

Abstract
Endoplasmic reticulum (ER) stress is involved in the pathogenesis of several diseases including Alzheimer disease and Parkinson disease. Many recent studies have shown that ER stress is related to the pathogenesis of diabetes mellitus, and with the death of pancreatic β-cells, insulin resistance, and the death of the vascular cells in the retina. Diabetic retinopathy is a major complication of diabetes and results in death of both neural and vascular cells. Because the death of the neurons directly affects visual function, the precise mechanism causing the death of neurons in early diabetic retinopathy must be determined. The ideal therapy for preventing the onset and the progression of diabetic retinopathy would be to treat the factors involved with both the vascular and neuronal abnormalities in diabetic retinopathy. In this review, we present evidence that ER stress is involved in the death of both retinal neurons and vascular cells in diabetic eyes, and thus reducing or blocking ER stress may be a potential therapy for preventing the onset and the progression of diabetic retinopathy.
Keywords: endoplasmic reticulum stress, diabetic retinopathy, vascular cell death, neuronal cell death
Introduction
Diabetic retinopathy is a major complication in patients with diabetes and it can lead to severe visual decrease in a high percentage of diabetic patients (Oshitari 2006; Oshitari and Roy 2007). Although the precise mechanism(s) for the onset and progression of diabetic retinopathy has still not been determined, recent studies have indicated that neuronal and vascular abnormalities are associated with the pathogenesis of early diabetic retinopathy (Barber et al 1998; Takano et al 1999; Asnaghi et al 2003; Martin et al 2004; Oshitari et al 2005). The neuronal abnormalities in the early stage of diabetic retinopathy are difficult to observe and evaluate by routine clinical tests, but ophthalmologists should consider neuronal abnormalities, including the death of retinal ganglion cell (RGCs), when evaluating eyes with diabetic retinopathy. This is important because the death of retinal neurons is irreversible and directly affects the visual function (Oshitari 2006; Oshitari and Roy 2007).
The endoplasmic reticulum (ER) is a critical intracellular organelle, which has several vital functions such as protein synthesis (Chevet et al 2001), protein transport (Palade 1975), and acts as a reservoir of Ca2+ (Nielsen and Podolsky 1972). The accumulation of unfolded proteins or an upset in the Ca2+ homeostasis in the ER will activate the ER stress response, eg, the unfolded protein response (UPR) and the ER overload response (Rao et al 2004; Lindholm et al 2006). Most importantly, ER stress activates several cell death pathways including the caspase-12-dependent pathway (Nakagawa et al 2000), apoptosis signal-regulating kinase 1 (ASK1) – c-Jun N-terminal kinase (JNK) pathway (Nishitoh et al 2002) and the PKR-like endoplasmic reticulum kinase (PERK) – C/ERB homologous protein (CHOP) pathway (Ma et al 2002) (Figure 1).
Figure 1.
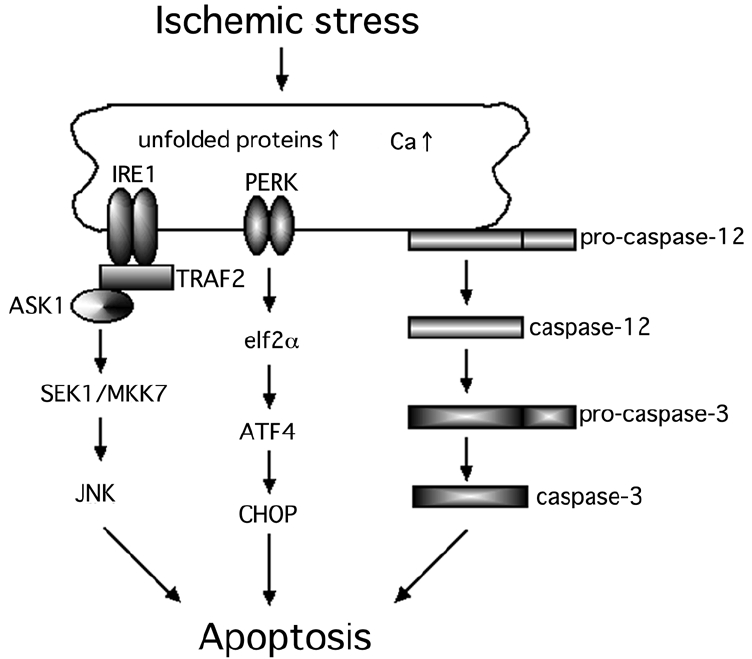
Hypothesized scheme of the ER stress-mediated cell death pathways. At least, three major cell death pathways are associated with ER stress-induced cell death.
Abbreviations: ER, endoplasmic reticulum; TRAF2, tumor necrosis factor receptor-associated factor 2; ASK1, apoptosis signal-regulating kinase 1; SEK1, SAPK/ERK kinase 1; MKK7, mitogen-activated protein kinase kinase 7; JNK, c-Jun N-terminal kinase; ATF4, activating transcription factor 4; PERK, PKR-like endoplasmic reticulum kinase; elF2α, a subunit of translation eukaryotic initiation factor 2; CHOP, C/ERB homologous protein.
The results of recent studies have shown that ER stress-mediated cell death is associated with the death of pancreatic β-cells in patients with diabetes (Oyadomari et al 2001, 2002; Laybutt et al 2007). In addition, a recent study reported that the ER stress-induced apoptosis is related to changes in the glucose concentration and results in the death of pericytes (Ikesugi et al 2006). Roybal and colleagues (2004) suggested that the ER stress-mediated activating transcriptional factor 4 (ATF4) activation was associated with an over-expression of vascular endothelial growth factor (VEGF). In addition, recent studies indicate that ER stress-induced apoptosis is involved in the death of neurons in the brain and retina under different physiological conditions (Guo et al 1997; Tobisawa et al 2003; Imai and Takahashi 2004; Larner et al 2004; Tajiri et al 2004; Wootz et al 2004; Hayashi et al 2005; Awai et al 2006; Shimazawa et al 2007).
One therapeutic strategy that might be used to prevent the development and progression of type 2 diabetes is the inhibition of ER stress. This would then block the ER stress-induced pancreatic β-cell death, and may also prevent the onset and progression of diabetic retinopathy. We shall discuss the possible role of ER stress in the pathogenesis of diabetic retinopathy and describe potential therapeutic strategies to block the development and the progression of diabetic retinopathy.
ER stress-mediated pancreatic β-cell death in diabetes
The ER is involved in the maintenance of cellular homeostasis by inducing the UPR. The UPR of mammals is mediated by at least three types of ER transmembrane proteins; IRE1 (protein kinase and site-specific endoribonuclease) (Tirasophon et al 1998; Wang et al 1998), PERK (Harding et al 2000), and ATF6 (Ye et al 2000). Under diabetic conditions, the pancreatic β-cells are continuously exposed to oxidative stress, eg, exposure to reactive oxygen species (ROS; Kaneto et al 2005) and to nitrous oxide (NO; Oyadomari et al 2001). The ER is highly developed in pancreatic β-cells because of the continuous insulin secretion. Thus, it seems that even under physiological conditions, a potential ER stress is present because there are many unfolded proteins and premature proteins in the ER of pancreatic β-cells (Harding et al 2001; Weir et al 2001). These unfolded and premature proteins can easily become targets of ROS and NO (Oyadomari et al 2001; Kaneto et al 2005). Once the ER stress is increased in pancreatic β-cells, the JNK- and CHOP-mediated cell death pathways are activated (Oyadomari et al 2001, 2002; Kaneto et al 2005). The activation of the JNK pathway under diabetic stress is known to induce the serine phosphorylation of insulin receptor substance 1, which in turn leads to insulin resistance (Özcan et al 2004). When the number of pancreatic β-cell is decreased, ER stress is increased in the remaining pancreatic β-cells to compensate for the reduced insulin secretion leading to pancreatic β-cell dysfunction (Weir et al 2001; Poitout et al 2002). Thus, ER stress-mediated pancreatic β-cell death is critical and a key alteration for the pathogenesis of type 2 diabetes.
ER stress involved in vascular abnormalities in eyes with diabetic retinopathy
The loss of pericytes from the microvessels in diabetic retinas is one of the characteristic pathological changes in the early stage of diabetic retinopathy. The results of a recent study indicated that the UPR, activated by ER stress, is induced in retinal pericytes by the changes in the glucose concentration (Ikesugi et al 2006). Thus, ER stress-mediated cell death is the common pathology in the death of pancreatic β-cells and pericytes in diabetes.
VEGF plays important roles in the pathogenesis of diabetic retinopathy (Shweiki et al 1992; Amin et al 1997; Lu et al 1998; Ishida et al 2000; Qaum et al 2001; El-Remessy et al 2003). The expression of VEGF is increased in diabetic retinas by the high-glucose, ischemia, and hypoxia, and this leads to the development of neovascularization and/or increased vascular permeability (Shweiki et al 1992; Amin et al 1997; Lu et al 1998; Ishida et al 2000; Qaum et al 2001; El-Remessy et al 2003). Abcouwer et al showed that the glutamine deprivation-induced ER stress is associated with an up-regulation of VEGF expression in human retinal pigment epithelial cells (Abcouwer et al 2002). Roybal and colleagues (2004) suggested that the homocysteine-induced ER stress is related to the over-expression of VEGF under the control of ATF4. Hyperglycemia has been suggested to increase the intracellular homocysteine levels in the retinal pigment epithelium cells. Thus, there is good evidence that the diabetic stress-induced ER stress is involved in vascular abnormalities such as pericyte loss and neovascularization.
ER-stress-mediated neuronal cell death
Recent studies have shown that RGCs die at the early stage of diabetes (Barber et al 1998; Asnaghi et al 2003; Oshitari and Roy 2005). The neuronal abnormalities, such as RGC death, are irreversible and may precede the vascular abnormalities including the increased vascular permeability in diabetic retinas. This is observed even in retinas with a clinical diagnosis of non-diabetic retinopathy, however neuronal abnormalities, such as the reduction of retinal nerve fiber thickness, can be detected by optical coherence tomography even at this stage (Sugimoto et al 2005).
Guo and colleagues (1997) suggest that an upset of the Ca2+ homeostasis in the ER caused by mutations in presenilin-1 is associated with the neuronal cell death in Alzheimer’s disease. Recently, many studies have reported that the ER stress-mediated neuronal cell death is related to the pathogenesis of various neuronal diseases in the brain and retina, eg, polyglutamine diseases (Nishito et al 2002), Parkinson’s disease (Imai and Takahashi 2004), amyotrophic lateral sclerosis (Tobisawa et al 2003; Wootz et al 2004), acute brain disorders (Larner et al 2004; Tajiri et al 2004; Hayashi et al 2005), and retinal ischemia (Awai et al 2006; Shimazawa et al 2007).
Other studies have shown that the PERK-CHOP pathway, one of the ER stress-mediated pathways that is induced in ischemic retinas, is related to neuronal cell death (Awai et al 2006; Shimazawa et al 2007). It is well-known that under ischemic stress, an increase of intracellular Ca2+ level disturbs the ER Ca2+ homeostasis which in turn leads to ER stress-induced neuronal degeneration (Verkhratsky and Toescu 2003). Unfolded proteins that accumulate in the ER in ischemic retinas become targets of ROS and NO. Thus, it seems reasonable that in ischemic retinas, the ER stress-mediated cell death pathways are related to the neuronal degeneration.
We have examined the IRE1-JNK pathway to determine if it is associated with the neuronal death in ischemic retinas using the ischemia-reperfusion injury model (unpublished data). Our results suggested that the expressions of IRE1α and tumor necrosis factor receptor-associated factor 2 (TRAF2) were significantly increased in the ischemic retinas compared to that in control retinas. In addition, we found that the expression of ASK1, SAPK/ERK kinase 1 (SEK1), and JNK were expressed in the same degenerating neurons of ischemic retinas (unpublished data). Thus, not only the PERK-CHOP pathway but also the IRE1-JNK pathway is associated with neuronal death in ischemic retinas.
The exact mechanism leading to the death of RGCs has not been conclusively determined especially at the early stage of diabetic retinopathy. One possible link between vascular abnormalities and neuronal abnormalities may be the changes in the glial cells at the early stage of diabetic retinopathy (Figure 2). Glial processes make contact with both the blood vessels and neurons, and form the blood-retinal barrier (Kim et al 2006) (Figure 2). Glial cells can maintain the blood-retinal barrier by expressing VEGF, and there are many studies that have shown interactions between glial cells and neuronal cells in the retina (Fruttiger et al 1996; Rauen et al 1999; Harada et al 2000). During the onset and the progression of diabetic retinopathy, Müller cells are changed, eg, up-regulation of glial fibrillary acidic protein (GFAP) and accumulation of gamma-aminobutyric acid (Ishikawa et al 1996; Lieth et al 1998; Barber et al 2000; Rungger-Brändle et al 2000). Pannicke and colleagues (2006) suggested that glial abnormalities, eg, swelling of the cell body, may lead to the retinal edema detected in diabetic retinas. Thus, neuro-glial interactions may be involved at the onset and the progression of diabetic retinopathy, and glial changes may be related to both neuronal and vascular abnormalities at the early stage of diabetic retinopathy (Figure 2).
Figure 2.
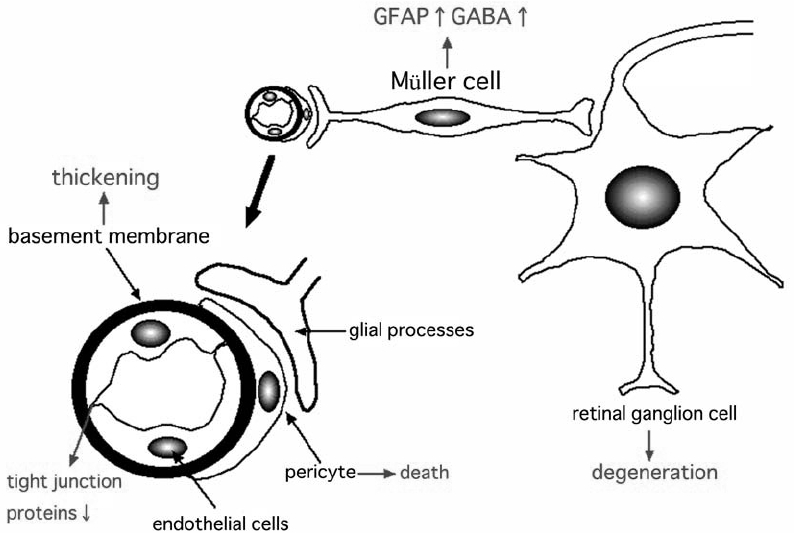
Hypothesized scheme of the pathogenesis of early diabetic retinopathy. The characteristic changes of early diabetic retinas may be glial changes, which in turn leads to vascular and neuronal abnormalities such as increased vascular permeability or neuronal cell death. The ideal therapies for diabetic retinopathy may be the improvement of both vascular and neuronal abnormalities.
Abbreviations: GFAP, glial fibrillary acidic protein; GABA, gamma-aminobutyric acid.
The glutamate levels are known to be elevated in the vitreous of diabetic patients (Ambati et al 1997), which could lead to neuronal cell death. Because an over-stimulation of neurons by glutamate upsets the Ca2+ homeostasis in the ER, ER stress may be present in degenerating neurons under diabetic stress.
Because there is very little evidence of a direct association of ER stress and the pathogenesis of diabetic retinopathy, ER stress may be an epiphenomenon and/or only one of the players, perhaps with a minor role, in the development of diabetic retinopathy. Thus, although ER-stress-related factors may be promising targets for the prevention and the progression of diabetic retinopathy, additional studies are needed to determine the relationship between ER stress and neuronal cell death in diabetic retinas.
Potential therapeutic strategies for diabetic retinopathy
An epidemiological study performed by the Japanese Ministry of Welfare in 2005 showed that diabetic retinopathy was the second most common eye disease to cause blindness in the Japanese. Over 3,000 patients with diabetic retinopathy lose their vision each year in Japan. This indicates that the current management and therapies for diabetic retinopathy are not sufficient to prevent the progression to blindness in patients with diabetic retinopathy. To reduce the number of patients who lose their vision from diabetic retinopathy, new therapeutic strategies must be established to prevent the onset and the progression of the diabetic retinopathy.
At present, the standard treatments for diabetic retinopathy include: control of the blood glucose levels and the blood pressure (Klein et al 1995; Diabetes Control and Complications Trial Research Group 1995a, 1995b; UK Prospective Diabetes Study (UKPDS) Group 1998; Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group 2000; Writing Team for the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group 2002; Matthews et al 2004), focal laser photocoagulation (Early Treatment Diabetic Retinopathy Study Research Group 1985, 1987, 1991a), pan-retinal laser photocoagulation (Diabetic Retinopathy Study Research Group 1978, 1981), and early vitrectomy (Diabetic Retinopathy Vitrectomy Study Research Group 1985a, 1985b, 1988a, 1988b, 1990). Although the diabetic retinopathy continues to progress in some patients in spite of good control of blood glucose and blood pressure, a tight control of blood glucose levels and blood pressure is the first choice for the treatment of diabetic retinopathy.
Focal laser treatment significantly decreased the risk of visual disturbances in diabetic patients with macular edema. However, side effects, eg, foveal burns, central visual field defects and retinal fibrosis, are not uncommon. Pan-retinal photocoagulation also has many side effects such as visual field constriction, night blindness, and exacerbation of macular edema. However, pan-retinal laser photocoagulation significantly decreased the risk of visual disturbances in diabetic patients with severe non-proliferative and proliferative retinopathy. Early vitrectomy reduced the risk of visual disturbances in diabetic patients with proliferative retinopathy and vitreous hemorrhage. Again, there are many side effects of vitrectomy including vitreous hemorrhage, retinal detachment, neovascular glaucoma, and infection.
Intravitreal or sub-Tenon injections of triamcinolone acetonide (TA) has been recently used to treat the macular edema common in diabetic patients, and during the early post-TA period, there were improvements in both the macular edema and visual acuity (Martidis et al 2002; Avitabile et al 2005; Sorbin and D’Amico 2005; Jonas et al 2006a). Steroids are known to up-regulate the expression of tight junctions proteins, eg, occludin and ZO-1, in endothelial cells, and this increase reduce the increased vascular permeability in the retina (Antonetti et al 2002). Unfortunately, repeated injections of TA are frequently required, and there are many side effects such as increased intraocular pressure, cataracts, and infections (Gillies et al 2004; Jonas et al 2005, 2006b; Westfall et al 2005). Although long-term follow-up studies of TA must be made, the treatments by TA may be considered together with laser treatments and vitrectomy in diabetic patients with severe macular edema.
There are many medical therapies that are being tried to prevent the development and progression of diabetic retinopathy, eg, aspirin (Early Treatment Diabetic Retinopathy Study Research Group 1991b; Chew et al 1995), anti-VEGF agents (Cunningham et al 2005; Avery et al 2006; Chun et al 2006; Spaide et al 2006), protein kinase C inhibitors (PKC-DRS Study Group 2005, 2006; PKC-DMES Study Group 2007), growth hormone inhibitors (Kirkegaard et al 1990), and aldose reductase inhibitors (Sorbinil Retinopathy Trial Research Group 1990). However, Mohamed and colleagues (2007) stated that these treatments cannot be recommended for routine use because evidence to support their use has not been published.
The most important factor to consider in the management and the treatment of diabetic retinopathy is the protection of visual function. Because the onset and progression of neuronal and vascular abnomralities lead to visual dysfunction, the targets of the therapeutic methods should be the factors that are common to both vascular and neuronal abnormalities of diabetic retinopathy. Thus, we have stated in an earlier review article that two of the candidates common to both vascular and neuronal abnormalities in diabetic retinopathy are tumor necrosis factor-alpha and Bax (Oshitari 2006). ER stress-related factors should also be considered as targets of new therapeutic strategies for diabetic retinopathy as well as the targets of new therapies for type 2 diabetes because ER stress is also related to pancreatic β-cell death and insulin resistance in patients with type 2 diabetes.
The results of a recent study showed that oral administration of chemical chaperones, 4-phenyl butyric acid or taurine-conjugated ursodeoxycholic acid, alleviated ER stress and improved the action of systemic insulin in diabetic animals (Özcan et al 2006). Although the precise mechanism involved in the improvement of type 2 diabetes is unclear, these chemical chaperones may stabilize protein conformation and improve the folding capacity of the ER, which in turn would reduce the ER stress in these diabetic animals. These chemical chaperones may become a standard treatment for type 2 diabetes because of their safety profiles in vivo (Maestri et al 1996; Kaplan and Gershwin 2005).
Recently, a Bax inhibitor-1 (BI-1) was identified to be an anti-apoptotic protein in mammals (Chae et al 2003), and this is relevant because BI-1 can regulate a cell death pathway involved in ER stress (Chae et al 2004). In addition, an over-expression of BI-1 can protect against the neuronal cell death induced by ER stress (Dohm et al 2006). Although the mechanism for this protective effect was not determined, a recent study showed that BI-1 can inhibit ER stress proteins such as CHOP, IRE1α or phospho-JNK (Lee et al 2007). The activity of BI-1 may provide clues on developing new ways to regulate the ER stress-induced apoptosis. For example, brain-derived neurotrophic factor (BDNF), which is known to reduce the neuronal degeneration of diabetic retinas in vivo (Seki et al 2004), prevents ER stress-mediated neuronal cell death by suppressing caspase-12 activation in vitro (Shimoke et al 2004).
Although these neuroprotective therapies may be promising, the prevention of neuronal cell death under chronic diabetic stress may have limitations. Even if one major cell death pathway is blocked, another cell death pathway may become activated. Even when apoptosis can be blocked, other types of cell death such as necrosis or autophagy may be induced (Koh et al 1995; Hartmann et al 2001; Vandenabeele et al 2006). Thus, even if we can establish neuroprotective therapies for diabetic retinopathy, the first choice of the treatment for diabetic retinopathy must still be the standard treatment of controlling blood glucose levels and blood pressure to reduce the causes of diabetic stress.
In conclusion, ER stress is involved in the pathogenesis of type 2 diabetes and diabetic retinopathy. The reduction of ER stress by chemical chaperones such as 4-phenyl butyric acid or taurine-conjugated ursodeoxycholic acid may become one of the standard treatments for type 2 diabetes. Such treatments may be helpful in preventing the development of diabetic retinopathy. Additional studies are required to determine the optimal methods to reduce ER stress in patients with diabetic retinopathy.
Acknowledgments
This study was supported by the grant from The Eye Research Foundation for the Aged. We thank Prof. Duco Hamasaki for editing this manuscript.
References
- Abcouwer SF, Marjon PL, Loper RK, et al. Response of VEGF expression to amino acid deprivation and inducers of endoplasmic reticulum stress. Invest Ophthalmol Vis Sci. 2002;43:2791–8. [PubMed] [Google Scholar]
- Ambati J, Chalam KV, Chawla DK, et al. Elevated gamma-aminobutyric acid, glutamate, and vascular endothelial growth factor levels in the vitreous of patients with proliferative diabetic retinopathy. Arch Ophthalmol. 1997;115:1161–6. doi: 10.1001/archopht.1997.01100160331011. [DOI] [PubMed] [Google Scholar]
- Amin RH, Frank RN, Kennedy A, et al. Vascular endothelial growth factor is present in glial cells of the retina and optic nerve of human subjects with nonproliferative diabetic retinopathy. Invest Ophthalmol Vis Sci. 1997;38:36–47. [PubMed] [Google Scholar]
- Antonetti DA, Wolpert EB, DeMaio L, et al. Hydrocortisone decreases retinal endothelial cell water and solute flux coincident with increased content and decreased phosphorylation of occludin. J Neurochem. 2002;80:667–77. doi: 10.1046/j.0022-3042.2001.00740.x. [DOI] [PubMed] [Google Scholar]
- Asnaghi V, Gerhardinger C, Hoehn T, et al. A role for the polyol pathway in the early neuroretinal apoptosis and glial changes induced by diabetes in the rat. Diabetes. 2003;52:506–11. doi: 10.2337/diabetes.52.2.506. [DOI] [PubMed] [Google Scholar]
- Avery RL, Pearlman J, Pieramici DJ, et al. Intravitreal bevacizumab (Avastin) in the treatment of proliferative diabetic retinopathy. Ophthalmology. 2006;113:1695. doi: 10.1016/j.ophtha.2006.05.064. [DOI] [PubMed] [Google Scholar]
- Avitabile T, Longo A, Reibaldi A. Intravitreal triamcinolone compared with macular laser grid photocoagulation for the treatment of cystoid macular edema. Am J Ophthalmol. 2005;140:695–702. doi: 10.1016/j.ajo.2005.05.021. [DOI] [PubMed] [Google Scholar]
- Awai M, Koga T, Inomata Y, et al. NMDA-induced retinal injury is mediated by an endoplasmic reticulum stress-related protein, CHOP/GADD153. J Neurochem. 2006;96:43–52. doi: 10.1111/j.1471-4159.2005.03502.x. [DOI] [PubMed] [Google Scholar]
- Barber AJ, Antonetti DA, Gardner TW. Altered expression of retinal occludin and glial fibrillary acidic protein in experimental diabetes. The Penn State Retina Research Group. Invest Ophthalmol Vis Sci. 2000;41:3561–8. [PubMed] [Google Scholar]
- Barber AJ, Lieth E, Khin SA, et al. Neural apoptosis in the retina during experimental and human diabetes. Early onset and effect of insulin. J Clin Invest. 1998;102:783–91. doi: 10.1172/JCI2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chae HJ, Ke N, Kim HR, et al. Evolutionarily conserved cytoprotection provided by Bax Inhibitor-1 homologs from animals, plants, and yeast. Gene. 2003;323:101–13. doi: 10.1016/j.gene.2003.09.011. [DOI] [PubMed] [Google Scholar]
- Chae HJ, Kim HR, Xu C, et al. BI-1 regulates an apoptosis pathway linked to endoplasmic reticulum stress. Mol Cell. 2004;15:355–66. doi: 10.1016/j.molcel.2004.06.038. [DOI] [PubMed] [Google Scholar]
- Chevet E, Cameron PH, Pelletier MF, et al. The endoplasmic reticulum: integration of protein folding, quality control, signaling and degradation. Curr Opin Struct Biol. 2001;11:120–4. doi: 10.1016/s0959-440x(00)00168-8. [DOI] [PubMed] [Google Scholar]
- Chew EY, Klein ML, Murphy RP, et al. Effects of aspirin on vitreous/preretinal hemorrhage in patients with diabetes mellitus. Early Treatment Diabetic Retinopathy Study report no. 20. Arch Ophthalmol. 1995;113:52–5. doi: 10.1001/archopht.1995.01100010054020. [DOI] [PubMed] [Google Scholar]
- Chun DW, Heier JS, Topping TM, et al. A pilot study of multiple intravitreal injections of ranibizumab in patients with center-involving clinically significant diabetic macular edema. Ophthalmology. 2006;113:1706–12. doi: 10.1016/j.ophtha.2006.04.033. [DOI] [PubMed] [Google Scholar]
- Cunningham ET, Jr, Adamis AP, Altaweel M, et al. A phase II randomized double-masked trial of pegaptanib, an anti-vascular endothelial growth factor aptamer, for diabetic macular edema. Ophthalmology. 2005;112:1747–57. doi: 10.1016/j.ophtha.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Diabetes Control and Complications Trial Research Group. Progression of retinopathy with intensive versus conventional treatment in the Diabetes Control and Complications Trial. Ophthalmology. 1995a;102:647–61. doi: 10.1016/s0161-6420(95)30973-6. [DOI] [PubMed] [Google Scholar]
- Diabetes Control and Complications Trial Research Group. The relationship of glycemic exposure (HbA1c) to the risk of development and progression of retinopathy in the diabetes control and complications trial. Diabetes. 1995b;44:968–83. [PubMed] [Google Scholar]
- Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Retinopathy and nephropathy in patients with type 1 diabetes four years after a trial of intensive therapy. N Engl J Med. 2000;342:381–9. doi: 10.1056/NEJM200002103420603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diabetic Retinopathy Study Research Group. Photocoagulation treatment of proliferative diabetic retinopathy: the second report of diabetic retinopathy study findings. Ophthalmology. 1978;85:82–106. doi: 10.1016/s0161-6420(78)35693-1. [DOI] [PubMed] [Google Scholar]
- Diabetic Retinopathy Study Research Group. Photocoagulation treatment of proliferative diabetic retinopathy. Clinical application of Diabetic Retinopathy Study (DRS) findings, DRS Report Number 8. Ophthalmology. 1981;88:583–600. [PubMed] [Google Scholar]
- Diabetic Retinopathy Vitrectomy Study Research Group. Two-year course of visual acuity in severe proliferative diabetic retinopathy with conventional management. Diabetic Retinopathy Vitrectomy Study (DRVS) report #1. Ophthalmology. 1985a;92:492–502. doi: 10.1016/s0161-6420(85)34002-2. [DOI] [PubMed] [Google Scholar]
- Diabetic Retinopathy Vitrectomy Study Research Group. Early vitrectomy for severe vitreous hemorrhage in diabetic retinopathy. Two-year results of a randomized trial. Diabetic Retinopathy Vitrectomy Study report 2. The Diabetic Retinopathy Vitrectomy Study Research Group. Arch Ophthalmol. 1985b;103:1644–52. [PubMed] [Google Scholar]
- Diabetic Retinopathy Vitrectomy Study Research Group. Early vitrectomy for severe proliferative diabetic retinopathy in eyes with useful vision. Results of a randomized trial – Diabetic Retinopathy Vitrectomy Study Report 3. The Diabetic Retinopathy Vitrectomy Study Research Group. Ophthalmology. 1988a;95:1307–20. doi: 10.1016/s0161-6420(88)33015-0. [DOI] [PubMed] [Google Scholar]
- Diabetic Retinopathy Vitrectomy Study Research Group. Early vitrectomy for severe proliferative diabetic retinopathy in eyes with useful vision. Clinical application of results of a randomized trial – Diabetic Retinopathy Vitrectomy Study Report 4. The Diabetic Retinopathy Vitrectomy Study Research Group. Ophthalmology. 1988b;95:1321–34. doi: 10.1016/s0161-6420(88)33014-9. [DOI] [PubMed] [Google Scholar]
- Diabetic Retinopathy Vitrectomy Study Research Group. Early vitrectomy for severe vitreous hemorrhage in diabetic retinopathy. Four-year results of a randomized trial: Diabetic Retinopathy Vitrectomy Study Report 5. Arch Ophthalmol. 1990;108:958–64. doi: 10.1001/archopht.1990.01070090060040. [DOI] [PubMed] [Google Scholar]
- Dohm CP, Siedenberg S, Liman J, et al. Bax inhibitor-1 protects neurons from oxygen-glucose deprivation. J Mol Neurosci. 2006;29:1–8. doi: 10.1385/JMN:29:1:1. [DOI] [PubMed] [Google Scholar]
- Early Treatment Diabetic Retinopathy Study Research Group. Photocoagulation for diabetic macular edema. Early Treatment Diabetic Retinopathy Study report number 1. Arch Ophthalmol. 1985;103:1796–806. [PubMed] [Google Scholar]
- Early Treatment Diabetic Retinopathy Study Research Group. Treatment techniques and clinical guidelines for photocoagulation of diabetic macular edema. Early Treatment Diabetic Retinopathy Study Report Number 2. Ophthalmology. 1987;94:761–74. doi: 10.1016/s0161-6420(87)33527-4. [DOI] [PubMed] [Google Scholar]
- Early Treatment Diabetic Retinopathy Study Research Group. Early Treatment Diabetic Retinopathy Study design and baseline patient characteristics. ETDRS report number 7. Ophthalmology. 1991a;98:741–56. doi: 10.1016/s0161-6420(13)38009-9. [DOI] [PubMed] [Google Scholar]
- Early Treatment Diabetic Retinopathy Study Research Group. Effects of aspirin treatment on diabetic retinopathy. ETDRS report number 8. Ophthalmology. 1991b;98:757–65. [PubMed] [Google Scholar]
- El-Remessy AB, Behzadian MA, Abou-Mohamed G, et al. Experimental diabetes causes breakdown of the blood-retina barrier by a mechanism involving tyrosine nitration and increases in expression of vascular endothelial growth factor and urokinase plasminogen activator receptor. Am J Pathol. 2003;162:1995–2004. doi: 10.1016/S0002-9440(10)64332-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fruttiger M, Calver AR, Kruger WH, et al. PDGF mediates a neuron-astrocyte interaction in the developing retina. Neuron. 1996;17:1117–31. doi: 10.1016/s0896-6273(00)80244-5. [DOI] [PubMed] [Google Scholar]
- Gillies MC, Simpson JM, Billson FA, et al. Safety of an intravitreal injection of triamcinolone: results from a randomized clinical trial. Arch Ophthalmol. 2004;122:336–40. doi: 10.1001/archopht.122.3.336. [DOI] [PubMed] [Google Scholar]
- Guo Q, Sopher BL, Furukawa K, et al. Alzheimer’s presenilin mutation sensitizes neural cells to apoptosis induced by trophic factor withdrawal and amyloid beta-peptide: involvement of calcium and oxyradicals. J Neurosci. 1997;17:4212–22. doi: 10.1523/JNEUROSCI.17-11-04212.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada T, Harada C, Nakayama N, et al. Modification of glial-neuronal cell interactions prevents photoreceptor apoptosis during light-induced retinal degeneration. Neuron. 2000;26:533–41. doi: 10.1016/s0896-6273(00)81185-x. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, Bertolotti A, et al. Perk is essential for translational regulation and cell survival during the unfolded protein response. Mol Cell. 2000;5:897–904. doi: 10.1016/s1097-2765(00)80330-5. [DOI] [PubMed] [Google Scholar]
- Harding HP, Zeng H, Zhang Y, et al. Diabetes mellitus and exocrine pancreatic dysfunction in perk-/- mice reveals a role for translational control in secretory cell survival. Mol Cell. 2001;7:1153–63. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- Hartmann A, Troadec JD, Hunot S, et al. Caspase-8 is an effector in apoptotic death of dopaminergic neurons in Parkinson’s disease, but pathway inhibition results in neuronal necrosis. J Neurosci. 2001;21:2247–55. doi: 10.1523/JNEUROSCI.21-07-02247.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T, Saito A, Okuno S, et al. Damage to the endoplasmic reticulum and activation of apoptotic machinery by oxidative stress in ischemic neurons. J Cereb Blood Flow Metab. 2005;25:41–53. doi: 10.1038/sj.jcbfm.9600005. [DOI] [PubMed] [Google Scholar]
- Ikesugi K, Mulhern ML, Madson CJ, et al. Induction of endoplasmic reticulum stress in retinal pericytes by glucose deprivation. Curr Eye Res. 2006;31:947–53. doi: 10.1080/02713680600966785. [DOI] [PubMed] [Google Scholar]
- Imai Y, Takahashi R. How do Parkin mutations result in neurodegeneration? Curr Opin Neurobiol. 2004;14:384–9. doi: 10.1016/j.conb.2004.04.002. [DOI] [PubMed] [Google Scholar]
- Ishida S, Shinoda K, Kawashima S, et al. Coexpression of VEGF receptors VEGF-R2 and neuropilin-1 in proliferative diabetic retinopathy. Invest Ophthalmol Vis Sci. 2000;41:1649–56. [PubMed] [Google Scholar]
- Ishikawa A, Ishiguro S, Tamai M. Accumulation of gamma-amino-butyric acid in diabetic rat retinal Muller cells evidenced by electron microscopic immunocytochemistry. Curr Eye Res. 1996;15:958–64. doi: 10.3109/02713689609017641. [DOI] [PubMed] [Google Scholar]
- Jonas JB, Degenring RF, Kreissig I, et al. Intraocular pressure elevation after intravitreal triamcinolone acetonide injection. Ophthalmology. 2005;112:593–8. doi: 10.1016/j.ophtha.2004.10.042. [DOI] [PubMed] [Google Scholar]
- Jonas JB, Kamppeter BA, Harder B, et al. Intravitreal triamcinolone acetonide for diabetic macular edema: a prospective, randomized study. J Ocul Pharmacol Ther. 2006a;22:200–7. doi: 10.1089/jop.2006.22.200. [DOI] [PubMed] [Google Scholar]
- Jonas JB, Kreissig I, Spandau UH, et al. Infectious and noninfectious endophthalmitis after intravitreal high-dosage triamcinolone acetonide. Am J Ophthalmol. 2006b;141:579–80. doi: 10.1016/j.ajo.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Kaneto H, Kawamori D, Matsuoka TA, et al. Oxidative stress and pancreatic beta-cell dysfunction. Am J Ther. 2005;12:529–33. doi: 10.1097/01.mjt.0000178773.31525.c2. [DOI] [PubMed] [Google Scholar]
- Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353:1261–73. doi: 10.1056/NEJMra043898. [DOI] [PubMed] [Google Scholar]
- Kim JH, Kim JH, Park JA, et al. Blood-neural barrier: intercellular communication at glio-vascular interface. J Biochem Mol Biol. 2006;39:339–45. doi: 10.5483/bmbrep.2006.39.4.339. [DOI] [PubMed] [Google Scholar]
- Kirkegaard C, Nφrgaard K, Snorgaard O, et al. Effect of one year continuous subcutaneous infusion of a somatostatin analogue, octreotide, on early retinopathy, metabolic control and thyroid function in Type I (insulin-dependent) diabetes mellitus. Acta Endocrinol (Copenh) 1990;122:766–72. doi: 10.1530/acta.0.1220766. [DOI] [PubMed] [Google Scholar]
- Klein BE, Klein R, Moss SE, et al. A cohort study of the relationship of diabetic retinopathy to blood pressure. Arch Ophthalmol. 1995;113:601–6. doi: 10.1001/archopht.1995.01100050069033. [DOI] [PubMed] [Google Scholar]
- Koh JY, Gwag BJ, Lobner D, et al. Potentiated necrosis of cultured cortical neurons by neurotrophins. Science. 1995;268:573–5. doi: 10.1126/science.7725105. [DOI] [PubMed] [Google Scholar]
- Larner SF, Hayes RL, McKinsey DM, et al. Increased expression and processing of caspase-12 after traumatic brain injury in rats. J Neurochem. 2004;88:78–90. doi: 10.1046/j.1471-4159.2003.02141.x. [DOI] [PubMed] [Google Scholar]
- Laybutt DR, Preston AM, Akerfeldt MC, et al. Endoplasmic reticulum stress contributes to beta cell apoptosis in type 2 diabetes. Diabetologia. 2007;50:752–63. doi: 10.1007/s00125-006-0590-z. [DOI] [PubMed] [Google Scholar]
- Lee GH, Kim HK, Chae SW, et al. Bax inhibitor-1 regulates endoplasmic reticulum stress-associated reactive oxygen species and heme oxygenase-1 expression. J Biol Chem. 2007;282:21618–28. doi: 10.1074/jbc.M700053200. [DOI] [PubMed] [Google Scholar]
- Lieth E, Barber AJ, Xu B, et al. Glial reactivity and impaired glutamate metabolism in short-term experimental diabetic retinopathy. Penn State Retina Research Group. Diabetes. 1998;47:815–20. doi: 10.2337/diabetes.47.5.815. [DOI] [PubMed] [Google Scholar]
- Lindholm D, Wootz H, Korhonen L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006;13:385–92. doi: 10.1038/sj.cdd.4401778. [DOI] [PubMed] [Google Scholar]
- Lu M, Kuroki M, Amano S, et al. Advanced glycation end products increase retinal vascular endothelial growth factor expression. J Clin Invest. 1998;101:1219–24. doi: 10.1172/JCI1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, Brewer JW, Diehl JA, et al. Two distinct stress signaling pathways converge upon the CHOP promoter during the mammalian unfolded protein response. Mol Biol. 2002;318:1351–65. doi: 10.1016/s0022-2836(02)00234-6. [DOI] [PubMed] [Google Scholar]
- Maestri NE, Brusilow SW, Clissold DB, et al. Long-term treatment of girls with ornithine transcarbamylase deficiency. N Engl J Med. 1996;335:855–9. doi: 10.1056/NEJM199609193351204. [DOI] [PubMed] [Google Scholar]
- Martidis A, Duker JS, Greenberg PB, et al. Intravitreal triamcinolone for refractory diabetic macular edema. Ophthalmology. 2002;109:920–7. doi: 10.1016/s0161-6420(02)00975-2. [DOI] [PubMed] [Google Scholar]
- Martin PM, Roon P, Van Ells TK, et al. Death of retinal neurons in streptozotocin-induced diabetic mice. Invest Ophthalmol Vis Sci. 2004;45:3330–6. doi: 10.1167/iovs.04-0247. [DOI] [PubMed] [Google Scholar]
- Matthews DR, Stratton IM, Aldington SJ, et al. Risks of progression of retinopathy and vision loss related to tight blood pressure control in type 2 diabetes mellitus: UKPDS 69. Arch Ophthalmol. 2004;122:1631–40. doi: 10.1001/archopht.122.11.1631. [DOI] [PubMed] [Google Scholar]
- Mohamed Q, Gillies MC, Wong TY. Management of diabetic retinopathy: a systematic review. JAMA. 2007;298:902–16. doi: 10.1001/jama.298.8.902. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Zhu H, Morishima N, et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98–103. doi: 10.1038/47513. [DOI] [PubMed] [Google Scholar]
- Nielsen SP, Petersen OH. Transport of calcium in the perfused submandibular gland of the cat. J Physiol. 1972;223:685–97. doi: 10.1113/jphysiol.1972.sp009869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishitoh H, Matsuzawa A, Tobiume K, et al. ASK1 is essential for endoplasmic reticulum stress-induced neuronal cell death triggered by expanded polyglutamine repeats. Genes Dev. 2002;16:1345–55. doi: 10.1101/gad.992302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oshitari T. Non-viral gene therapy for diabetic retinopathy. Drug Dev Res. 2006;67:835–41. [Google Scholar]
- Oshitari T, Roy S. Diabetes: a potential enhancer of retinal injury in rat retinas. Neurosci Lett. 2005;390:25–30. doi: 10.1016/j.neulet.2005.07.057. [DOI] [PubMed] [Google Scholar]
- Oshitari T, Roy S. Common therapeutic strategies for diabetic retinopathy and glaucoma. Curr Drug Ther. 2007;2:224–32. [Google Scholar]
- Oyadomari S, Koizumi A, Takeda K, et al. Targeted disruption of the Chop gene delays endoplasmic reticulum stress-mediated diabetes. J Clin Invest. 2002;109:525–32. doi: 10.1172/JCI14550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari S, Takeda K, Takiguchi M, et al. Nitric oxide-induced apoptosis in pancreatic beta cells is mediated by the endoplasmic reticulum stress pathway. Proc Natl Acad Sci USA. 2001;98:10845–50. doi: 10.1073/pnas.191207498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Özcan U, Cao Q, Yilmaz E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–61. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- Özcan U, Yilmaz E, Özcan L, et al. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–40. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palade G. Intracellular aspects of the process of protein synthesis. Science. 1975;189:867. doi: 10.1126/science.189.4206.867-b. [DOI] [PubMed] [Google Scholar]
- Pannicke T, Iandiev I, Wurm A, et al. Diabetes alters osmotic swelling characteristics and membrane conductance of glial cells in rat retina. Diabetes. 2006;55:633–9. doi: 10.2337/diabetes.55.03.06.db05-1349. [DOI] [PubMed] [Google Scholar]
- PKC-DMES Study Group. Effect of ruboxistaurin in patients with diabetic macular edema: thirty-month results of the randomized PKC-DMES clinical trial. Arch Ophthalmol. 2007;125:318–24. doi: 10.1001/archopht.125.3.318. [DOI] [PubMed] [Google Scholar]
- PKC-DRS Study Group. The effect of ruboxistaurin on visual loss in patients with moderately severe to very severe nonproliferative diabetic retinopathy: initial results of the Protein Kinase C beta Inhibitor Diabetic Retinopathy Study (PKC-DRS) multicenter randomized clinical trial. Diabetes. 2005;54:2188–97. doi: 10.2337/diabetes.54.7.2188. [DOI] [PubMed] [Google Scholar]
- PKC-DRS2 Group. Aiello LP, Davis MD, et al. Effect of ruboxistaurin on visual loss in patients with diabetic retinopathy. Ophthalmology. 2006;113:2221–30. doi: 10.1016/j.ophtha.2006.07.032. [DOI] [PubMed] [Google Scholar]
- Poitout V, Robertson RP. Minireview: Secondary beta-cell failure in type 2 diabetes – a convergence of glucotoxicity and lipotoxicity. Endocrinology. 2002;143:339–42. doi: 10.1210/endo.143.2.8623. [DOI] [PubMed] [Google Scholar]
- Qaum T, Xu Q, Joussen AM, et al. VEGF-initiated blood-retinal barrier breakdown in early diabetes. Invest Ophthalmol Vis Sci. 2001;42:2408–13. [PubMed] [Google Scholar]
- Rao RV, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Cell Death Differ. 2004;11:372–80. doi: 10.1038/sj.cdd.4401378. [DOI] [PubMed] [Google Scholar]
- Rauen T, Fischer F, Wiessner M. Glia-neuron interaction by high-affinity glutamate transporters in neurotransmission. Adv Exp Med Biol. 1999;468:81–95. doi: 10.1007/978-1-4615-4685-6_7. [DOI] [PubMed] [Google Scholar]
- Roybal CN, Yang S, Sun CW, et al. Homocysteine increases the expression of vascular endothelial growth factor by a mechanism involving endoplasmic reticulum stress and transcription factor ATF4. J Biol Chem. 2004;279:14844–52. doi: 10.1074/jbc.M312948200. [DOI] [PubMed] [Google Scholar]
- Rungger-Brändle E, Dosso AA, Leuenberger PM. Glial reactivity, an early feature of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2000;41:1971–80. [PubMed] [Google Scholar]
- Seki M, Tanaka T, Nawa H, et al. Involvement of brain-derived neurotrophic factor in early retinal neuropathy of streptozotocin-induced diabetes in rats: therapeutic potential of brain-derived neurotrophic factor for dopaminergic amacrine cells. Diabetes. 2004;53:2412–9. doi: 10.2337/diabetes.53.9.2412. [DOI] [PubMed] [Google Scholar]
- Shimazawa M, Inokuchi Y, Ito Y, et al. Involvement of ER stress in retinal cell death. Mol Vis. 2007;13:578–87. [PMC free article] [PubMed] [Google Scholar]
- Shimoke K, Utsumi T, Kishi S, et al. Prevention of endoplasmic reticulum stress-induced cell death by brain-derived neurotrophic factor in cultured cerebral cortical neurons. Brain Res. 2004;1028:105–11. doi: 10.1016/j.brainres.2004.09.005. [DOI] [PubMed] [Google Scholar]
- Shweiki D, Itin A, Soffer D, et al. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–5. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- Sobrin L, D’Amico DJ. Controversies in intravitreal triamcinolone acetonide use. Int Ophthalmol Clin. 2005;45:133–41. doi: 10.1097/01.iio.0000176353.56990.3b. [DOI] [PubMed] [Google Scholar]
- Sorbinil Retinopathy Trial Research Group. A randomized trial of sorbinil, an aldose reductase inhibitor, in diabetic retinopathy. Arch Ophthalmol. 1990;108:1234–44. doi: 10.1001/archopht.1990.01070110050024. [DOI] [PubMed] [Google Scholar]
- Spaide RF, Fisher YL. Intravitreal bevacizumab (Avastin) treatment of proliferative diabetic retinopathy complicated by vitreous hemorrhage. Retina. 2006;26:275–8. doi: 10.1097/00006982-200603000-00004. [DOI] [PubMed] [Google Scholar]
- Sugimoto M, Sasoh M, Ido M, et al. Detection of early diabetic change with optical coherence tomography in type 2 diabetes mellitus patients without retinopathy. Ophthalmologica. 2005;219:379–85. doi: 10.1159/000088382. [DOI] [PubMed] [Google Scholar]
- Takano M, Sango K, Horie H, et al. Diabetes alters neurite regeneration from mouse retinal explants in culture. Neurosci Lett. 1999;275:175–8. doi: 10.1016/s0304-3940(99)00768-5. [DOI] [PubMed] [Google Scholar]
- Tajiri S, Oyadomari S, Yano S, et al. Ischemia-induced neuronal cell death is mediated by the endoplasmic reticulum stress pathway involving CHOP. Cell Death Differ. 2004;11:403–15. doi: 10.1038/sj.cdd.4401365. [DOI] [PubMed] [Google Scholar]
- Tirasophon W, Welihinda AA, Kaufman RJ. A stress response pathway from the endoplasmic reticulum to the nucleus requires a novel bifunctional protein kinase/endoribonuclease (Ire1p) in mammalian cells. Genes Dev. 1998;12:1812–24. doi: 10.1101/gad.12.12.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobisawa S, Hozumi Y, Arawaka S, et al. Mutant SOD1 linked to familial amyotrophic lateral sclerosis, but not wild-type SOD1, induces ER stress in COS7 cells and transgenic mice. Biochem Biophys Res Commun. 2003;303:496–503. doi: 10.1016/s0006-291x(03)00353-x. [DOI] [PubMed] [Google Scholar]
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33) Lancet. 1998;352:837–53. [PubMed] [Google Scholar]
- Vandenabeele P, Vanden Berghe T, Festjens N. Caspase inhibitors promote alternative cell death pathways. Sci STKE. 2006;358:44. doi: 10.1126/stke.3582006pe44. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Toescu EC. Endoplasmic reticulum Ca2+ homeostasis and neuronal death. J Cell Mol Med. 2003;7:351–61. doi: 10.1111/j.1582-4934.2003.tb00238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XZ, Harding HP, Zhang Y, et al. Cloning of mammalian Ire1 reveals diversity in the ER stress responses. EMBO J. 1998;17:5708–17. doi: 10.1093/emboj/17.19.5708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir GC, Laybutt DR, Kaneto H, et al. Beta-cell adaptation and decompensation during the progression of diabetes. Diabetes. 2001;50:S154–9. doi: 10.2337/diabetes.50.2007.s154. [DOI] [PubMed] [Google Scholar]
- Westfall AC, Osborn A, Kuhl D, et al. Acute endophthalmitis incidence: intravitreal triamcinolone. Arch Ophthalmol. 2005;123:1075–7. doi: 10.1001/archopht.123.8.1075. [DOI] [PubMed] [Google Scholar]
- Wootz H, Hansson I, Korhonen L, et al. Caspase-12 cleavage and increased oxidative stress during motoneuron degeneration in transgenic mouse model of ALS. Biochem Biophys Res Commun. 2004;322:281–6. doi: 10.1016/j.bbrc.2004.07.118. [DOI] [PubMed] [Google Scholar]
- Writing Team for the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Research Group. Effect of intensive therapy on the microvascular complications of type 1 diabetes mellitus. JAMA. 2002;287:2563–9. doi: 10.1001/jama.287.19.2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, Rawson RB, Komuro R, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6:1355–64. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]