

Abstract
Low-density lipoprotein-cholesterol (LDL-C) is a well established coronary heart disease (CHD) risk factor. However, the ability of this metabolic risk factor alone to identify individuals at rigk for future CHD events is limited. The raised triglycerides-low high-density lipoprotein-cholesterol (HDL-C) dyslipidaemia was shown to be an important cardiovascular risk factor independently of LDL-C levels. Fibric acid derivatives (fibrates) have been used in clinical practice for more than 2 decades as a class of agents known to decrease triglyceride levels while substantially increasing HDL-C levels. Through peroxisome proliferator-activated α-receptors, fibrates have a significant impact on the synthesis of several apolipoproteins and enzymes of lipoprotein metabolism as well as on the expression of several genes involved in fibrinolysis and inflammation. Data from recent primary and secondary prevention clinical trials demonstrate the efficacy of fibrate therapy in patients with the raised triglycerides-low HDL-C dyslipidaemia. This review summarizes current data regarding mechanism of action and the metbolic effects of fibrates, as well as results from major clinical trials on the efficacy of this mode of lipid lowering therapy. In addition, recent data from subgroup analyses of the Bezafibrate Infarction Prevention trial, demonstrating several important metabolic and long-term cardiovascular effects of bezafibrate therapy, are detailed.
Keywords: fibrates, high-density lipoprotein-cholsterol, metabolic syndrome, peroxisome proliferator-activated α-receptors, cardiac events
Introduction
Considerable progress in the understanding of coronary heart disease (CHD) risk factors has been made in the past decades, leading to the development of strategies to reduce CHD event rate. The ability of HMG-CoA reductase inhibitors (statins) to reduce the occurrence of CHD in individuals with hypercholesterolaemia has been reported in large primary and secondary prevention trials. Intervention trials using statins to lower low-density lipoprotein cholesterol (LDL-C) have consistently shown substantial reductions in major cardiovascular events in the treated groups (Shepard et al 1995; Sacks et al 1996; Downs et al 1998; Sever et al 2003; Baigent et al 2005), that was shown to be a function of the extent of LDL-C lowering (Baigent et al 2005). However, risk reduction with statin therapy is limited to about 30%, and the ability to adequately identify individuals at high risk for the development of CHD by total or LDL-C levels is limited by the fact that almost 50% of CHD patients have serum cholesterol levels in the normal range (Genest et al 1992). Thus, additional factors appear to modulate the risk of CHD independently LDL-C level.
Early data had shown that low high-density lipoprotein cholesterol (HDL-C) levels are inversely related to CHD incidence in both men and women (Yaari et al 1981; Gordon et al 1997). It has also been demonstrated that the risk associated with HDL-C is independent of LDL-C levels (Castelli et al 1987). Furthermore, a recent post hoc analysis from the Treating to New Targets (TNT) study (Barter et al 2007), has shown that HDL-C levels are predictive of major cardiovascular events even in patients who maintain LDL-C levels below 70 mg/dL while on statin therapy. In the majority of patients with low HDL-C who develop CHD, this lipoprotein alteration is most often accompanied by elevated triglyceride levels (Manninem et al 1992; Jeppesen et al 1997; Gotto et al 1998). This raised triglycerides-low HDL-cholesterol dyslipidemic state is a common feature of the altered risk profile of patients with type 2 diabetes, and is largely the consequence of an insulin-resistant state found in viscerally obese patients with the metabolic syndrome (Howard et al 1987; Laakso et al 1990; Bieman et al 1992). Thus, even in the absence of hyperglycemia or of type 2 diabetes, overweight/obese patients with the features of the metabolic syndrome and the raised triglycerides-low HDL-cholesterol dyslipidemic state have a substantially increased risk of CHD, despite LDL-C levels that are presumably within the ‘normal’ range (Lammarch et al 1998). Such dyslipidemia is also characterized by increased levels of atherogenic lipoproteins including, elevated apo B, and small, dense LDL and HDL particles (Tchernof et al 1996).
It had been known for decades that treatment with fibrates results in substantial decreases in triglyceride levels associated with a moderate increase in HDL-C levels and usually a small reduction in LDL-C levels. This class of drugs has been particularly useful in patients with primary hypertriglyceridemia, mixed hyperlipidemia, and in patients with type 2 diabetes with the raised triglycerides-low HDL-C dyslipidemia (Fruchart et al 1998). In recent years, significant progress has been made in understanding the mode of action of fibrates. These data have shown that fibrates activate specific transcription factors belonging to the nuclear hormone receptor superfamily, termed peroxisome proliferator-activated receptors (PPAR). Activation of these receptors alters the transcription rate of target genes which play a key role in the development of atherosclerosis. Recent data from the Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial ([VA-HIT] Rubins et al 1999), and subgroup analyses from the Bezafibrate Infarction Prevention trial ([BIP] The BIP Study Group 2000) have demonstrated that fibrate therapy improves both insulin sensitivity and the blood lipid profile, and significantly attenuates the risk of long-term major cardiovascular events (Tennenbaum et al 2005; Goldenberg et al 2006; Goldenberg et al 2007). In this review, the pharmacology of the fibrates is discussed, together with data from recent clinical trials that indicate the potential of the fibrates in general, and bezafibrate specifically, in managing dyslipidemia, with a focus on the use of these drugs in the atherogenic dyslipidemia commonly observed in obese, insulin-resistant and diabetic individuals.
Mechanism of action
Fibrates act as synthetic ligands for PPARα, which are mainly located in the liver, skeletal muscle, heart muscle, and cells in the arterial wall that play a key role in the development of atherosclerosis (Vamecq et al 1999; Tennenbaum et al 2003; Berger et al 2005). PPAR bind as heterodimers with another nuclear receptor, the retinoid X receptor, to specific response elements termed peroxisome proliferator response elements and alter the transcription rate of target genes. A summary of PPARα-mediated gene regulation by fibrates is provided in Table 1.
Table 1.
PPARα-mediated gene regulation by fibrates
| Target gene | Function of gene product | Gene expression |
|---|---|---|
| Lipoprotein lipase | Lipolysis, clearance of triglyceride-rich lipoproteins | Increase |
| Apolipoprotein CIII | Inhibits VLDL clearance | Decrease |
| Apolipoprotein AI | HDL-C formation, reverse cholesterol transport | Increase |
| Apolipoprotein AII | HDL-C formation, reverse cholesterol transport | Increase |
| Acyl CoA synthetase | Fatty acid activation, acyl CoA esters | Increase |
| ABCA1 | HDL-C formation, cellular cholesterol efflux | Increase |
| Fatty acid binding protein | Cellular fatty acid uptake | Increase |
| b-oxidation pathway | Fatty acid oxidation | Increase |
| Acetyl CoA carboxylase | Fatty acid synthesis | Decrease |
| Fibrinogen | Blood clotting | Decrease |
| C-reactive protein | Acute phase reactant | Decrease |
| Interleukin 6 | Acute phase reactant | Decrease |
| Cyclooxygenase-2 | Fatty acid metabolism | Decrease |
| VCAM-1 | Adhesion molecule | Decrease |
Abbreviations: ABCA, ATP binding cassette transporter A1; VCAM-1, vascular cell adhesion molecule-1.
Effect on plasma lipids and lipoproteins
Fibrates reduce plasma levels of triglycerides by 30%–50% and typically increase levels of HDL-C by 5%–15%, depending on lipid phenotype and baseline concentration. Fibrates may also reduce LDL-C by up to 15%–20% although the effect is variable, depending on the underlying lipid abnormality and baseline lipid phenotype (Chapman et al 2006). Recent investigations have revealed that the lipid effects of fibrates are mediated, at least partly, through the transcription of genes encoding for proteins that control lipoprotein metabolism (Figure 1 and Table 1). Activation of PPARα results in increased lipolysis and plasma clearance of atherogenic triglyceride-rich lipoproteins via the activation of lipoprotein lipase and ApoAV and reduced production of the lipoprotein lipase inhibitor ApoCIII (Haubenwallner et al 1995; Staels et al 1995a; Stael et al 1998; Chapman et al 2006). Fibrate therapy also promotes the β-oxidation of fatty acids, thus reducing the availability of free fatty acids for triglyceride synthesis (Schoonjans et al 1996). In addition, de novo fatty acid synthesis is inhibited through reductions in acetyl-CoA carboxylase and fatty acid synthase activity; thereby reducing the availability of fatty acids for triglyceride synthesis (Schoonjans et al 1996a; Schoonjans et al 1996b).
Figure 1.
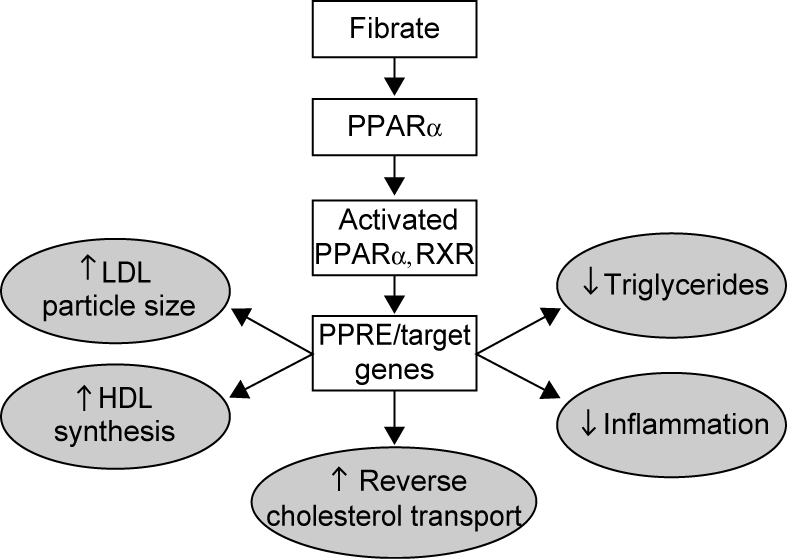
Mechanism of action of fibrates.
Abbreviations: HDL, high-density lipoprotein; LDL, low-density lipoprotein; PPAEα, peroxisome proliferator-activated receptor-α; PPRE, peroxisome proliferator response elements; RXR, retinoid X receptor; ↑ indicates increase; ↓ indicates decrease.
PPARα activation also increases ApoAI and ApoAII synthesis (the major proteins in HDL) (Schoonjans et al 1996a; Schoonjans et al 1996b). Cholesteryl ester transfer protein (CETP) activity was found to be reduced with fibrate therapy (Kiyanagi et al 2006); and the reduction in CETP-mediated transfer of lipid from HDL-C to very (V) LDL may also contribute to the observed increase in HDL-C levels.
Effects on LDL and HDL particle subclasses
Evidence suggests that LDL particle subclasses may differ in atherogenicity, with small, dense LDL associated with greater CHD risk than large, buoyant LDL. The larger particles have a high binding affinity for cellular LDL receptors, and are less susceptible to oxidation. Individuals with the metabolic syndrome and the raised triglycerides-low HDL-C dyslipidemia characteristically have LDL particles that are smaller and contain less cholesterol than average (Berneis et al 2002). As a result, the number of atherogenic LDL particles is frequently elevated in this population, even though LDL-C levels are not. Cardiovascular risk was found to be better explained by concentrations of LDL particles or plasma levels of apolipoprotein B (ApoB), than by LDL-C (Berneis et al 2002; Blake et al 2002; Rizzo et al 2007). Fibrate therapy was shown to alter LDL subclass distribution among normolipemic subjects with predominantly dense LDL-C (Superko et al 2005), resulting in a reduction in small dense LDL and an opposite effect on HDL subclass distribution (Guérin et al 1996). In a substudy of VA-HIT (Otvos et al 2006), treatment with gemfibrozil was associated with an increase LDL size and a reduction in the number of LDL particles by 5%, while raising the number of HDL particles by approximately 10% and of small HDL subclass particles by 21%. Furthermore, concentrations of these LDL and HDL particles achieved with gemfibrozil were shown to be significant, independent, predictors of major cardiac events and helped explain the demonstrated benefit of fibrate therapy in this population. Other studies have shown that treatment with fenofibrate, ciprofibrate, bezafibrate also exert a beneficial effect on LDL subclass distribution. Notably, among the different agents, fenofibrate has been found to be particularly effective in modulating LDL size and subclasses (Rizzo et al 2007).
Non-lipid effects
Fibrates were shown to exert pleiotropic effects in the artery wall. PPARα is involved in the control of the anti-inflammatory response, via inhibition of the transcription factor NFκB (Chapman 2003), and to attenuate the production of pro-inflammatory stimuli such as interleukin 6 and various prostaglandins, as well as the acute phase proteins, including fibrinogen and C-reactive protein (Devchand et al 1996; Staels et al 1998).
Fibrates have favorable effects on coagulation and fibrinolysis. Bezafibrate was shown to reduce levels of fibrinogen by up to 20% (Schonfeld et al 1994), and ciprofibrate and fenofibrate have been shown to increase fibrinolysis and attenuate platelet hyperaggregability in hypercholesterolemic subjects (Simpson et al 1998).
Adverse effects and precautions
Common adverse reactions of fibrate therapy include gastrointestinal (epigastric distress, flatulence, nausea, diarrhea, constipation) and dermatologic (pruritus, urticaria, or erythema) symptoms, whereas reactions related to the musculoskeletal (muscular weakness, pain, and muscle cramps) and neurological (headache, dizziness) systems are less common. Several specific precautions are listed below.
Hematologic changes
Mild hemoglobin, leukocyte and platelet decreases have occurred occasionally following initiation of bezafibrate therapy. However, these levels stabilize during long-term administration. Periodic blood counts are recommended during the first 12 months of administration.
Liver function
Abnormal liver function tests have been observed occasionally during fibrate administration, including elevated transaminases, and decreased or, rarely, increased alkaline phosphatase. However, these abnormalities are reversible upon discontinuation of the drug. Therefore, periodic liver function tests (AST, ALT, and GGT [if originally elevated]) in addition to other baseline tests are recommended after 3–6 months and at least yearly thereafter. Fibrate therapy should be terminated if drug-related abnormalities persist.
Cholelithiasis
Fibrates may increase cholesterol excretion into the bile, and may lead to cholelithiasis. If such is suspected, gallbladder studies are indicated. Fibrate therapy should be discontinued if gallstones are found.
Hepatobiliary disease
In patients with a past history of jaundice or hepatic disorder, fibrates should be used with caution.
Skeletal muscle
Treatment with drugs of the fibrate class has been associated on rare occasions with myositis or rhabdomyolysis, usually in patients with impaired renal function. Myopathy should be considered in any patient with diffuse myalgias, muscle tenderness/weakness, or marked elevations in creatinine phosphokinase (CPK) levels. Patients should be advised to report unexplained muscle pain, tenderness or weakness promptly, particularly if accompanied by malaise or fever. Levels of CPK should be assessed in patients reporting these symptoms, and fibrate therapy should be discontinued if markedly elevated CPK levels (10 times the upper limit of normal) occur or myopathy is diagnosed.
Children
Limited experience is available in children. Therefore, in the absence of adequate information concerning the long-term safety, fibrates should be used with caution in treating this population.
Pregnancy and lactation
Strict birth control procedures must be exercised by women of childbearing potential. If pregnancy occurs despite birth control procedures, fibrate therapy should be discontinued. Women planning a pregnancy should discontinue fibrate therapy several months prior to conception. In the absence of data concerning the presence of fibrates in human breast milk, the drugs should not be used by nursing mothers.
Clinical trials
Fibrates have been in clinical use for over 30 years. Clofibrate was introduced in the 1970s and was followed by gemfibrozil, bezafibrate, fenofibrate and ciprofibrate. Although there have been fewer intervention studies with fibrates than with statins, the majority of available evidence indicate long-term beneficial effects in high-risk patients with the triglyceride-low HDL-C dyslipidemia of the metabolic syndrome, both in primary and secondary intervention settings. A summary of data from major randomized trials of fibrate therapy is provided in Table 2.
Table 2.
Summary of major randomized intervention trials involving fibrate therapy
| Trial | Drug | No. of patients | Baseline lipids mg/dL | End point | Outcome | ||
|---|---|---|---|---|---|---|---|
| TC | TG | HDL-C | |||||
| Primary prevention | |||||||
| WHO | Clofibrate | 10627 | 248 | NR | NR | Nonfatal MI | 25% reduction (p < 0.05) |
| Helsinki Heart Study | Gemfibrozil | 4081 | 270 | 176 | 47 | Nonfatal MI/CHD death | 34% reduction (p < 0.05) |
| Secondary prevention | |||||||
| Newcastle | Clofibrate | 497 | 246 | NR | NR | Fatal/nonfatal MI | 14% reduction (p > 0.10) |
| Edinburgh | Clofibrate | 717 | 265 | NR | NR | CHD events | 53% reduction (p < 0.05) |
| CDP | Clofibrate | 3892 | 250 | NR | NR | Nonfatal MI/CHD death | 8% reduction (p > 0.10) |
| Stockholm Ischaemic Heart Disease | Clofibrate | 555 | 249 | 209 | 48 | Total death | 26% reduction (p < 0.05) |
| VA-HIT | Gemfibrozil | 2531 | 175 | 161 | 32 | Nonfatal MI/CHD death | 22% reduction (p < 0.05) |
| BIP | Bezafibrate | 3090 | 212 | 145 | 35 | Fatal/nonfatal MI/sudden death | 9% reduction (p > 0.10) |
| Leader | Bezafibrate | 1568 | 216 | 189 | 43 | Nonfatal MI/CHD death/stroke | 4% reduction (p > 0.10) |
| Primary/secondary prevention | |||||||
| FIELD | Fenofibrate | 2795 | 195 | 119 | 42 | Nonfatal MI/CHD death | 11% reduction (p > 0.10) |
Notes: See text for abbreviations of study names.
Abbreviations: CHD, coronary heart disease; MI, myocardial infarction; NR, not reported.
Primary prevention
Clofibrate and gemfibrozil have been investigated in a primary prevention setting in the general population (Table 2). In the WHO Clofibrate Study (Committee of Principal Investigators 1978), 10,627 men without CHD treated with clofibrate for 5 years showed a 25% reduction in myocardial infarction (MI) and a 20% reduction in first major coronary event. These beneficial results were, however, overshadowed by the reported increase in mortality from non-cardiovascular disease, particularly cancer (Committee of Principal Investigators 1978), although subsequent follow-up showed a smaller difference in cancer incidence between the clofibrate and control groups (Committee of Principal Investigators 1978).
The Helsinki Heart Study (Frick et al 1987) was a placebo-controlled study, including more than 4,000 men at moderate risk of CHD with a range of lipid patterns, but generally with an elevated LDL-C level. The study showed that treatment with gemfibrozil (1,200 mg/day), resulted in an 11% decrease in LDL-C, a 35% decrease in triglycerides and an 11% increase in HDL-C, compared with placebo. These lipid changes were associated with a 34% reduction in major coronary events at five years (p < 0.02), as well as a 37% reduction in non-fatal MI (p < 0.05). No significant difference in overall mortality was observed between the two groups.
Secondary prevention
The value of fibrate therapy in the secondary prevention setting is supported by numerous studies (Table 2). Two small trials of clofibrate therapy, Newcastle (Trial of clofibrate in the treatment of ischemic heart disease 1971) and Edinburgh (Trial of clofibrate in the treatment of ischemic heart disease), failed to find a significant reduction in CHD deaths, but did find that clofibrate prevented nonfatal-MI to a variable extent. The Coronary Drug Project (CDP) (Clofibrate and niacin in coronary heart disease 1975) was a larger secondary prevention trial that showed a non-significant reduction in CHD events with clofibrate therapy. However, a late lipid-lowering therapy effect was observed in an extended follow-up of the trial (Canner et al 1986), nine years after termination of the trial. The authors attributed this to the transformation of an early favorable effect of niacin in decreasing nonfatal reinfarction, into a mortality benefit over subsequent years. In the Stockholm Ischaemic Heart Disease Secondary Prevention Study (Carlson et al 1988), combination treatment with clofibrate (2 g/day) and nicotinic acid (3 g/day) was investigated in 555 post-MI patients (279 received active treatment). Compared with a control group, active treatment reduced triglycerides by 19% and significantly reduced total and CHD mortality by 26% (p < 0.05) and 36% (p < 0.01), respectively. Notably, CHD risk reduction was only evident in patients with triglyceride levels higher than the median value at baseline; subjects in whom triglyceride levels were reduced by more than 30% (over 40% of treated patients) experienced a 60% reduction in CHD mortality (p < 0.01). Subsequently, VA-HIT (Rubins et al 1999), involving 2,531 men with established CHD, acceptable LDL-C levels, low HDL-C, and elevated triglycerides (Table 2), showed that treatment with gemfibrozil produced a significant 22% reduction in the risk of the primary end point of CHD death or non-fatal MI, as well as CHD mortality. It is noteworthy that 25% of patients enrolled in VA-HIT had diagnosed diabetes and over 50% of patients had features of the metabolic syndrome. The BIP study (The BIP Study Group 2000; also described separately in the following section) comprised 3090 patients allocated to bezafibrate (400 mg/day) or placebo. A significant reduction in plasma triglyceride levels and an increase in HDL-C were observed in this study although there was no significant difference in the five-year coronary event rate compared with placebo (9.4% reduction, p = 0.26). However, bezafibrate was shown to be particularly beneficial among patients with elevated triglycerides (>200 mg/dL) at baseline, in whom a 40% reduction in coronary risk at five years was observed. The Lower Extremity Arterial Disease Event Reduction (LEADER) trial (Meade et al 2002) enrolled 1568 elderly men with lower extremity arterial disease, and showed that bezafibrate (400 mg/day) produced a 40% reduction in non-fatal coronary events (p = 0.05), although there was no significant reduction in the combined end point of CHD and stroke.
Angiographic studies have demonstrated the beneficial effect of fibrate therapy on atherosclerosis progression. The Bezafibrate Coronary Atherosclerosis Intervention Trial ([BECAIT] Ericsson et al 1996), was a relatively small trial designed to evaluate the effects of treatment with bezafibrate on angiographic disease in 92 young male MI survivors. The study showed a significant reduction in the progression of coronary atherosclerosis, as assessed by the change in minimum lumen diameter, in patients treated with bezafibrate compared with placebo. In the Lopid Coronary Angiography Trial (Frick et al 1997), 395 men with low HDL-C and LDL-C levels who had previously undergone CABG, were treated with gemfibrozil (1200 mg/day). The study medication was associated with significantly decreased progression of coronary artery disease in native coronary segments compared with placebo.
Type 2 diabetes
The pharmacological profile of the fibrates, described above, suggests that these agents may prove useful in correcting the atherogenic dyslipidemia typically associated with type 2 diabetes and the metabolic syndrome.
Subgroup analyses of data from the Helsinki Heart Study (Koskinen et al 1992) and VA-HIT (Rubins et al 2002) showed that among patients with type 2 diabetes, treatment with gemfibrozil was associated with a significant reduction in the risk of coronary events that ranged from 68% (Koskinen et al 1992) in the primary intervention setting to 32% (Rubins et al 2002) in the secondary prevention setting. In the St. Mary’s, Ealing, Northwick Park Diabetes Cardiovascular Prevention (SENDCAP) study (Elkeles et al 1998), 164 type 2 diabetes patients without a history of CHD, treated with bezafibrate (400 mg/day) for three years, had a lower CHD event rate compared with placebo (7% vs. 23%, p = 0.01). There was, however, no difference between the groups in progression of carotid and femoral artery atherosclerosis, as assessed by changes in intima-media thickness.
More recently, the Diabetes Atherosclerosis Intervention Study ([DAIS] Vakkilainen et al 2003), involving 418 patients with type 2 diabetes has provided clear evidence of the value of fibrate therapy in diabetic patients. Treatment with micronised fenofibrate, at a dose of 200 mg/day for at least 3 years, was associated with a 40% reduction in angiographic progression of coronary artery disease, as well as a 23% reduction in the cardiovascular event rate, compared with placebo. These beneficial effects were associated with reduction in the levels of triglycerides (by 29%) and LDL-C (by 5%), and an increase in HDL-C (by 7%) and LDL particle size (Vakkilainen et al 2003).
The Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study (The FIELD study investigators 2005) evaluated the role of fibrate therapy in preventing cardiovascular events in patients with type 2 diabetes, considered at increased risk for CHD. After a mean follow-up of 5 years, therapy with fenofibrate was associated wuth a non-significant 11% reduction in the primary end point of nonfatal MI or CHD death. However, total cardiovascular events, corresponding to the secondary end point, were significantly reduced with fenofibrate by 11% (p = 0.035), mainly due to significant reductions in non-fatal MI (by 24%, p = 0.01) and coronary revascularization (by 21%, p = 0.003). Treatment effects were larger in patients without cardiovascular disease than in those with a history of cardiovascular disease. Notably, end points that concerned microangiographic manifestations of diabetes were significantly improved by fenofibrate treatment; these included a reduction in the need for laser treatment for retinopathy and reduction in the progression of albuminuria.
Bezafibrate therapy: recent data from the BIP trial
Bezafibrate was the first clinically tested pan – (alpha, beta/delta, gamma) PPAR activator (Peters et al 2000; The BIP Study Group 2000; Cobrero et al 2001; Poirier 2001; Vazquez 2001), and is a sole pan PPAR activator with more than a quarter of a century of a therapeutic experience with a good safety profile. The BIP study was designed to determine whether bezafibrate would reduce CHD mortality and nonfatal MI in CHD patients with low HDL-C and LDL-C in a moderate range (The BIP Study Group 2000). The study demonstrated fewer cardiac events with bezafibrate (13.6%) as compared with placebo (15.0%) during a 6.2-year mean follow-up period. However, event rate was not significantly reduced (p = 0.26) despite reductions of 5% in LDL-C, 16% in triglyceride, and 10% in fibrinogen levels, together with an increase of 14% in HDL-C levels compared with placebo. A post hoc analysis in patients with high baseline triglycerides (200 mg/dL) revealed that the reduction in the cumulative probability of the primary end point by bezafibrate was 39.5% (p = 0.02) in that subgroup.
There are several explanations for the differences in outcome between VA-HIT and BIP in addition to the difference in the type of fibrate drug. First, LDL-C levels in BIP were in a moderately high range at baseline (averaging 149 mg/dL), necessitating a relatively large prescription rate of “off-trial” lipid lowering therapies, mainly in the placebo group (15% of placebo-allocated patients received active lipid therapy by the end of the study), thereby attenuating the margin of difference in outcome between the placebo and active treatment groups. Second, differences in patient characteristics might have affected outcome in the 2 trials: BIP excluded individuals with diabetes and higher levels of blood glucose as well as recruited fewer individuals with components of the metabolic syndrome, in whom the efficacy of fibrate therapy is enhanced, resulting in a lower event rate during follow-up. Despite these differences in trial design, recent subgroup analyses from BIP revealed several important findings regarding the long-term cardiovascular effects of bezafibrate that are described below.
Relationship between on-treatment increments in serum HDL-cholesterol levels and cardiac mortality
In a subanalysis of BIP we evaluated the relationship between changes in serum levels of HDL-C under bezafibrate therapy and cardiac mortality in enrolled subjects (Goldenberg et al 2006). Long-term follow-up revealed that cardiac mortality was significantly reduced with increasing increments of on-treatment HDL-C change (9.5%, 6.6%, and 6.3% of patients died of cardiac causes in tertiles 1, 2, and 3 of HDL-C increase, respectively [p for trend = 0.02]); Figure 2). The magnitude of on-treatment HDL-C increment in the trial was shown to be independently associated with reduced risk of cardiac death: patients who did not respond to bezafibrate therapy by increasing serum HDL-C levels (tertile 1 HDL-C change) did not experience a survival benefit with bezafibrate therapy (hazard ratio: 1.05 [95% CI 0.74–1.47]), whereas responders (tertile 3 HDL-C increase) experienced a significant 35% reduction in the risk of cardiac death with bezafibrate (HR = 0.65 [95% CI 0.43–0.97]). Furthermore, the risk of cardiac mortality was reduced by 27% for every 5 mg/dL increase in on-treatment HDL-C change (p < 0.001). Thus, these findings demonstrate an independent association between a rise in HDL-C with bezafibrate therapy and a reduction in cardiac mortality, and suggest that monitoring short-term response to bezafibrate therapy through HDL-C change may indicate the potential long-term benefit of the drug.
Figure 2.
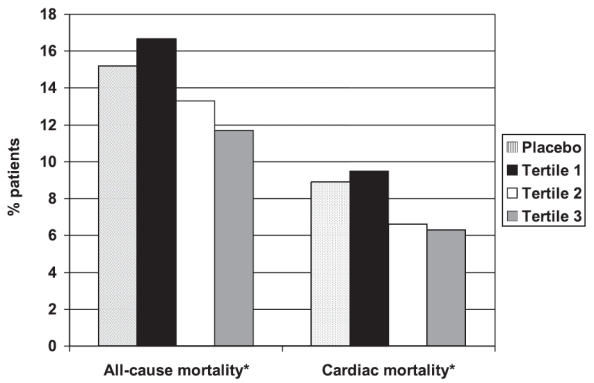
All-cause and cardiac mortality rates in tertiles of HDL-C change in the bezafibrate group compared to the placebo group.
*p for trend < 0.05
Reprinted with permission from Goldenberg et al (2006).
Long-term cardioprotective effects of bezafibrate therapy
We have recently completed an evaluation of the effect of bezafibrate therapy over an extended follow-up period of BIP (Goldenberg et al 2007). After discontinuation of study medication, patients were observed for coronary events for an additional period, bringing the total follow-up time in the current analysis to a mean of 8.2 years (median: 7.9 years; interquartile range: 7.2–8.7 years). Our data demonstrate that during long-term follow-up bezafibrate therapy was associated with a significant 17% reduction (p = 0.03) in the combined end point of cardiac death or nonfatal MI (Figure 3). Using interaction-term analysis, we have also shown that the benefit of bezafibrate therapy was pronounced (18% risk reduction; p = 0.03) without- or before- treatment with off-trial lipid lowering therapies was initiated during follow-up, and attenuated (HR = 1.05; p = 0.85) after therapy with off-trial lipid lowering therapies was initiated during the observation period. These new data suggest that bezafibrate therapy is associated with significant long-term cardiovascular protection that may extend beyond the period of active drug therapy.
Figure 3.
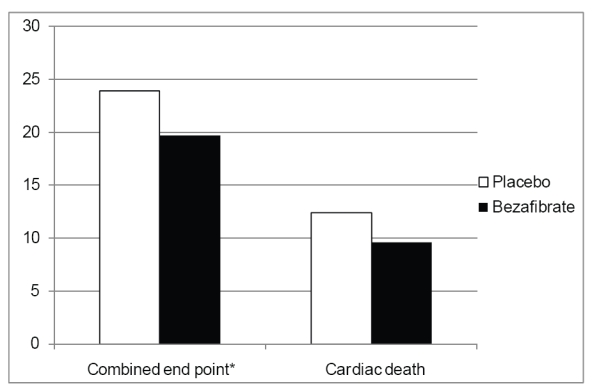
Kaplan-Meier cumulative probability of outcome the combined endpoint of cardiac death or nonfatal myocardial infarction, and cardiac death separately, during an extended follow-up of the BIP trial.
*p < 0.05.
Effect of bezafibrate therapy in patients with the metabolic syndrome
A post hoc analysis of BIP evaluated the effect of bezafibrate on the incidence of MI during long-term follow-up in enrolled patients with metabolic syndrome (Tenenbaum et al 2005). Patients who displayed at least 3 of the following 5 risk factors were considered to have metabolic syndrome: (1) a fasting glucose level of 110 mg/dL (6.11 mmol/L); (2) a triglyceride level of 150 mg/dL (1.70 mmol/L); (3) HDL-C level less than 40 mg/dL in men or less than 50 mg/dL in women; (4) a systolic blood pressure of 130 mm Hg or diastolic blood pressure of 85 mm Hg; and (5) a body mass index of 28.0 kg/m2. During the course of the extended follow-up period bezafibrate was associated with a significant reduction in the risk of MI, and of the combined primary endpoint of BIP, which consisted of nonfatal MI or sudden cardiac death, among patients with at least 3 risk factors for the metabolic syndrome (Figure 4). Furthermore, the benefit of bezafibrate therapy was shown to be enhanced in patients with augmented features (at least 4 risk factors) of the metabolic syndrome (56% reduction of cardiac mortality during 8-year follow-up), whereas the benefit of bezafibrate was attenuated in patients without features of the metabolic syndrome.
Figure 4.
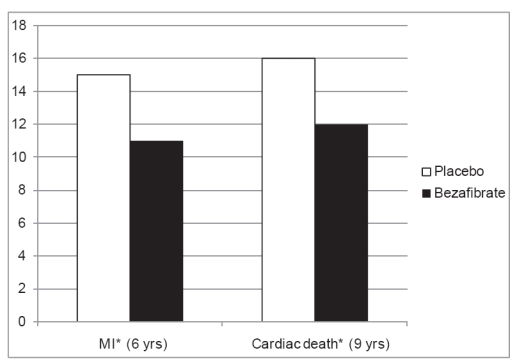
Outcome of patients with the metabolic syndrome in BIP.
*p < 0.05.
Abbreviation: MI, myocardial infarction.
Bezafibrate therapy for the prevention of type 2 diabetes mellitus in patients with coronary artery disease
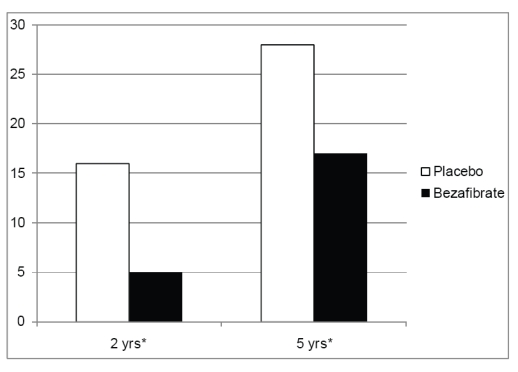
In a recent study from BIP we evaluated whether pharmacological intervention with bezafibrate that influences primary lipid metabolism can also delay development of type 2 diabetes (Tenenbaum et al 2004). The study comprised 303 non-diabetic study patients with a fasting blood glucose level of 110 to 125 mg/dL, who were allocated to either 400 mg bezafibrate (n = 156) or placebo (n = 147 patients) once a day. During follow-up, development of new-onset diabetes occurred at a significantly lower rate among bezafibrate allocated patients as compared with placebo (Figure 5). The mean time until onset of new diabetes was significantly delayed among the former subgroup as compared with the latter: 4.6 ± 2.3 versus 3.8 ± 2.6 years, respectively, (p = 0.004), and bezafibrate therapy was shown to be associated with a significant 30% reduction in the risk of new diabetes development. These findings suggest that treatment with fibrates may reduce the incidence and delay the onset of type 2 diabetes in patients with impaired fasting glucose, and need to be validated in future prospective studies.
Figure 5.
Two- and five-year Kaplan-Meier cumulative probability of diabetes incidence (in accordance with time of diagnosis after annual fasting blood glucose measurements) in the bezafibrate and placebo groups of the BIP trial.
*p < 0.05.
Conclusions and future perspectives
With the epidemic proportions of obesity, especially high-risk abdominal obesity, type 2 diabetes and the metabolic syndrome, it is important to consider lipid lowering therapies that affect the dyslipidemic profile found in these patients, independently of LDL-C. Data from recent major clinical trials have shown that fibrates produce beneficial effects on atherogenic lipoproteins through activation of PPARα, and confer long-term cardiovascular protection, both in the primary- and secondary- prevention setting. Therefore, this mode of lipid-lowering therapy should be considered part of an optimal management plan in a significant proportion of patients with a high-risk metabolic profile.
Recent data from the BIP trial have demonstrated several important findings regarding the benefit of this fibric acid derivative in CHD patients, including (1) long-term cardioprotective effects that extend beyond the period of active drug therapy; (2) enhanced efficacy in “responders” (patients in whom HDL-C levels increase in response to bezafibrate therapy; (3) enhanced efficacy in patients with elevated triglycerides and those with clinical and laboratory features suggestive of the metabolic syndrome; and (4) a significant reduction in the incidence of new onset of type 2 diabetes in patients with impaired fasting glucose.
Further studies are needed to compare the benefit of statin versus fibrate therapy, or the combination of both classes of drugs, in different subpopulations of patients with a high-risk metabolic profile.
References
- Baigent C, Keech A, Kearney PM, et al. Efficacy and safety of cholesterol – lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. 2005;366:1267–8. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- Barter P, Gotto AM, LaRosa JC, et al. Treating to New Targets Investigators. HDL cholesterol, very low levels of LDL cholesterol, and cardiovascular events. N Engl J Med. 2007;357:1301–10. doi: 10.1056/NEJMoa064278. [DOI] [PubMed] [Google Scholar]
- Berger JP, Akiyama TE, Meinke PT. PPARs: Therapeutic targets for metabolic disease. Trends Pharmacol Sci. 2005;26:244–51. doi: 10.1016/j.tips.2005.03.003. [DOI] [PubMed] [Google Scholar]
- Berneis KK, Krauss RM. Metabolic origins and clinical significance of LDL heterogeneity. J Lipid Res. 2002;43:1363–79. doi: 10.1194/jlr.r200004-jlr200. [DOI] [PubMed] [Google Scholar]
- Bierman EL. Atherogenesis in diabetes. Arterioscler Thromb Vasc Biol. 1992;12:647–56. doi: 10.1161/01.atv.12.6.647. [DOI] [PubMed] [Google Scholar]
- Blake GJ, Otvos JD, Rifai N, Ridker PM. LDL particle concentration and size as determined by NMR spectroscopy as predictors of cardiovascular disease in women. Circulation. 2002;106:1930–7. doi: 10.1161/01.cir.0000033222.75187.b9. [DOI] [PubMed] [Google Scholar]
- Cabrero A, Alegret M, Sanchez RM, et al. Bezafibrate reduces mRNA levels of adipocyte markers and increases fatty acid oxidation in primary culture of adipocytes. Diabetes. 2001;50:1883–90. doi: 10.2337/diabetes.50.8.1883. [DOI] [PubMed] [Google Scholar]
- Canner PL, Berge KG, Wenger NK, et al. Fifteen year mortality in Coronary Drug Project patients: long-term benefit with niacin. J Am Coll Cardiol. 1986;8:1245–55. doi: 10.1016/s0735-1097(86)80293-5. [DOI] [PubMed] [Google Scholar]
- Carlson LA, Rosenhamer G. Reduction of mortality in the Stockholm Ischaemic Heart Disease Secondary Prevention Study by combined treatment with clofibrate and nicotinic acid. Acta Med Scand. 1988;223:405–18. doi: 10.1111/j.0954-6820.1988.tb15891.x. [DOI] [PubMed] [Google Scholar]
- Castelli WP. Cholesterol and lipids in the risk of coronary artery disease: the Framingham Heart Study. Can J. 1988;4(Suppl A):5A–10A. [PubMed] [Google Scholar]
- Chapman MJ. Fibrates: therapeutic review. Br J Diabetes Vasc Dis. 2006;6:11–18. [Google Scholar]
- Clofibrate and niacin in coronary heart disease. JAMA. 1975;231:360–81. [PubMed] [Google Scholar]
- Committee of Principal Investigators. A co-operative trial in the primary prevention of ischaemic heart disease using clofibrate. 1978. Report from the Committee of Principal Investigators. Br Heart J. 1978;40:1069–118. doi: 10.1136/hrt.40.10.1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Committee of Principal Investigators. WHO cooperative trial on primary prevention of ischemic heart disease using clofibrate to lower serum cholesterol: final mortality follow-up. Lancet. 1984;ii:600–04. [PubMed] [Google Scholar]
- Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. JAMA. 1998;279:1615–22. doi: 10.1001/jama.279.20.1615. [DOI] [PubMed] [Google Scholar]
- Devchand P, Keller H, Peters J, et al. The PPARα-leucotriene B4 pathway to inflammation control. Nature. 1996;384:39–43. doi: 10.1038/384039a0. [DOI] [PubMed] [Google Scholar]
- Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. Lancet. 2005;366:1849–61. doi: 10.1016/S0140-6736(05)67667-2. [DOI] [PubMed] [Google Scholar]
- Elkeles RS, Diamond JR, Poulter C, et al. Cardiovascular outcomes in type 2 diabetes. A double-blind placebo-controlled study of bezafibrate: the St. Mary’s, Ealing, Northwick Park Diabetes Cardiovascular Prevention (SENDCAP) Study. Diabetes Care. 1998;21:641–8. doi: 10.2337/diacare.21.4.641. [DOI] [PubMed] [Google Scholar]
- Ericsson CG, Hamsten A, Nilsson J, et al. Angiographic assessment of bezafibrate on progression of coronary artery disease in young male postinfarction patients. Lancet. 1996;347:849–53. doi: 10.1016/s0140-6736(96)91343-4. [DOI] [PubMed] [Google Scholar]
- Frick MH, Syvanne M, Nieminen MS, et al. Lopid Coronary Angiography Trial (LOCAT) Study Group. Prevention of angiographic progression of coronary and vein-graft atherosclerosis by gemfibrozil after coronary bypass surgery in men with low levels of HDL cholesterol. Circulation. 1997;96:2137–43. doi: 10.1161/01.cir.96.7.2137. [DOI] [PubMed] [Google Scholar]
- Fruchart JC, Brewer HB, Jr, Leitersdorf E Fibrate Consensus Group. Consensus for the use of fibrates in the treatment of dyslipoproteinemia and coronary heart disease. Am J Cardiol. 1998;81:912–7. doi: 10.1016/s0002-9149(98)00010-1. [DOI] [PubMed] [Google Scholar]
- Genest JJ, McNamara JR, Ordovas JM, et al. Lipoprotein cholesterol, apolipoprotein A-I and B and lipoprotein (a) abnormality in men with premature coronary heart disease. J Am Coll Cardiol. 1992;19:792–802. doi: 10.1016/0735-1097(92)90520-w. [DOI] [PubMed] [Google Scholar]
- Goldenberg I, Goldbourt U, Boyco V, et al. Relationship between on-treatment increments in serum HDL levels and cardiac events in patients with coronary heart disease: an extended follow-up of the Bezafibrate Infarction Prevention Trial. Am J Cardiol. 2006;97:466–71. doi: 10.1016/j.amjcard.2005.09.078. [DOI] [PubMed] [Google Scholar]
- Goldenberg I, Benderly M, Goldbourt U. Secondary prevention with bezafibrate therapy for the treatment of dyslipidemia. An extended follow-up of the Bezafbrate Infarction Prevention Study. J Am Coll Cardiol. 2007 doi: 10.1016/j.jacc.2007.09.048. In press. [DOI] [PubMed] [Google Scholar]
- Gordon T, Castelli WP, Hjortland MC, et al. High density lipoprotein as a protective factor against coronary heart disease: the Framingham Study. Am J Med. 1977;62:707–14. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- Gotto AM., Jr Triglyceride as a risk factor for coronary artery disease. Am J Cardiol. 1998;82:22Q–5Q. doi: 10.1016/s0002-9149(98)00770-x. [DOI] [PubMed] [Google Scholar]
- Guérin M, Bruckert E, Dolphin PJ, et al. Fenofibrate reduces plasma cholesteryl ester transfer from HDL to VLDL and normalizes the atherogenic, dense LDL profile in combined hyperlipidemia. Arterioscler Thromb Vasc Biol. 1996;6:763–72. doi: 10.1161/01.atv.16.6.763. [DOI] [PubMed] [Google Scholar]
- Haubenwallner S, Essenburg AD, Barnett BC, et al. Hypolipidemic activity of select fibrates correlates to changes in hepatic apolipoprotein C-III expression: a potential physiologic basis for their mode of action. J Lipid Res. 1995;12:2541–51. [PubMed] [Google Scholar]
- Howard BV. Lipoprotein metabolism in diabetes mellitus. J Lipid Res. 1987;28:613–28. [PubMed] [Google Scholar]
- Jeppesen J, Hein HO, Suadicani P, et al. Relation of high TG-low HDL cholesterol and LDL cholesterol to the incidence of ischemic heart disease: an 8-year follow-up in the Copenhagen Male Study. Arterioscler Thromb Vasc Biol. 1997;17:1114–20. doi: 10.1161/01.atv.17.6.1114. [DOI] [PubMed] [Google Scholar]
- Kiyanagi T, Miyazaki T, Kume A, et al. Decrease in CETP activity by fenofibrate may increase LDL particle size measured by HPLC method in patients with coronary artery disease. Atherosclerosis. 2006;7(3 Suppl):559. [Google Scholar]
- Koskinen P, Manttari M, Manninen V, et al. Coronary heart disease in NIDDM patients in the Helsinki Heart Study. Diabetes Care. 1992;15:820–5. doi: 10.2337/diacare.15.7.820. [DOI] [PubMed] [Google Scholar]
- Laakso M, Sarlund H, Mykkanen L. Insulin resistance is associated with lipid and lipoprotein abnormalities in subjects with varying degrees of glucose tolerance. Arteriosclerosis. 1990;10:223–31. doi: 10.1161/01.atv.10.2.223. [DOI] [PubMed] [Google Scholar]
- Lamarche B, Tchernof A, Mauriège P, et al. Fasting insulin and apolipoprotein B levels and low-density lipoprotein particle size as risk factors for ischemic heart disease. JAMA. 1998;279:1955–61. doi: 10.1001/jama.279.24.1955. [DOI] [PubMed] [Google Scholar]
- Manninen V, Tenkanen L, Koshinen P, et al. Joint effects of serum triglyceride and LDL cholesterol and HDL cholesterol concentrations on coronary heart disease risk in the Helsinki Heart Study: implications for treatment. Circulation. 1992;85:37–45. doi: 10.1161/01.cir.85.1.37. [DOI] [PubMed] [Google Scholar]
- Meade T, Zuhrie R, Cook C, et al. Bezafibrate in men with lower extremity arterial disease: randomised controlled trial. BMJ. 2002;25:1139–44. doi: 10.1136/bmj.325.7373.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, Aoyama T, Burns AM, et al. Bezafibrate is a dual ligand for PPARalpha and PPARbeta: studies using null mice. Biochim Biophys Acta. 2005;1632:80–9. doi: 10.1016/s1388-1981(03)00065-9. [DOI] [PubMed] [Google Scholar]
- Poirier H, Niot I, Monnot MC, et al. Differential involvement of peroxisome-proliferator-activated receptors alpha and delta in fibrate and fatty-acid-mediated inductions of the gene encoding liver fatty-acid-binding protein in the liver and the small intestine. Biochem J. 2001;355:481–8. doi: 10.1042/0264-6021:3550481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzo M, Berneis K. The clinical significance of the size of low-density-lipoproteins and the modulation of subclasses by fibrates. Curr Med Res Opin. 2007;23:1103–11. doi: 10.1185/030079907x187892. [DOI] [PubMed] [Google Scholar]
- Rizzo M, Rini GB, Berneis K. Effects of statins, fibrates, rosuvastatin and ezetimibe beyond cholesterol: the modulation of LDL size and subclasses in high-risk patients. Adv Ther. 2007;24:575–82. doi: 10.1007/BF02848780. [DOI] [PubMed] [Google Scholar]
- Rubins HB, Robins SJ, Collins D, et al. Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial Study Group. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. N Engl J Med. 1999;341:410–8. doi: 10.1056/NEJM199908053410604. [DOI] [PubMed] [Google Scholar]
- Rubins HB, Robins SJ, Collins D, et al. Diabetes, plasma insulin, and cardiovascular disease. Subgroup analysis from the Department of Veterans Affairs High-density Lipoprotein Intervention Trial (VA-HIT) Arch Intern Med. 2002;162:2597–604. doi: 10.1001/archinte.162.22.2597. [DOI] [PubMed] [Google Scholar]
- Sacks FM, Pfeffer MA, Moye LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. N Engl J Med. 1996;335:1001–9. doi: 10.1056/NEJM199610033351401. [DOI] [PubMed] [Google Scholar]
- Schoonjans K, Staels B, Auwerx J. The peroxisome proliferator activated receptors (PPARs) and their effects on lipid metabolism and adipocyte differentiation. Biochim Biophys Acta. 1996a;1302:93–109. doi: 10.1016/0005-2760(96)00066-5. [DOI] [PubMed] [Google Scholar]
- Schoonjans K, Staels B, Auwerx J. Role of the peroxisome proliferator-activated receptor (PPAR) in mediating the effects of fibrates and fatty acids on gene expression. J Lipid Res. 1996b;37:907–25. [PubMed] [Google Scholar]
- Schonfeld G. The effects of fibrates on lipoprotein and hemostatic coronary risk factors. Atherosclerosis. 1994;111:161–74. doi: 10.1016/0021-9150(94)90090-6. [DOI] [PubMed] [Google Scholar]
- Scottish Society of Physicians. Ischaemic heart disease: a secondary prevention trial using clofibrate. Report by a research committee of the Scottish Society of Physicians. BMJ. 1971;4:775–84. [PMC free article] [PubMed] [Google Scholar]
- Simpson IA, Lorimer AR, Walker ID, et al. Effect of ciprofibrate on platelet aggregation and fibrinolysis in patients with hypercholesterolaemia. Thromb Haemostat. 1989;54:442–4. [PubMed] [Google Scholar]
- Sever PS, Dahlöf B, Poulter NR, et al. Prevention of coronary and stroke events with atorvastatin in hypertensive patients who have average or lower-than-average cholesterol concentrations, in the Anglo-Scandinavian Cardiac Outcomes Trial-Lipid Lowering Arm (ASCOT-LLA): a multicentre randomised controlled trial. Lancet. 2003;361:1149–58. doi: 10.1016/S0140-6736(03)12948-0. [DOI] [PubMed] [Google Scholar]
- Shepherd J, Cobbe SM, Ford I, et al. Prevention of coronary heart disease with pravastatin in men with hypercholesterolemia. N Engl J Med. 1995;333:1301–7. doi: 10.1056/NEJM199511163332001. [DOI] [PubMed] [Google Scholar]
- Staels B, Koenig W, Habib A, et al. Activation of human aortic smooth muscle cells is inhibited by PPARα but not by PPARγ activators. Nature. 1998;393:790–3. doi: 10.1038/31701. [DOI] [PubMed] [Google Scholar]
- Superko RH, Berneis KK, Williams P, et al. Gemfibrozil reduces small low-density lipoprotein more in normolipemic subjects classified as low-density lipoprotein pattern B compared with pattern A. Am J Cardiol. 2005;96:1266–72. doi: 10.1016/j.amjcard.2005.06.069. [DOI] [PubMed] [Google Scholar]
- Tchernof A, Lamarche B, Prud’homme D, et al. The dense LDL phenotype: association with plasma lipoprotein levels, visceral obesity, and hyperinsulinemia in men. Diabetes Care. 1996;19:629–37. doi: 10.2337/diacare.19.6.629. [DOI] [PubMed] [Google Scholar]
- Tenenbaum A, Fisman EZ, Motro M. Metabolic syndrome and type 2 diabetes mellitus: focus on peroxisome proliferator activated receptors (PPAR) Cardiovasc Diabetol. 2003;2:4. doi: 10.1186/1475-2840-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tenenbaum A, Motro M, Fisman, et al. Bezafibrate for the secondary prevention of myocardial infarction in patients with metabolic syndrome. Arch Intern Med. 2005;165:1154–60. doi: 10.1001/archinte.165.10.1154. [DOI] [PubMed] [Google Scholar]
- Tenenbaum A, Motro M, Fisman EZ, et al. Peroxisome proliferator-activated receptor ligand bezafibrate for prevention of type 2 diabetes mellitus in patients with coronary artery disease. Circulation. 2004;109:2197–202. doi: 10.1161/01.CIR.0000126824.12785.B6. [DOI] [PubMed] [Google Scholar]
- The BIP Study Group. Secondary prevention by raising HDL cholesterol and reducing triglycerides in patients with coronary artery disease. Circulation. 2000;102:21–7. doi: 10.1161/01.cir.102.1.21. [DOI] [PubMed] [Google Scholar]
- The FIELD study investigators. Effect of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomized controlled trial. Lancet. 2005;67:667–2. doi: 10.1016/S0140-6736(05)67667-2. [DOI] [PubMed] [Google Scholar]
- Trial of clofibrate in the treatment of ischaemic heart disease. Five-year study by a group of physicians of the Newcastle upon Tyne region. BMJ. 1971;4:767–75. [PMC free article] [PubMed] [Google Scholar]
- Vakkilainen J, Steiner G, Ansquer JC, et al. Relationships between low-density lipoprotein particle size, plasma lipoproteins, and progression of coronary artery disease: the Diabetes Atherosclerosis Intervention Study (DAIS) Circulation. 2003;107:1733–7. doi: 10.1161/01.CIR.0000057982.50167.6E. [DOI] [PubMed] [Google Scholar]
- Vamecq J, Latruffe N. Medical significance of peroxisome proliferator-activated receptors. Lancet. 1999;354:141–8. doi: 10.1016/S0140-6736(98)10364-1. [DOI] [PubMed] [Google Scholar]
- Vazquez M, Roglans N, Cabrero A, et al. Bezafibrate induces acyl-CoA oxidase mRNA levels and fatty acid peroxisomal beta-oxidation in rat white adipose tissue. Mol Cell Biochem. 2001;216:71–8. doi: 10.1023/a:1011060615234. [DOI] [PubMed] [Google Scholar]
- Yaari S, Goldbourt U, Even-Zohar S, et al. Associations of serum high density lipoprotein and total cholesterol with total cardiovascular and cancer mortality in a 7-year prospective study of 10,000 men. Lancet. 1981;1:1011–5. doi: 10.1016/s0140-6736(81)92184-x. [DOI] [PubMed] [Google Scholar]