

Abstract
Since the pioneering work of John Gofman in the 1950s, our understanding of high density lipoprotein cholesterol (HDL-C) and its relationship to coronary heart disease (CHD) has grown substantially. Numerous clinical trials since the Framingham Study in 1977 have demonstrated an inverse relationship between HDL-C and one’s risk of developing CHD. Over the past two decades, preclinical research has gained further insight into the nature of HDL-C metabolism, specifically regarding the ability of HDL-C to promote reverse cholesterol transport (RCT). Recent attempts to harness HDL’s ability to enhance RCT have revealed the complexity of HDL-C metabolism. This review provides a detailed update on HDL-C as an evolving therapeutic target in the management of cardiovascular disease.
Keywords: high density lipoprotein cholesterol (HDL-C), coronary, atherosclerosis, reverse cholesterol transport
Introduction
In the early 1900s, a German chemist named Adolph Windaus determined atheromatous plaque from human aortas contained 20-fold higher concentrations of cholesterol than normal aortas. Soon after, the Russian pathologist Nikolai Anitschov strengthened this observation by feeding rabbits a high-cholesterol diet and creating the first animal model of atherosclerosis (Khavkin et al 1975; Vance et al 2000). In 1955, a biophysicist named John Gofman used ultracentrifugation to separate plasma lipoproteins by density and correlated risk of myocardial infarction (MI) with elevated low-density lipoprotein cholesterol (LDL-C) levels. Henceforth, the ‘lipid hypothesis’ proposed that elevated LDL-C, elevated triacylglycerols (TG), and low levels of high density lipoprotein cholesterol (HDL-C) were causally associated with an increased risk of coronary heart disease (CHD) (Soloff 1998).
In 1977, the Framingham Study showed that elevated LDL-C and reduced HDL-C levels independently predict one’s risk for developing cardiovascular disease (Gordon et al 1977). Since the first Adult Treatment Panel (ATP) recommendations in 1988 [Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel II) 1993], guidelines have increasingly focused on aggressive management of elevated LDL-C in populations at risk for CHD. According to the National Cholesterol Education Panel (NCEP) ATP III guidelines published in 2001, patients with established CHD, non-coronary atherosclerosis, diabetes mellitus, or greater than two major cardiac risk factors with a calculated Framingham risk score of greater than 20% should have an LDL-C goal of less than 100 mg/dL [Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) 2001]. Subsequently, the Heart Protection Study (HPS) (MRC/BHF Heart Protection Study 1999) and Pravastatin or Atorvastatin Evaluation and Infection Therapy (PROVE-IT) (Cannon et al 2004) trial reported incremental 22% and 16% reductions, respectively, in the risk of cardiovascular events with LDL-C levels lowered below 100 mg/dL. Based on these data, an “optional” target of therapy focusing on aggressive LDL-C lowering less than 70 mg/dL for patients with known coronary disease or CHD equivalent status has been proposed (Grundy et al 2004).
However, despite lowering LDL-C to levels recommended by the NCEP in 90% of treated patients, an overall reduction of only 20%–35% in the rate of cardiovascular events has been observed in randomized trials (Shah et al 2002). Moreover, nearly 15% of patients with acute myocardial infarction (MI) have LDL-C levels less than 100 mg/dL at presentation (Forrester et al 2005), suggesting that previously cited LDL-C targets remain too high or the benefit of very aggressive LDL-C lowering is quite limited. The Treating to New Targets (TNT) trial (LaRosa et al 2005) showed a 2.2% absolute and a 22% relative risk reduction for major cardiovascular events in patients receiving high dose compared to low dose atorvastatin. The mean LDL-C levels were 77 mg/dL in the high dose statin group versus 100 mg/dL in the low dose statin group. Thus, while great emphasis has been placed on the importance of LDL-C lowering in CVD risk reduction, there is growing interest directed at raising HDL-C levels for further risk reduction.
At present, no approved therapies increase HDL-C levels by any comparable magnitude to therapies designed to lower LDL-C levels. This review provides a detailed update on HDL-C as a therapeutic target for CVD risk reduction.
HDL-C and coronary heart disease
Substantial epidemiologic evidence suggests a negative linear correlation between HDL-C levels and the incidence of CHD. First proposed by Barr and colleagues in 1951 (Barr et al 1951), an inverse relationship between HDL-C and cardiovascular disease was not well established until the Framingham study in the 1970s (Gordon et al 1997). This analysis of 2815 men and women aged 49–82 years identified HDL-C as a powerful risk factor inversely associated with the incidence of CHD. Similarly, in the early 1980s, the Prospective Cardiovascular Munster (PROCAM) (Kannel 1983) study evaluated 4559 male participants aged 40–64 years and found a strong negative linear correlation between the incidence of CAD and HDL-C levels (CHD risk ratio of 4.0 for HDL-C <25 mg/dL versus 1.0 for HDL-C >65 mg/dL, p < 0.001).
Data from four studies (Framingham Heart Study, the Lipid Research Clinic Prevalence Mortality Follow-up Study, Lipid Research Clinic Primary Prevention Trial, and Multiple Risk Factor Intervention Trial) estimate a 2% reduction in cardiovascular risk for every 1 mg/dL increase in serum HDL-C (Gordon et al 1989; Castelli et al 1992; Multiple Risk Factor Intervention Trial 1982). Individuals with low HDL-C (<40 mg/dL in men and <50 mg/dL in women) are at increased risk of CVD (Miller et al 1977), restenosis following coronary balloon angioplasty (Shah and Amin 1992) and cardiovascular death (Wilson et al 1988).
One of the first trials demonstrating the benefit of raising HDL-C and lowering TG levels in individuals with low baseline HDL-C levels was the Helsinki Heart Study (HHS) (Manninen et al 1992), in which 4081 men with dyslipidemia were randomized to receive gemfibrozil (600 mg twice daily) or placebo. At 5 years of follow-up, gemfibrozil therapy increased HDL-C levels by 11% and reduced total cholesterol (TC), LDL-C, and TG levels by 10%, 11%, and 35% respectively. Gemfibrozil therapy reduced the primary end point of cardiac death or non-fatal MI by 34% (27.3 vs 41.4/1,000, p < 0.02) with the greatest reduction found in patients with low HDL-C and high TG at baseline (Manttari et al 1990).
The Air Force/Texas Coronary Atherosclerosis Prevention Study (AFCAPS/TexCAPS) (Downs et al 1998) compared treatment with lovastatin versus placebo for the prevention of a first major coronary event in adults with average TC and LDL-C levels, but low baseline levels of HDL-C. Lovastatin decreased both TC and LDL-C levels by 18% and 25%, respectively, while increasing HDL-C levels by 6%. After more than 5 years of follow-up, the absolute risk in the primary composite end point of fatal or non-fatal MI, unstable angina, or sudden cardiac death was reduced in absolute terms by 2.2% in men and 1.2% in women with a relative risk reduction of 37%. This study was the first primary prevention study to show that individuals with HDL-C <40 mg/dL received the greatest benefit, suggesting the lower cutpoint HDL-C of 35 mg/dL in ATP II should be raised to 40 mg/dL in ATP III. These clinical trials confirm an increased risk associated with low serum levels of HDL-C and the beneficial effects of pharmacotherapy in adults with low HDL-C in primary prevention.
Patients with manifest CHD also benefit from raising HDL-C and lowering TG. The Veterans Affairs High-Density Lipoprotein Cholesterol Intervention Trial (VA-HIT) compared treatment with gemfibrozil versus placebo in more than 2500 men with established CHD, average LDL-C levels (<140 mg/dL), and low HDL-C levels (<40 mg/dL). After a mean follow-up of 5 years, gemfibrozil decreased TG levels by 31% and increased HDL-C levels by 6%, while levels of LDL-C remained quantitatively unchanged; there was a relative risk reduction of 22% (17.3% vs 21.7% p < 0.006) in CHD death and non-fatal MI in the treatment group. Gemfibrozil therapy was associated with a 24% relative risk reduction in the composite end point of nonfatal MI, stroke, and CHD death (p < 0.001) (Rubins et al 2001).
The Scandanavian Simvastatin Survival Study Group (4S) was a large, randomized, placebo-controlled trial evaluating simvastatin (20–40 mg/day) in 4444 men and women aged 35–70 years over a median follow-up period of 5.4 years. Simvastatin therapy decrease TC and LDL-C (25% and 35%, respectively) and increased HDL-C by 8% compared to placebo. Simvastatin treatment resulted in a 30% relative risk reduction in overall mortality (8.2% vs 11.5%, p = 0.0003) and reduced non-fatal MI, ischemic heart disease death, and coronary revascularization (Scandinavian Simvastatin Survival Study Group 1994).
The Bezafibrate Infarction Prevention Study (BIPS) was a blinded, placebo-controlled trial of bezafibrate therapy in 3122 patients with previous MI or angina pectoris and baseline LDL-C <180 mg/dL, HDL-C <45 mg/dL, and TG <300 mg/dL. At 5–7 years of follow-up, bezafibrate increased HDL-C more than 15% and decreased TG levels by 25%. The overall relative reduction in the primary end point of 9% was not statistically significant; however, a 40% relative risk reduction was observed in patients with baseline TG >200 mg/dL, suggesting fibrates may be beneficial in patients with known CAD and elevated TG levels (Kaplinsky 1998).
Based on the epidemiologic data available, the NCEP ATP III guidelines raised the cut-point for low HDL-C levels from 35 mg/dL to 40 mg/dL, thereby identifying a larger number of adults at risk for developing CHD. Current guidelines define “high” HDL-C levels as above 60 mg/dL (Grundy et al 2004). The definition of “optimal” HDL-C will likely undergo further modification as data becomes available.
HDL-C metabolism and reverse cholesterol transport (RCT)
By transporting excess cholesterol from peripheral cells to the liver for excretion in a process known as reverse cholesterol transport (RCT), HDL-C may retard the progression of atherosclerosis. In animal studies, exogenous infusions of HDL-C or apolipoprotein A-1 (Apo AI), the major apolipoprotein associated with HDL-C, prevents atherosclerosis from developing or progressing (Badimon et al 1990; Duverger et al 1996). Similar findings have been reported in animal models of Apo AI gene over-expression (Dimayuga et al 1999). Harnessing this unique ability of HDL-C requires further insight into the basic mechanisms of HDL-C metabolism.
HDL-C metabolism involves numerous enzymes and unique nuclear transcription regulatory proteins intimately linked to other lipoproteins. HDL-C is a macromolecule containing lipids and proteins that transport water-insoluble fats in blood. A phospholipid (PL) monolayer containing free cholesterol (FC) and apolipoproteins (Apo) surrounds a non-polar lipid core containing FC and TG. Apo AI and AII are the major protein components of HDL-C. Apo AI is produced by the liver and intestines and constitutes 70% of HDL-C protein content. Apo AII is produced only by the liver and constitutes 20% of HDL-C protein content. While Apo AI is ubiquitously associated with HDL-C, Apo AII is found in about 60% of HDL-C molecules (Lewis et al 2005). Apolipoproteins serve as receptor ligands and enzymatic catalysts for all circulating liporoteins.
Clinical observations of premature CHD in patients with inherited disorders of metabolism resulting in low circulating HDL-C levels, such as homozygous Apo AI/CIII deficiency and hypoalphaproteinemia support the association between low serum HDL-C levels and atherosclerosis (Forte TM 1984). Conversely, individuals with inherited defects resulting in low serum HDL-C levels have variable or no premature CHD (eg, Tangier disease, Apo AI Milano, and familial lecithin:cholesterol acyltransferase (LCAT) deficiency), highlighting the complexity of HDL-C metabolism (Miller et al 1990; Elkhalil et al 1997). Characterized in 1968 by Glomset, RCT can be divided into four phases (Glomset 1968) (Figure 1).
Figure 1.
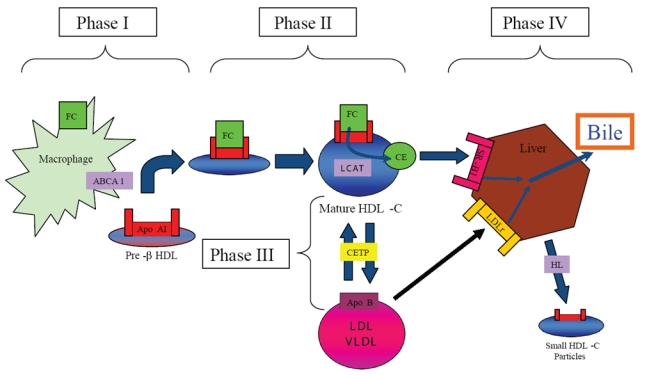
HDL-C mediated reverse cholesterol transport. Reverse cholesterol transport (RCT) can be divided into four phases. 1) transfer of free cholesterol (FC) to pre-b HDL via ABCA1, 2) esterification of surface-associated FC by the enzyme Lecithin:acyl CoA Transferase (LCAT), 3) transfer of FC and triglycerides (TG) between HDL-C and Apo B-containing lipoproteins mediated by the enzyme cholesteryl ester transfer protein (CETP), and 4) uptake by the scavenger receptor B1 (SR-B1) and catabolism of mature HDL-C into bile or small HDL-C particles by hepatic lipase (HL). Apo B-containing lipoproteins can be acquired by the LDL-receptor (LDLr) for hepatic catabolism.
Phase one: nascent HDL-C acquires free cholesterol
The nascent form of circulating HDL-C rich in Apo AI, termed discoidal pre-β HDL, removes FC and PL from peripheral cells throughout the body by interacting with a membrane associated protein ubiquitously expressed in peripheral tissues, known as ATP-binding cassette transporter 1 (ABCA1). Pre-β HDL is rich in Apo AI and serves as a template for the generation of lipid-rich HDL-C (Sviridov et al 2002). Pre-β HDL is generated by either de novo secretion from hepatocytes or the intestinal mucosa, direct dissociation from chylomicrons and very low density lipoprotein (VLDL) mediated by lipoprotein lipase (LL), or as a by-product of HDL-C particle interconversion (Kwiterovich 1998).
Once generated, pre-β HDL receives PL and FC from peripheral cells by associating with the surface protein ABCA1 (Oram and Lawn 2001), which is expressed by the liver and intestinal mucosa. Patients with Tangier Disease, an autosomal recessive disorder characterized by two non-functional ABCA1 alleles and extremely low levels of HDL-C, exemplify the significance of ABCA1 in HDL-C metabolism (Bodzioch et al 1999). Heterozygous individuals with a partial reduction in functional ABCA1 have a corresponding 50% decrease in serum HDL-C levels (Marcil et al 1999).
Animal models further support the critical role of ABCA1 in RCT. ABCA1-deficient mice generated by targeted gene ablation in DBA-1J embryonic stem cells demonstrated a 99.5% and 99.8% reduction in serum HDL-C and Apo AI levels, respectively. In this model, loss of ABCA1 correlated with increased accumulation of lipid-laden macrophages, an integral component of atherosclerotic plaque (McNeish et al 2000). Conversely, overexpression of ABCA1 in transgenic mice is associated with increased TC, HDL-C levels, and Apo AI with enhanced cholesterol efflux and reduced levels of atherogenesis (Brewer et al 2004). Finally, crossing transgenic mice overexpressing ABCA1 with athero-susceptible transgenic mice, such as LDL receptor (LDLr) or Apolipoprotein E knockout (KO) models, reduced atheromatous progression (Joyce et al 2003).
ABCG1 is another member of the ATP-binding cassette family that promotes efflux of PL and FC from macrophages to mature HDL-C rather than pre-β HDL (Kennedy et al 2005) Macrophages deficient in ABCG1 also have impaired FC efflux and accumulate excess cholesterol (Out R 2006). Taken together, these data suggest that both ABCA1 and ABCG1 are potential therapeutic targets to raise HDL-C and promote RCT.
Transcription of both ABCA1 and ABCG1 is regulated by members of a steroid superfamily of nuclear receptors known as the Liver X receptor/Retinoid X receptor (LXR/RXR) heterodimer. When activated by oxysterols from FC this heterodimer stimulates ABCA1 and ABCG1 gene expression, thereby enhancing cholesterol efflux (Vaughan and Oram 2005; Venkateswaran et al 2000). The heterodimer is also regulated by the activity of peroxisome proliferator-activated receptors (PPAR) α and γ, which are closely linked to insulin resistance and the metabolic syndrome (Anderson et al 2004). PPAR-α and PPAR-γ agonists have been shown to upregulate LXR and ABCA1 expression and promote macrophage cholesterol efflux (Schmitz et al 2002; Chawla et al 2001). Synthetic LXR agonists also promote cholesterol efflux in vivo and promote regression of atherosclerosis in mice (Bruemmer and Law 2005).
Phase two: lecithin: acyl CoA transferase (LCAT) esterifies free cholesterol
As FC is acquired from peripheral tissues, pre-β HDL matures from an incipient discoidal form into a larger spherical molecule. Newly acquired FC undergoes esterfication to form cholesteryl esters (CE) which migrate to the center of the discoidal pre-beta HDL molecule. The spherical morphology of mature α-HDL-C promotes further HDL-C metabolism and cholesterol efflux (Wang and Briggs 2004). Cholesterol esterification prevents transfer of FC back to the periphery, thereby potentiating further RCT.
Mediating this necessary step in HDL-C maturation is the enzyme LCAT, which is synthesized by the liver. Circulating LCAT esterifies lecithin and FC on both HDL-C and Apo B lipoproteins. The phospholipid component of HDL-C appears to mediate binding to LCAT, while the apolipoprotein component activates the enzyme (Furbee et al 2002). Recent evidence suggests that increased glycation of Apo A1 in subjects with diabetes progressively decreases the rate of LCAT-mediated cholesterol esterification (Nobecourt et al 2007).
Familial LCAT deficiency (FLD) and fish-eye disease (FED) are distinct inherited syndromes associated with absent or impaired LCAT expression, respectively. Low HDL-C levels and corneal opacification characterize both diseases, while individuals with FLD also suffer from anemia and renal failure. Variable degrees of premature atherosclerosis have been observed in these populations (Funke et al 1991). Conversely, over expression of human LCAT in transgenic animal models correlates with a 7-fold increase in serum HDL-C, increases in Apo AI levels, and a marked reduction in atheromatous plaque burden (Francone et al 1990). These findings suggest that LCAT over expression may serve as a novel therapeutic target in the prevention and treatment of CHD.
Phase three: cholesterol ester transfer protein (CETP) mediates exchange of cholesterol esters between HDL-C and Apo B lipoproteins
The enzyme cholesteryl ester transfer protein (CETP) exchanges CE from HDL-C for TG in LDL-C and VLDL. Newly acquired CE in VLDL and LDL-C is then taken up by the hepatic LDLr for excretion as bile (Morton and Greene 1997). Found predominantly in the liver, spleen, and adipose tissue, expression of CETP is enhanced by hypercholesterolemic diets via activation of a sterol regulatory protein and through binding of oxysterols to the LXR/RXR receptor (De Grooth et al 2004).
Heritable CETP deficiencies in the Japanese population are associated with significant increases in large HDL-C particles and smaller LDL-C particles with a lower affinity for LDL-C receptors (Inazu et al 1990). Heterozygotes with a 40% decrease in CETP levels had a mean increase in HDL-C of 30% and no significant change in LDL-C levels, while homozygotes with complete loss of CETP levels had a greater than 100% increase in HDL-C and 40% decrease in LDL-C and Apo B levels (Inazu et al 1990; Koizumi et al 1991). Despite these findings the correlation between low CETP levels and reduced CHD risk remained elusive as heterozygotes deficient in CETP continue to manifest atherosclerotic coronary disease (Hirano et al 1995).
Two population studies have associated high CETP levels with an increased risk of CHD. A nested case control study known as the European Prospective Investigation into Cancer and nutrition (EPIC)-Norfolk cohort study suggested an increased CHD risk in patients with elevated TG and elevated CETP levels (Boekholdt et al 2004). Data from the Regression Growth Evaluation Statin Study (REGRESS) (Klerkx et al 2004) study also associated high CETP levels and rapid progression of established CHD in men. In this study, treatment with pravastatin significantly improved lipid and angiographic parameters in patients with high baseline CETP independent of baseline lipids, suggesting plasma CETP levels may be a marker of response to statin therapy.
The role of CETP in modulating atherogenesis is complicated by dual pro-atherogenic and anti-atherogenic effects of transferring CE and TG between lipoproteins (Figure 2). Potential proatherogenic properties of CETP activity include: 1) TG-laden HDL-C particles may undergo enhanced renal excretion thereby reducing total circulating HDL-C levels and RCT, 2) CE-laden VLDL and LDL-C particles may be acquired by peripheral macrophages and promote atherosclerosis, and 3) CETP also transfers TG from VLDL to LDL-C thereby generating small atherogenic LDL particles (Hirano et al 2000; Barter et al 2003).
Figure 2.
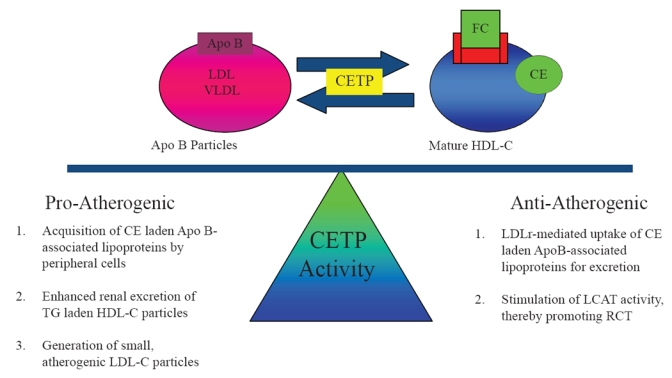
Dual nature of CETP activity. By shuttling cholesteryl esters (CE) and triglycerides (TG) between HDL-C and Apo B-associated lipoproteins, the enzyme cholesteryl ester transfer protein (CETP) creates substrate for both pro-atherogenic and anti-atherogenic pathways.
However, complete abolition of CETP activity results in large, cholesterol-laden, dysfunctional HDL-C with reduced cholesterol efflux RCT capacity (Yamashita et al 1988; Sakai et al 1991; Ikewaki et al 1995). Furthermore, CETP activity may be anti-atherogenic if CE-laden lipoproteins are bound by the LDLr for hepatic uptake and excretion. CETP activity may promote RCT by stimulating LCAT activity and regenerating pre-β HDL (Brewer et al 2004). For this reason, partial inhibition of CETP activity has been the focus of novel therapeutic strategies employing CETP inhibition as discussed below.
Phase four: HDL-C catabolism
As CE accumulate in its central core, pre-β HDL-C matures into larger HDL-C particles known as HDL-3 and HDL-2. These larger molecules undergo hepatic catabolism and excretion in bile. HDL-C catabolism is mediated by 4 mechanisms: 1) hepatic uptake of larger HDL-C particles via hepatic scavenger receptor B1 (SR-B1) receptors for excretion as bile, 2) metabolism of mature HDL-C by hepatic lipase (HL) to smaller particles devoid of lipid and rich in Apo AI, 3) renal uptake of smaller HDL-C particles mediated by apo-E receptors such as cubulin, or 4) LDLr-mediated hepatic uptake of LDL-C and VLDL-C after acquiring CE via CETP activity (Moestrup and Koz 2000; Lewis 2006).
Pleiotropic effects of HDL-C: beyond RCT
The vasoprotective properties of HDL-C extend beyond its ability to initiate RCT (Figure 3). Substantial evidence supports the contention that HDL-C and Apo AI prevent oxidative damage, inhibit systemic inflammation, promote vascular reactivity and integrity, and prevent thrombosis (Hayek et al 1995; Watson 1995; Navab et al 1996; Bonnefant-Rousselot et al 1999; Mineo et al 2006).
Figure 3.
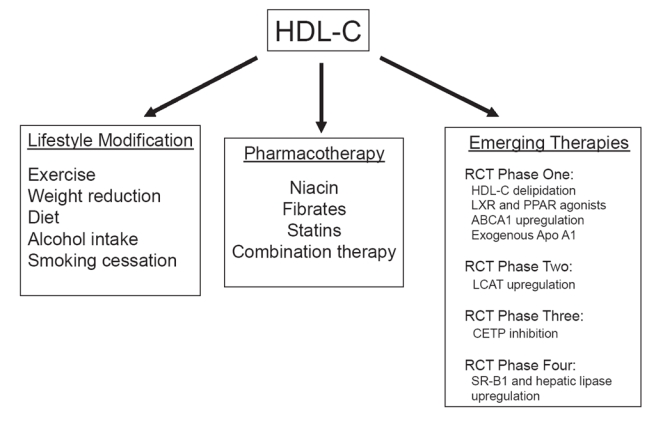
Approaches to enhance HDL-C levels and activity.
Antioxidant effects of HDL-C
The major anti-oxidant effects of HDL-C are mediated by two associated enzymes paroxonase (PON) and platelet-activating factor acetylhydrolase (PAFAH) (Graham et al 1997). PON, an arylesterase enzyme carried by Apo AI, inhibits oxidation of LDL-C (Mackness et al 2000). LDLr-null mice lacking PON are susceptible to organophosphate toxicity and manifest accelerated atherosclerosis (Shih et al 1998). In epidemiologic studies, genetic polymorphisms of the PON gene may be associated with an increased risk of CHD (Hegele 1999).
Platelet activating factor (PAF) is a potent phospholipid released by activated endothelial cells. PAF promotes cell adhesion, platelet aggregation, and vascular permeability. HDL-C inhibits PAF production by endothelial cells in dose-dependent manner (Sugatani et al 1996) via hydrolysis of acetyl residues mediated by PAFAH. Genetic polymorphisms of the enzyme PAFAH have been associated with an increased risk of acute MI (Liu et al 2006). PAFAH also degrades oxidized LDL-C and inhibit oxidation of LDL-C (Mackness et al 2004; Arakawa et al 2005).
Anti-inflammatory effects of HDL-C
The role of inflammation in atherogenesis has been well-established by a number of studies demonstrating accumulation of macrophages derived from circulating monocytes in atheromatous plaques. Anti-inflammatory effects of HDL-C include: 1) neutralization of lipopolysaccharide-induced tumor necrosis factor alpha (TNF-α) release, 2) inhibition of complement activation, 3) inhibition of vascular cell adhesion molecules (VCAM) and monocyte chemotactic protein (MCP-1), which are known to mediate monocyte-endothelial cell interaction, and 4) induced expression of the anti-inflammatory cytokine transforming growth factor-beta 2 by HDL-3 (Dimayuga et al 1999; Shah et al 2001; Calabresi et al 2003; Barter et al 2004; Norata et al 2005).
Effects of HDL-C on endothelial function and integrity
In patients with known CAD, elevation of HDL-C levels via pharmacologic therapy improves endothelial function (O’Connell et al 2001). In patients with CHD, HDL-C levels correlate positively with coronary vasomotor tone (Zeiher et al 1994). In vitro, HDL-C enhances endothelial nitric oxide synthase (eNOS) activity (Kuvin et al 2002). The mechanism of HDL-C mediated eNOS activation remains unknown, however may involve an interaction between endothelial SR-B1 and Apo AI (Yuhanna et al 2001). In a murine model of myocardial infarction, exogenous administration of human HDL-C increases myocardial perfusion as measured by 99mTc-MIBI uptake via NOS-dependent mechanisms (Levkau et al 2004).
HDL-C also enhances endothelial integrity. HDL-C inhibits endothelial apoptosis induced by TNF-α in a dose-dependent manner by inhibiting caspase 3 activity (Sugano et al 2000). HDL-associated lysosphingolipids also suppress mitochondrial pathways of apoptosis by activating the anti-apoptotic serine/threonine protein kinase, Akt (Nofer et al 2001). HDL-C also promotes endothelial cell proliferation via a mechanism involving increased phospholipase C activity (Darbon et al 1986; Honda et al 1999).
Antithrombotic effects of HDL-C
HDL-C is also associated with anti-thrombotic and profibrinolytic effects. HDL-C inhibits platelet aggregation by blocking thromboxane-A2 (TXA2) and PAF activity, while stimulating nitric oxide (NO) and PGI2 synthesis (Saku et al 1985; Naqvi et al 1999). In the Atherosclerosis Risk in Communities (ARIC) study, HDL-C levels inversely correlated with circulating von Willebrand factor (vWF) levels, suggesting that HDL-C may prevent synthesis of this pro-thrombotic protein. HDL-C also enhances the anti-thrombotic activity of protein C and protein S (Griffin et al 1999). HDL-C may also attenuate the activity of tissue factor, a potent stimulant of the extrinsic coagulant pathway (Carson 1981).
Taken together, the pleiotropic effects of HDL-C suggest that its use either as a supplement or an infusion may not be limited to chronic therapy, but may eventually play a role in the acute management of vascular disorders. To date, exogenous infusions of synthetic HDL-C have been effective in animal models of atherosclerosis (Shah et al 2001), restenosis after angioplasty (Ameli et al 1994), vascular thrombosis (Li et al 1999), myocardial ischemia-reperfusion injury (Calabresi et al 2003), and septic shock (McDonald et al 2003). Approaches to raise HDL-C levels and subsequently promote RCT include lifestyle modifications, standard pharmacologic therapy, and several emerging therapeutics based on metabolic targets involved in RCT (Figure 3).
Approaches to raising HDL-C levels: lifestyle modifictions
Exercise and weight loss
Numerous studies associate excess body weight with higher TC, LDL-C, and TG levels and lower HDL-C levels. Active weight loss improves HDL-C levels, while decreasing LDL-C levels (8% decrease for every 1 kg of weight lost). A meta-analysis of 70 studies examining the effects of weight reduction on lipid profiles published between 1966 and 1989 demonstrated a 1 mg/dL increase in HDL-C for every 3 kg of weight lost (Dattilo et al 1992). A one-year randomized controlled study evaluating weight loss on plasma lipid profiles in 131 overweight sedentary men demonstrated a significant increase in plasma HDL-C levels (44 mg/dL with exercise, 47 mg/dL with diet, versus 40 mg/dL in controls; p < 0.01), while LDL-C levels remained unchanged (Wood et al 1998).
As a means to reducing weight, regular aerobic exercise increases HDL-C by 10%–20% on average in sedentary adults (Williams 1997). Previous studies report an increase in HDL-C levels by 1 mg/dL for every 4 to 5 miles run per week (ie, 49 mg/dL with 5 miles (8 km) run per week, 51 mg/dL with 9 miles (15) run per week, 53 mg/dL with 12 miles (20 km) run per week, and 57 mg/dL with 31 miles (50 km) run per week; p < 0.001 versus non-runners) (Kokkinos et al 1995). While exercise quantity and intensity differ between studies, the duration of aerobic exercise rather than intensity appears to have a greater impact on HDL-C levels (Durstine et al 2001).
Variable changes in HDL-C have been observed in response to exercise. Some individuals significantly increase HDL-C levels after 8 weeks of regular aerobic exercise (running), while other individuals may not manifest changes in HDL-C for nearly 2 years (Durstine et al 2001). In another study, no significant change in HDL-C was observed in adults with low HDL-C and moderately elevated LDL-C after 6 weeks of walking or jogging 10 miles (16 km) per week (Stefanick et al 1998). Moreover, women appear to experience greater improvement in HDL-C with cardiac rehabilitation than men (Savage et al 2004). In general, HDL-C increases with exercise supporting the recommendation of a program of regular, brisk aerobic exercise program most days of the week (US Department of Health and Human Services 1999). The mechanisms, by which exercise and reduced weight increase HDL-C likely involves enchanced lipoprotein lipase (LL) activity, increased RCT, and increased levels of pre-β HDL (Gupta et al 1993; Sviridov et al 2003).
Dietary modifications
Major dietary influences on HDL-C levels include total fat intake (independent of fat type), trans – fatty acids, and alcohol intake (Thornton et al 1983; Rossner and Bjor 1987; Lichtenstein 1999). A low saturated fat diet lowers both LDL-C and HDL-C. In a recent study, 11 healthy volunteers were randomized to either a low fat diet (19% fat) or a high fat diet (50% fat) (Meksawan et al 2004). Individuals consuming a low fat diet had a significantly (p < 0.05) lower HDL-2 subpopulation (54 ± 3 vs 63 ± 3 mg/dL) and Apo AI (118 ± 4 vs 127 ± 3 mg/dL) compared with subjects consuming a high fat diet. In addition, reduced dietary fat has been shown to significantly decrease HDL-C2 fractions, which has been linked to antiatherogenic effects of HDL-C (Berglund et al 1999). This suggests that low fat diets may adversely affect the most antiatherogenic HDL subpopulation. However, a simultaneous decrease in LDL-C with low fat diets appears to be more clinically important than the reduction in HDL-C levels.
High consumption of n-3 polyunsaturated fatty acids observed in Native Chukot Peninsula residents is associated with higher HDL-C/Apo AI ratios and increased cholesterol efflux from cellular membranes to HDL-C (Gerasimova et al 1991). Consumption of foods high in n-3 polyunsaturated fats (cold-water fish, some shellfish, as well as flax seed, canola, soybean oils and walnuts) increase HDL-C. However, the ability of n-3 polyunsaturated fats to raise HDL-C maybe influenced by TG levels. Dietary modification with omega-3 fatty acids (fish oil), such as eicosapentaenoic acid and docosahexaenoic acid, leads to significant reductions in VLDL-C (25%–30%) and triglyceride levels, yet exerts only a modest effect on HDL-C levels (0 to 3% increase) in patients with TG levels above approximately 175 mg/dL (Kris-Etherton et al 2002). This suggests that optimization of an individuals TG must occur before a clinically significant increase in HDL-C in response to a diet high in n-3 polyunsaturated fats is observed.
Moderate alcohol consumption has been shown to elevate HDL-C levels (Ellison RC 2004). Mechanisms by which alcohol consumption increases HDL-C may involve changes in Apo AI synthesis and transportation, inhibition of CETP activity and stimulation of early steps in RCT (Van der Gaag et al 2001). A meta-analysis of 25 studies found that consumption of 30 g of alcohol per day increases HDL-C by about 4 mg/dl, irrespective of the type of alcohol consumed. With weighted regression, this represents a 0.133 mg/dL increase in HDL-C per gram of alcohol consumed per day, an 8% increase from pre-treatment levels (Rimm et al 1999). Similarly, in a review of 340 MI patients presenting with MI, alcohol consumption was strongly associated with increased HDL-C and a significantly reduced relative risk of MI in the two highest consumption categories (≥1 drink/day and 3 drinks/day) (Gaziano et al 1993). Mild to moderate alcohol consumption (1–2 alcoholic beverages several days a week) is reasonable for those individuals with low HDL-C. Caution should be used, however, when recommending alcohol consumption as a therapeutic mechanism in populations at risk for alcohol abuse.
A recent study suggests that the greatest improvement in HDL-C for both men and women in response to weight loss, exercise, and alcohol consumption was seen in individuals within the highest percentiles of HDL-C at baseline, with lower levels of baseline HDL-C being more resistant to lifestyle modifications (Williams 2004). Separating the effect of one lifestyle modification from another on HDL-C is difficult. For instance, increases in HDL-C with exercise may be due to the resultant weight loss or gene-environmental interactions (Miller et al 2003).
Smoking cessation
Cigarette smoking is associated with lower HDL-C levels (Craig et al 1989). The mechanisms by which cigarette smoking lowers HDL-C remain unclear. Cigarette smokers have significantly lower LCAT activity (Imamura et al 2002) and exhibit changes in lipid transfer proteins and CETP activity compared to non-smokers (Freeman et al 1998; Mero et al 1998). In a meta-analysis of 29 studies HDL-C levels were significantly increased after smoking cessation, more so in women and to a greater extent in individuals with higher baseline HDL-C (>1.2 mmol/L) (Maeda et al 2003). Moffatt and colleagues demonstrated women smokers have 15%–20% lower HDL-C levels compared to non-smokers (p < 0.05); the HDL-C values improved to normal levels within 30–60 days of smoking cessation (Moffatt 1988). Using a comprehensive approach to smoking cessation (wellbutrin/nicotine-replacement/counseling) remains an important recommendation for those individuals with low HDL-C. Despite the complexity of lifestyle modifications on HDL-C, patients with low HDL-C should be encouraged to become physically active on a regular basis, stop smoking, obtain stable weight reduction with a BMI <25, and increase consumption of polyunsaturated and monounsaturated fats.
Approaches to raising HDL-C levels: standard pharmacotherapy
While dietary and lifestyle modifications can raise HDL-C levels, their effect on cardiovascular outcomes may result from beneficial effects on non-HDL-C lipid components such as LDL-C. At present, standard pharmacotherapy to raise HDL-C levels includes niacin, fibrates, and statins.
Niacin
Since 1955, the B-vitamin niacin (nicotinic acid) has been used in the treatment of dyslipidemia (Altschul et al 1955). Niacin is the most useful pharmacologic therapy for raising HDL-C levels; it has been shown to increase HDL-C by 35%, while lowering TG levels by 20%–50% and LDL-C levels by 5%–25% (Szapary and Rader 2004). Niacin raises HDL-C levels by reducing the fractional catabolic rate of Apo AI containing HDL-C particles, decreasing hepatic removal of lipoprotein A-I (LpA-I) (a cardioprotective subfraction of HDL-C without Apo AII), and inhibiting removal of Apo AI without affecting HDL cholesterol ester (Jin et al 1997); resulting in an increase of Apo AI enriched, pre-β HDL particles (Ganji et al 2003). Using carotid intima-medial thickness (CIMT) as a measure of subclinical atherosclerosis, a randomized, placebo-controlled study of extended release niacin in addition to statin therapy in 167 patients with known CAD and low serum HDL-C (<45 mg/dL) showed a significantly reduced rate of IMT progression in individuals without insulin resistance (p = 0.026) (Taylor et al 2004). An upcoming study known as ARBITER 6-HALTS (HDL and LDL Treatment Strategies) will randomize 400 subjects with coronary heart disease to HDL-C (extended-release niacin) and LDL-C (ezetimibe) focused strategies of lipid therapy and will measure changes in mean CIMT after 14 months (Devine et al 2007).
Fibrates
Fibric acid derivatives (fibrates) reduce CHD risk in patients with baseline LDL:HDL-C ratios of >5.0 (Huttunen 1991). Fibrates slow the progression of coronary atherosclerosis and reduce coronary events (Ericsson et al 1996; Frick et al 1997). Fibrates induce a 5%–20% increase in HDL-C, with generally modest reductions in LDL-C and a pronounced reduction in triglyceride-rich lipoproteins (Despres 2001).
Fibrate therapy increases HDL-C levels by activating PPARα and by enhancing expression of Apo AI and AII, LL, and ABCA1, which collectively enhance RCT (Tilly-Kiesi et al 1992). By inducing LL activity, fibrates also increase hepatic fatty acid uptake, enhance removal of LDL particles, and reduce lipid exchange between VLDL and HDL (Staels et al 1998). The hypotriglyceridemic effects of fibrate therapy result from enhanced LL activity and inhibition of Apo CIII gene expression by fibrate-mediated PPARα activation (Staels et al 1995; Motojima et al 1997).
Depending on baseline lipid profiles and the potency of individual fibrates, variable effects on HDL-metabolism have been observed. Despite a greater than 15% increase in HDL-C levels with bezafibrate therapy, the Bezafibrate Infarction Prevention (BIP) failed to demonstrate a significant reduction in the primary composite end point of fatal or nonfatal MI or sudden death (Goldbourt et al 1993). In contrast, in the VA-HIT study, gemfibrozil increased HDL-C on average by 7.5% with a 2% reduction in risk correlated with every 1% increase in HDL-C (Rubenset al 2001). Similarly, in the Lopid Coronary Angiography Trial (LOCAT), gemfibrozil slowed progression of coronary atherosclerosis and the formation of bypass graft lesions.
Statins
Statins inhibit HMG-CoA reductase, the rate-limiting step in cholesterol biosynthesis, resulting in increased LDLr density with decreases in LDL, IDL, and VLDL particle synthesis (Farnier 1998; Segrest et al 2000). Subsequent reductions in LDL-C and TG concentrations are the primary antiatherogenic properties of statins (Harper and Jacobsen 1999). Statins reduce LDL-C levels by 25%–35% at low to moderate doses, which correlates with a reduction in relative risk of coronary events by 25%–37% (Sacks et al 1996; West of Scotland Coronary Prevention Group 1996; Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group 1998).
Statins increase HDL-C by 5%–15% and decrease TG levels by 7%–30% (Belalcazar et al 1998). The mechanism of statin-induced increases in HDL-C remains incompletely understood. Some studies suggest increased HDL-C results from a decreased fractional catabolic rate of Apo AI and an increased production of Apo AI induced by inhibiting HMG-Co A reductase (Schaefer et al 1999). Statins have also been shown to increase Apo AI levels by inhibiting the Rho-A kinase signal transduction pathway, resulting in activation of PPARα (Martin et al 2001).
Statins may also reduce hepatic lipase activity, resulting in enhanced synthesis of mature HDL-C. Another potential mechanism for increased HDL-C levels in response to statin therapy is by inhibiting CETP activity. A study of patients with the “B1” variant of the CETP gene showed high levels of baseline CETP activity and low levels of HDL-C with corresponding progression of atherosclerosis. Treatment with pravastatin abolished the progression in atheromatous burden and non-significantly increased HDL-C levels in patients with the “B1” variant (Kuivenhoven et al 1998).
Of the available statins, simvastatin, rosuvastatin, and fluvastatin more effectively raise HDL-C levels compared with atorvastatin at doses that lead to similar reductions in LDL-C. In a 36-week, multicenter, double-blind, dose titration study, 826 patients with LDL-C >160 mg/dL and triglyceride <350 mg/dL were randomized to receive titrated doses of simvastatin (maximum 40 mg/day) or atorvastatin (maximum 40 mg/day) over 6–12 weeks. Significantly greater increases in HDL-C and Apo AI with simvastatin compared to atorvastatin (HDL-C: 9% vs 7% p < 0.001; Apo AI: 6% vs 3%, p < 0.001) were observed (Kastelein et al 2000). Independent clinical studies have shown that 40 mg/day of simvastatin increase HDL-C by approximately 7%–9% versus a 4%–5% increase with 20 mg/day of atorvastatin (Heinonen et al 1996; Crouse et al 1999). Generally niacin is more effective than statins alone in raising HDL-C levels.
Combination therapy
Combination therapy using statins with niacin or fibrates has been evaluated in a number of small clinical trials (Davidson 2002). The HDL Atherosclerosis Treatment Study (HATS) studied the combination of statin plus extended-release niacin in 160 patients with known CAD, low serum HDL-C levels (<35 mg/dL in males and <40 mg/dL in females), LDL-C levels <145 mg/dL, and TG levels <400 mg/dL. After 3 years of follow-up, combination therapy increased HDL-C levels by 26%, reduced LDL-C levels by 42%, induced regression of the average coronary stenosis by 0.4% (p < 0.001) and reduced cardiovascular events by greater than 60% (Brown et al 2001).
Reflecting the ability of statins and fibrates to synergistically activate PPARα, statin-fibrate combinations have been evaluated in a number of clinical trials (Ellen and McPherson 1998; Papadakis et al 1999; Farnier et al 2000; Athyros et al 2002; Vega et al 2003;). A multi-center, double-blind trial of 333 patients using fluvastatin compared to bezafibrate versus a combination of the two agents for 24 weeks showed a significant decrease in LDL-C levels in treatment groups receiving fluvastatin compared to bezafibrate alone. Furthermore, HDL-C levels were significantly elevated with bezafibrate alone or in combination with fluvastatin compared with fluvastatin alone (p < 0.001). The combination of fluvastatin and bezafibrate was well tolerated and significantly increased HDL-C (22%), decreased LDL-C (24%), and decreased TG levels (38%) compared with fluvastatin alone (p < 0.001) (Pauciullio et al 2000).
Recently, the Comparative Effects on Lipid Levels (COMPELL) trial determined the relative efficacy of combination therapy with a statin and niacin or ezetimibe compared with a statin alone over 12 weeks. Coadministration of niacin (500 mg up-titrated to 2000 mg) with atorvastatin (20–40 mg, n = 60) or rosuvastatin (10–20 mg, n = 65) decreased LDL-C by 56% and 51% and increased HDL-C by 22% and 24% respectively (p = NS). While simvastatin plus ezetimibe decreased LDL-C by 57%, HDL-C only increased by 10% compared to baseline. Rosuvastatin (10–40 mg) monotherapy decreased LDL-C by 53% and raised HDL-C by 7% (Jones 2006). Future studies involving rosuvastatin/fenofibrate combination therapy and the recently announced combination of rosuvastatin with a next generation fenofibrate (ABT-335) will provide further insight into the efficacy of dual-targeted therapy.
Approaches to raising HDL-C levels: emerging therapeutics
Based on preclinical data, multiple strategies to enhance the beneficial effects of HDL-C are being considered. HDL-C delipidation therapy (Kostner et al 2002), exogenous Apo AI mimetics (Navab et al 2004), CETP inhibition (Brousseau et al 2004), LXR/RXR agonists (Brewer et al 2004), selective and non-selective PPAR agonists (Oliver et al 2001; Schmitz et al 2002), and drugs targeting HDL-C catabolism (Mezdour et al 1997; Jansen et al 2004) are among some of the novel emerging therapies harnessing the anti-atherogenic, anti-oxidant, anti-inflammatory, and pro-endothelial functions of HDL-C.
HDL-C delipidation therapy
Selective HDL-C delipidation therapy utilizes plasmapheresis whereby extracted plasma is mixed with a delipidating agent and separated into an inorganic and organic phase. The organic component contains a high concentration of delipidated HDL, similar to lipid-poor pre-β HDL produced by the liver, which is then returned to the circulation. In a series of animal studies delipidation therapy has been shown to markedly increase circulating pre-β HDL levels and subsequently increase ABCA1-mediated cholesterol efflux from peripheral cells without exerting a significant effect on LDL-C metabolism Animal studies evaluating delipidation therapy followed by intravascular ultrasound assessment of vascular plaque progression or regression are ongoing (Shah 2007).
Exogenous administration of Apo AI and Apo AI mimetics
Exogenous administration of Apo AI directly enhances RCT via the ABCA1 pathway (Zhang et al 2003; Navab et al 2004; Arakawa et al 2004). Treating normal human LDL-C with exogenous Apo AI in vitro reduces levels of oxidized lipids by 50%–60% and prevents monocyte chemotactic activity, a primary step in atherogenesis (Poon et al 1997). Exogenous administration of Apo AI-associated lecithin discs reduces the ability of LDL to induce monocyte chemotaxis, increases concentrations of pre-β HDL, and stimulates RCT in human subjects (Nanjee et al 2001). Apo AI infusions also modulate phospholipids transfer protein (PLTP), LCAT, and CETP activity, all of which potentially contribute to RCT (Kujiraoka et al 2003).
Recently, Tardiff and colleagues administered 4 weekly infusions of a mixture of human wild-type Apo AI and soybean phosphatidylcholine (CSL-111; 40 mg/kg or 80 mg/kg) or volume-matched placebo to 183 patients presenting with an acute coronary syndrome as part of the Effect of reconstituted HDL on Atherosclerosis – Safety and Efficacy (ERASE) study. Two weeks after the last infusion, intravascular ultrasound (IVUS) and quantitative coronary angiographic (QCA) measurements were compared to baseline. Patients receiving CSL-111 experienced a −3.4% change in atheroma volume (p = 0.48 vs placebo; p < 0.001 vs baseline) with an absolute change of −5.3 mm3 (p = 0.39 vs placebo; p < 0.001 vs baseline). Notable, transient liver function abnormalities were observed in the CSL-111 group versus placebo. One patient developed a 100-fold increase in ALT levels in the high dose (80 mg/kg) infusion group (Tardif et al 2007) While the primary endpoint of the study was negative, the data presented suggest a potential benefit for inducing plaque regression. This study highlights the complexity surrounding exogenous Apo AI therapy.
Based on observations in a family with low HDL-C and a lack of atherosclerotic disease from Limone sul Garda, Italy, a variant form of Apo AI, known as Apo AI Milano (AIM), was identified in 1980 (Franceschini et al 1980). A cysteinearginine substitution at position 173 in the amino acid sequence allows the mutant protein to form disulfide bonds with other Apo AI molecules and Apo AII. AIM homodimers and heterodimers may enhance cholesterol efflux thereby augmenting RCT (Chiesa et al 2002).
Administration of recombinant Apo AI Milano (rAIM) reduces plaque cross sectional area compared to saline-placebo by up to 40% in rabbit carotid models of atherosclerosis (Ameli et al 1994; Ibanez 2007). Similar results have been demonstrated in balloon injured arteries in hypercholesterolemic rabbits, Apo E-deficient mice, and in transgenic mouse models of Apo AI over-expression (Rubin et al 1991; Shah et al 1998). Exogenous HDL-C or Apo AI administration also enhance fecal steroid excretion, increase serum pre-β HDL, and enhance RCT in humans (Westman et al 1995; Eriksson et al 1999).
In 2003 a landmark study using rAIM (ETC-216) quantified coronary plaque volume as a response to pharmacologic intervention with intravascular ultrasound (IVUS). This study evaluated the effect of exogenous administration of ETC-216 on coronary atherosclerosis in patients with acute coronary syndromes. ETC-216 reduced total atheroma volume by 1.3% (39.7–38.4) and 0.7% (37.2–36.6) in the moderate and high dose treatment groups respectively, while a 0.14% (34.8–34.9) increase was noted in the placebo group (Nissen et al 2003). This “proof-of-concept” study demonstrated the ability of Apo AI mimetic peptides to halt progression and potentially induce regression of atheromatous plaque.
A series of Apo AI mimetic peptides are currently under investigation. ETC-642 is a second generation Apo AI synthetic peptide containing three charged residues in a 22 amino-acid sequence, rendering the peptide more hydrophobic (Navab et al 2005). Within hours of treatment with ETC-642 increased HDL-C serum levels have been observed in rabbit models. Increased CE content in HDL-C indicates concomitant LCAT activation by ETC-642. This rapid elevation of HDL-C levels suggests a possible future role for Apo AI mimetic peptides in the management of acute coronary syndromes or in the setting of ischemia-reperfusion injury (Marchesi et al 2004).
Another Apo AI mimetic peptide known as D4F reduces atherosclerosis in mouse models (Garber et al 2001). Peptide D4F contains 18 amino acids in a class A amphipathic helix with polar and non-polar faces yielding high lipid affinity (Datta et al 2001). D4F enhances the anti-inflammatory properties of HDL-C, reduces LDL-mediated monocyte chemotaxis, reduces macrophage migration into atheromatous plaques, and reduces atherosclerosis in Apo E KO mice alone or in combination with statin therapy. Both oral and intraperitoneal administration of D4F significantly reduced evolving atherosclerotic lesions in vein grafts but not established atherosclerotic lesions in the aortic sinus, suggesting specific types of atherosclerotic lesions may modulate the beneficial effects of Apo AI mimetic peptides (Li et al 2004).
Taking advantage of the amphipathic helical structure common to apolipoproteins, numerous Apo AI mimetic peptides are being developed. Unique helical configuration with opposing hydrophobic and hydrophilic faces enhances interaction between lipid surfaces and apolipoproteins for the removal of membrane bound cholesterol. Some novel agents under development include: ETC-588 (large unilamellar vesicles – LUV), ETC-1001 (small molecule investigational product), helical peptides (Esperion 24218), and trimeric Apo-A (Proteopharma/Borean pharma) (Navab et al 2006).
Nuclear regulation of RCT: LXR and PPAR agonists
Liver X-receptors (LXR) are nuclear receptors that sense excess intracellular cholesterol (Wang and Briggs 2004). Hydroxylated cholesterol stimulates LXR-mediated transcription of ABCA1, which subsequently enhances RCT from peripheral tissues (Lund et al 2006). Two types of LXR receptors exist, LXRα and LXRβ. LXRα has been identified in liver, intestine, macrophages and adipose tissues, while LXRβ is ubiquitously expressed similar to ABCA1 (Lala et al 2005).
LXR agonists prevent development of atherosclerosis by modulating metabolic and inflammatory gene expression in rodent models. Non-selective LXR agonists increase ABCA1 synthesis with a gradual increase in HDL-C serum levels (Lund et al 2006). In a mouse LXRα knockout model, treatment with a non-selective LXR agonist increased HDL-C by day 7 with a less significant increase in hepatic TG content (Joseph et al 2002). Similarly, treating LDLr KO mice with the LXR ligand, T-0901317, reduced atherosclerotic lesion development without affecting plasma total cholesterol levels (Terasaka et al 2003). Recently administration of the LXR agonist GW3965 to mice increased the rate of RCT from macrophages to feces in vivo (Naik et al 2006).
A major concern associated with LXR agonists is the development of hepatic steatosis. Since LXR agonists induce genes that stimulate lipogenesis, including the sterol response element binding protein (SREBP1-c) and fatty acid synthetase (FAS). The induction of these genes in the liver cause increased hepatic triglyceride synthesis, hypertriglyceridemia, and hepatic steatosis. Current research has focused on selective LXR modulators that may circumvent this adverse effect on hepatic function (Miao et al 2004).
First identified in rodent models of fibrate-induced hepatic peroxisome proliferation, peroxisome proliferators activated receptors (PPARs) are another family of nuclear receptors closely linked to HDL-C metabolism (Everett et al 2000). Acting as synthetic ligands for PPARα activation, fibrates increase circulating levels of HDL-C, enhance RCT and reduce vascular inflammation and thrombogenicity (Barbier et al 2002; Gervois et al 2007). PPARα agonists enhance gene expression of SR BI, Apo AI, Apo AII, LPL, and ABCA1 (Toth 2005). Statins also enhance PPARα activity and may enhance cholesterol efflux (Martin et al 2001; Inoue et al 2002).
Agents for the management of Type 2 diabetes such as thiazolidinediones are known PPARγ agonists and enhance ABCA1 mediated RCT and increase HDL-C levels in primates (Oliver et al 2001). PPARα and γ mediate activation of the LXR/RXR heterodimer, which in turn regulates cholesterol efflux via ABCA1 and ABCG1 activation (Schmitz et al 2002). Unfortunately, the development of novel PPAR agonists to date, particularly PPARγand PPARα/γ, have been halted due to preclinical and clinical adverse effects (Rubenstrunk et al 2007).
Cholesterol exchange transfer protein (CETP) inhibition
The complex relationship between CETP activity and atherosclerotic disease has been illustrated by several recent studies evaluating CETP inhibition therapy. In the past, antisense oligodeoxynucleotides and antibodies against CETP increased HDL-C levels and reduced aortic atherosclerotic burden in cholesterol-fed rabbits (Sugano et al 1998; Rittershaus et al 2000). In a phase II, randomized, placebo-controlled trial, 148 statin-naïve patients with mild hyperlipidemia were treated with a CETP inhibitor known as JTT-705. In this study, a 37% reduction in CETP activity correlated with a 34% increase in HDL levels and 7% decrease in LDL levels (De Grooth et al 2002).
Recently, the CETP inhibitor, torcetrapib, has been extensively studied evaluated in a number of human trials. In a phase I, multi-dose study, torcetrapib was administered to 40 normolipidemic individuals in doses of 10, 30, 60, and 120 mg per day and 120 mg twice daily. Significant increases in serum HDL-C levels ranging from 16% to 91% (10 mg daily versus 120 mg twice daily) with a decrease in LDL-C from 7% to 42% (60 mg daily versus 120 mg twice daily) without changing TC levels was observed. At the highest dosing regimen, Apo AI and Apo E increased by 27% and 66% respectively, while Apo B decreased by 26% (Clark et al 2004).
Another single-blinded study compared torcetrapib alone at variable doses versus torcetrapib in combination with 20 mg of atorvastatin for one month. HDL-C levels increased by 46%, 61%, and 106%, while LDL-C levels decreased by 17%, 7.5%, and 17% after treatment with 120 mg daily, 120 mg daily plus 20 mg atorvastatin, and 120 mg twice daily respectively. No major adverse events were reported (Brousseau et al 2004).
Based on these promising findings, torcetrapib was evaluated in a large international clinical trial known as the Investigation of Lipid Level Management to Understand Its Impact in Atherosclerotic Events (ILLUMINATE). In this study, 15,000 patients at high risk for CHD received torcetrapib with atorvastatin versus atorvastatin alone. In December 2006, this study was prematurely terminated due to an excess of deaths in the torcetrapib/atorvastatin versus atorvastatin groups (81 vs 51, respectively). Additionally, patients receiving torcetrapib had a higher incidence of heart failure, angina, and revascularization procedures. The adverse outcomes of the ILLUMINATE study may have been related to off-target effects of torcetrapib, such as an increase in systolic blood pressure (limited to 1–2 mmHg) or low levels of CETP inhibition and reduced RCT (Honey 2007). Notably, inherited deficiencies of CETP and other CETP inhibitor formulations did not increase systolic blood pressure, suggesting that the chemical structure of torcetrapib itself may have contributed to poor outcomes (Tall et al 2007).
Two separate studies evaluated the impact of torcetrapib on atherosclerotic progression. The Investigation of Lipid Level Management Using Coronary Ultrasound to Assess Reduction of Atherosclerosis by CETP Inhibition and HDL Elevation (ILLUSTRATE) studied 1,188 patients with coronary disease in a prospective, randomized, double-blinded fashion. Patients initially received atorvastatin monotherapy until LDL-C levels were below 100 mg/dL, after which time they were randomized to either atorvastatin (10–80 mg daily) monotherapy or atorvastatin plus 60mg torcetrapib daily. After 24 months, the torcetrapib/atorvastatin group had a 61% relative increase in HDL-C and 20% decrease in LDL-C. However, no significant reduction in percent atheroma volume was observed between atorvastatin monotherapy and torcetrapib combination therapy (0.19% vs 0.12%, respectively). Torcetrapib therapy was again associated with a mean 4.6 mmHg increase in systolic blood pressure (Nissen et al 2007).
Further substantiating these findings, the Rating of Atherosclerotic Disease Change by Imaging with a New CETP Inhibitor (RADIANCE 1 and 2) trials, demonstrated no further reduction of atherosclerosis progression in carotid intima-media thickness after 24 months of therapy with atorvastatin versus atorvastatin/torcetrapib combination (Bots et al 2007; Kastelein et al 2007).
Summary
A considerable body of evidence supports the correlation between HDL-C levels and cardiovascular risk. However, trials evaluating HDL-C targeted therapies are limited, in part due to a lack of pharmacologic agents specifically designed to raise HDL-C and our limited ability to measure HDL-C effectiveness. As a result, there is not enough data to support guidelines recommending aggressive increases in HDL-C levels. With this in mind, evaluating the clinical efficacy of emerging HDL-C targeted therapies will be of paramount importance.
Given the complexity of HDL-C metabolism, serum levels of HDL-C may not be an adequate indicator of efficacy. At present, plasma HDL-C measurements have a ± 10% margin of error, which could lead to errors in measurement of up to 4 mg/dL (Friedewald et al 2007). The functional properties of circulating HDL-C levels, the kinetics of HDL-C metabolism, and the variable effects of HDL-C subfractions on atherogenesis are ignored by current laboratory measures of HDL-C (Forrester et al 2005). While measuring HDL-C subfractions are not recommended at present, recent data suggests that increased Apo AI plasma levels and Apo AI:Apo B ratios correlate with a reduced risk of myocardial infarction and stroke (Qureshi et al 2002).
As a crude marker of RCT, measuring sterol content in fecal matter indirectly reflects the amount of cholesterol excreted by the liver as bile. Emerging therapies such as Apo AI mimetics (Eriksson et al 1999) and LXR agonists (Plosch et al 2002) increase fecal sterol excretion (FSE), while CETP inhibition with torcetrapib fails to affect fecal sterol content (Brousseau et al 2005). The clinical significance of FSE as a marker of RCT remains uncertain.
At present, volumetric IVUS in combination with standard HDL-C measurement is our best measure of plaque stabilization or regression. Novel approaches using high-speed multislice CT, cardiac MRI, and intracoronary MRI may replace IVUS. Clinical trials using both surrogate endpoints such as volumetric IVUS and carotid IMT in combination with hard clinical endpoints may be required to evaluate the efficacy of emerging HDL-C based therapies.
During the next 10 years, many clinical trials will evaluate the effects of HDL-modification therapy. While the past decade has focused on lowering levels of LDL-C and non-HDL-C to reduce atherosclerotic progression and CVD events, the next decade may prove that HDL-focused therapy induces regression of luminal atherosclerosis and improves CVD outcomes. We eagerly await the completion of the ongoing clinical trials employing a variety of novel strategies to raise HDL-C and possibly halt the progression of atherosclerotic vascular disease.
References
- Altschul R, Hoffer A, Stephen JD. Influence of Nicotinic Acid on Serum Cholesterol in Man. Arch Biochem Biophys. 1955;54:558–9. doi: 10.1016/0003-9861(55)90070-9. [DOI] [PubMed] [Google Scholar]
- Ameli S, Hultgardh-Nilsson A, Cercek B, et al. Recombinant apolipoprotein A-I Milano reduces intimal thickening after balloon injury in hypercholesterolemic rabbits. Circulation. 1994;90:1935–41. doi: 10.1161/01.cir.90.4.1935. [DOI] [PubMed] [Google Scholar]
- Anderson SP, Dunn C, Laughter A, et al. Overlapping transcriptional programs regulated by the nuclear receptors peroxisome proliferator-activated receptor alpha, retinoid X receptor, and liver X receptor in mouse liver. Mol Pharmacol. 2004;66:1440–52. doi: 10.1124/mol.104.005496. [DOI] [PubMed] [Google Scholar]
- Arakawa R, Hayashi M, Remaley AT, et al. Phosphorylation and stabilization of ATP binding cassette transporter A1 by synthetic amphiphilic helical peptides. J Biol Chem. 2004;279(8):6217–20. doi: 10.1074/jbc.C300553200. [DOI] [PubMed] [Google Scholar]
- Arakawa H, Qian JY, Baatar D, et al. Local expression of platelet-activating factor-acetylhydrolase reduces accumulation of oxidized lipoproteins and inhibits inflammation, shear stress-induced thrombosis, and neointima formation in balloon-injured carotid arteries in nonhyperlipidemic rabbits. Circulation. 2005;111:3302–9. doi: 10.1161/CIRCULATIONAHA.104.476242. [DOI] [PubMed] [Google Scholar]
- Athyros VG, Papageorgiou AA, Demitriades DS, et al. Atorvastatin versus four statin-fibrate combinations in patients with familial combined hyperlipidaemia. J Cardiovasc Risk. 2002;9:33–9. doi: 10.1177/174182670200900105. [DOI] [PubMed] [Google Scholar]
- Barbier O, Torra IP, Duguay Y, et al. Pleiotropic actions of peroxisome proliferator-activated receptors in lipid metabolism and atherosclerosis. Arterioscler Thromb Vasc Biol. 2002;22:717–26. doi: 10.1161/01.atv.0000015598.86369.04. [DOI] [PubMed] [Google Scholar]
- Barr DP, Russ EM, Eder HA. Protein-lipid relationships in human plasma. Am J Med. 1951;11:480. doi: 10.1016/0002-9343(51)90183-0. [DOI] [PubMed] [Google Scholar]
- Barter PJ, Brewer HB, Chapman MJ, et al. Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler Thromb Vasc Biol. 2003;23:160–7. doi: 10.1161/01.atv.0000054658.91146.64. [DOI] [PubMed] [Google Scholar]
- Barter PJ, Nicholls S, Rye KA, et al. Antiinflammatory properties of HDL. Circ Res. 2004;8:764–72. doi: 10.1161/01.RES.0000146094.59640.13. [DOI] [PubMed] [Google Scholar]
- Badimon JJ, Badimon L, Fuster V. Regression of atherosclerotic lesions by high density lipoprotein plasma fraction in the cholesterol-fed rabbit. J Clin Invest. 1990;85:1234–41. doi: 10.1172/JCI114558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belalcazar LM, Ballantyne CM. Defining specific goals of therapy in treating dyslipidemia in the patient with low high-density lipoprotein cholesterol. Prog Cardiovasc Dis. 1998;41:151–74. doi: 10.1016/s0033-0620(98)80010-8. [DOI] [PubMed] [Google Scholar]
- Berglund L, Oliver EH, Fontanez N, et al. HDL-subpopulation patterns in response to reductions in dietary total and saturated fat intakes in healthy subjects. Am J Clin Nutr. 1999;70:992–1000. doi: 10.1093/ajcn/70.6.992. [DOI] [PubMed] [Google Scholar]
- Boekholdt SM, Kuivenhoven JA, Wareham NJ, et al. Plasma levels of cholesteryl ester transfer protein and the risk of future coronary artery disease in apparently healthy men and women: the prospective EPIC (European Prospective Investigation into Cancer and nutrition)-Norfolk population study. Circulation. 2004;110:1418–23. doi: 10.1161/01.CIR.0000141730.65972.95. [DOI] [PubMed] [Google Scholar]
- Bodzioch M, Orso E, Klucken J, et al. The gene encoding ATP-binding cassette transporter 1 is mutated in Tangier disease. Nat Genet. 1999;22:347–51. doi: 10.1038/11914. [DOI] [PubMed] [Google Scholar]
- Bonnefont-Rousselot D, Therond P, Beaudeux JL, et al. High density lipoproteins (HDL) and the oxidative hypothesis of atherosclerosis. Clin Chem Lab Med. 1999;37:939–48. doi: 10.1515/CCLM.1999.139. [DOI] [PubMed] [Google Scholar]
- Brewer HB, Jr, Remaley AT, Neufeld EB, et al. Regulation of plasma high-density lipoprotein levels by the ABCA1 transporter and the emerging role of high-density lipoprotein in the treatment of cardiovascular disease. Arterioscler Thromb Vasc Biol. 2004;24:1755–60. doi: 10.1161/01.ATV.0000142804.27420.5b. [DOI] [PubMed] [Google Scholar]
- Brousseau ME, Diffenderfer MR, Millar JS, et al. Effects of cholesteryl ester transfer protein inhibition on high-density lipoprotein subspecies, apolipoprotein A-I metabolism, and fecal sterol excretion. Arterioscler Thromb Vasc Biol. 2005;25:1057–64. doi: 10.1161/01.ATV.0000161928.16334.dd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brousseau M, Schaefer EJ, Wolfe ML, et al. Effects of an inhibitor of cholesteryl ester transfer protein on HDL cholesterol. N Engl J Med. 2004;350:1505–15. doi: 10.1056/NEJMoa031766. [DOI] [PubMed] [Google Scholar]
- Brown BG, Zhao XQ, Chait A, et al. Simvastatin and niacin, anti-oxidant vitamins, or the combination for the prevention of coronary disease. N Engl J Med. 2001;345:1583–92. doi: 10.1056/NEJMoa011090. [DOI] [PubMed] [Google Scholar]
- Bruemmer D, Law RE. Liver x receptors: potential novel targets in cardiovascular diseases. Curr Drug Targets Cardiovasc Haematol Disord. 2005;5:533–40. doi: 10.2174/156800605774961988. [DOI] [PubMed] [Google Scholar]
- Calabresi L, Gomaraschi M, Franceschini G. Endothelial protection by high-density lipoproteins: from bench to bedside. Arterioscler Thromb Vasc Biol. 2003;23:1724–31. doi: 10.1161/01.ATV.0000094961.74697.54. [DOI] [PubMed] [Google Scholar]
- Cannon CP, Braunwald E, McCabe CH, et al. Intensive versus moderate lipid lowering with statins after acute coronary syndromes. N Engl J Med. 2004;350:1495–504. doi: 10.1056/NEJMoa040583. [DOI] [PubMed] [Google Scholar]
- Carson SD. Plasma high density lipoproteins inhibit the activation of coagulation factor X by factor VIIa and tissue factor. FEBS Lett. 1981;132:37–40. doi: 10.1016/0014-5793(81)80422-x. [DOI] [PubMed] [Google Scholar]
- Castelli WP, Anderson K, Wilson PW, et al. Lipids and risk of coronary heart disease. The Framingham Study. Ann Epidemiol. 1992;2:23–8. doi: 10.1016/1047-2797(92)90033-m. [DOI] [PubMed] [Google Scholar]
- Chawla A, Boisvert WA, Lee CH, et al. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell. 2001;7:161–71. doi: 10.1016/s1097-2765(01)00164-2. [DOI] [PubMed] [Google Scholar]
- Chiesa G, Monteggia E, Marchesi M, et al. Recombinant apolipoprotein A-I (Milano) infusion into rabbit carotid artery rapidly removes lipid from fatty streaks. Circ Res. 2002;90:974–80. doi: 10.1161/01.res.0000018422.31717.ee. [DOI] [PubMed] [Google Scholar]
- Clark RW, Sutfin TA, Ruggeri RB, et al. Raising high-density lipoprotein in humans through inhibition of cholesteryl ester transfer protein: an initial multidose study of torcetrapib. Arterioscler Thromb Vasc Biol. 2004;24:490–7. doi: 10.1161/01.ATV.0000118278.21719.17. [DOI] [PubMed] [Google Scholar]
- Craig WY, Palomaki GE, Haddow JE. Cigarette smoking and serum lipid and lipoprotein concentrations: an analysis of published data. BMJ. 1989;298:784–8. doi: 10.1136/bmj.298.6676.784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouse JR, Frohlich J, Ose L, et al. Effects of high doses of simvastatin and atorvastatin on high-density lipoprotein cholesterol and apolipoprotein A-I. Am J Cardiol. 1999;83:1476–7. A7. doi: 10.1016/s0002-9149(99)00153-8. [DOI] [PubMed] [Google Scholar]
- Darbon JM, Tournier JF, Tauber JP, et al. Possible role of protein phosphorylation in the mitogenic effect of high density lipoproteins on cultured vascular endothelial cells. J Biol Chem. 1986;261:8002. [PubMed] [Google Scholar]
- Datta G, Chaddha M, Hama S, et al. Effects of increasing hydrophobicity on the physical-chemical and biological properties of a class A amphipathic helical peptide. J Lipid Res. 2001;42:1096–104. [PubMed] [Google Scholar]
- Dattilo AM, Kris-Etherton PM. Effects of weight reduction on blood lipids and lipoproteins: a meta-analysis. Am J Clin Nutr. 1992;56:320–8. doi: 10.1093/ajcn/56.2.320. [DOI] [PubMed] [Google Scholar]
- Davidson MH. Combination therapy for dyslipidemia: safety and regulatory considerations. Am J Cardiol. 2002;90:50K–60K. doi: 10.1016/s0002-9149(02)02970-3. [DOI] [PubMed] [Google Scholar]
- De Grooth GJ, Klerkx AH, Stroes ES, et al. A review of CETP and its relation to atherosclerosis. J Lipid Res. 2004;45:1967–74. doi: 10.1194/jlr.R400007-JLR200. [DOI] [PubMed] [Google Scholar]
- De Grooth GJ, Kuivenhoven JA, Stalenhoef AF, et al. Efficacy and safety of a novel cholesteryl ester transfer protein inhibitor, JTT-705, in humans: a randomized phase II dose-response study. Circulation. 2002;105:2159–65. doi: 10.1161/01.cir.0000015857.31889.7b. [DOI] [PubMed] [Google Scholar]
- Despres JP. Increasing high-density lipoprotein cholesterol: an update on fenofibrate. Am J Cardiol. 2001;88:30N–6N. doi: 10.1016/s0002-9149(01)02150-6. [DOI] [PubMed] [Google Scholar]
- Devine PJ, Turco MA, Taylor AJ. Design and Rationale of the ARBITER 6 Trial (Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol)-6-HDL and LDL Treatment Strategies in Atherosclerosis (HALTS) Cardiovasc Drugs Ther. 2007;21:221–5. doi: 10.1007/s10557-007-6020-8. [DOI] [PubMed] [Google Scholar]
- Dimayuga P, Zhu J, Oguchi S, et al. Reconstituted HDL containing human apolipoprotein A-1 reduces VCAM-1 expression and neointima formation following periadventitial cuff-induced carotid injury in apoE null mice. Biochem Biophys Res Commun. 1999;264:465–8. doi: 10.1006/bbrc.1999.1278. [DOI] [PubMed] [Google Scholar]
- Downs JR, Clearfield M, Weis S, et al. Primary prevention of acute coronary events with lovastatin in men and women with average cholesterol levels: results of AFCAPS/TexCAPS. Air Force/Texas Coronary Atherosclerosis Prevention Study. JAMA. 1998;279:1615–222. doi: 10.1001/jama.279.20.1615. [DOI] [PubMed] [Google Scholar]
- Durstine JL, Grandjean PW, Davis PG, et al. Blood lipid and lipoprotein adaptations to exercise: a quantitative analysis. Sports Med. 2001;31:1033–62. doi: 10.2165/00007256-200131150-00002. [DOI] [PubMed] [Google Scholar]
- Duverger N, Kruth H, Emmanuel F, et al. Inhibition of atherosclerosis development in cholesterol-fed human apolipoprotein A-I-transgenic rabbits. Circulation. 1996;94:713–7. doi: 10.1161/01.cir.94.4.713. [DOI] [PubMed] [Google Scholar]
- Elkhalil L, Majd Z, Bakir R, et al. Fish-eye disease: structural and in vivo metabolic abnormalities of high-density lipoproteins. Metabolism. 1997;46:474–83. doi: 10.1016/s0026-0495(97)90180-3. [DOI] [PubMed] [Google Scholar]
- Ellen RL, McPherson R. Long-term efficacy and safety of fenofibrate and a statin in the treatment of combined hyperlipidemia. Am J Cardiol. 1998;81:60B–65B. doi: 10.1016/s0002-9149(98)00040-x. [DOI] [PubMed] [Google Scholar]
- Ellison RC, Zhang Y, Qureshi MM, et al. Lifestyle determinants of high-density lipoprotein cholesterol: the National Heart, Lung, and Blood Institute Family Heart Study. Am Heart J. 2004;147:529–35. doi: 10.1016/j.ahj.2003.10.033. [DOI] [PubMed] [Google Scholar]
- Ericsson CG, Hamsten A, Nilsson J, et al. Angiographic assessment of effects of bezafibrate on progression of coronary artery disease in young male postinfarction patients. Lancet. 1996;347:849–53. doi: 10.1016/s0140-6736(96)91343-4. [DOI] [PubMed] [Google Scholar]
- Eriksson M, Carlson LA, Miettinen TA, et al. Stimulation of fecal steroid excretion after infusion of recombinant proapolipoprotein A-I. Potential reverse cholesterol transport in humans. Circulation. 1999;100:594–8. doi: 10.1161/01.cir.100.6.594. [DOI] [PubMed] [Google Scholar]
- Everett L, Galli A, Crabb D. The role of hepatic peroxisome proliferator-activated receptors (PPARs) in health and disease. Liver. 2000;20:191–9. doi: 10.1034/j.1600-0676.2000.020003191.x. [DOI] [PubMed] [Google Scholar]
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Summary of the second report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel II) JAMA. 1993;269:3015–23. [PubMed] [Google Scholar]
- Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults. Executive summary of the third report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) JAMA. 2001;285:2486–97. doi: 10.1001/jama.285.19.2486. [DOI] [PubMed] [Google Scholar]
- Farnier M. Cerivastatin in the treatment of mixed hyperlipidemia: the RIGHT study. The Cerivastatin Study Group. Cerivastatin Gemfibrozil Hyperlipidemia Treatment. Am J Cardiol. 1998;82:47J–51J. doi: 10.1016/s0002-9149(98)00437-8. [DOI] [PubMed] [Google Scholar]
- Farnier M, Portal JJ, Maigret P. Efficacy of atorvastatin compared with simvastatin in patients with hypercholesterolemia. J Cardiovasc Pharmacol Ther. 2000;5:27–32. doi: 10.1177/107424840000500104. [DOI] [PubMed] [Google Scholar]
- Forrester JS, Makkar R, Shah PK. Increasing high-density lipoprotein cholesterol in dyslipidemia by cholesteryl ester transfer protein inhibition: an update for clinicians. Circulation. 2005;111:1847–54. doi: 10.1161/01.CIR.0000160860.36911.BD. [DOI] [PubMed] [Google Scholar]
- Forte TM, Nichols AV, Krauss RM, et al. Familial apolipoprotein AI and apolipoprotein CIII deficiency. Subclass distribution, composition, and morphology of lipoproteins in a disorder associated with premature atherosclerosis. J Clin Invest. 1984;74:1601–13. doi: 10.1172/JCI111576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschini G, Sirtori CR, Capurso A, 2, et al. A-IMilano apoprotein. Decreased high density lipoprotein cholesterol levels with significant lipoprotein modifications and without clinical atherosclerosis in an Italian family. J Clin Invest. 1980;66:892–900. doi: 10.1172/JCI109956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francone OL, Fielding CJ, Fielding PE. Distribution of cell-derived cholesterol among plasma lipoproteins: a comparison of three techniques. J Lipid Res. 1990;31:2195–200. [PubMed] [Google Scholar]
- Freeman DJ, Caslake MJ, Griffin BA, et al. The effect of smoking on post-heparin lipoprotein and hepatic lipase, cholesteryl ester transfer protein and lecithin:cholesterol acyl transferase activities in human plasma. Eur J Clin Invest. 1998;28:584–91. doi: 10.1046/j.1365-2362.1998.00328.x. [DOI] [PubMed] [Google Scholar]
- Frick MH, Syvanne M, Nieminen MS, et al. Prevention of the angiographic progression of coronary and vein-graft atherosclerosis by gemfibrozil after coronary bypass surgery in men with low levels of HDL cholesterol. Lopid Coronary Angiography Trial (LOCAT) Study Group. Circulation. 1997;96:2137–43. doi: 10.1161/01.cir.96.7.2137. [DOI] [PubMed] [Google Scholar]
- Friedewald VE, Brewer HB, Grundy SM, et al. The Editor’s Roundtable: High-Density Lipoprotein Cholesterol. Am J Cardiol. 2007;99:1698–705. doi: 10.1016/j.amjcard.2007.03.020. [DOI] [PubMed] [Google Scholar]
- Funke H, von Eckardstein A, Pritchard PH, et al. A molecular defect causing fish eye disease: an amino acid exchange in lecithin-cholesterol acyltransferase (LCAT) leads to the selective loss of alpha-LCAT activity. Proc Natl Acad Sci. 1991;88:4855–9. doi: 10.1073/pnas.88.11.4855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furbee JW, Jr, Francone O, Parks JS. In vivo contribution of LCAT to apolipoprotein B lipoprotein cholesteryl esters in LDL receptor and apolipoprotein E knockout mice. J Lipid Res. 2002;43:428–37. [PubMed] [Google Scholar]
- Ganji SH, Kamanna VS, Kashyap ML. Niacin and cholesterol: role in cardiovascular disease (review) J Nutr Biochem. 2003;14:298–305. doi: 10.1016/s0955-2863(02)00284-x. [DOI] [PubMed] [Google Scholar]
- Garber DW, Datta G, Chaddha M, et al. A new synthetic class A amphipathic peptide analogue protects mice from diet-induced atherosclerosis. J Lipid Res. 2001;42:545–52. [PubMed] [Google Scholar]
- Gaziano JM, Buring JE, Breslow JL, et al. Moderate alcohol intake, increased levels of high-density lipoprotein and its subfractions, and decreased risk of myocardial infarction. N Engl J Med. 1993;329:1829–34. doi: 10.1056/NEJM199312163292501. [DOI] [PubMed] [Google Scholar]
- Gerasimova E, Perova N, Ozerova I, et al. The effect of dietary n-3 polyunsaturated fatty acids on HDL cholesterol in Chukot residents vs muscovites. Lipids. 1991;26:261–5. doi: 10.1007/BF02537134. [DOI] [PubMed] [Google Scholar]
- Gervois P, Fruchart JC, Staels B. Drug Insight: mechanisms of action and therapeutic applications for agonists of peroxisome proliferator-activated receptors. Nat Clin Pract Endocrinol Metab. 2007;3:145–56. doi: 10.1038/ncpendmet0397. [DOI] [PubMed] [Google Scholar]
- Glomset JA. The plasma lecithin:cholesterol acyltransferase reaction. J Lipid Res. 1968;9:155–67. [PubMed] [Google Scholar]
- Goldbourt U, Behar S, Reicher-Reiss H, et al. Rationale and design of a secondary prevention trial of increasing serum high-density lipoprotein cholesterol and reducing triglycerides in patients with clinically manifest atherosclerotic heart disease (the Bezafibrate Infarction Prevention Trial) Am J Cardiol. 1993;71:909–915. doi: 10.1016/0002-9149(93)90905-r. [DOI] [PubMed] [Google Scholar]
- Gordon DJ, Probstfield JL, Garrison RJ, et al. High-density lipoprotein cholesterol and cardiovascular disease. Four prospective American studies. Circulation. 1977;79:8–15. doi: 10.1161/01.cir.79.1.8. [DOI] [PubMed] [Google Scholar]
- Gordon T, Castelli WP, Hjortland MC, et al. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am J Med. 62(5):707–14. doi: 10.1016/0002-9343(77)90874-9. [DOI] [PubMed] [Google Scholar]
- Graham A, Hassall DG, Rafique S, et al. Evidence for a paraoxonase-independent inhibition of low-density lipoprotein oxidation by high-density lipoprotein. Atherosclerosis. 1997;135:193–204. doi: 10.1016/s0021-9150(97)00162-7. [DOI] [PubMed] [Google Scholar]
- Griffin JH, Kojima K, Banka CL, et al. High-density lipoprotein enhancement of anticoagulant activities of plasma protein S and activated protein C. J Clin Invest. 1999;103:219–27. doi: 10.1172/JCI5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grundy SM, Cleeman JI, Merz CN, et al. Implications of recent clinical trials for the National Cholesterol Education Program Adult Treatment Panel III Guidelines. J Am Coll Cardiol. 2004;44:720–32. doi: 10.1016/j.jacc.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Gupta AK, Ross EA, Myers JN, et al. Increased reverse cholesterol transport in athletes. Metabolism. 1993;42:684–90. doi: 10.1016/0026-0495(93)90233-e. [DOI] [PubMed] [Google Scholar]
- Joyce C, Brewer BH, Freeman L. Study of ABCA1 function in transgenic mice. Arterioscler Thromb Vasc Biol. 2003;23:965–71. doi: 10.1161/01.ATV.0000055194.85073.FF. [DOI] [PubMed] [Google Scholar]
- Harper CR, Jacobson TA. New perspectives on the management of low levels of high-density lipoprotein cholesterol. Arch Intern Med. 1999;159:1049–57. doi: 10.1001/archinte.159.10.1049. [DOI] [PubMed] [Google Scholar]
- Hayek T, Oiknine J, Dankner G, et al. HDL apolipoprotein A-I attenuates oxidative modification of low density lipoprotein: studies in transgenic mice. Eur J Clin Chem Clin Biochem. 1995;33:721–5. doi: 10.1515/cclm.1995.33.10.721. [DOI] [PubMed] [Google Scholar]
- Hegele RA. Paraoxonase genes and disease. Ann Med. 1999;31:217–24. doi: 10.3109/07853899909115981. [DOI] [PubMed] [Google Scholar]
- Heinonen TM, Stein E, Weiss SR, et al. The lipid-lowering effects of atorvastatin, a new HMG-CoA reductase inhibitor: results of a randomized, double-masked study. Clin Ther. 1996;18:853–63. doi: 10.1016/s0149-2918(96)80045-2. [DOI] [PubMed] [Google Scholar]
- Hirano K, Yamashita S, Kuga Y, et al. Atherosclerotic disease in marked hyperalphalipoproteinemia. Combined reduction of cholesteryl ester transfer protein and hepatic triglyceride lipase. Arterioscler Thromb Vasc Biol. 1995;15:1849–56. doi: 10.1161/01.atv.15.11.1849. [DOI] [PubMed] [Google Scholar]
- Hirano K, Yamashita S, Matsuzawa Y. Pros and cons of inhibiting cholesteryl ester transfer protein. Curr Opin Lipidol. 2000;11:589–96. doi: 10.1097/00041433-200012000-00004. [DOI] [PubMed] [Google Scholar]
- Honda HM, Leitinger N, Frankel M, et al. High-density lipoprotein increases intracellular calcium levels by releasing calcium from internal stores in human endothelial cells. Atherosclerosis. 1999;2:299–306. doi: 10.1016/s0021-9150(98)00302-5. [DOI] [PubMed] [Google Scholar]
- Honey K. Drug designed to raise HDL levels falls down. J Clin Invest. 2007;117:282. doi: 10.1172/JCI31253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huttunen J. The Helsinki Heart Study: central findings and clinical implications. Ann Med. 1991;23:155–9. doi: 10.3109/07853899109148041. [DOI] [PubMed] [Google Scholar]
- Ibanez B. Short-term treatment with Apo AI Milano indues atherosclerotic plaque regression and signs of plaque stabilization: in vivo MRI study. Paper presented at: Annual Meeting of the American College of Cardiology; March 24-27; New Orleans, LA. 2007. [Google Scholar]
- Ikewaki K, Nishiwaki M, Sakamoto T, et al. Increased catabolic rate of low density lipoproteins in humans with cholesteryl ester transfer protein deficiency. J Clin Invest. 1995;96:1573–81. doi: 10.1172/JCI118196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura H, Teshima K, Miyamoto N, et al. Cigarette smoking, high-density lipoprotein cholesterol subfractions, and lecithin: cholesterol acyltransferase in young women. Metabolism. 2002;51:1313–6. doi: 10.1053/meta.2002.34040. [DOI] [PubMed] [Google Scholar]
- Inazu A, Brown ML, Hesler CB, et al. Increased high-density lipoprotein levels caused by a common cholesterylester transfer protein gene mutation. N Engl J Med. 1990;323:1234–8. doi: 10.1056/NEJM199011013231803. [DOI] [PubMed] [Google Scholar]
- Inoue I, Itoh F, Aoyagi S, et al. Fibrate and statin synergistically increase the transcriptional activities of PPARalpha/RXRalpha and decrease the transactivation of NFkappaB. Biochem Biophys Res Commun. 2002;290:131–9. doi: 10.1006/bbrc.2001.6141. [DOI] [PubMed] [Google Scholar]
- Jansen H. Hepatic lipase: friend or foe and under what circumstances? Curr Atheroscler Rep. 2004;6:343–7. doi: 10.1007/s11883-004-0044-3. [DOI] [PubMed] [Google Scholar]
- Jin FY, Kamanna VS, Kashyap ML. Niacin decreases removal of high-density lipoprotein apolipoprotein A-I but not cholesterol ester by Hep G2 cells. Implication for reverse cholesterol transport. Arterioscler Thromb Vasc Biol. 1997;17:2020–8. doi: 10.1161/01.atv.17.10.2020. [DOI] [PubMed] [Google Scholar]
- Jones P. International Symposium on Atherosclerosis; June 18-22; Rome, Italy. 2006. [Google Scholar]
- Joseph SB, McKilligin E, Pei L, et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci USA. 2002;99:7604–9. doi: 10.1073/pnas.112059299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannel WB. High-density lipoproteins: epidemiologic profile and risks of coronary artery disease. Am J Cardiol. 1983;52:9B–12B. doi: 10.1016/0002-9149(83)90649-5. [DOI] [PubMed] [Google Scholar]
- Kaplinsky E. Data presented at European Society of Cardiology Meeting; Vienna, Austria. 1998. [Google Scholar]
- Kastelein JJ, Isaacsohn JL, Ose L, et al. Comparison of effects of simvastatin versus atorvastatin on high-density lipoprotein cholesterol and apolipoprotein A-I levels. Am J Cardiol. 2000;86:221–3. doi: 10.1016/s0002-9149(00)00942-5. [DOI] [PubMed] [Google Scholar]
- Kennedy MA, Barrera GC, Nakamura K, et al. ABCG1 has a critical role in mediating cholesterol efflux to HDL and preventing cellular lipid accumulation. Cell Metab. 2005;1:121–31. doi: 10.1016/j.cmet.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Khavkin TN. Nikolai Nikolaevich Anitschkow. Beitr Pathol. 1975;156:301–12. doi: 10.1016/s0005-8165(75)80042-4. [DOI] [PubMed] [Google Scholar]
- Klerkx AH, de Grooth GJ, Zwinderman AH, et al. Cholesteryl ester transfer protein concentration is associated with progression of atherosclerosis and response to pravastatin in men with coronary artery disease (REGRESS) Eur J Clin Invest. 2004;34:21–8. doi: 10.1111/j.1365-2362.2004.01281.x. [DOI] [PubMed] [Google Scholar]
- Koizumi J, Inazu A, Yagi K, et al. Serum lipoprotein lipid concentration and composition in homozygous and heterozygous patients with cholesteryl ester transfer protein deficiency. Atherosclerosis. 1991;90:189–96. doi: 10.1016/0021-9150(91)90114-i. [DOI] [PubMed] [Google Scholar]
- Kokkinos PF, Holland JC, Narayan P, et al. Miles run per week and high-density lipoprotein cholesterol levels in healthy, middle-aged men. A dose-response relationship. Arch Intern Med. 1995;155:415–20. [PubMed] [Google Scholar]
- Kostner KM, Smith JL, Smith BE. Presentation. A novel extracorporeal plasma delipidation procedure for the treatment of atherosclerosis. 73rd Eur Athero Soc Meeting.2002. [Google Scholar]
- Kris-Etherton PM, Harris WS, Appel LJ, et al. Fish consumption, fish oil, omega-3 fatty acids, and cardiovascular disease. Circulation. 2002;106:2747–57. doi: 10.1161/01.cir.0000038493.65177.94. [DOI] [PubMed] [Google Scholar]
- Kuivenhoven JA, Jukema JW, Zwinderman AH, et al. The role of a common variant of the cholesteryl ester transfer protein gene in the progression of coronary atherosclerosis. The Regression Growth Evaluation Statin Study Group. N Engl J Med. 1998;338:86–93. doi: 10.1056/NEJM199801083380203. [DOI] [PubMed] [Google Scholar]
- Kujiraoka T, Nanjee MN, Oka T, et al. Effects of intravenous apolipoprotein A-I/phosphatidylcholine discs on LCAT, PLTP, and CETP in plasma and peripheral lymph in humans. Arterioscler Thromb Vasc Biol. 2003;23:1653–9. doi: 10.1161/01.ATV.0000089328.23279.3F. [DOI] [PubMed] [Google Scholar]
- Kuvin JT, Ramet ME, Patel AR, et al. A novel mechanism for the beneficial vascular effects of high-density lipoprotein cholesterol: enhanced vasorelaxation and increased endothelial nitric oxide synthase expression. Am Heart J. 2002;144:165–72. doi: 10.1067/mhj.2002.123145. [DOI] [PubMed] [Google Scholar]
- Kwiterovich PO., Jr The antiatherogenic role of high-density lipoprotein cholesterol. Am J Cardiol. 1998;82:13Q–21Q. doi: 10.1016/s0002-9149(98)00808-x. [DOI] [PubMed] [Google Scholar]
- Lala DS. The liver X receptors. Curr Opin Investig Drugs. 2005;6:934–43. [PubMed] [Google Scholar]
- LaRosa JC, Grundy SM, Waters DD, et al. Intensive Lipid Lowering with Atorvastatin in Patients with Stable Coronary Disease. N Engl J Med. 2005;352:1425–35. doi: 10.1056/NEJMoa050461. [DOI] [PubMed] [Google Scholar]
- Levkau B, Hermann S, Theilmeier S, et al. High-density lipoprotein stimulates myocardial perfusion in vivo. Circulation. 2004;110:3355–9. doi: 10.1161/01.CIR.0000147827.43912.AE. [DOI] [PubMed] [Google Scholar]
- Lewis GF. Determinants of plasma HDL concentrations and reverse cholesterol transport. Curr Opin Cardiol. 2006;21:345–52. doi: 10.1097/01.hco.0000231405.76930.a0. [DOI] [PubMed] [Google Scholar]
- Lewis GF, Rader DJ. New insights into the regulation of HDL metabolism and reverse cholesterol transport. Circ Res. 2005;96:1221–32. doi: 10.1161/01.RES.0000170946.56981.5c. [DOI] [PubMed] [Google Scholar]
- Li X, Chyu KY, Neto JR, et al. Differential effects of apolipoprotein A-I-mimetic peptide on evolving and established atherosclerosis in apolipoprotein E-null mice. Circulation. 2004;110:1701–5. doi: 10.1161/01.CIR.0000142857.79401.69. [DOI] [PubMed] [Google Scholar]
- Lichtenstein AH, Ausman LM, Jalbert SM, et al. Effects of different forms of dietary hydrogenated fats on serum lipoprotein cholesterol levels. N Engl J Med. 1999;340:1933–40. doi: 10.1056/NEJM199906243402501. [DOI] [PubMed] [Google Scholar]
- Liu PY, Li YH, Wu HL, et al. Platelet-activating factor-acetylhydrolase A379V (exon 11) gene polymorphism is an independent and functional risk factor for premature myocardial infarction. J Thromb Haemost. 2006;4:1023–8. doi: 10.1111/j.1538-7836.2006.01895.x. [DOI] [PubMed] [Google Scholar]
- Long-Term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group. Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med. 1998;339:1349–57. doi: 10.1056/NEJM199811053391902. [DOI] [PubMed] [Google Scholar]
- Lund EG, Peterson LB, Adams AD, et al. Different roles of liver X receptor alpha and beta in lipid metabolism: effects of an alpha-selective and a dual agonist in mice deficient in each subtype. Biochem Pharmacol. 2006;71:453–63. doi: 10.1016/j.bcp.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Mackness MI, Durrington P, Mackness B. Paraoxonase 1 activity, concentration and genotype in cardiovascular disease. Curr Opin Lipidol. 2004;15:399–404. doi: 10.1097/01.mol.0000137227.54278.29. [DOI] [PubMed] [Google Scholar]
- Mackness MI, Durrington PN, Mackness B. How high-density lipoprotein protects against the effects of lipid peroxidation. Curr Opin Lipidol. 2000;11:383–8. doi: 10.1097/00041433-200008000-00007. [DOI] [PubMed] [Google Scholar]
- Maeda K, Noguchi Y, Fukui T. The effects of cessation from cigarette smoking on the lipid and lipoprotein profiles: a meta-analysis. Prev Med. 2003;37:283–90. doi: 10.1016/s0091-7435(03)00110-5. [DOI] [PubMed] [Google Scholar]
- Manninen V, Tenkanen L, Koskinen P, et al. Joint effects of serum triglyceride and LDL cholesterol and HDL cholesterol concentrations on coronary heart disease risk in the Helsinki Heart Study. Implications for treatment. Circulation. 1992;85:37–45. doi: 10.1161/01.cir.85.1.37. [DOI] [PubMed] [Google Scholar]
- Manttari M, Huttunen JK, Koskinen P, et al. Lipoproteins and coronary heart disease in the Helsinki Heart Study. Eur Heart J. 1990;11(Suppl H):26–31. doi: 10.1093/eurheartj/11.suppl_h.26. [DOI] [PubMed] [Google Scholar]
- Marcil M, Yu L, Krimbou L, et al. Cellular cholesterol transport and efflux in fibroblasts are abnormal in subjects with familial HDL deficiency. Arterioscler Thromb Vasc Biol. 1999;19:159–69. doi: 10.1161/01.atv.19.1.159. [DOI] [PubMed] [Google Scholar]
- Marchesi M, Booth EA, Davis T, et al. Apolipoprotein A-IMilano and 1-palmitoyl-2-oleoyl phosphatidylcholine complex (ETC-216) protects the in vivo rabbit heart from regional ischemia-reperfusion injury. J Pharmacol Exp Ther. 2004;311:1023–31. doi: 10.1124/jpet.104.070789. [DOI] [PubMed] [Google Scholar]
- Martin G, Duez H, Blanquart C, et al. Statin-induced inhibition of the Rho-signaling pathway activates PPARalpha and induces HDL apoA-I. J Clin Invest. 2001;107:1423–32. doi: 10.1172/JCI10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meksawan K, Pendergast DR, Leddy JJ, et al. Effect of low and high fat diets on nutrient intakes and selected cardiovascular risk factors in sedentary men and women. J Am Coll Nutr. 2004;23:131–40. doi: 10.1080/07315724.2004.10719353. [DOI] [PubMed] [Google Scholar]
- Mero N, Van Tol A, Scheek LM, et al. Decreased postprandial high density lipoprotein cholesterol and apolipoproteins A-I and E in nor-molipidemic smoking men: relations with lipid transfer proteins and LCAT activities. J Lipid Res. 1998;39:1493–502. [PubMed] [Google Scholar]
- Mezdour H, Jones R, Dengremont C, et al. Hepatic lipase deficiency increases plasma cholesterol but reduces susceptibility to atherosclerosis in apolipoprotein E-deficient mice. J Biol Chem. 1997;272:13570–5. doi: 10.1074/jbc.272.21.13570. [DOI] [PubMed] [Google Scholar]
- McDonald MC, Dhadly P, Cockerill GW, et al. Reconstituted high-density lipoprotein attenuates organ injury and adhesion molecule expression in a rodent model of endotoxic shock. Shock. 2003;20:551–7. doi: 10.1097/01.shk.0000097249.97298.a3. [DOI] [PubMed] [Google Scholar]
- McNeish J, Aiello RJ, Guyot D, et al. High density lipoprotein deficiency and foam cell accumulation in mice with targeted disruption of ATP-binding cassette transporter-1. Proc Nat Acad Sci USA. 2000;97:4245–50. doi: 10.1073/pnas.97.8.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao B, Zondlo S, Gibbs S, et al. Raising HDL cholesterol without inducing hepatic steatosis and hypertriglyceridemia by a selective LXR modulator. J Lipid Res. 2004;45:1410–7. doi: 10.1194/jlr.M300450-JLR200. [DOI] [PubMed] [Google Scholar]
- Miller M, Kwiterovich PO. Isolated low HDL-cholesterol as an important risk factor for coronary heart disease. Eur Heart J. 1990;11(Suppl H):9–14. doi: 10.1093/eurheartj/11.suppl_h.9. [DOI] [PubMed] [Google Scholar]
- Miller NE, Thelle DS, Forde OH, et al. The Tromso heart-study. High-density lipoprotein and coronary heart-disease: a prospective case-control study. Lancet. 1977;1:965–8. doi: 10.1016/s0140-6736(77)92274-7. [DOI] [PubMed] [Google Scholar]
- Mineo C, Deguchi H, Griffin JH, et al. Endothelial and antithrombotic actions of HDL. Circ Res. 2006;98:1352–64. doi: 10.1161/01.RES.0000225982.01988.93. [DOI] [PubMed] [Google Scholar]
- Moestrup SK, Kozyraki R. Cubilin, a high-density lipoprotein receptor. Curr Opin Lipidol. 2000;11:133–40. doi: 10.1097/00041433-200004000-00005. [DOI] [PubMed] [Google Scholar]
- Moffatt RJ. Effects of cessation of smoking on serum lipids and high density lipoprotein-cholesterol. Atherosclerosis. 1988;74:85–9. doi: 10.1016/0021-9150(88)90194-3. [DOI] [PubMed] [Google Scholar]
- Morton RE, Greene DJ. Suppression of lipid transfer inhibitor protein activity by oleate. A novel mechanism of cholesteryl ester transfer protein regulation by plasma free fatty acids. Arterioscler Thromb Vasc Biol. 1997;17:3041–8. doi: 10.1161/01.atv.17.11.3041. [DOI] [PubMed] [Google Scholar]
- Motojima K, Peters JM, Gonzalez FJ. PPAR alpha mediates peroxisome proliferator-induced transcriptional repression of nonperoxisomal gene expression in mouse. Biochem Biophys Res Commun. 1997;230:155–8. doi: 10.1006/bbrc.1996.5906. [DOI] [PubMed] [Google Scholar]
- MRC/BHF. Heart Protection Study of cholesterol-lowering therapy and of antioxidant vitamin supplementation in a wide range of patients at increased risk of coronary heart disease death: early safety and efficacy experience. Eur Heart J. 1996;20:725–41. doi: 10.1053/euhj.1998.1350. [DOI] [PubMed] [Google Scholar]
- Multiple Risk Factor Intervention Trial. Risk factor changes and mortality results. Multiple Risk Factor Intervention Trial Research Group. JAMA. 1982;248:1465–77. [PubMed] [Google Scholar]
- Naik SU, Wang X, Da Silva JS, et al. Pharmacological activation of liver X receptors promotes reverse cholesterol transport in vivo. Circulation. 2006;113:90–7. doi: 10.1161/CIRCULATIONAHA.105.560177. [DOI] [PubMed] [Google Scholar]
- Nanjee MN, Cooke CJ, Garvin R, et al. Intravenous apoA-I/lecithin discs increase pre-beta-HDL concentration in tissue fluid and stimulate reverse cholesterol transport in humans. J Lipid Res. 2001;42:1586–93. [PubMed] [Google Scholar]
- Naqvi TZ, Shah PK, Ivey PA, et al. Evidence that high-density lipoprotein cholesterol is an independent predictor of acute platelet-dependent thrombus formation. Am J Cardiol. 1999;9:1011–7. doi: 10.1016/s0002-9149(99)00489-0. [DOI] [PubMed] [Google Scholar]
- Navab M, Anantharamaiah GM, Reddy ST, et al. Apolipoprotein A-I mimetic peptides and their role in atherosclerosis prevention. Nat Clin Pract Cardiovasc Med. 2006;3:540–7. doi: 10.1038/ncpcardio0661. [DOI] [PubMed] [Google Scholar]
- Navab M, Anantharamaiah GM, Reddy ST, et al. Human apolipoprotein A-I and A-I mimetic peptides: potential for atherosclerosis reversal. Curr Opin Lipidol. 2004;15:645–9. doi: 10.1097/00041433-200412000-00004. [DOI] [PubMed] [Google Scholar]
- Navab M, Berliner JA, Watson AD, et al. The Yin and Yang of oxidation in the development of the fatty streak. A review based on the 1994 George Lyman Duff Memorial Lecture. Arterioscler Thromb Vasc Biol. 1996;16:831–42. doi: 10.1161/01.atv.16.7.831. [DOI] [PubMed] [Google Scholar]
- Nissen SE, Tardif JC, Nicholls SJ, et al. Effect of torcetrapib on the progression of coronary atherosclerosis. N Engl J Med. 2007;356:1304–16. doi: 10.1056/NEJMoa070635. [DOI] [PubMed] [Google Scholar]
- Nissen SE, Tsunoda T, Tuzcu EM, et al. Effect of recombinant ApoA-I Milano on coronary atherosclerosis in patients with acute coronary syndromes: a randomized controlled trial. JAMA. 2003;290:2292–300. doi: 10.1001/jama.290.17.2292. [DOI] [PubMed] [Google Scholar]
- Nobecourt E, Davies MJ, Brown BE, et al. The impact of glycation on apolipoprotein A-I structure and its ability to activate lecithin: cholesterol acyltransferase. Diabetologia. 2007;50:643–53. doi: 10.1007/s00125-006-0574-z. [DOI] [PubMed] [Google Scholar]
- Nofer JR, Levkau B, Wolinska I, et al. Suppression of endothelial cell apoptosis by high density lipoproteins (HDL) and HDL-associated lysosphingolipids. J Biol Chem. 2001;276:34480–5. doi: 10.1074/jbc.M103782200. [DOI] [PubMed] [Google Scholar]
- Norata GD, Callegari E, Marchesi M, et al. High-density lipoproteins induce transforming growth factor-beta2 expression in endothelial cells. Circulation. 2005;111:2805–11. doi: 10.1161/CIRCULATIONAHA.104.472886. [DOI] [PubMed] [Google Scholar]
- O’Connell BJ, Denis M, Genest J. High-density lipoproteins and endothelial function. Circulation. 2001;104:1978–83. doi: 10.1161/hc3901.096667. [DOI] [PubMed] [Google Scholar]
- Oliver WR, Shenk JL, Snaith MR, et al. A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport. Proc Natl Acad Sci. 2001;98:5306–11. doi: 10.1073/pnas.091021198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oram JF, Lawn RM. ABCA1: The gatekeeper for eliminating excess tissue cholesterol. J Lipid Res. 2001;42:1173–9. [PubMed] [Google Scholar]
- Out R, Hoekstra M, Hildebrand RB, et al. Macrophage ABCG1 deletion disrupts lipid homeostasis in alveolar macrophages and moderately influences atherosclerotic lesion development in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2006;26:2295–300. doi: 10.1161/01.ATV.0000237629.29842.4c. [DOI] [PubMed] [Google Scholar]
- Papadakis JA, Ganotakis ES, Jagroop IA, et al. Statin + fibrate combination therapy fluvastatin with bezafibrate or ciprofibrate in high risk patients with vascular disease. Int J Cardiol. 1999;3:237–44. doi: 10.1016/s0167-5273(99)00013-3. [DOI] [PubMed] [Google Scholar]
- Pauciullo P, Borgnino C, Paoletti R, et al. Efficacy and safety of a combination of fluvastatin and bezafibrate in patients with mixed hyperlipidaemia (FACT study) Atherosclerosis. 2000;150:429–36. doi: 10.1016/s0021-9150(00)00379-8. [DOI] [PubMed] [Google Scholar]
- Plosch T, Kok T, Bloks VW, et al. Increased hepatobiliary and fecal cholesterol excretion upon activation of the liver X receptor is independent of ABCA1. J Biol Chem. 2002;277:33870–7. doi: 10.1074/jbc.M206522200. [DOI] [PubMed] [Google Scholar]
- Poon M, Zhang X, Dunsky K, et al. Apolipoprotein(a) induces monocyte chemotactic activity in human vascular endothelial cells. Circulation. 1997;96:2514–9. doi: 10.1161/01.cir.96.8.2514. [DOI] [PubMed] [Google Scholar]
- Qureshi AI, Giles WH, Croft JB, et al. Apolipoproteins A-1 and B and the likelihood of non-fatal stroke and myocardial infarction – data from The Third National Health and Nutrition Examination Survey. Med Sci Monit. 2002;8:CR311–6. [PubMed] [Google Scholar]
- Rimm EB, Williams P, Fosher K, et al. Moderate alcohol intake and lower risk of coronary heart disease: meta-analysis of effects on lipids and haemostatic factors. BMJ. 1999;319:1523–8. doi: 10.1136/bmj.319.7224.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittershaus CW, Miller DP, Thomas LJ, et al. Vaccine-induced antibodies inhibit CETP activity in vivo and reduce aortic lesions in a rabbit model of atherosclerosis. Arterioscler Thromb Vasc Biol. 2000;20:2106–12. doi: 10.1161/01.atv.20.9.2106. [DOI] [PubMed] [Google Scholar]
- Rossner S, Bjorvell H. Early and late effects of weight loss on lipoprotein metabolism in severe obesity. Atherosclerosis. 1987;64:125–30. doi: 10.1016/0021-9150(87)90237-1. [DOI] [PubMed] [Google Scholar]
- Rubenstrunk A, Hanf R, Hum DW, et al. Safety issues and prospects for future generations of PPAR modulators. Biochim Biophys Acta. 2007 doi: 10.1016/j.bbalip.2007.02.003. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Rubin EM, Krauss RM, Spangler EA, et al. Inhibition of early atherogenesis in transgenic mice by human apolipoprotein AI. Nature. 1991;353:265–7. doi: 10.1038/353265a0. [DOI] [PubMed] [Google Scholar]
- Rubins HB, Davenport J, Babikian V, et al. Reduction in stroke with gemfibrozil in men with coronary heart disease and low HDL cholesterol. The Veterans Affairs HDL Intervention Trial (VA-HIT) Circulation. 2001;103:2828–33. doi: 10.1161/01.cir.103.23.2828. [DOI] [PubMed] [Google Scholar]
- Sacks FM, Pfeffer MA, Moye LA, et al. The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. Cholesterol and Recurrent Events Trial investigators. N Engl J Med. 1996;335:1001–9. doi: 10.1056/NEJM199610033351401. [DOI] [PubMed] [Google Scholar]
- Sakai N, Matsuzawa Y, Hirano K, et al. Detection of two species of low density lipoprotein particles in cholesteryl ester transfer protein deficiency. Arterioscler Thromb. 1991;11:71–9. doi: 10.1161/01.atv.11.1.71. [DOI] [PubMed] [Google Scholar]
- Saku K, Ahmad M, Glas-Greenwalt P, et al. Activation of fibrinolysis by apolipoproteins of high density lipoproteins in man. Thromb Res. 1985;39:1–8. doi: 10.1016/0049-3848(85)90116-1. [DOI] [PubMed] [Google Scholar]
- Savage PD, Brochu M, Ades PA. Gender alters the high-density lipoprotein cholesterol response to cardiac rehabilitation. J Cardiopulm Rehabil. 2004;24:248–54. doi: 10.1097/00008483-200407000-00007. [DOI] [PubMed] [Google Scholar]
- Scandinavian Simvastatin Survival Study Group. Randomized trial of cholesterol lowering in 4,444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S) Lancet. 1994;344:1383–9. [PubMed] [Google Scholar]
- Schaefer JR, Schweer H, Ikewaki K, et al. Metabolic basis of high density lipoproteins and apolipoprotein A-I increase by HMG-CoA reductase inhibition in healthy subjects and a patient with coronary artery disease. Atherosclerosis. 1999;144:177–84. doi: 10.1016/s0021-9150(99)00053-2. [DOI] [PubMed] [Google Scholar]
- Schmitz G, Drobnik W. ATP-binding cassette transporters in macrophages: promising drug targets for treatment of cardiovascular disease. Curr Opin Investig Drugs. 2002;3:853–8. [PubMed] [Google Scholar]
- Schmitz G, Langmann T. High-density lipoproteins and ATP-binding cassette transporters as targets for cardiovascular drug therapy. Curr Opin Investig Drugs. 2005;6:907–19. [PubMed] [Google Scholar]
- Segrest JP, Li L, Anantharamaiah GM, et al. Structure and function of apolipoprotein A-I and high-density lipoprotein. Curr Opin Lipidol. 2000;11:105–15. doi: 10.1097/00041433-200004000-00002. [DOI] [PubMed] [Google Scholar]
- Shah PK. Emerging HDL-based therapies for atherothrombotic vascular disease. Curr Treat Options Cardiovasc Med. 2007;9:60–70. doi: 10.1007/s11936-007-0052-z. [DOI] [PubMed] [Google Scholar]
- Shah PK. Low-density lipoprotein lowering and atherosclerosis progression: does more mean less? Circulation. 2002;106:2039–40. doi: 10.1161/01.cir.0000037742.25731.3d. [DOI] [PubMed] [Google Scholar]
- Shah PK, Amin J. Low high density lipoprotein level is associated with increased restenosis rate after coronary angioplasty. Circulation. 1992;85:1279–85. doi: 10.1161/01.cir.85.4.1279. [DOI] [PubMed] [Google Scholar]
- Shah PK, Kaul S, Nilsson J, et al. Exploiting the vascular protective effects of high-density lipoprotein and its apolipoproteins: an idea whose time for testing is coming, part II. Circulation. 2001;104:2498–502. doi: 10.1161/hc4501.098468. [DOI] [PubMed] [Google Scholar]
- Shah PK, Nilsson J, Kaul S, et al. Effects of recombinant apolipoprotein A-I(Milano) on aortic atherosclerosis in apolipoprotein E-deficient mice. Circulation. 1998;97:780–5. doi: 10.1161/01.cir.97.8.780. [DOI] [PubMed] [Google Scholar]
- Shih DM, Gu L, Xia YR, et al. Mice lacking serum paraoxonase are susceptible to organophosphate toxicity and atherosclerosis. Nature. 1998;394:284–7. doi: 10.1038/28406. [DOI] [PubMed] [Google Scholar]
- Soloff LA. Intermediate lipoproteins, atherosclerosis, and Gofman. Circulation. 1998;97:708. [PubMed] [Google Scholar]
- Staels B, Dallongeville J, Auwerx J, et al. Mechanism of action of fibrates on lipid and lipoprotein metabolism. Circulation. 1998;98:2088–93. doi: 10.1161/01.cir.98.19.2088. [DOI] [PubMed] [Google Scholar]
- Staels B, Vu-Dac N, Kosykh VA, et al. Fibrates downregulate apolipoprotein C-III expression independent of induction of peroxisomal acyl coenzyme A oxidase. A potential mechanism for the hypolipidemic action of fibrates. J Clin Invest. 1995;95:705–12. doi: 10.1172/JCI117717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanick ML, Mackey S, Sheehan M, et al. Effects of diet and exercise in men and postmenopausal women with low levels of HDL cholesterol and high levels of LDL cholesterol. N Engl J Med. 1998;339:12–20. doi: 10.1056/NEJM199807023390103. [DOI] [PubMed] [Google Scholar]
- Sugano M, Makino N, Sawada S, et al. Effect of antisense oligonucleotides against cholesteryl ester transfer protein on the development of atherosclerosis in cholesterol-fed rabbits. J Biol Chem. 1998;273:5033–6. doi: 10.1074/jbc.273.9.5033. [DOI] [PubMed] [Google Scholar]
- Sugano M, Tsuchida K, Makino N. High-density lipoproteins protect endothelial cells from tumor necrosis factor-alpha-induced apoptosis. Biochem Biophys Res Commun. 2000;272:872–6. doi: 10.1006/bbrc.2000.2877. [DOI] [PubMed] [Google Scholar]
- Sugatani J, Miwa M, Komiyama Y, et al. High-density lipoprotein inhibits the synthesis of platelet-activating factor in human vascular endothelial cells. J Lipid Mediat Cell Signal. 1996;13:73–88. doi: 10.1016/0929-7855(95)00047-x. [DOI] [PubMed] [Google Scholar]
- Sviridov D, Kingwell B, Hoang A, et al. Single session exercise stimulates formation of pre beta 1-HDL in leg muscle. J Lipid Res. 2003;44:522–6. doi: 10.1194/jlr.M200436-JLR200. [DOI] [PubMed] [Google Scholar]
- Sviridov D, Miyazaki O, Theodore K, et al. Delineation of the role of pre-beta 1-HDL in cholesterol efflux using isolated pre-beta 1-HDL. Arterioscler Thromb Vasc Biol. 2002;22:1482–8. doi: 10.1161/01.atv.0000029120.44088.fe. [DOI] [PubMed] [Google Scholar]
- Szapary PO, Rader DJ. The triglyceride-high-density lipoprotein axis: an important target of therapy? Am Heart J. 2004;148:211–21. doi: 10.1016/j.ahj.2004.03.037. [DOI] [PubMed] [Google Scholar]
- Tall AR, Yvan-Charvet L, Wang N. The failure of torcetrapib: was it the molecule or the mechanism? Arterioscler Thromb Vasc Biol. 2007;27:257–60. doi: 10.1161/01.ATV.0000256728.60226.77. [DOI] [PubMed] [Google Scholar]
- Tardif JC. Effect of reconstituted high density lipoprotein infusions on coroanry atherosclerosis. Paper presented at Annual Meeting of the American College of Cardiology; March 24–27; New Orleans, LA. 2007. [Google Scholar]
- Taylor AJ, Sullenberger LE, Lee HJ, et al. Arterial Biology for the Investigation of the Treatment Effects of Reducing Cholesterol (ARBITER) 2: a double-blind, placebo-controlled study of extended-release niacin on atherosclerosis progression in secondary prevention patients treated with statins. Circulation. 2004;110:3512–7. doi: 10.1161/01.CIR.0000148955.19792.8D. [DOI] [PubMed] [Google Scholar]
- Terasak N, Hiroshima A, Koieyama T, et al. T-0901317, a synthetic liver X receptor ligand, inhibits development of atherosclerosis in LDL receptor-deficient mice. FEBS Lett. 2003;536:6–11. doi: 10.1016/s0014-5793(02)03578-0. [DOI] [PubMed] [Google Scholar]
- Thornton J, Symes C, Heaton K. Moderate alcohol intake reduces bile cholesterol saturation and raises HDL cholesterol. Lancet. 1983;2:819–22. doi: 10.1016/s0140-6736(83)90738-9. [DOI] [PubMed] [Google Scholar]
- Tilly-Kiesi M, Tikkanen MJ. Effects of lovastatin and gemfibrozil on high-density lipoprotein subfraction density and composition in patients with familial hypercholesterolemia. J Lab Clin Med. 1992;120(1):103–10. [PubMed] [Google Scholar]
- Toth PP. High-density lipoprotein as a therapeutic target: clinical evidence and treatment strategies. Am J Cardiol. 2005;96:50K–58K. doi: 10.1016/j.amjcard.2005.08.008. discussion 34K–35K. [DOI] [PubMed] [Google Scholar]
- US Department of Health and Human Services. Physical activity and health: A report of the Surgeon General: Adolescents and young adults fact sheet. 1999 http://www.cdc.gov/nccdphp/sgr/adoles.htm.
- Vance DE, Van den Bosch H. Cholesterol in the year 2000. Biochim Biophys Acta. 2000;1529:1–8. doi: 10.1016/s1388-1981(00)00133-5. [DOI] [PubMed] [Google Scholar]
- Wilson PW, Abbott RD, Castelli WP. High density lipoprotein cholesterol and mortality. The Framingham Heart Study. Arteriosclerosis. 1988;8:737–41. doi: 10.1161/01.atv.8.6.737. [DOI] [PubMed] [Google Scholar]
- Van der Gaag MS, van Tol A, Vermunt SH, et al. Alcohol consumption stimulates early steps in reverse cholesterol transport. J Lipid Res. 2001;42:2077–83. [PubMed] [Google Scholar]
- Vaughan AM, Oram JF. ABCG1 redistributes cell cholesterol to domains removable by high density lipoprotein but not by lipid-depleted apolipoproteins. J Biol Chem. 2005;280:30150–7. doi: 10.1074/jbc.M505368200. [DOI] [PubMed] [Google Scholar]
- Vega GL, Ma PT, Cater NB, et al. Effects of adding fenofibrate (200 mg/day) to simvastatin (10 mg/day) in patients with combined hyperlipidemia and metabolic syndrome. Am J Cardiol. 2003;91:956–60. doi: 10.1016/s0002-9149(03)00111-5. [DOI] [PubMed] [Google Scholar]
- Venkateswaran A, Laffitte BA, Joseph SB, et al. Control of cellular cholesterol efflux by the nuclear oxysterol receptor LXR alpha. Proc Natl Acad Sci. 2000;97:12097–102. doi: 10.1073/pnas.200367697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Briggs MR. HDL: the metabolism, function, and therapeutic importance. Chem Rev. 2004;104(1):119–37. doi: 10.1021/cr020466v. [DOI] [PubMed] [Google Scholar]
- Watson AD, Berliner JA, Hama SY, et al. Protective effect of high density lipoprotein associated paraoxonase. Inhibition of the biological activity of minimally oxidized low density lipoprotein. J Clin Invest. 1995;96:2882–91. doi: 10.1172/JCI118359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West of Scotland Coronary Prevention Group. West of Scotland Coronary Prevention Study: identification of high-risk groups and comparison with other cardiovascular intervention trials. Lancet. 1996;348:1339–1342. [PubMed] [Google Scholar]
- Westman J, Roobol C, Carlson LA, et al. In vitro reverse cholesterol transport from THP-1-derived macrophage-like cells with synthetic HDL particles consisting of proapolipoprotein A1 or apolipoprotein A1 and phosphatidylcholine. Scand J Clin Lab Invest. 1995;55:23–33. doi: 10.3109/00365519509075375. [DOI] [PubMed] [Google Scholar]
- Williams PT. Interactive effects of exercise, alcohol, and vegetarian diet on coronary artery disease risk factors in 9242 runners: the National Runners’ Health Study. Am J Clin Nutr. 1997;66:1197–206. doi: 10.1093/ajcn/66.5.1197. [DOI] [PubMed] [Google Scholar]
- Williams PT. The relationships of vigorous exercise, alcohol, and adiposity to low and high high-density lipoprotein-cholesterol levels. Metabolism. 2004;53:700–9. doi: 10.1016/j.metabol.2004.01.004. [DOI] [PubMed] [Google Scholar]
- Wood PD, Stefanick ML, Dreon DM, et al. Changes in plasma lipids and lipoproteins in overweight men during weight loss through dieting as compared with exercise. N Engl J Med. 1988;319:1173–9. doi: 10.1056/NEJM198811033191801. [DOI] [PubMed] [Google Scholar]
- Yamashita S, Matsuzawa Y, Okazaki M, et al. Small polydisperse low density lipoproteins in familial hyperalphalipoproteinemia with complete deficiency of cholesteryl ester transfer activity. Atherosclerosis. 1988;70:7–12. doi: 10.1016/0021-9150(88)90094-9. [DOI] [PubMed] [Google Scholar]
- Yuhanna IS, Zhu Y, Cox BE, et al. High-density lipoprotein binding to scavenger receptor-BI activates endothelial nitric oxide synthase. Nat Med. 2001;7:853–7. doi: 10.1038/89986. [DOI] [PubMed] [Google Scholar]
- Zeiher AM, Schachlinger B, Hohnloser SH, et al. Coronary atherosclerotic wall thickening and vascular reactivity in humans. Elevated high-density lipoprotein levels ameliorate abnormal vasoconstriction in early atherosclerosis. Circulation. 1994;89:2525–32. doi: 10.1161/01.cir.89.6.2525. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zanotti I, Reilly MP, et al. Overexpression of apolipoprotein A-I promotes reverse transport of cholesterol from macrophages to feces in vivo. Circulation. 2003;108:661–3. doi: 10.1161/01.CIR.0000086981.09834.E0. [DOI] [PubMed] [Google Scholar]