

Abstract
The distribution and evolution of ultraconserved elements (UCEs, DNA stretches that are perfectly identical in primates and rodents) were examined in genomes of 3 primate species (human, chimpanzee, and rhesus macaque). It was found that the number of UCEs has decreased throughout primate evolution. At least 26% of ancestral UCEs have diverged in hominoids, whereas an additional 17% have accumulated one or more single nucleotide polymorphisms in the human genome. Sequence polymorphism analyses indicate that mutation fixation within an UCE can trigger a relaxation in the selective constraint on that element. Homogeneous mutation accumulations in UCEs served as a template by which purifying selection acted more effectively on protein-coding UCEs. Gene ontology annotation suggests that UCE sequence variation, primarily occurring in noncoding regions, might be linked to the reprogramming of the expression pattern of transcription factors and developmentally important genes. Many of these genes are expressed in the central nervous system. Finally, UCE sequence variability within human populations has been identified, including population-specific nonsynonymous changes in protein-coding regions.
Keywords: evolution, ultraconservation, gene regulation, comparative genomics
Introduction
Evolutionary sequence conservation is a reliable indicator of biological function in the analysis of anonymous sequences of genomes (Loots et al. 2000; Dermitzakis et al. 2003; Woolfe et al. 2005). Through whole-genome sequencing and analysis, the rate of neutral evolution in mammalian genomes has been defined. It is estimated that ∼5% of the human genome is under selective pressure (Waterston et al. 2002). Stemming from these observations, a unique set of “ultraconserved elements” (UCEs) has been identified. UCEs were originally defined as elements greater than 200 base pairs (bps) in length and are completely identical among human, mouse, and rat species (Bejerano et al. 2004). UCEs primarily cluster in the vicinity of developmental genes and appear to be free of population variation.
Only 6 single nucleotide polymorphisms (SNPs) have been reported across ∼126 kb of UCE sequences, suggesting that these elements are under extreme evolutionary pressure and possibly harbor critical biological functions (Bejerano et al. 2006). UCEs anchored at exon–intron boundaries are specifically linked to splicing regulation and have been associated with unproductive splicing (Lareau et al. 2007; Ni et al. 2007). In addition, noncoding RNA transcription from an intergenic UCE has been reported (Feng et al. 2006). In vivo characterization of noncoding UCEs revealed their preponderance to function as positive regulators of transcription or enhancers. The majority of tested UCEs are expressed in the central and peripheral nervous systems (Bejerano et al. 2006; Pennacchio et al. 2006). A few preliminary attempts to link mutations in UCEs to human neural disorders, including multiple sclerosis, mental retardation, and autism have been unsuccessful, thus raising the question of the importance of these elements in the proper expression and function of their surrounding genes (Ban et al. 2005; Richler et al. 2006; Bottani et al. 2007).
The preservation of UCEs over more than 100 Myr of evolution is a conundrum of genomics that contradicts recent observations that the selective pressure governing the evolution of gene regulatory elements in the primate lineage is significantly relaxed (Kryukov et al. 2005). Moreover, cis-regulatory elements in hominids might be diverging at a neutral rate (Keightley et al. 2005). In contrast, many conserved noncoding sequences, including noncoding UCEs, are under selective constraint in hominoids (Bush and Lahn 2005; Drake et al. 2006; Chen et al. 2007). Most recently, it was reported that UCEs might reside at the extremes of purifying selection by being “ultraselected” (Katzman et al. 2007). This might explain their presence in the genomes of modern humans. However, highly elevated levels of purifying selection should serve as indication of biological importance. Thus, it is puzzling that the homozygous deletion of 4 noncoding UCEs resulted in viable mice (Ahituv et al. 2007).
In an effort to delineate the evolutionary history of ultraconservation and to better understand the significance of ultraconservation, UCEs have been subjected to a 3-tier analysis of mutation accumulation, within primates, within hominoids, and within the human population.
In this study, indications of a widespread divergence of the ultraconservation data set have been observed in primates. A single mutation within an UCE results in disruption of its ideal sequence identity. Accumulation of fixed mutations in primate UCEs resulted in a decrease in the number of UCEs from approximately 1,000 in ancestral primates to 635 in modern humans. UCE divergence has been occurring in primates despite the strong purifying selection associated with UCEs. Genetic drift and population bottlenecks are possible explanations for mutation fixations in UCEs. However, the ultraconservation state might be fragile, with few fixed mutations resulting in decreased purifying selection strength. Additionally, human polymorphism data indicate that a large number of SNPs are found in UCEs. This is a significant departure from previous reports (Bejerano et al. 2004; Richler et al. 2006). Results also reveal differential evolutionary forces that act on coding and noncoding UCEs, inviting speculation about the biological consequences of UCE divergence in hominoids.
Methods
The University of California, Santa Cruz (UCSC) Genome Browser collection of genomic data sets (Kuhn et al. 2007) was used for this study. The following genomes were downloaded from the UCSC Genome Browser database: Human NCBI Build 36.1 (hg18; March 2006), Chimpanzee Build 2 Version 1 (panTro2; March 2006), Rhesus macaque draft assembly v.1.0 from the Baylor College of Medicine Human Genome Sequencing Center (rheMac2; January 2006), and Mouse Build 36 (mm8; February 2006). Their pairwise genome alignments were downloaded from the ECR Browser (Ovcharenko et al. 2004). The following annotation tables were downloaded from the UCSC Genome Browser in tabular format: RefSeq, “UCSC known” (Kuhn et al. 2007), and Ensembl (Hubbard et al. 2007) gene annotation; mRNA and expressed sequence tag (EST) annotation; as well as GNF expression data linked to UCSC known genes.
Allele fixation within the human population was studied using SNP data (version 21a), downloaded from the HapMap 2 project Web site (HapMapConsortium 2005). Only SNPs with minor allele frequencies of at least 0.01 in 1 of 3 populations (YRI, CEU, and ASN) were included in the analysis. These SNPs were then overlaid onto the human/chimp and human/rhesus alignment table from the UCSC Genome Browser (snp126orthoPanTro2RheMac2) to define the ancestral allele for each SNP. Finally, HapMap SNPs were projected onto the nonrepetitive sequence of the human genome to calculate the baseline genome average derived allele frequency (DAF) distribution. For 0.98% of HapMap SNPs mapped onto the nonrepetitive part of the human genome, the chimp allele did not match either 1 of 2 human alleles. This anchor control count is in agreement with the previously published data (Drake et al. 2006; Eberle et al. 2006).
Estimates of the number of expected coding SNPs in UCEs were made using the fraction of coding HapMap SNPs (0.0132) in the pool of all HapMap SNPs originating from the nonrepetitive sequence of the human genome (2.59 million SNPs). A correction for the difference in the coding content of UCEs versus the rest of the nonrepetitive sequence of the human genome was made to account for the 5.4- and 8.0-fold enrichment in coding DNA sequence in UCEs and short UCEs (sUCEs), respectively. These calculations resulted in 3.69 and 36.4 expected coding SNPs in UCEs and sUCEs, respectively.
Binning of UCEs into different genic categories was based on overlap of these elements with coding exons and untranslated regions (UTRs) of annotated genes and mRNA/EST structures (supplementary table S1, Supplementary Material online). Elements overlapping exons of RefSeq, Ensembl, and/or UCSC known genes were classified as coding, 5′ UTR, or 3′ UTR. An overlap of at least a 10 bp with a gene feature, was required to annotate an element to a particular category. Priority was given first to coding exons, then to UTRs, and then to promoters. Remaining elements were juxtaposed to mRNA and spliced EST structures to identify potentially translated elements (called putatively coding). All other elements were split into intronic and intergenic sets, depending on their attribution to genes.
Statistical analyses of the observation of multiple mutations in UCEs were done using the μ = 2.5 × 10−8 mammalian mutation rate per nucleotide site per generation (Nachman and Crowell 2000). Assuming an average 20-year generation time in primates and 0.5-year generation time in rodents (Li et al. 1996), the H/C and Chimpanzee and mouse (C/M) divergence can be estimated at 600,000 (2 × 6 Myr/20) and 164,000,000 (80 Myr/20 + 80 Myr/0.5) generations, respectively. Given 173 kb of the C/M UCE sequence, one can expect about SC/M = 711,000 mutations emerging in C/M UCEs throughout 80 Myr of mammalian divergence. Assuming the independent nature of these mutations, a probability of observing zero nucleotide changes in these elements of less than 0.05 would imply the probability of mutation fixation px in C/M UCEs to be less than 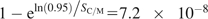 .
.
Using the same approach, the number of expected H/C random mutations in C/M UCEs, SHC, can be estimated at ∼2,600. Testing the null hypothesis that the probability of mutation fixation in hominoid and nonhominoid lineages is the same, the probability of observing at least 343 fixed nucleotide changes in human homologs (HHs) of C/M UCEs is approximately px343, which is almost zero. Either 100-fold greater or 100-fold smaller mutation rate would not affect the significance of this observation.
Results
Widespread Divergence of UCEs in Primates
Despite the invariant nature of ultraconserved sequences in human, mouse, and rat genomes, the invariance in UCEs does not extend to distantly related vertebrates. Only 6% (29 of 481) of UCEs have been reported to preserve their complete sequence identity in the avian lineage, whereas no UCEs have been completely preserved in fish lineages (Bejerano et al. 2004). It was also suggested that examining ultraconservation in humans and rodents provides a static and narrow snapshot into the mammalian evolution of a much larger set of well-conserved and similarly acting elements (Visel et al. 2008).
To address the evolutionary dynamics of ultraconservation and to investigate the details of ultraconservation evolution, ultraconserved sequences (200 bps and longer stretches of perfect sequence identity) in either “Rhesus macaque” and mouse (R/M) or Chimpanzee and mouse (C/M) were compared with their human (H) homologs to detect UCE sequence changes that occurred within primate and hominoid lineages. Changes in the human population assessed through the analysis of HHs of R/M and C/M UCEs, as well as H/M UCEs themselves, were also examined using SNP data from single nucleotide polymorphism database (dbSNP) and HapMap databases (Sherry et al. 1999; HapMapConsortium 2005).
The analysis was designed such that sequencing errors in the primate and mouse genomes would preclude the identification of the complete list of C/M and R/M UCEs but would not count as evolutionary sequence changes. This approach also ensured that the identified UCEs corresponded to the ancestral primate/rodent species and that the differences between these elements and their HHs represent evolutionary changes specific to either the primate or hominoid lineages. By omitting additional comparisons with the rat genome while still preserving the requirement of 100% sequence identity between a primate and a rodent, it was possible to focus on primate, rather than rodent, effects on UCE evolution.
In several cases, it was necessary to utilize an expanded set of UCEs, including shorter UCEs (shorter than 200 bps, but at least 100 bps in length, dubbed short UCEs or sUCEs) to reach statistical significance for some observations. It is important to note that sUCEs have been recently shown to have similar biological functions as UCEs (Visel et al. 2008). All trends in this study were identical between UCEs and sUCEs. The ability to achieve statistical significance using sUCEs is associated with their prevalence; these elements are about 10 times more abundant in primates than UCEs (supplementary table S4, Supplementary Material online).
There are 695, 653, and 635 UCEs in rhesus, chimp, and human genomes, respectively, as determined by pairwise comparisons with the mouse genome (table 1). Only 459 UCEs (70% of C/M UCEs) are shared by all 3 data sets, whereas 543 (83% of C/M UCEs) are shared by hominoid sets. This demonstrates a noticeable divergence of UCEs in primates. There is a gradual 8.8% decrease in the number of UCEs following the evolutionary separation of primates, starting from rhesus and shifting first to chimp and then to human. Given similar pairwise branch lengths between each primate and the mouse and the high quality of human genomic sequence data, this gradual reduction in UCEs suggests an elevated rate of UCE disappearance in hominoids, especially humans.
Table 1.
UCEs in Primates
| UCE | |||
| Human (%) | Chimp (%) | Rhesus (%) | |
| Total (N) | 635 | 653 | 695 |
| Diverged + SNPs | - | 12.1 | 14.8 |
| Diverged, no SNPs | - | 14.1 | 23.5 |
| Identical + SNPs | 26.3 | 16.8 | 15.1 |
| Identical, no SNPs | 73.7 | 57.0 | 46.6 |
| Total diverged and identical with SNPs | 26.3 | 43.0 | 53.4 |
NOTE.—SNP annotation of H/M UCEs and HHs of C/M and R/M UCEs was performed using human dbSNP data (Sherry et al. 1999).
It should be noted that the reduction in the number of UCEs corresponds to a limited number of nucleotide substitutions that simply disrupt the definition of UCEs, effectively reclassifying an UCE into a highly conserved element category. These substitutions might not have a profound phenotypic effect (Ahituv et al. 2007; Visel et al. 2008) but preclude the identification of these elements using the standard ultraconservation filter (Bejerano et al. 2004).
To investigate the nature of evolutionary forces impacting ultraconservation, the divergence of UCE sequences in primates was juxtaposed to known human variations. A large number of HHs of C/M and R/M UCEs have either diverged from the ancestral allele or harbored one or more SNPs, with a total of 43% of C/M UCEs and 53% of R/M UCEs (table 1). Only one-third of these elements are invariant in interprimate comparisons, yet contain SNPs (representing recent, human-specific evolutionary changes). For the majority of sequences (61% of C/M and 72% of R/M diverged elements), the human reference sequence differed from the ancestral ultraconserved allele. This observation led to the conclusion that UCEs diverge broadly in primates as well as in hominoids, with many changes already fixed in the human population. However, the extent to which a purifying selection counteracts mutational events in UCEs, as well as the differential strength of selection of different classes of UCEs, remains unknown. These important questions will be addressed in the following sections.
Hominoid and Primate UCEs Are Selectively Constrained
Over 25% of UCEs harbor one or more SNP. Approximately 200 UCEs contain SNPs (table 1). This relatively large number of SNPs superimposed over the HapMap information on population-specific allele frequency (HapMapConsortium 2005) permits estimating the rate of mutation fixation and the strength of the selective constraint within UCEs in humans. HapMap analysis groups individual variation into 3 population panels by their ancestry: Nigerian (Yoruba, YRI), European (Utah residents, CEU), and Asian (Japanese and Han Chinese, JTP + CHB or ASN). The number of UCE SNPs differs between populations. There is also a noticeable discrepancy in SNP distributions among different populations. For example, only 24 H/M UCE SNPs (52.2% of YRI or 75.0% ASN sets) are shared by all 3 panels (fig. 1A). Differences in SNP density and allele frequencies between populations are well documented (HapMapConsortium 2005). However, the population-specific discrepancy in SNP distributions is lower across all noncoding sequences in the human genome than in the UCE data set, with 66.1% of YRI SNPs (or 78.9% of ASN SNPs) shared by all 3 panels.
FIG. 1.—

Total number of SNPs in H/M UCEs and HHs of C/M and R/M UCEs (A). Derived allele frequency (DAF) distributions in UCEs (red) compared with the nonrepetitive human genome sequence (blue)—YRI population (B), CEU population (C), ASN population (D). Standard deviation (σ) of UCE DAF binning (B–D) was estimated using the binomial distribution as , where p represented the fraction of SNPs in a particular bin and n represented the total number of SNPs in that distribution.
A hypergeometric distribution–based analysis partially rejects the null hypothesis that the increased discrepancy in SNP distributions in UCEs is due to the small number of observations (P = 0.036 for YRI UCE SNPs, P< < 0.01 for both YRI and ASN sUCE SNP comparisons, statistical significance was not achieved for ASN UCE SNPs). The elevated discrepancy in UCE SNPs across these 3 populations, combined with the decreased SNP density, comparing UCEs to the nonrepetitive part of the human genome (table 2), and the enrichment in ancestral alleles in UCEs (fig. 1B–D) further strengthen the notion that a purifying selection is acting on these elements (Katzman et al. 2007).
Table 2.
HapMap SNP Densities per Kilobase of Genome Sequence Averaged across YRI, CEU, and ASN Populations
| UCEs | Genome Average | ||
| H/M | HHs of C/M | HHs of R/M | |
| 0.24 | 0.32 | 0.34 | 1.20 |
NOTE.—The genome average corresponds to a nonrepetitive DNA sequence. A Poisson distribution was utilized to assess the significance of SNP density change. The change in SNP density was highly significant comparing UCEs with the genome average (P < < 10−6), significant comparing HHs of either C/M or R/M UCEs with H/M UCEs (P < 0.015) and not significant comparing HHs of R/M with HHs of C/M (P = 0.4).
A purifying selection, which was previously speculated to reach the level of ultraselection in UCEs (Katzman et al. 2007), is likely the main evolutionary force preventing UCEs from accumulating mutations. Quantitatively, the fingerprint of the purifying selection can be observed as a shift in the DAF toward ancestral alleles (Drake et al. 2006). DAF binning within UCEs confirms strong purifying selection signals within all human populations under consideration (fig. 1B–D). Additionally, population-specific differences in the level of constraint were observed in this study. For example, the most profound DAF shift corresponds to the YRI population, with none of 62 YRI UCE SNPs possessing a DAF over 70% (fig. 1B). The strength of the purifying selection is somewhat decreased in the CEU population and more decreased in the ASN population, as shown by the higher incidence of SNPs with lower DAF in YRU than CEU and ASN (fig. 1B–D).
SNP density in HHs of C/M UCEs is higher than in H/M UCEs. SNP density in HHs of R/M UCEs is also higher than either in HHs of C/M UCEs or human UCEs (table 2). The trend is consistent across all 3 human populations (fig. 1A). As these are 3 sets of human-specific sequences that differ only by their mapping to a particular set of UCEs, this leads to the conclusion that there is a relaxation in selective constraint in humans, corresponding to HHs of UCEs from more distant species. Additionally, it was observed that HHs of UCEs corresponding to more distant species are associated with an elevated level of fixed (and recent) mutations.
Juxtaposition of these 2 observations suggests that HHs of UCEs that diverged in primates are associated with a decreased human-specific purifying selection. These observations imply that once a mutation within an UCE is fixed, this element becomes prone to accumulating additional variation. Thus, ultraconservation appears to be fragile in nature. A mutation fixed within an UCE either reflects or triggers a relaxation in the purifying selective constraint on that UCE, leading to elevated accumulation of additional sequence variation within that DNA element as compared with invariant UCEs. It should be noted, however, that there is no evidence of a flip from purifying to positive selection following UCE mutation fixation. Only a decrease in purifying selection was observed (table 2). Additional support for this hypothesis is given by the decreased fraction of ancestral alleles (DAF < 0.2) in HHs of C/M and R/M UCEs comparing to H/M UCEs, observed for all 3 populations (table 3, supplementary fig. S2 and table S6, Supplementary Material online). The fragility of ultraconservation may partially explain why there are few human UCEs ultraconserved in chicken and other distantly related vertebrates.
Table 3.
Fraction of SNPs with DAF < 0.2 (ancestral alleles)
| YRI | CEU | ASN | |
| H/M UCEs | 0.63 | 0.56 | 0.43 |
| HHs of C/M UCEs | 0.58 | 0.42 | 0.32 |
| HHs of R/M UCEs | 0.56 | 0.41 | 0.33 |
| Regular conserved | 0.47 | 0.38 | 0.37 |
| Nonrepetitive genome | 0.45 | 0.37 | 0.36 |
NOTE.—The regular conserved subset corresponds to evolutionary conserved regions of the human genome obtained using the standard 70% identity, with a length threshold of 100 bps.
HapMap studies have an inherent ascertainment bias originating from the dbSNP collection. Many SNPs are missing from these studies, whereas those that are present are nonhomogeneously distributed across chromosomes. Additional ascertainment bias relates to the deficiency of low-frequency alleles in limited panel sampling. Although it is not possible to reject the possibility of ascertainment bias influencing this study, there are indications that its impact is limited. First, there is no association between the location of UCEs and SNP ascertainment. Thus, ascertained SNPs are expected to have a random distribution within the UCE data set, as a randomly chosen haplotype is largely unaffected by the ascertainment bias (Nielsen et al. 2007).
Second, the ascertainment bias is unlikely to result in false positive inferences (Clark et al. 2005). The ascertainment bias also has a limited effect on the DAF comparisons of noncoding sequences (Drake et al. 2006). Finally, it will be also shown in the next section that the analysis of SNPs from coding and noncoding UCE subgroups confirms little or no impact of the ascertainment bias on this study.
Purifying Selection Is Stronger in Coding UCEs
Over 50% of the originally reported UCEs are noncoding, lacking any evidence of transcription (Bejerano et al. 2004). These elements often cluster around genes encoding transcription factors and developmental genes (Bejerano et al. 2004). Presumably, these UCEs function as transcriptional regulatory elements, with the majority of tested UCEs exhibiting enhancer properties in vivo (Pennacchio et al. 2006). To investigate the imbalance in selection acting on UCEs that encode protein sequences versus those with regulatory roles, the genic origin of UCEs was superimposed onto polymorphism and evolutionary data. Approximately 13% of H/M UCEs overlap a coding exon of RefSeq, Ensembl, and/or UCSC known genes. An additional 10% of H/M UCEs can be classified as putatively coding (see Methods), with 68.2% of putatively coding H/M UCEs being intronic (supplementary table S2, Supplementary Material online). Remaining 77% of H/M UCEs consist of noncoding elements originating from promoter, 3′ UTR, 5′ UTR, intron, and intergenic categories (supplementary table S1, Supplementary Material online). Binning of H/M UCEs into different categories did not uncover a dependence of SNP accumulation on UCE genic type (supplementary fig. S4, Supplementary Material online). This demonstrates the negligible effects of ascertainment bias on this study and reveals the homogeneous mutation accumulation within different classes of UCEs.
The fraction of coding HHs of C/M UCEs with fixed mutations in the human population is present at a 3-fold decreased level, compared with the complete distribution of HHs of C/M UCEs (P < 10−4; the same 3-fold decrease was observed for coding sUCEs, P < 10−26) (fig. 2), suggesting that this category is less prone to mutation fixation. The asymmetry in mutation fixation, combined with the divergence data, suggests that whereas mutation accumulation is homogeneous with respect to genic features, the purifying selection is strongly biased toward coding UCEs. This results in a higher fraction of coding elements being retained within UCEs throughout evolution.
FIG. 2.—

Comparing diverged and identical UCEs corresponding to different genic categories. HHs of C/M UCE were binned into 7 categories: coding, promoter, 5′ UTR, 3′ UTR, intronic, intergenic, and putatively coding. Statistically significant differences (assessed using hypergeometric distribution) are indicated by an asterisk.
A statistically significant 3-fold decrease in the fraction of diverged UCEs was observed for 3′ UTR UCEs. This might be an indication of the involvement of UCEs in posttranscriptional regulation that is also under strong purifying selection.
A stronger purifying selection that acts preferentially on coding UCEs should result in an elevated fraction of ancestral SNP alleles in these elements. There are 52 HapMap SNPs in the full set of 206 UCE SNPs, for which DAF information is available. Notably, there are no HapMap SNPs in coding UCEs (P = 0.025—Poisson distribution with 3.69 expected SNPs; see Methods) and 64% less than expected in coding sUCEs (13 with 36.4 expected, P < 10−5). The complete absence of coding HapMap SNPs in UCEs further strengthens the hypothesis that an elevated purifying selection acts on the coding data set. However, this precludes a DAF analysis for coding versus noncoding elements.
Moreover, it is possible to perform this analysis using 26 genic (coding and UTR) and 319 nongenic (intronic and intergenic) HapMap sUCE SNPs, providing further evidence of elevated purifying selections acting on coding sUCEs (supplementary fig. S3, Supplementary Material online). Although the difference was not statistical significant (likely due to the small number of coding sUCE SNP), the number of coding SNPs with derived alleles in sUCEs was twice as low as than expected (P = 0.12).
There are only 2 coding sUCE SNPs that have predominantly derived alleles in human populations, out of 13 total, characterized using the DAF 0.2 threshold. One derived sUCE SNP maps to the HOXA7 gene, another to the CNOT4 gene. Both SNPs result in a nonsynonymous substitution. HOXA7 and CNOT4 SNPs are fixed in CEU and ASN populations, but only the CNOT4 SNP is also fixed in the YRI population. An additional fixed coding SNP present in the CEU population leads to a nonsynonymous mutation in the CALU gene. Another 2 fixed SNPs specific to the YRI population are synonymous mutations in the CDH7 and ENST00000314238 genes.
The HOXA7 UCE mutation that leads to Alanine/Threonine substitution has a DAF of 85% in CEU and 84% in ASN populations but only 6.7% in the YRI population (supplementary table S3, Supplementary Material online). It is interesting that the original Alanine is preserved in the HOXA7 peptide in rodents, dogs, opossums, and even in frogs. Alanine is mutated only in chicken and fish. Therefore, one can speculate that this very recent mutation fixation in the UCE of the HOXA7 gene, which is associated with ovarian cancer (Ota et al. 2007) and acute myeloid leukemia (Wang et al. 2007), might have had a phenotypic effect, specific in CEU and ASN human populations.
Functional Characteristics of Diverged and Preserved Ultraconservation
With less than 20% of all UCEs and less than 10% of diverged UCEs being coding (fig. 2), it is clear that the evolution of ultraconservation is primarily noncoding in nature. Noncoding UCEs are likely to function as transcriptional regulatory elements, and over 100 noncoding UCEs have been shown to act as enhancers in vivo (Pennacchio et al. 2006). Functional sequence changes in noncoding UCEs are expected to modulate gene expression. Presumably, some of these changes have a phenotypic effect, given that strong purifying selection has kept these elements intact throughout almost 100 Myr of mammalian evolution. Gene Ontology (GO) annotation (Ashburner et al. 2000) and GNF Novartis tissue-specificity profiling of gene expression (Su et al. 2002) allowed functional characterization of evolutionary changes in UCE. To characterize recent, hominoid-specific evolutionary effects shaping the ultraconservation architecture of the human genome, the analysis was based on HHs of C/M UCEs.
Despite common strong associations between diverged and identical UCEs with regulation of transcription, development, and metabolism (P < 10−35, P < 10−11, and P < 10−10, respectively), there were distinct differences in the GO annotation detected between diverged and identical elements (fig. 3). Four RNA-specific functions (RNA splicing, RNA binding, RNA processing, and RNA metabolism) were significantly enriched in the identical UCE data set (P < 10−6), whereas the number of genes in these categories from the diverged UCE data set was at the level expected by random chance. This clearly indicates that RNA-related functions of UCEs are selectively and strongly preserved throughout the evolution of ultraconservation.
FIG. 3.—

.—Differential enrichment in GO categories of UCEs that remained identical in hominoids (red) and diverged (blue). Four nonredundant GO categories with the highest (left side) and lowest (right side) ratios of identical to diverged GO category enrichment are presented. Only well-populated GO categories with more than 5 genes per category in either the identical or diverged data set were included in the analysis.
Alternatively, some GO categories are much more strongly enriched in the diverged data set of UCEs than in the identical data set of UCEs, suggesting the presence of a characteristic speciation trend in the divergence of UCEs. Whereas basic gene regulation and developmental processes are characteristic of all UCEs, gene enrichment in these categories was much more profound in the diverged category (fig. 2; P < 0.05). These data show that the divergence of ultraconservation might preferentially target regulation of transcription factors and developmental genes.
UCE Tissue Specificity
The fixation of sequence variation in human UCEs, most of which act as cis-regulatory sequences, implies that some of these mutations may serve as substrates for human-specific differences in gene regulation. To address the tissue specificity of UCEs as a class, the GNF Novartis microarray profiling of human gene expression was utilized to characterize genes linked to noncoding UCEs, using HHs of C/M UCEs for the analysis. Given the difficulty in correctly assigning a gene as a target for an intergenic regulatory element, the analysis was based on intronic, promoter, and UTR UCEs. Analysis of tissue-specific differences between genes harboring noncoding UCEs and the genome average revealed that in 9 out of 10 tissues, these signals originate from different compartments of the central nervous system (CNS) (fig. 4). This indicates that noncoding UCEs may be involved into the regulation of CNS gene expression.
FIG. 4.—

Tissues with the 10 largest increases (top) or decreases (bottom) in the fraction of positively expressed genes harboring noncoding UCEs (blue), as compared with the genome average (gray). The positive/negative gene expression value was extracted from the gnfAtlas2 UCSC Genome Browser table data, in which individual transcript expression was averaged across all 79 tissues and transformed logarithmically. A Poisson distribution was used to estimate the standard deviation of gene counts , where p represented the genome average fraction of positively expressed genes and n represented the total number of expressed genes harboring noncoding UCEs.
Discussion
Adaptation and survival of species are facilitated by the accumulation of advantageous mutations and the removal of deleterious mutations. Ultraconservation can be simply defined as long stretches of DNA that remained absolutely identical since the speciation point of primates and rodents. This definition reflects the indispensable nature of UCEs. However, an alternative to this hypothesis might be that there were once many more UCEs in ancestral primates than currently observed, which have been removed by a gradual process of mutation accumulation. Either stochastic or targeted accumulation of mutations in UCEs results in the accumulation of a potentially small number of sequence changes, but each such change, when fixed, immediately disqualifies the segment from being an UCE.
The current study suggests that the second hypothesis might be more reasonable. By tracking C/M UCEs through just the last 6 Myr of hominoid evolution since the separation of humans and chimpanzee (Gibbs et al. 2007), it was possible to detect sequence variation in 43% of these elements, with about 40% of this variation emerging in the human population. It is difficult to define an appropriate null hypothesis for determining the statistical significance of this effect. Any sequence variation model for UCEs would interfere with the definition of ultraconservation. However, a null hypothesis that the same UCE mutation fixation rate had occurred in hominoid and nonhominoid lineages can be tested, using the average mutation rate for mammals (Nachman and Crowell 2000). This null hypothesis was rejected, as the probability to observe 343 fixed mutations in HHs of C/M UCEs and zero in the original C/M UCEs is less than 10−2000 (see Methods).
There are some intriguing consequences of the presence of large-scale UCE polymorphisms within the human population. Despite the ultimate levels of sequence identity used in the original UCE definition (Bejerano et al. 2004), the resulting UCE set is rather variable depending on the reference human genome. Up to 26% of original UCEs could be selected differently, if a different reference human genome had been chosen (table 1). This UCE data set variability will increase even further, if the polymorphism in rodent populations is considered. Thus, different studies of ultraconservation might be more informative if they allow some minimal variation, as represented by sUCEs or a more inclusive data set of highly conserved elements (Visel et al. 2008).
In addition to the widespread UCE divergence (but limited number of sequence changes), there are indications of fragility associated with the state of ultraconservation. There is a positive correlation between mutation fixation and an increased density of SNPs in UCEs, indicating that a mutation fixed within an UCE triggers a relaxation in the strength of the purifying selection, effectively promoting the accumulation of additional mutations. This can partially explain why there are only 635 H/M UCEs today, although the total number of UCEs in primates may have been close to 1,000. There are 893 unique H/M, C/M, and R/M UCEs (fig. 5); additional inclusion of UCEs diverged in all 3 primate lineages will further increase the estimated number of primate/rodent UCEs in the ancestral primate genome.
FIG. 5.—

UCE divergence in the primate lineage. Venn diagram represents an overlap of 893 H/M, C/M, and R/M UCEs.
Several lines of evidence indicate that mutations continually target UCEs but are countered by strong purifying selection that rejects the population fixation of mutations in UCEs. Nevertheless, the small population size and the relatively large generation time characteristic of primates often override the purifying forces that aim to keep UCEs unchanged (Chen et al. 2007). UCE divergence is biased toward noncoding sequences, the majority of which supposedly function as cis-regulatory elements. This implies that the evolution of ultraconservation primarily influences gene regulation. It is quite intriguing that these diverged noncoding UCEs have been found to be associated with transcription factors and developmental gene categories. The latter suggests that UCE divergence might have an impact on remodeling the spatial and temporal expression patterns of key members of gene regulatory and signaling networks.
Noncoding UCEs have been associated with transcription factors and developmental genes. Some of these are predominantly expressed in different morphological structures of the CNS, thus positioning them as key candidates for controlling gene expression during the development. Additionally, noncoding UCEs are the most common type of UCEs and accumulate mutations much faster than coding UCEs. Therefore, if a fraction of fixed mutations in UCEs had functional consequences, it is intriguing to speculate that they were important in gene regulatory speciation within primate lineages.
In summary, this study reveals that the apparent imperviousness of ultraconservation has frequently been compromised during the primate evolution. Ultraconservation has been further challenged throughout the evolution of hominoids, resulting in profound changes in the ultraconservation genome architecture. Extensive variation detected in primate UCEs, coupled with the strong purifying selection applied to these elements, suggests that UCEs combined with other sets of deeply conserved elements that have accumulated recent evolutionary changes may be important in the investigation of biological innovations that have emerged in humans.
Supplementary Material
Supplementary tables S1–S6 and figures S1–S4 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Supplementary Material
Acknowledgments
I am indebted to Marcelo Nobrega and Gabriela Loots for numerous helpful discussions and their critical reading of the manuscript. I also would like to thank the anonymous reviewers of this manuscript for their useful comments and suggestions. This study was supported by the Intramural Research Program of the National Institutes of Health, National Library of Medicine.
References
- Ahituv N, Zhu Y, Visel A, Holt A, Afzal V, Pennacchio LA, Rubin EM. Deletion of ultraconserved elements yields viable mice. PLoS Biol. 2007;5:e234. doi: 10.1371/journal.pbio.0050234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, et al. (20 co-authors) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban M, Maranian M, Yeo TW, Gray J, Compston A, Sawcer S. Ultraconserved regions in multiple sclerosis. Eur J Hum Genet. 2005;13:998–999. doi: 10.1038/sj.ejhg.5201457. [DOI] [PubMed] [Google Scholar]
- Bejerano G, Lowe CB, Ahituv N, King B, Siepel A, Salama SR, Rubin EM, Kent WJ, Haussler D. A distal enhancer and an ultraconserved exon are derived from a novel retroposon. Nature. 2006;441:87–90. doi: 10.1038/nature04696. [DOI] [PubMed] [Google Scholar]
- Bejerano G, Pheasant M, Makunin I, Stephen S, Kent WJ, Mattick JS, Haussler D. Ultraconserved elements in the human genome. Science. 2004;304:1321–1325. doi: 10.1126/science.1098119. [DOI] [PubMed] [Google Scholar]
- Bottani A, Chelly J, de Brouwer AP, Pardo B, Barker M, Capra V, Bartoloni L, Antonarakis SE, Conrad B. Sequence variation in ultraconserved and highly conserved elements does not cause X-linked mental retardation. Am J Med Genet A. 2007;143:888–890. doi: 10.1002/ajmg.a.31651. [DOI] [PubMed] [Google Scholar]
- Bush EC, Lahn BT. Selective constraint on noncoding regions of hominid genomes. PLoS Comput Biol. 2005;1:e73. doi: 10.1371/journal.pcbi.0010073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CT, Wang JC, Cohen BA. The strength of selection on ultraconserved elements in the human genome. Am J Hum Genet. 2007;80:692–704. doi: 10.1086/513149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AG, Hubisz MJ, Bustamante CD, Williamson SH, Nielsen R. Ascertainment bias in studies of human genome-wide polymorphism. Genome Res. 2005;15:1496–1502. doi: 10.1101/gr.4107905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dermitzakis ET, Reymond A, Scamuffa N, Ucla C, Kirkness E, Rossier C, Antonarakis SE. Evolutionary discrimination of mammalian conserved non-genic sequences (CNGs) Science. 2003;302:1033–1035. doi: 10.1126/science.1087047. [DOI] [PubMed] [Google Scholar]
- Drake JA, Bird C, Nemesh J, et al. (11 co-authors) Conserved noncoding sequences are selectively constrained and not mutation cold spots. Nat Genet. 2006;38:223–227. doi: 10.1038/ng1710. [DOI] [PubMed] [Google Scholar]
- Eberle MA, Rieder MJ, Kruglyak L, Nickerson DA. Allele frequency matching between SNPs reveals an excess of linkage disequilibrium in gene regions of the human genome. PLoS Genet. 2006;2:e142. doi: 10.1371/journal.pgen.0020142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Bi C, Clark BS, Mady R, Shah P, Kohtz JD. The Evf-2 noncoding RNA is transcribed from the Dlx-5/6 ultraconserved region and functions as a Dlx-2 transcriptional coactivator. Genes Dev. 2006;20:1470–1484. doi: 10.1101/gad.1416106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs RA, Rogers J, Katze MG, et al. (136 co-authors) Evolutionary and biomedical insights from the rhesus macaque genome. Science. 2007;316:222–234. doi: 10.1126/science.1139247. [DOI] [PubMed] [Google Scholar]
- HapMapConsortium. A haplotype map of the human genome. Nature. 2005;437:1299–1320. doi: 10.1038/nature04226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard TJ, Aken BL, Beal K, et al. (58 co-authors) Ensembl 2007. Nucleic Acids Res. 2007;35:D610–D617. doi: 10.1093/nar/gkl996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katzman S, Kern AD, Bejerano G, Fewell G, Fulton L, Wilson RK, Salama SR, Haussler D. Human genome ultraconserved elements are ultraselected. Science. 2007;317:915. doi: 10.1126/science.1142430. [DOI] [PubMed] [Google Scholar]
- Keightley PD, Lercher MJ, Eyre-Walker A. Evidence for widespread degradation of gene control regions in hominid genomes. PLoS Biol. 2005;3:e42. doi: 10.1371/journal.pbio.0030042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryukov GV, Schmidt S, Sunyaev S. Small fitness effect of mutations in highly conserved non-coding regions. Hum Mol Genet. 2005;14:2221–2229. doi: 10.1093/hmg/ddi226. [DOI] [PubMed] [Google Scholar]
- Kuhn RM, Karolchik D, Zweig AS, et al. (25 co-authors) The UCSC genome browser database: update 2007. Nucleic Acids Res. 2007;35:D668–D673. doi: 10.1093/nar/gkl928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lareau LF, Inada M, Green RE, Wengrod JC, Brenner SE. Unproductive splicing of SR genes associated with highly conserved and ultraconserved DNA elements. Nature. 2007;446:926–929. doi: 10.1038/nature05676. [DOI] [PubMed] [Google Scholar]
- Li WH, Ellsworth DL, Krushkal J, Chang BH, Hewett-Emmett D. Rates of nucleotide substitution in primates and rodents and the generation-time effect hypothesis. Mol Phylogenet Evol. 1996;5:182–187. doi: 10.1006/mpev.1996.0012. [DOI] [PubMed] [Google Scholar]
- Loots GG, Locksley RM, Blankespoor CM, Wang ZE, Miller W, Rubin EM, Frazer KA. Identification of a coordinate regulator of interleukins 4, 13, and 5 by cross-species sequence comparisons. Science. 2000;288:136–140. doi: 10.1126/science.288.5463.136. [DOI] [PubMed] [Google Scholar]
- Nachman MW, Crowell SL. Estimate of the mutation rate per nucleotide in humans. Genetics. 2000;156:297–304. doi: 10.1093/genetics/156.1.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni JZ, Grate L, Donohue JP, Preston C, Nobida N, O'Brien G, Shiue L, Clark TA, Blume JE, Ares M., Jr Ultraconserved elements are associated with homeostatic control of splicing regulators by alternative splicing and nonsense-mediated decay. Genes Dev. 2007;21:708–718. doi: 10.1101/gad.1525507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Hellmann I, Hubisz M, Bustamante C, Clark AG. Recent and ongoing selection in the human genome. Nat Rev Genet. 2007;8:857–868. doi: 10.1038/nrg2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota T, Gilks CB, Longacre T, Leung PC, Auersperg N. HOXA7 in epithelial ovarian cancer: interrelationships between differentiation and clinical features. Reprod Sci. 2007;14:605–614. doi: 10.1177/1933719107307781. [DOI] [PubMed] [Google Scholar]
- Ovcharenko I, Nobrega MA, Loots GG, Stubbs L. ECR Browser: a tool for visualizing and accessing data from comparisons of multiple vertebrate genomes. Nucleic Acids Res. 2004;32:W280–W286. doi: 10.1093/nar/gkh355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchio LA, Ahituv N, Moses AM, et al. (19 co-authors) In vivo enhancer analysis of human conserved non-coding sequences. Nature. 2006;444:499–502. doi: 10.1038/nature05295. [DOI] [PubMed] [Google Scholar]
- Richler E, Reichert JG, Buxbaum JD, McInnes LA. Autism and ultraconserved non-coding sequence on chromosome 7q. Psychiatr Genet. 2006;16:19–23. doi: 10.1097/01.ypg.0000180683.18665.ef. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward M, Sirotkin K. dbSNP-database for single nucleotide polymorphisms and other classes of minor genetic variation. Genome Res. 1999;9:677–679. [PubMed] [Google Scholar]
- Su AI, Cooke MP, Ching KA, et al. (14 co-authors) Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci USA. 2002;99:4465–4470. doi: 10.1073/pnas.012025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Prabhakar S, Akiyama JA, Shoukry M, Lewis KD, Holt A, Plajzer-Frick I, Afzal V, Rubin EM, Pennacchio LA. Ultraconservation identifies a small subset of extremely constrained developmental enhancers. Nat Genet. 2008;40:158–160. doi: 10.1038/ng.2007.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GG, Cai L, Pasillas MP, Kamps MP. NUP98-NSD1 links H3K36 methylation to Hox-A gene activation and leukaemogenesis. Nat Cell Biol. 2007;9:804–812. doi: 10.1038/ncb1608. [DOI] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, et al. (223 co-authors) Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Woolfe A, Goodson M, Goode DK, et al. (16 co-authors) Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 2005;3:e7. doi: 10.1371/journal.pbio.0030007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.