequence. Exp Gerontol. 2007;42:385–390. doi: 10.1016/j.exger.2006.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rossi DJ, Bryder D, Zahn JM, Ahlenius H, Sonu R, Wagers AJ, Weissman IL. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc Natl Acad Sci U S A. 2005;102:9194–9199. doi: 10.1073/pnas.0503280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suner D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, Carlsson E, Poulsen P, Vaag A, Stephan Z, Spector TD, Wu YZ, Plass C, Esteller M. Epigenetic differences arise during the lifetime of monozygotic twins. Proc Natl Acad Sci U S A. 2005;102:10604–10609. doi: 10.1073/pnas.0500398102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Eckhardt F, Lewin J, Cortese R, Rakyan VK, Attwood J, Burger M, Burton J, Cox TV, Davies R, Down TA, Haefliger C, Horton R, Howe K, Jackson DK, Kunde J, Koenig C, Liddle J, Niblett D, Otto T, Pettett R, Seemann S, Thompson C, West T, Rogers J, Olek A, Berlin K, Beck S. DNA methylation profiling of human chromosomes 6, 20 and 22. Nat Genet. 2006;38:1378–1385. doi: 10.1038/ng1909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Laird PW. Cancer epigenetics. Hum Mol Genet. 2005;14 Spec No 1:R65–R76. doi: 10.1093/hmg/ddi113. [DOI] [PubMed] [Google Scholar]
- 110.Campisi J. Cancer and ageing: rival demons? Nat Rev Cancer. 2003;3:339–349. doi: 10.1038/nrc1073. [DOI] [PubMed] [Google Scholar]
- 111.Williams GC. Pleiotropy, natural selection and the evolution of senescence. Evolution. 1957;11:398–411. [Google Scholar]
- 112.Cairns P. Gene methylation and early detection of genitourinary cancer: the road ahead. Nat Rev Cancer. 2007;7:531–543. doi: 10.1038/nrc2170. [DOI] [PubMed] [Google Scholar]
- 113.Kopelovich L, Crowell JA, Fay JR. The epigenome as a target for cancer chemoprevention. J Natl Cancer Inst. 2003;95:1747–1757. doi: 10.1093/jnci/dig109. [DOI] [PubMed] [Google Scholar]
- 114.Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, Jin L, Boss O, Perni RB, Vu CB, Bemis JE, Xie R, Disch JS, Ng PY, Nunes JJ, Lynch AV, Yang H, Galonek H, Israelian K, Choy W, Iffland A, Lavu S, Medvedik O, Sinclair DA, Olefsky JM, Jirousek MR, Elliott PJ, Westphal CH. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712–716. doi: 10.1038/nature06261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A, Prabhu VV, Allard JS, Lopez-Lluch G, Lewis K, Pistell PJ, Poosala S, Becker KG, Boss O, Gwinn D, Wang M, Ramaswamy S, Fishbein KW, Spencer RG, Lakatta EG, Le Couteur D, Shaw RJ, Navas P, Puigserver P, Ingram DK, de Cabo R, Sinclair DA. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337–342. doi: 10.1038/nature05354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Bordone L, Guarente L. Calorie restriction, SIRT1 and metabolism: understanding longevity. Nat Rev Mol Cell Biol. 2005;6:298–305. doi: 10.1038/nrm1616. [DOI] [PubMed] [Google Scholar]
- 117.Borra MT, Smith BC, Denu JM. Mechanism of human SIRT1 activation by resveratrol. J Biol Chem. 2005;280:17187–17195. doi: 10.1074/jbc.M501250200. [DOI] [PubMed] [Google Scholar]
- 118.Kaeberlein M, McDonagh T, Heltweg B, Hixon J, Westman EA, Caldwell SD, Napper A, Curtis R, DiStefano PS, Fields S, Bedalov A, Kennedy BK. Substrate-specific activation of sirtuins by resveratrol. J Biol Chem. 2005;280:17038–17045. doi: 10.1074/jbc.M500655200. [DOI] [PubMed] [Google Scholar]