

Summary
To understand the functions of SPG6, mutated in the neurodegenerative disease hereditary spastic paraplegia, and of ichthyin, mutated in autosomal recessive congenital ichthyosis, we have studied their Drosophila ortholog, spichthyin (Spict). Spict is found on early endosomes. Loss of Spict leads to upregulation of BMP signaling and expansion of the neuromuscular junction. BMP signaling is also necessary for a normal microtubule cytoskeleton and axonal transport; analysis of loss and gain-of-function phenotypes suggests that Spict antagonizes this function of BMP signaling. Spict interacts with BMP receptors and promotes their internalization from the plasma membrane, suggesting that it inhibits BMP signaling by regulating BMP receptor traffic. This is the first demonstration of a role for an SPG protein or ichthyin family member in a specific signaling pathway, and suggests disease mechanisms for hereditary spastic paraplegia that involve dependence of the microtubule cytoskeleton on BMP signaling.
Keywords: Neuromuscular junction, hereditary spastic paraplegia, SPG6, endosome, TGF-beta, Ichthyin
Introduction
Axonal abnormalities, including impairment of transport, are a hallmark of many neurological and neurodegenerative diseases1. These include the hereditary spastic paraplegias (HSPs), a heterogeneous set of diseases characterized by degeneration of corticospinal tract axons and spasticity of the lower extremities2,3. Different forms of the disease are termed either pure or complicated, depending on whether other mainly neurological symptoms are present. The mechanisms of degeneration in HSPs are unknown, but over twenty causative loci (SPG loci) have been mapped and thirteen cloned. Some SPG products are implicated in microtubule function or transport, including the microtubule motor protein kinesin, and the microtubule-severing protein spastin2,3. Since microtubules are the route for fast axonal transport, the most distal portions of axons are likely to be most sensitive to impairments of microtubule function. A second class of SPG products are mitochondrial proteins2,3, but it is not known how mutations in these cause axonal degeneration. A third class of SPG products are apparently associated with endosomes, judged by immunolocalization or the presence of domains such as MIT or FYVE4-8. HSP is also caused by some mutations in the amyotrophic lateral sclerosis gene ALS2, which encodes alsin, a guanine-nucleotide-exchange-factor for the early endosomal GTPase Rab59. However, the mechanism by which impairment of endosomal membrane traffic might cause axonal degeneration is unknown.
One membrane protein encoded by an SPG gene is SPG6, mutations in which (Fig. 1a) cause a dominant pure form of HSP, and which is widely expressed, although enriched in brain tissue10-13. SPG6 is a member of a protein family (Pfam: DUF803) predicted to have between seven and nine transmembrane (TM) domains14,15. Three different amino-acid substitutions are known, one of which is found in ethnically disparate families and another caused by different nucleotide substitutions in the same codon10-13, suggesting a dominant gain-of-function disease mechanism that can be mediated by only a few mutations in the protein. This protein family includes another human disease protein, ichthyin, mutations in which cause autosomal recessive congenital ichthyosis (ARCI), a skin disorder whose cellular basis is not understood16. Ichthyin is widely expressed, although with high expression in keratinocytes, and little or no expression in brain, and at least six recessive alleles are known that cause substitutions of mainly conserved amino acids in different parts of the protein. In summary, little is known of the cellular roles of the SPG6 and ichthyin family.
Figure 1. SPG6 homologs and Drosophila spict gene.

(a) Multiple alignment of Drosophila and human SPG6 and ichthyin homologs, generated using ClustalW50. White letters on a black background represent residues that are identical in the sequences aligned. White letters on a grey background are the residuals that are similar. The sites of the mutations T45R, A100T and G106R, identified in human SPG6 patients, are marked by arrows. Transmembrane (TM) domains predicted from the Drosophila protein sequence are highlighted by lines below the alignment. The NCBI protein accession numbers are: Fly, NP_609586; hSPG6, DAA01477; hNIPA2, DAA01509; hNPAL1, NP_997213; hICHTHYIN, XP_371777. (b) Map of the spict gene, showing the insertion site of P element EP(2)2202, 45-52 bp upstream of the spict coding region. Parentheses indicate the boundaries of the deletion in the imprecise excision strain, spictmut. The deletion extends 1753 bp to the right of the insertion site of EP(2)2202, deleting 44 bp of the 69-bp 5' UTR, the entire 1155 bp of the spict coding sequence (interrupted by an intron of 498 bp), and 56 bp of the 236-bp 3' UTR.
To understand the normal role of the SPG6 and ichthyin protein family, and how changes in their function might lead to cellular defects, we have studied their Drosophila homolog, spichthyin (Spict). Spict shows preferential localization on early endosomes. It regulates growth of the neuromuscular junction (NMJ) presynaptically, by inhibition of BMP (Bone Morphogenic Protein)/TGF-β (Transforming Growth Factor-β) signaling. BMP signaling regulates synaptic growth, function and stabilization at the NMJ17-21. We show a novel role for BMP signaling in maintenance of microtubules and axonal transport, and that this function is also inhibited by Spict. Our data suggest that Spict inhibits BMP signaling by regulating BMP receptor traffic. Our findings provide a cellular role for the Spict family of proteins, and suggest potential mechanisms for the pathology of HSPs and ARCI that include dependence of microtubules on BMP signaling.
Results
Identification of Drosophila spichthyin
A BLASTP search using human SPG6 identified one Drosophila homolog, CG12292. A search using CG12292 identified four predicted human proteins that were 40-50% identical to it: SPG6 (NIPA1), NIPA2, ichthyin and NPAL1 (Fig. 1a). Two more distantly related human proteins, NPAL2 and NPAL3 (data not shown) are more closely related to plant and fungal homologs than to CG12292, and probably represent a subfamily lost from the Drosophila lineage. Since Drosophila CG12292 appears orthologous to both SPG6 and ichthyin, we designated it spichthyin (spict).
To generate spict mutant flies, we used transposase-mediated imprecise excision of a P element, EP(2)2202, inserted in the spict 5′ untranslated region (Fig. 1b). One imprecise excision, spictmut, had lost the entire coding region, and was therefore a null allele of spict. We also recovered several precise excision events; one of these was used as a wild-type control in most subsequent experiments, and is referred to as spict+. Homozygous spictmut flies were viable and fertile, and took about a day longer than spict+ flies to reach adulthood.
Drosophila Spict is widely expressed and is localized on early endosomes
To determine where Spict might act, we examined its expression pattern and subcellular localization. spict mRNA was found ubiquitously during embryogenesis, with elevated expression in some tissues, including CNS and muscles (Fig. 2a). EGFP-Spict and Spict-EGFP fusions both showed punctate distributions in Drosophila S2 cells, that overlapped substantially with the early endosome compartment detected using anti-Rab5 (Fig. 2b), but showed no striking overlap with the late endosomal/multivesicular body marker Hook, the recycling endosomal marker Rab11, or the late endosomal/lysosomal markers Spinster and LysoTracker (Supplementary Fig. 1). A Spict-mRFP fusion protein also showed a punctate cytoplasmic distribution in wild-type (data not shown) and spictmut third instar larvae, which also overlapped substantially with Rab5, but not with late endosomal/lysosomal markers, in muscles and NMJs (Fig. 2c; Supplementary Fig. 1). Trypsin digestion of N-terminally and C-terminally tagged Spict, redistibuted to the plasma membrane by blockage of endocytosis, suggested that the N-terminus of Spict is in the endosome lumen, and its C-terminus in the cytosol (Supplementary Fig. 2). This result is consistent with previous suggestions that Spict family members might either have nine transmembrane domains14, or be divergent members of the 7-TM superfamily15. Attempts to raise an antibody that recognized endogenous Spict in immunomicroscopy were unsuccessful. However, since Spict-EGFP and EGFP-Spict fusions had apparently identical localizations in S2 cells, the Spict-mRFP fusion could rescue a spictmut phenotype and cause the same overexpression phenotypes as wild type Spict (see below and data not shown), these fusions are likely to have the same localization as endogenous Spict.
Figure 2. Drosophila spict expression and localization.

(a) Whole mount spict in situ hybridizations. (i-iii) Lateral views of spict+ embryos at stages 11, 13 and 14, respectively. E, epidermis, M, muscle precursors, FG, foregut, HG, hindgut. (iv) Ventral view of a stage 13 embryo showing spict mRNA in the CNS (arrows: segmental neuromeres). (v) No signal is detected in a spictmut embryo. Anterior is on the left. Scale bars, (i-iii, v), 100 μm; (iv), 50 μm. (b) EGFP-Spict or Spict-EGFP fusions (rows i and ii respectively) were expressed transiently in S2 cells. In row iii, anti-Rab5 was omitted. Row iv shows an untransfected cell. (c) Localization of Spict-mRFP and Rab5 at the NMJ (top row) and muscle (bottom row) of spictmut third instar larvae, in which Spict-mRFP is driven by elav-GAL4 and MHC-GAL4, respectively. Examples of colocalization are shown with arrows. (d) Rab5 staining at muscle 6/7 NMJs of third instar larvae. spictrescue is spictmut UAS-spict Actin5C-GAL4. Presynaptic boutons are stained by anti-CSP. (e) Quantification of Rab5 levels in type Ib boutons (marked by CSP) normalized to a control channel (anti-CSP) at muscle 6/7. *, P < 0.05; **, P < 0.01, ***, P < 0.001, error bars are S.E.M., for all figures. n=20 boutons from 5 larvae. The mean intensity of CSP is not significantly different among the three genotypes (P = 0.96, n=20 boutons from 5 larvae, one-way ANOVA). Panels in b-d are single confocal sections. Scale bars, 5 μm.
Since tagged Spict proteins localized with Rab5, we tested whether Rab5 staining is normal when Spict is lacking. Rab5 staining was less intense in spictmut NMJ boutons compared to wild-type; these phenotypes were rescued by ubiquitous expression of UAS-spict (Fig. 2d-e). Rab5 staining was also reduced in muscles but not obviously affected in neuronal cell bodies and axons of spictmut larvae, or in S2 cells treated by spict RNAi (data not shown). Therefore, Spict is essential for a normal Rab5 compartment at the NMJ, but not in all situations.
Spict regulates synaptic growth at the NMJ
To test whether loss of Spict at the NMJ affected neuronal signaling, we compared the morphology of the NMJ in spictmut and spict+ larvae. Significant NMJ overgrowth was found in spictmut third instar larvae. At muscles 6/7 from segment A3, bouton numbers of spictmut larvae were approximately double those of spict+ controls (Fig. 3a, b). spictmut larvae also had NMJs that were significantly longer and had more branches than spict+ (Fig. 3a; Supplementary Fig. 3). NMJ overgrowth was found throughout third instar larvae (Supplementary Fig. 3 and data not shown), and was not obviously more severe in more posterior segments (compare Supplementary Fig. 3d, e with Fig. 3a, b). Average bouton sizes were not significantly affected in spictmut larvae: at the NMJ of muscles 6/7 of A3 in spictmut and spict+ larvae, type Ib boutons had cross-sectional areas of 7.5 ± 0.6 μm2 and 6.1 ± 0.2 μm2 respectively (P = 0.6, n=33 NMJs) and type Is boutons had cross-sectional areas of 2.1 ± 0.1 μm2 and 1.9 ± 0.1 μm2 respectively (P = 0.11, n=40 NMJs). NMJ overgrowth phenotypes were fully rescued by expression of either Spict or Spict-mRFP in neurons, but not in muscles (Fig. 3a, b; Supplementary Fig. 3). Therefore, Spict acts in wild-type neurons to inhibit synaptic growth at the NMJ.
Figure 3. spict null mutants cause BMP-dependent NMJ overgrowth.
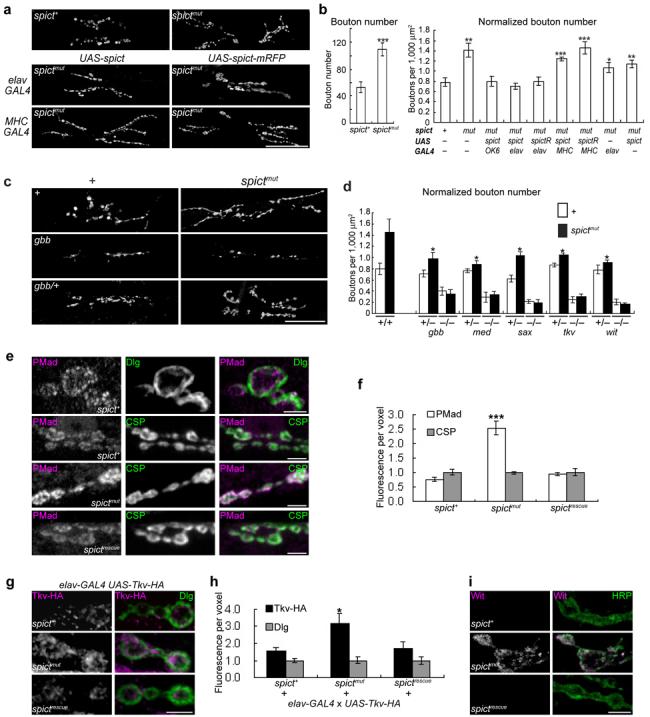
(a) NMJ phenotypes (visualized with anti-synaptotagmin) at muscles 6/7 of hemisegment A3 (used for all panels in this and subsequent figures, except where otherwise indicated). (b) Bouton numbers, both raw (left histogram) and normalized to the areas of both muscles (right histogram), in genotypes shown in a and additional genotypes. spictR represents spict-mRFP. Comparisons with spict+. n=8-20 larvae. (c) NMJs of BMP and spict mutants. (d) Normalized bouton numbers in genotypes shown in c and additional genotypes. *, P < 0.05 for all spictmut and heterozygous BMP-pathway double mutants, compared to spictmut. (e) PMad levels at the NMJ. (f) Quantification of fluorescence intensities shown in e. n=8. (g) Tkv-HA levels in NMJ boutons. Most Tkv-HA staining is close to the mainly postsynaptic marker Dlg. (h) Quantification of fluorescence intensities shown in g. n=8. (i) Wit levels at the NMJ. Most Wit puncta are close to the plasma membrane, as visualized with the neuronal surface marker anti-HRP. The Wit antibody used is not sensitive enough to detect endogenous Wit at the NMJ under standard staining conditions17. a and c are projections of confocal sections; e, g and i are single confocal sections. In e-i, spictrescue is spictmut UAS-spict elav-GAL4. The mean intensity of CSP (f) or Dlg (h) labeling is not significantly different between the three genotypes (P > 0.3, n=8, one-way ANOVA) and is assigned as 1 for each genotype. Scale bars, (a, c), 50 μm; (e, g, i), 5 μm.
BMP signaling is essential for the NMJ expansion of spictmut
One of the signaling pathways with the largest effects on synaptic size at the Drosophila NMJ is the BMP pathway17-20, which stimulates synaptic growth. The expanded NMJ phenotype of spictmut is similar to that of spinster (also known as benchwarmer), which also shows defects in endosomal-lysosomal trafficking and requires an active BMP/TGF-β signaling pathway for NMJ expansion22. It is also similar to the increase in bouton number of highwire NMJs20,23. Highwire encodes a putative E3 ubiquitin ligase that appears to affect multiple signaling pathways including JNK and BMP20,24. To determine whether the synaptic overgrowth of spictmut larvae requires BMP signaling, we genetically removed key BMP signaling components from spictmut larvae. Mutations affecting the type I receptor subunits Tkv (Thickvein) and Sax (Saxophone), the type II receptor subunit Wit (Wishful Thinking), the type II receptor ligand Gbb (Glass Bottom Boat), or the co-Smad Med (Medea) all suppressed the NMJ overgrowth of spictmut larvae (Fig. 3c, d; Supplementary Fig. 4). In all cases, the synaptic undergrowth in larvae that were doubly homozygous for spictmut and BMP pathway mutations was indistinguishable from that of homozygous BMP pathway mutations alone. In addition, all heterozygous BMP pathway mutations tested partly suppressed the NMJ expansion of spictmut larvae, but had no effect on NMJ bouton number in a wild type background. Therefore, BMP signaling is essential for the excessive NMJ growth of spictmut larvae.
The contrasting phenotypes of spictmut and loss of BMP signaling, and the genetic interactions between spict and BMP signaling mutants, suggest that Spict antagonizes BMP signaling in the control of NMJ growth. Nevertheless, alternative models are possible: for example, highwire mutations interact with BMP signaling mutations, but Highwire affects synaptic size primarily through a MAPK signaling pathway24. However, our evidence strongly supports a direct effect of Spict on BMP signaling. During BMP signaling in neurons, the R-Smad protein Mad is phosphorylated by active BMP receptors, and phosphorylated Mad (PMad) is then translocated to the nucleus and acts as a transcription factor17-21. At the NMJ, PMad overlaps mainly with the presynaptic marker cysteine string protein (CSP), but also with the largely postsynaptic marker Discs-large (Dlg) (Fig. 3e). PMad is also found in cell body nuclei in the larval CNS (Supplementary Fig. 4; refs 21, 24). PMad levels were significantly higher in spictmut than in spict+ larvae, both at the NMJ and in CNS cell bodies, and this phenotype was fully rescued by neuronal expression of UAS-spict (Fig. 3e, f; Supplementary Fig. 4). Therefore, BMP signaling is upregulated at spictmut neurons, in contrast to highwire neurons24. We next tested for upregulation of BMP receptors at spictmut NMJs. HA-tagged Tkv was found mainly in a punctate distribution in the periphery of synaptic boutons, at or close to the plasma membrane, and at higher levels in spictmut than in spict+ boutons (Fig. 3g, h). Wit was barely detectable in spict+ boutons, but was present at higher levels in spictmut boutons, also in a punctate pattern mainly at or close to the plasma membrane (Fig. 3i). The effect of spictmut on Tkv-HA and Wit levels was rescued by neuronal expression of UAS-spict (Fig. 3g-i). We found no effect of spictmut on levels of other neuronal membrane proteins (Fasciclin II, Syntaxin), or on the neuronal surface antigen recognized by anti-Horseradish Peroxidase (HRP) at the NMJ (Supplementary Fig. 4). Therefore, Spict action specifically lowers the levels of BMP receptors at the presynaptic NMJ.
Spict and BMP signaling regulate microtubules
Some HSPs may be caused by axonal transport defects, and presynaptic microtubules are affected by loss or overexpression of another HSP homolog, spastin, in Drosophila25,26. We therefore investigated whether Spict and BMP signaling had a role in maintenance of axonal microtubules. Microtubules were visualized using antibodies against α-tubulin (recognizes free tubulin and microtubules), acetylated-α-tubulin (recognizes stable microtubules but not free tubulin27), tyrosinated-α-tubulin (YL1/2; recognizes free tubulin and newly polymerized microtubules28), and the microtubule-associated protein Futsch29, homologous to mammalian MAP1B, which binds to tyrosinated microtubules30. Both α-tubulin and acetylated-α-tubulin staining were significantly reduced in tkv mutant axons and presynaptic boutons, suggesting a loss of microtubules in mutant neurons. These reductions were rescued by motor neuron expression of Tkv-HA (Fig. 4; Supplementary Fig. 4). By contrast, YL1/2 and Futsch staining were not affected in tkv mutant axons (P = 0.39 and 0.24 respectively compared to w1118, n=6 larvae; data not shown); however, Futsch was lost from more distal boutons in tkv mutant NMJs, leaving significantly more boutons that lacked Futsch than in wild-type NMJs (Fig. 4c,ii, Supplementary Fig. 4). Therefore, BMP signaling is required for a normal axonal and NMJ microtubule cytoskeleton. Consistent with the model of Spict as an inhibitor of BMP signaling, α-tubulin and acetylated-α-tubulin staining were also significantly decreased in spict-overexpressing neurons, but increased in spictmut neurons (Fig 4; Supplementary Fig. 4). The loss of both total tubulin and microtubules in axons and the NMJ caused by Spict overexpression was suppressed by constitutively active Tkv, expression of which alone had no significant effect (Fig. 4; Supplementary Fig. 4), suggesting that Spict regulates maintenance of microtubules via BMP signaling.
Figure 4. Spict regulates microtubules by inhibiting BMP signaling.
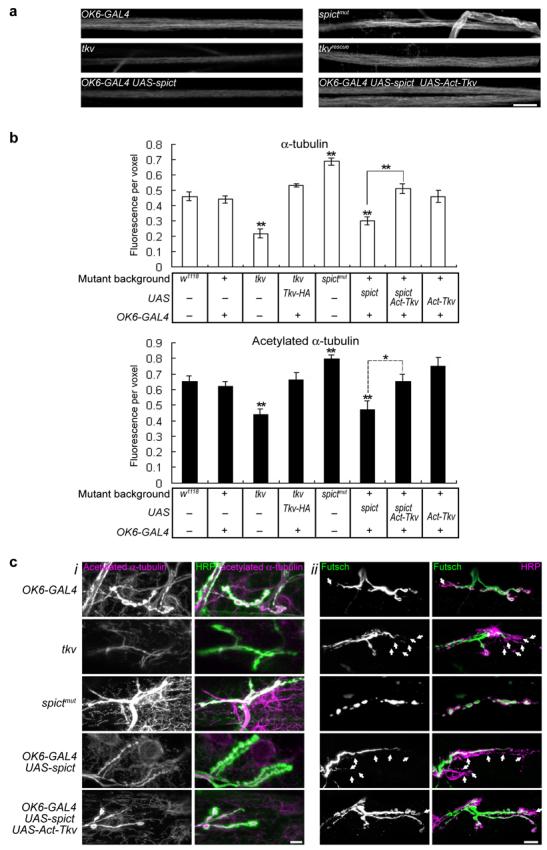
(a) Acetylated α-tubulin staining in segmental nerves passing through segment A4. (b) Quantification of levels of α-tubulin or acetylated-α-tubulin in 200-μm lengths of segmental nerve. Fluorescence intensity per voxel is normalized to a control channel (anti-HRP). Levels of anti-HRP are not significantly different among the genotypes in each panel (P > 0.05, n=6 larvae, one-way ANOVA). All comparisons are with w1118 unless indicated.. (c,i) Acetylated α-tubulin at NMJs of third instar larvae. Note that spictmut and tkv mutations affect both neuronal microtubules (overlapping with anti-HRP, green) and the muscle microtubule network visualized using anti-acetylated-α-tubulin (outside the anti-HRP-labeled area). Transgenes driven by the motor neuron-expressing OK6-GAL4 affect only presynaptic microtubules. (c,ii) Futsch is lost from distal boutons in tkv and in UAS-spict OK6-GAL4 NMJs (arrows show boutons labeled only with anti-HRP but not with anti-Futsch). Quantification is in Supplementary Fig. 4. In all panels, tkv mutant genotype is tkv7/tkv16713, tkvrescue is tkv7,OK6-GAL4/tkv16713; UAS-Tkv-HA. All images are projections of confocal sections. Scale bars, 10 μm.
To test whether BMP signaling is also required for fast axonal transport, which occurs along microtubules, and whether Spict affects this transport via BMP signaling, we tested whether the synaptic vesicle protein synaptotagmin (Syt) accumulated in axons due to a failure to transport it properly. tkv mutant axons accumulated significantly more Syt than control genotypes; this could be rescued by motor neuron expression of Tkv-HA (Fig. 5a, b; Supplementary Fig. 4). Axonal Syt accumulation was also found in other BMP mutants including sax, gbb and wit (ref. 17 and data not shown). In live tkv axons, anterograde and retrograde fast axonal transportation of Syt-eGFP were impaired compared to controls, and many large nonmotile Syt-eGFP accumulations were found (Fig. 5c; Supplementary Videos 1-2). Therefore, presynaptic BMP signaling is essential for normal axonal transport.
Figure 5. Spict-overexpression and tkv mutants impair fast axonal transport.

(a) Accumulation of synaptotagmin (arrows) in segmental nerves passing through segment A4. tkv genotypes are as in Fig. 4. (b) Quantification of synaptotagmin puncta in 100-μm lengths of segmental nerves of genotypes shown in a and e. Comparisons are with OK6-GAL4 (left bar) unless indicated. n=5 larvae. (c) Average velocities of 100 Syt-eGFP puncta, recorded over 60 seconds. Anterograde movements are positive, retrograde movements negative. Average speeds of labeled vesicles are significantly lower in tkv (P < 0.001) and Spict-overexpressing (P < 0.001), but not in spictmut larvae (P = 0.4) compared to in wild-type larvae (n=100 vesicles from 10 larvae in all genotypes). (d) The top row shows one frame from a time-lapse movie of Syt-eGFP-labeled vesicle transport in a segmental nerve of each genotype. For generation of kymographs, the segmental nerve was divided into substacks numbered 1-3 (second row). Kymographs of synaptic vesicle transport in the individual substacks are shown in the third to fifth rows. Moving vesicles appear as diagonal lines, and stationary vesicles as vertical lines. Individual vesicles can be identified by their characteristic intensities and sizes. The vertical scale bar represents 10 seconds. The lower three rows show lines drawn by hand to illustrate the types of movement analyzed. (e) Effects of BMP signaling on the axonal accumulation of synaptotagmin caused by Spict overexpression. Quantification is in b. Scale bars, (a), 50 μm; (d, e), 10 μm.
Larval spictmut axons did not accumulate Syt (Fig. 5a), and showed normal motility of Syt-eGFP puncta (Fig. 5c; Supplementary Videos 1, 3), suggesting that fast axonal transport is normal in the absence of Spict. However, overexpression of Spict caused an increase in axonal levels of Syt and a severe axonal transport defect (Fig. 5a-d; Supplementary Fig. 4; Supplementary Videos 1, 4), compared to control genotypes. Axonal Syt accumulation was not more severe in the posterior segments (Supplementary Fig. 4). Furthermore, accumulation of Syt in Spict-overexpressing axons was suppressed by constitutively activated Tkv, and exacerbated by a heterozygous loss-of-function tkv mutant; neither activated Tkv nor heterozygous tkv had any obvious effect on axonal accumulation of Syt in the absence of Spict overexpression (Fig. 5b, e; Supplementary Fig. 4). These data suggest that BMP signaling is required for normal axonal transport, and that Spict overexpression impairs axonal transport by inhibiting BMP signaling.
To test whether the perturbed microtubule cytoskeleton was the cause of synaptotagmin accumulation in tkv mutant and Spict-overexpressing axons, we treated dissected live larvae with the microtubule-stabilizing drug Taxol at a concentration that had no obvious effect on wild type control phenotypes (Fig. 6; ref. 26). Taxol treatment significantly ameliorated the synaptotagmin accumulation and loss of acetylated α-tubulin in these axons (Fig. 6), suggesting that loss of microtubules is the underlying cause of the axonal blockages and transport defects.
Figure 6. Spict overexpression and reduction of BMP signaling impairs axonal transport by disrupting the microtubule cytoskeleton.

(a) A 60-minute Taxol treatment ameliorated the axonal blockage (i) and stable microtubule loss (ii) phenotypes of Spict-overexpressing (OK6-GAL4 UAS-spict) or tkv (tkv7/tkv16713) mutant axons. Panels are projections of confocal sections. Scale bar, 10 μm. (b) Taxol treatment caused a significant decrease of synaptotagmin puncta in 100-μm lengths of Spict-overexpressing or tkv mutant segmental nerves (P = 0.008 and 0.008, respectively; n=5 larvae; left panel), and a significant increase in levels of acetylated-α-tubulin in Spict overexpressing or tkv mutant axons (P = 0.002 and 0.026, respectively; n=5 larvae; right panel), compared to controls without Taxol treatment. No significant differences are seen in levels of α-tubulin (P > 0.1; n=5 larvae; middle panel). Fluorescence intensity per voxel is normalized to a control channel (anti-HRP). The mean intensity of anti-HRP is not significantly different among the three genotypes, or without and with Taxol treatment in each panel (P > 0.1, n=6 larvae, one-way ANOVA).
Spict antagonizes BMP signaling by regulating BMP receptor trafficking
How does Spict regulate BMP signaling? Given the endosomal localization of Spict, and the widespread regulation of receptor signaling through endocytosis, Spict could determine BMP receptor levels by regulating their trafficking. This model predicts that Spict and BMP receptors should interact physically, and show some colocalization. Indeed, we could co-immunoprecipitate Wit (but not a number of other neuronal membrane or vesicular components) using anti-Spict and extracts from spict+, but not from spictmut flies. Conversely, we could co-immunoprecipitate endogenous Spict with Wit-HA, precipitated from S2 cell lysates using anti-HA (Fig. 7a, b). We also detected partial colocalization of Spict-mRFP puncta with either Tkv-HA or Wit in NMJ boutons (Fig. 7c), and substantial colocalization of Spict-EGFP puncta with Wit in S2 cells (see below), suggesting a direct effect of Spict on BMP receptors.
Figure 7. Involvement of Spict in BMP signaling.

(a) Western blots of whole fly extracts (Input) and anti-Spict immunoprecipitations (IP). Input lanes contained one tenth of the amounts immunoprecipitated. (b) Western blots of cell lysates (Input) or anti-HA immunoprecipitations (IP), with or without Wit-HA transfection. Input lanes contained one quarter of the amounts immunoprecipitated. The arrow indicates the predicted size of Wit-HA. (c) Overlap of Spict-mRFP with Tkv-HA and Wit in NMJ boutons. (d) PMad levels in S2 cells transfected with Wit-HA, Mad-Flag. Levels of β-tubulin, Mad and Wit were constant across the three genotypes (P > 0.4, n=4, one-way ANOVA). Levels of PMad normalized to β-tubulin were 146 ± 16% in spict-RNAi treated cells, 48 ± 6% in Spict-EGFP transfected cells, compared to control cells (P = 0.03 and 0.03 respectively, n=4). (e) A control (middle row) and a spict RNAi-treated S2 cell (top row) show Wit localization on the cell surface. Spict-EGFP (bottom row) redistributes Wit to Spict-EGFP puncta inside the cell. (f) In S2 cells, Spict-EGFP expression (bottom row) redistributes Wit from the cell surface (no Spict-EGFP expression, top row) to early endosomes (anti-Rab5). Arrows show examples of triple colocalization. Broken lines show approximate cell boundaries. (g) Partial loss of the posterior crossvein (arrow) in a spictmut wing. Note the smaller size of the spictmut wing. Panels are single confocal sections in c, e, f. Scale bars, (c, e, f), 5 μm; (g), 0.5 mm. Additional data for this figure are in Supplementary Fig. 5.
Also in agreement with this model, we detected upregulation of Wit at the NMJ presynaptic plasma membrane in spictmut NMJ boutons (Fig. 3i). We found no obvious effect of spictmut on Wit levels in whole larval or adult extracts (Supplementary Fig. 5 and data not shown).
Since the small size of NMJ boutons limits the resolution with which receptor trafficking can be studied, we analyzed the effects on Wit of manipulating Spict levels in S2 cells. We expressed Wit together with the R-Smad protein Mad by transient transfection. Levels of Wit were not significantly changed by spict knockdown or Spict-EGFP overexpression; however, levels of PMad were significantly increased by spict knockdown, and significantly decreased by Spict-EGFP overexpression (Fig. 7d), indicating that Spict regulates BMP signaling probably without directly affecting synthesis or degradation of Wit in S2 cells. We next tested whether Spict affects the subcellular localization of Wit. spict knockdown significantly increased the proportion of total Wit at the plasma membrane (Fig. 7e; Supplementary Fig. 5). Conversely, overexpression of Spict-EGFP significantly reduced the proportion of total Wit at the plasma membrane and redistributed it to intracellular puncta that mainly contained Spict-EGFP (Fig. 7e, f; Supplementary Fig. 5). Most intracellular Wit puncta overlapped with Rab5 (69%), and some also overlapped with Hook, Rab11, and Spinster (19%, 7% and 2% respectively; n=48 cells from 2 transfections). Therefore, our data suggest that Spict inhibits BMP signaling by stimulating trafficking of BMP receptors from the plasma membrane to early endosomes, but does not affect degradation of Wit.
To test whether BMP signaling was affected at locations other than the NMJ in spict mutants, we examined flies for phenotypes that might be caused by perturbation of BMP signaling. Some spictmut flies (26.9 ± 0.9%, n=3 vials with over 100 flies per vial) had interrupted posterior crossveins in both wings (Fig. 7g); this phenotype was never seen in spict+ wings (P < 0.001 for comparison to spictmut), and was rescued completely by expressing UAS-spict using Actin5C-GAL4. Wing size was significantly smaller in spictmut crossveinless flies than in spict+ flies (70.4 ± 2.4% of spict+ wing size, P = 0.002), but not in spictmut flies that had normal crossveins (97.4 ± 2.0% of spict+ wing size, P = 0.5). This size decrease was specific for wings; the body size of spictmut crossveinless flies was not significantly reduced (99.8 ± 2.1% of spict+, P = 0.97). The posterior crossveinless phenotype is suggestive of perturbed BMP signaling during pupal development31,32. However we found no obvious difference in PMad levels or localization in the posterior crossvein region of pupal wing discs (Supplementary Fig. 5). No obvious increases in PMad levels were seen in early embryos or in third instar wing discs from a spictmut stock (Supplementary Fig. 5 and data not shown).
Discussion
The opposing effects of Spict and BMP signaling on NMJ and neuronal microtubules suggest that Spict is a novel antagonist of BMP signaling. BMP signaling acts both presynaptically16-20 and postsynaptically33 at the NMJ; our rescue experiments show that Spict acts presynaptically to regulate NMJ expansion. Our data suggest a direct effect of Spict on the presynaptic BMP signaling machinery. First, elevated levels of PMad and BMP receptors are seen at spictmut NMJs. Second, Spict can be co-immunoprecipitated with Wit. Third, Spict shows partial colocalization with the BMP receptors Tkv-HA or Wit at NMJ boutons. Fourth, Spict promotes relocalization of Wit from the surface of S2 cells to the Rab5 early endosomal compartment. Therefore, our data suggest strongly that Spict antagonizes BMP signaling by regulating its receptor traffic. This is in contrast to Highwire - while synaptic overgrowth in highwire mutants can be suppressed by BMP signaling mutants, the highwire phenotype is more completely suppressed by loss of the Wallenda MAP kinase kinase kinase, and there is no apparent upregulation of PMad in highwire mutants24.
The posterior crossveinless phenotype in some spictmut adult wings is also typical of reduced BMP signaling in pupal wing discs31,32. At first sight a crossveinless phenotype is inconsistent with Spict being an antagonist of BMP signaling. However, lowered BMP signaling in the posterior crossvein primordium could be due not only to direct downregulation of signaling, but also to upregulation of receptors that reduces diffusion of BMP ligands31,32. We have not detected any changes in the level of BMP signaling about the time when the posterior crossvein primordium develops, but this could be due to either the partial penetrance of the phenotype, or the robustness of the regulatory and feedback mechanisms that translate smooth gradients of BMP ligands into more sharply defined developmental features31,32.
How might an endosomal protein regulate BMP signaling? Membrane trafficking from the plasma membrane to lysosomes regulates many signaling pathways including BMP/TGF-β. For example, mutations that impair endosome to lysosome traffic cause an increase in BMP signaling, in at least some cases accompanied by increased levels of Tkv22,34,35. However, the predominant localization of Spict on early endosomes, and its ability to internalize Wit to this compartment suggest that Spict functions at some step of plasma membrane to endosome traffic. First, Rab5 compartments fail to accumulate at spictmut NMJs, rather than enlarge as in Hrs mutants34. Second, Spict overexpression in S2 cells redistributes Wit mainly to early endosomes, rather than to late endosomes or lysosomes. Third, there is no obvious degradation of Wit in Spict-overexpressing cells that internalize Wit, suggesting that Spict does not directly target Wit for degradation, at least in S2 cells. While levels of BMP receptors are elevated locally in NMJ boutons that lack Spict (Fig. 3g-i), this could be either to altered trafficking or degradation, and BMP signaling in S2 cells can be affected by Spict, without detectable changes in levels of BMP receptors (Fig. 7d,e). Therefore, Spict might inhibit BMP signaling by internalizing vacant receptors and thus preventing them from responding to ligand; since clathrin RNAi treatment redistributes Spict to the plasma membrane (Supplementary Fig. 2), Spict probably appears at least transiently at the plasma membrane. However, more complex models are possible. For example, Spict might sequester BMP receptors in a compartment from which they cannot signal; Notch receptors apparently have to reach a specific endosomal compartment before they can signal35,36.
By studying Spict, we have identified a role for BMP signaling in maintenance of axonal microtubules. Notably, local loss of presynaptic microtubules has also been seen in loss of BMP signaling at the NMJ21,37, and apical microtubule arrays are eliminated in tkv mutant clones in wing imaginal discs38. Since BMP signaling promotes synaptic growth and synaptic strength at the NMJ, it would be logical for it also to stimulate the additional transport of materials and organelles that a larger more active synapse requires.
If human SPG6 alleles are dominant gain-of-function, then the HSP that they cause would resemble the situation of Spict overexpression in Drosophila, and axonal degeneration in HSP could then be caused by inhibition of BMP signaling, loss of axonal microtubules, and impaired axonal transport. Given the effect of BMP signaling on axonal microtubules, other HSP gene products apart from SPG6 may affect BMP signaling and thus maintenance of axonal microtubules.
In contrast to SPG6, ARCI appears to be caused by loss of ichthyin function15. Our identification of a role for the ichthyin ortholog Spict in inhibiting BMP signaling suggests upregulation of BMP signaling as a possible disease mechanism in ARCI. Indeed, the BMP-like ligand TGF-β1 has complex roles in maintenance of skin, and its overexpression can cause psoriasis, a condition that bears some resemblance to ichthyosis39. Inhibitors of BMP signaling may therefore be candidates for therapeutic purposes in ARCI or similar conditions.
In conclusion, we have established a cellular role for the SPG6 and ichthyin family of proteins, thus identifying a novel group of players in BMP signaling, and providing a framework for future understanding of diseases caused by mutations that affect these proteins.
Methods
Fly stocks
P{Actin5C-GAL4}17bFO1, P{MHC-GAL4}82, P{elav-GAL4}3A4, OK6-GAL417, P{UAS-Tkv-HA}1A3, P{UAS-Act-Tkv}8B3, P{UAS-wit}2A and P{UAS-syt-eGFP}3 were used for targeted expression. The following alleles were tested for interactions with spictmut: sax4/Df(2R)cn7969, tkv7/tkv16713, gbb1/gbb4, witA12/witB11, medea5, or heterozygous sax4, tkv7, gbb1, witA12, medea5. Heterozygous mutant stocks were maintained balanced over either CyO, Kr-GAL4 UAS-GFP40 or TM6B, Tb. Mutant alleles and transgenic lines that are not referenced here are documented in FlyBase (www.flybase.org).
Generation of a spict null allele
Excisions of EP(2)2202 were identified by loss of the w+ marker, and were screened by PCR using the forward primer S60507 (−539 to −517, relative to the first nucleotide of the coding region) and a series of reverse primers about 1 kb apart, A61352 (+285 to +306), A62327 (+1259 to +1281), A63284 (+2216 to +2238), and A64290 (+3223 to +3244). One imprecise excisions, spict41, removed the entire coding region and is referred to as spictmut (Fig. 1b). One precise excision, spict+48, was confirmed by sequencing and used as a spict+ allele in most experiments. All rescue strains and doubly homozygous mutants for spictmut and BMP were generated by crossing parents homozygous for spictmut. The genotypes of both parental stocks were confirmed by PCR.
Molecular biology
A rescue construct was generated by subcloning cDNA GM13388 (MRC Geneservice, Cambridge, UK) into pUAST41. S2 expression vectors pMT-spict-EGFP and pMT-EGFP-spict were generated by subcloning a PCR fragment from GM13388, containing all but the last codon of the spict coding region, into pMT-EGFP-V5-His (gift of J. Rocha), either upstream or downstream of EGFP . pUAST-spict-mRFP was generated by cloning the same spict fragment into a Gateway destination clone pUAST-DEST15, C-terminal mRFP (gift of Frederik Wirtz-Peitz). pUAST-wit (pMBO203917), pAcpA-GAL419, pAcpA-wit-HA19, and pAcpA-mad-flag19 were used in S2 cells.
Single-strand antisense RNAs labeled by digoxigenin were generated using a DIG RNA Labeling Kit (Roche Applied Science, Mannheim, Germany), from nucleotides 1079 to 1199 of spict cDNA (AY089521). In situ hybridization was performed as described42. Double-stranded RNAs were synthesized using a MEGAscript kit (Ambion, Austin, TX) from nucleotides 2038 to 2511 of Chc cDNA (Z14133), or 81 to 1199 of spict cDNA (AY089521). 1.89 kb sense RNA of the Xenopus elongation factor 1 gene (X55324) from pTRI-Xef1 provided with the MEGAscript kit was synthesized for control treatment .
Generation of an antibody, immunoprecipitation and immunoblot
Rabbit anti-Spict was raised against two peptides from the N- and C-termini of Spict protein (21-LPVEANQSPETAPL-34; 372-RRLSYGSSDIFRKA-385) by CovaLAb (Cambridge, UK). Details of immunoprecipitation and immunoblotting are in Supplementary Methods. Membranes were incubated overnight with anti-Spict at 1:200, anti-Wit (23C7)17 at 1:5, anti-synaptotagmin43 at 1:1000, anti-Spinster22 at 1:200, rabbit anti-HA (Abcam, Cambridge, UK) at 1:250, anti-Fasciclin II (1D4, Developmental Studies Hybridoma Bank, DSHB, IO) at 1:100 , anti-Syntaxin (8C3, DSHB) at 1:50, anti-PMad (PS1)44 at 1:500, anti-Flag (M2, Sigma, Poole, UK) at 1:500, and mouse anti-β-tubulin (Sigma, Poole, UK) at 1:500.
Histology and Microscopy
Third instar larvae were dissected in chilled Ca2+-free HL3 solution45, and fixed in 4% formaldehyde in PBS for 30 minutes. Pupal wing dissection, fixation and staining were performed as described32. Freshly collected embryos were devitellinized and fixed in 7% formaldehyde in PBS for 20 minutes before immunolabeling. Primary antibodies were used as follows: anti-synaptotagmin43 at 1:1000, anti-Rab546 at 1:25, anti-Hook47 at 1:200, anti-Discs-large (4F3)48 at 1:500, rabbit anti-HA (Abcam, Cambridge, UK) at 1:250, mouse anti-Rab11 (BD Biosciences Pharmingen, San Diego, CA) at 1:25, anti-Spinster22 at 1:500, anti-Wit (23C7)17 at 1:5, anti-cysteine string protein49 at 1:200, anti-PMad (PS1)44 at 1:400, anti-α-tubulin (DM1A, Sigma, Poole, UK) at 1:500, anti-acetylated-α-tubulin (6-11B-1, Sigma, Poole, UK) at 1:250, anti-tyrosinated-α-tubulin (YL1/2, Oxford Biotechnology, UK) at 1:50, anti-Futsch (22C10)29 at 1:250, anti-Fasciclin II (1D4, DSHB) at 1:100, anti-Syntaxin (8C3, DSHB) at 1:50. Secondary antibodies were Alexa-594-, FITC- or Cy5-labeled (Molecular Probes, Eugene, OR), used at 1:250. 1 μM LysoTracker (Molecular Probes, Eugene, OR) was incubated with dissected larvae for 60 minutes before fixation. For Taxol treatments, dissected larvae were incubated in HL3 containing 0.2% DMSO either alone (control), or with Taxol (50 μm, Sigma) for 60 minutes before fixation. Transfection and microscopy of cultured cells is described in Supplementary Methods.
Fixed preparations were mounted in Vectashield (Vector Laboratories, Burlingame, CA) and viewed using an MRC BioRad 1024 confocal microscope mounted on a Nikon Eclipse E800. Images were captured using 40x/1.3NA or 60x/1.4NA objectives, and exported using LaserSharp software (BioRad, Hemel Hempstead, UK). Brightness, contrast, levels and color channels were adjusted using Photoshop (Adobe, San Jose, CA). Analysis and quantification of images is described in Supplementary Methods.
Live imaging
Third instar larvae were dissected in chilled Ca2+-free HL3, and viewed using a 63x/0.95NA water-immersion objective on a Zeiss Axioskop 2 Microscope at room temperature (20-23°C). Image series were captured at 0.5-second intervals using a grayscale CCD camera (Hamamatsu Orca, C4742-95, Hamamatsu, Japan) and Openlab Software (Improvision, Coventry, UK).
Supplementary Material
Movies are played back in real time (2 frames per second). Scale bar is 10 μm. Anterograde movement is to the right, retrograde to the left. Both anterograde and retrograde movements can be observed.
Movies are played back in real time (2 frames per second). Scale bar is 10 μm. Anterograde movement is to the right, retrograde to the left. Large nonmotile bright aggregates are observed within the nerve.
Movies are played back in real time (2 frames per second). Scale bar is 10 μm. Anterograde movement is to the right, retrograde to the left. Both anterograde and retrograde movements can be observed.
Movies are played back in real time (2 frames per second). Scale bar is 10 μm. Anterograde movement is to the right, retrograde to the left. Large nonmotile bright aggregates are observed within the nerve.
Acknowledgements
We thank H. Aberle, D. Coulson, J. Drummond, V. Korolchuk, S. Sweeney and the Bloomington and Szeged Drosophila Stock Centers for fly stocks, S. Eaton, G. Margués, M. O'Connor, J. Rocha, K. Wang and F. Wirtz-Peitz G for DNA constructs, M. González-Gaitán, H. Krämer, T. Littleton, S. Sweeney, P. ten Dijke, A. Tolkovsky, T. Wucherpfennig, and the Developmental Studies Hybridoma Bank for antibodies, and L. Masuda-Nakagawa, D. Rubinsztein, and O'Kane lab members for helpful discussions. X.W. was supported by scholarships from Cambridge Overseas Trust and a UK Government ORS award. H.T.H.T. was supported by Tom Wahlig Stiftung, Croucher Foundation, Cambridge Overseas Trust, and a UK Government ORS award. E.R. is a Wellcome Trust Advanced Clinical Fellow. C.J.O'K. was supported by a Research Development Fellowship from the Biotechnology and Biological Sciences Research Council.
Footnotes
Publisher's Disclaimer: This is a pre-copy-editing, author-produced PDF of an article accepted for publication in Nature Neuroscience following peer review. The definitive publisherauthenticated version (Nature Neuroscience 10, 177 - 185 (2007)) is available online at: http://www.nature.com/neuro/journal/v10/n2/full/nn1841.html
References
- 1.Stokin GB, Goldstein LSB. Axonal Transport and Alzheimer's Disease. Annu. Rev. Biochem. 2006;75:607–627. doi: 10.1146/annurev.biochem.75.103004.142637. [DOI] [PubMed] [Google Scholar]
- 2.Reid E. Science in motion: common molecular pathological themes emerge in the hereditary spastic paraplegias. J Med Genet. 2003;40:81–86. doi: 10.1136/jmg.40.2.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fink JK. Hereditary spastic paraplegia. Curr. Neurol. Neurosci. Rep. 2006;6:65–76. doi: 10.1007/s11910-996-0011-1. [DOI] [PubMed] [Google Scholar]
- 4.Zeitlmann L, Sirim P, Kremmer E, Kolanus W. Cloning of ACP33 as a Novel Intracellular Ligand of CD4. J. Biol. Chem. 2001;276:9123–9132. doi: 10.1074/jbc.M009270200. [DOI] [PubMed] [Google Scholar]
- 5.Simpson MA, et al. Maspardin is mutated in mast syndrome, a complicated form of hereditary spastic paraplegia associated with dementia. Am J Hum Genet. 2003;73:1147–1156. doi: 10.1086/379522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bakowska JC, Jenkins R, Pendleton J, Blackstone C. The Troyer syndrome (SPG20) protein spartin interacts with Eps15. Biochem. Biophys. Res. Comm. 2005;334:1042–1048. doi: 10.1016/j.bbrc.2005.06.201. [DOI] [PubMed] [Google Scholar]
- 7.Reid E, et al. The hereditary spastic paraplegia protein spastin interacts with the ESCRT-III complex-associated endosomal protein CHMP1B. Hum. Mol. Genet. 2005;14:19–38. doi: 10.1093/hmg/ddi003. [DOI] [PubMed] [Google Scholar]
- 8.Mannan AU, et al. ZFYVE27 (SPG33), a novel spastin-binding protein, is mutated in hereditary spastic paraplegia. Am J Hum Genet. 2006;79:351–357. doi: 10.1086/504927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Otomo A, et al. ALS2, a novel guanine nucleotide exchange factor for the small GTPase Rab5, is implicated in endosomal dynamics. Hum. Mol. Genet. 2003;12:1671–1687. doi: 10.1093/hmg/ddg184. [DOI] [PubMed] [Google Scholar]
- 10.Rainier S, Chai JH, Tokarz D, Nicholls RD, Fink JK. NIPA1 Gene mutations cause autosomal dominant hereditary spastic paraplegia (SPG6) Am J Hum Genet. 2003;73:967–971. doi: 10.1086/378817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen S, et al. Distinct novel mutations affecting the same base in the NIPA1 gene cause autosomal dominant hereditary spastic paraplegia in two Chinese families. Human Mutation. 2005;25:135–141. doi: 10.1002/humu.20126. [DOI] [PubMed] [Google Scholar]
- 12.Reed JA, et al. A novel NIPA1 mutation associated with a pure form of autosomal dominant hereditary spastic paraplegia. Neurogenetics. 2005;6:79–84. doi: 10.1007/s10048-004-0209-9. [DOI] [PubMed] [Google Scholar]
- 13.Kaneko S, et al. Novel SPG6 mutation p.A100T in a Japanese family with autosomal dominant form of hereditary spastic paraplegia. Mov Disord. 2006;21:1531–1533. doi: 10.1002/mds.21005. [DOI] [PubMed] [Google Scholar]
- 14.Chai JH, et al. Identification of four highly conserved genes between breakpoint hotspots BP1 and BP2 of the Prader-Willi/Angelman Syndromes deletion region that have undergone evolutionary transposition mediated by flanking duplicons. Am J Hum Genet. 2003;73:898–925. doi: 10.1086/378816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lefèvre C, et al. Mutations in ichthyin a new gene on chromosome 5q33 in a new form of autosomal recessive congenital ichthyosis. Hum. Mol. Gen. 2004;13:2473–2482. doi: 10.1093/hmg/ddh263. [DOI] [PubMed] [Google Scholar]
- 16.DiGiovanna JJ, Robinson-Bostom L. Ichthyosis: etiology, diagnosis, and management. Am. J. Clin. Dermatol. 2003;4:81–95. doi: 10.2165/00128071-200304020-00002. [DOI] [PubMed] [Google Scholar]
- 17.Aberle H, et al. Wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron. 2002;33:545–558. doi: 10.1016/s0896-6273(02)00589-5. [DOI] [PubMed] [Google Scholar]
- 18.Marqués G, et al. The Drosophila BMP type II receptor Wishful Thinking regulates neuromuscular synapse morphology and function. Neuron. 2002;33:529–543. doi: 10.1016/s0896-6273(02)00595-0. [DOI] [PubMed] [Google Scholar]
- 19.McCabe BD, et al. The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron. 2003;39:241–254. doi: 10.1016/s0896-6273(03)00426-4. [DOI] [PubMed] [Google Scholar]
- 20.McCabe BD, et al. Highwire regulates presynaptic BMP signaling essential for synaptic growth. Neuron. 2004;41:891–905. doi: 10.1016/s0896-6273(04)00073-x. [DOI] [PubMed] [Google Scholar]
- 21.Eaton BA, Davis GW. LIM Kinase1 controls synaptic stability downstream of the type II BMP receptor. Neuron. 2005;47:695–708. doi: 10.1016/j.neuron.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 22.Sweeney ST, Davis GW. Unrestricted synaptic growth in spinster—a late endosomal protein implicated in TGF-β-mediated synaptic growth regulation. Neuron. 2002;36:403–416. doi: 10.1016/s0896-6273(02)01014-0. [DOI] [PubMed] [Google Scholar]
- 23.Wan HI, et al. Highwire regulates synaptic growth in Drosophila. Neuron. 2000;26:313–329. doi: 10.1016/s0896-6273(00)81166-6. [DOI] [PubMed] [Google Scholar]
- 24.Collins CA, Wairkar YP, Johnson SL, DiAntonio A. Highwire restrains synaptic growth by attenuating a MAP kinase signal. Neuron. 2006;51:57–69. doi: 10.1016/j.neuron.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 25.Sherwood NT, Sun Q, Xue M, Zhang B, Zinn K. Drosophila spastin regulates synaptic microtubule networks and is required for normal motor function. PLoS Biol. 2004;2:e429. doi: 10.1371/journal.pbio.0020429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trotta N, Orso G, Rossetto MG, Daga A, Broadie K. The hereditary spastic paraplegia gene, spastin, regulates microtubule stability to modulate synaptic structure and function. Curr. Biol. 2004;14:1135–1147. doi: 10.1016/j.cub.2004.06.058. [DOI] [PubMed] [Google Scholar]
- 27.Webster DR, Borisy GG. Microtubules are acetylated in domains that turn over slowly. J. Cell Sci. 1989;92:57–65. doi: 10.1242/jcs.92.1.57. [DOI] [PubMed] [Google Scholar]
- 28.Vadlamudi RK, et al. p21-Activated Kinase 1 Regulates Microtubule Dynamics by Phosphorylating Tubulin Cofactor B. Mol Cell Biol. 2005;25:3726–3736. doi: 10.1128/MCB.25.9.3726-3736.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roos J, Hummel T, Ng N, Klambt C, Davis GW. Drosophila Futsch regulates synaptic microtubule organization and is necessary for synaptic growth. Neuron. 2000;26:371–382. doi: 10.1016/s0896-6273(00)81170-8. [DOI] [PubMed] [Google Scholar]
- 30.Goold RG, Owen R, Gordon-Weeks PR. Glycogen synthase kinase 3β phosphorylation of microtubule-asociated protein 1B regulates the stability of microtubules in growth cones. J. Cell Sci. 1999;112:3373–3384. doi: 10.1242/jcs.112.19.3373. [DOI] [PubMed] [Google Scholar]
- 31.O'Connor MB, Umulis D, Othmer H, Blair SS. Shaping BMP morphogen gradients in the Drosophila embryo and pupal wings. Development. 2006;133:183–193. doi: 10.1242/dev.02214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ralston A, Blair SS. Long-range Dpp signaling is regulated to restrict BMP signaling to a crossvein competent zone. Dev. Biol. 2005;280:187–200. doi: 10.1016/j.ydbio.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 33.Dudu V, et al. Postsynaptic Mad signaling at the Drosophila neuromuscular junction. curr. Biol. 2006;16:625–635. doi: 10.1016/j.cub.2006.02.061. [DOI] [PubMed] [Google Scholar]
- 34.Jékely J, Rørth P. Hrs mediates downregulation of multiple signalling receptors in Drosophila. EMBO Reps. 2003;4:1163–1168. doi: 10.1038/sj.embor.7400019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thompson BJ, et al. Tumor suppressor properties of the ESCRT-II complex component Vps25 in Drosophila. Devel. Cell. 2005;9:711–720. doi: 10.1016/j.devcel.2005.09.020. [DOI] [PubMed] [Google Scholar]
- 36.Lu H, Bilder D. Endocytic control of epithelial polarity and proliferation in Drosophila. Nat. Cell Biol. 2005;7:1232–1242. doi: 10.1038/ncb1324. [DOI] [PubMed] [Google Scholar]
- 37.Eaton BA, Fetter RD, Davis GW. Dynactin is necessary for synapse stabilization. Neuron. 2002;34:729–741. doi: 10.1016/s0896-6273(02)00721-3. [DOI] [PubMed] [Google Scholar]
- 38.Gibson MC, Perrimon N. Extrusion and death of DPP/BMP-compromised epithelial cells in the developing Drosophila wing. Science. 2005;307:1785–1789. doi: 10.1126/science.1104751. [DOI] [PubMed] [Google Scholar]
- 39.Li AG, Wang D, Feng X-H, Wang X-J. Latent TGFβ1 overexpression in keratinocytes results in a severe psoriasis-like skin disorder. EMBO J. 2004;23:1770–1781. doi: 10.1038/sj.emboj.7600183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Casso D, Ramirez-Weber FA, Kornberg TB. GFP-tagged balancer chromosomes for Drosophila melanogaster. Mech. Devel. 2000;91:451–454. doi: 10.1016/s0925-4773(00)00248-3. [DOI] [PubMed] [Google Scholar]
- 41.Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- 42.Brummel T, et al. The Drosophila activin receptor baboon signals through dSmad2 and controls cell proliferation but not pattering during larval development. Genes Dev. 1999;13:98–111. doi: 10.1101/gad.13.1.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Littleton JT, Bellen HJ, Perin MS. Expression of synaptotagmin in Drosophila reveals transport and localization of synaptic vesicles to the synapse. Development. 1993;118:1077–1088. doi: 10.1242/dev.118.4.1077. [DOI] [PubMed] [Google Scholar]
- 44.Persson U, et al. The L45 loop in type I receptors for TGF-beta family members is a critical determinant in specifying Smad isoform activation. FEBS Lett. 1998;434:83–87. doi: 10.1016/s0014-5793(98)00954-5. [DOI] [PubMed] [Google Scholar]
- 45.Stewart BA, Atwood HL, Renger JJ, Wang J, Wu C-F. Improved stability of Drosophila larval neuromuscular preparations in haemolymph-like physiological solutions. J. Comp. Physiol. A. 1994;175:179–191. doi: 10.1007/BF00215114. [DOI] [PubMed] [Google Scholar]
- 46.Wucherpfennig T, Wilsch-Brauninger M, Gonzalez-Gaitan M. Role of Drosophila Rab5 during endosomal trafficking at the synapse and evoked neurotransmitter release. J. Cell Biol. 2003;161:609–624. doi: 10.1083/jcb.200211087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Krämer H, Phistry M. Mutations in the Drosophila hook gene inhibit endocytosis of the boss transmembrane ligand into multivesicular bodies. J. Cell Biol. 1996;133:1205–1215. doi: 10.1083/jcb.133.6.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Parnas D, Haghighi AP, Fetter RD, Kim SW, Goodman CS. Regulation of postsynaptic structure and protein localization by the Rho-type guanine nucleotide exchange factor dPix. Neuron. 2001;32:415–424. doi: 10.1016/s0896-6273(01)00485-8. [DOI] [PubMed] [Google Scholar]
- 49.Zinsmaier KE, et al. A cysteine-string protein is expressed in retina and brain of Drosophila. J. Neurogenet. 1990;7:15–29. doi: 10.3109/01677069009084150. [DOI] [PubMed] [Google Scholar]
- 50.Chenna R, et al. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movies are played back in real time (2 frames per second). Scale bar is 10 μm. Anterograde movement is to the right, retrograde to the left. Both anterograde and retrograde movements can be observed.
Movies are played back in real time (2 frames per second). Scale bar is 10 μm. Anterograde movement is to the right, retrograde to the left. Large nonmotile bright aggregates are observed within the nerve.
Movies are played back in real time (2 frames per second). Scale bar is 10 μm. Anterograde movement is to the right, retrograde to the left. Both anterograde and retrograde movements can be observed.
Movies are played back in real time (2 frames per second). Scale bar is 10 μm. Anterograde movement is to the right, retrograde to the left. Large nonmotile bright aggregates are observed within the nerve.