

Abstract
Aging is associated with progressive decline in energetic reserves compromising cardiac performance and tolerance to injury. Although deviations in mitochondrial functions have been documented in senescent heart, the molecular bases for the decline in energy metabolism are only partially understood. Here, high-throughput transcription profiles of genes coding for mitochondrial proteins in ventricles from adult (6-months) and aged (24-months) rats were compared using microarrays. Out of 614 genes encoding for mitochondrial proteins, 94 were differentially expressed with 95% downregulated in the aged. The majority of changes affected genes coding for proteins involved in oxidative phosphorylation (39), substrate metabolism (14) and tricarboxylic acid cycle (6). Compared to adult, gene expression changes in aged hearts translated into a reduced mitochondrial functional capacity, with decreased NADH-dehydrogenase and F0F1-ATPase complex activities and capacity for oxygen-utilization and ATP synthesis. Expression of genes coding for transcription co-activator factors involved in the regulation of mitochondrial metabolism and biogenesis were downregulated in aged ventricles without reduction in mitochondrial density. Thus, aging induces a selective decline in activities of oxidative phosphorylation complexes I and V within a broader transcriptional downregulation of mitochondrial genes, providing a substrate for reduced energetic efficiency associated with senescence.
Keywords: Myocardium, Aging, gene expression, mitochondria, oxidative phosphorylation, ATP, transcription co-activator factors
1. Introduction
Aging is associated with a progressive decline in physiological reserves that reduces the capacity for work (Chantler et al., 2006; Goldspink, 2005) and increases the susceptibility to injury (Abete et al., 1996; Juhaszova et al., 2005; Lesnefsky et al., 1994; Maggioni et al., 1993). A case in point is the myocardium, an aerobic tissue with high energy demands dependent on mitochondrial oxidative phosphorylation (OxPhos) where aging precipitates organ failure during stress. Although age-associated reduction in cardiac mitochondrial function and ATP generation (Jahangir et al., 2001; Pimentel et al., 2003; Wallace, 2001) has been recognized as a major factor contributing to the diminished responsiveness of the aging heart to metabolic demands (Goldspink, 2005), the molecular basis for senescence-related mitochondrial dysfunction remains poorly understood.
The purpose of this study was to determine age-related changes at the transcriptional level of genes regulating mitochondrial energetics in the heart. The expression profile of genes encoding proteins for mitochondrial substrate metabolism and OxPhos, including subunits of the electron transport chain (ETC) complexes and mitochondrial F0F1 ATP synthase were characterized in ventricles from 6 and 24 month old rats using high throughput microarray and verified by RT-PCR. In parallel, functional activities of the ETC and F0F1 ATPase complexes were compared using enzymatic assays and the overall capacity of mitochondria from adult and aged heart to utilize oxygen and synthesize ATP by functional assays. In addition, expression levels of major transcription co-activator factors regulating mitochondrial biogenesis (Finck and Kelly, 2006; Lehman et al., 2000), along with the number and morphology of mitochondria in the adult and aged heart were assessed. We report specific transcriptome patterns associated with compromised mitochondrial function providing molecular evidence for aging-related impairment in myocardial metabolism.
2. Materials and methods
2.1. Total RNA isolation and quantification
Ventricles were removed from anesthetized (sodium pentobarbital 50mg/kg, intraperitoneal injection) adult (6 month old, n=5) and aged (24 month old, n=5) male Fischer-344 rats (National Institute of Aging colonies) that were maintained on a standard chow diet and housed in a controlled environment for one week before being sacrificed. Immediately after harvesting, ventricular tissue was rapidly sliced (2mm thick), frozen in liquid nitrogen and stored at −80°C until further gene and protein expression analysis. Frozen tissue (~20 mg) was homogenized in TRIzol® Reagent (Invitrogen Corporation, Carlsbad, CA) using a Pellet Pestle® motor homogenizer (Kimble-Kontes, Vineland, NJ). Total RNA was isolated following the TRIzol® Reagent protocol (Invitrogen Corporation, Carlsbad, CA) and then further purified using an affinity resin column (QIAGEN Inc., Valencia, CA). Quantification of the concentration of total RNA was performed using spectrophotometric analysis (abs-emission A260/A280). No change in the amount of total RNA recovered was observed in the aging ventricles (11±4 µg of RNA) compared to adults (9±2 µg of RNA). The integrity of the samples was assessed qualitatively on an Agilent 2100 Bioanalyzer (Affymetrix, Santa Clara, CA). Each sample was then divided into two aliquots, one for microarray labeling and the other for verification of selected genes by RT-PCR.
2.2. Gene expression profiling
Total isolated RNA (100 ng) from each adult (n=5) and aged (n=5) sample was converted to cDNA following instructions provided in the Two-Cycle cDNA synthesis kit and Target Labeling Protocols (Affymetrix, Santa Clara, CA). The obtained cDNA was purified by phase lock gel (Eppendorf, Westbury, NY) with phenol/chloroform extraction and used as a template for the in vitro transcription reaction. Biotinylated cRNA was then synthesized using an RNA transcript labeling reagent (Affymetrix, Santa Clara, CA). Labeled cRNA was fragmented and hybridized onto the GeneChip® Rat Genome 230 2.0 arrays (Affymetrix, Santa Clara, CA) according to manufacturer’s protocol. Briefly, appropriate amounts of fragmented cRNA and control oligonucleotide B2 were added along with control cRNA (BioB, BioC, BioD), herring sperm DNA, and BSA to the hybridization buffer. The hybridization mixture was heated at 99°C for 5 min followed by incubation at 45°C for 5 min, before sample was injected into the microarray. Hybridization was carried out at 45°C for 16 hours, mixing on a rotisserie at 60 rpm. After hybridization, solutions were removed and arrays were washed and stained with streptavidin-phycoerythrin (Molecular Probes, Invitrogen Corporation, Carlsbad, CA). After washes, probe arrays were scanned using the GeneChip 3000 scanner (Affymetrix, Santa Clara, CA). The quality of the fragmented biotin-labeled cRNA in each experiment was evaluated before hybridization onto the GeneChip® Rat Genome 230 2.0 array by hybridizing onto a test-3 array, and analyzed as a measure of quality control (Sreekumar et al., 2002).
2.3. Microarray data analysis
GeneChip 3000 scanner (Affymetrix, Santa Clara, CA) was used to scan and quantitatively analyze images of hybridized microarrays. Intensity values for each probe cell in the arrays were calculated by GeneChip® software and flags were assigned to each probe set declaring Present, Marginal or Absent call (Detection Call Algorithm). Probe cell intensities were used to calculate an average intensity for each set of probe pairs representing a gene, which directly correlated with the amount of cRNA. Data analysis and normalization was performed by GeneSpring GX 7.3 bioinformatics software (Agilent Technologies, Palo Alto, CA), excluding the probe sets with absent call in all arrays. After applying quality filtering to diminish background noise created by non-significant gene probes, One-way ANOVA test with Multiple Testing Correction (Benjamini and Hochberg False Discovery Rate) was applied to the filtered gene list resulting in group of genes with significant p-values. Significantly down and upregulated genes were then clustered among the two conditions (a and b) using the following cosine correlation (r) equation:
Furthermore, the list of mitochondrial proteins with their molecular function and encoding genes were gathered using the MitoP2 database (http://www.mitop.de/). Functional annotation and pathway analysis of mitochondrial genes were performed using NetAffx (www.affymetrix.com/analysis/index.affx) and Ingenuity Pathway Analysis (Ingenuity® Systems, Redwood City, CA).
2.4. Real-time polymerase chain reaction
Taqman® Real-Time polymerase chain reaction (RT-PCR) was performed to quantify gene expression levels (Applied Biosystems, Foster City, CA). In brief, 1 µg of total RNA was added to the RT master mix in a 100 µl reaction volume and the combination incubated at 25°C for 10 min, then at 37°C for 120 min. Gene transcript levels were subsequently measured according to instructions for the Applied Biosystems Assay on Demand Gene Expression Product (Foster City, CA). The 20X Gene Expression Assay mix was added to 2X Taqman® Universal PCR Master Mix and 1 µg of cDNA was then added to create a final concentration for each reagent of 1X and a reaction volume of 20 µl. Mixtures were incubated at 50°C for 2 min, then at 95°C for 10 min, before thermal cycling 40 times at 95°C for 15 sec and 60°C for 1 min.
Gene expression assays for Citrate Synthase (CS), Cytochrome-c Oxidase Subunit Va (COX5a), Cytochrome-c Oxidase Subunit Vb (COX5b) and ATP synthase beta (ATP5B) were selected using TaqMan® Gene Expression Assays On-Demand (Applied Biosystems, Foster City, CA). Results were normalized to a pre-designed TaqMan® Endogenous Control, Eukaryotic 18s rRNA Endogenous Control (Applied Biosystems, Foster City, CA) which was used to normalize against differences in RNA isolation, RNA degradation and in the efficiencies of the reverse transcription and PCR reactions. All samples were run in triplicate and quantitated by normalizing signals with the 18s signal. Final quantitation was achieved by a relative standard curve.
2.5. Mitochondrial isolation
Following thoracotomy, ventricles were removed from adult (6 month old, n=6) and aged (24 month old, n=6) Fischer-344 male rats and placed into the isolation buffer (in mM): Sucrose 50, Mannitol 200, KH2PO4 5, EGTA 1, MOPS 5, 0.2% BSA (pH 7.3) and then homogenized using a PT10/35 Polytron (Brinkmann Instruments, Westburry, NY). Mitochondria were separated from membranes and nuclei by differential centrifugation (Sorvall RC5C, Kendro Laboratory Products, Newtown, CT) as previously described (Jahangir et al., 2001), washed with isolation buffer (containing no EGTA and BSA) and protein concentration determined with a DC-Protein Assay kit (BIO-RAD Laboratories, Hercules, CA). All mitochondrial parameters were determined within 4 hours of isolation.
2.6. Mitochondrial enzymatic activity
The enzymatic activity assay of the OxPhos complexes were carried out in mitochondria isolated from adult and aged hearts disrupted by three rapid freeze-thaw cycles and treated with n-dodecyl-β-D-maltoside (1 mM). Complex I enzymatic activity was measured by the rotenone-sensitive reduction of ubiquinone-1 (Abs 340 nm) and specific activity was calculated using an extinction coefficient of 6.81/mM/cm (Darley-Usmar et al., 1987). Complex II activity was measured as the rate of reduction of ubiquinone-2 by succinate (Abs 600 nm) and calculated using the extinction coefficient of 21/mM/cm (Darley-Usmar et al., 1987). Complex III activity was measured by following the increase in absorbance at 550 nm due to the reduction of ferricytochrome c after adding 6 µl of decylubiquinol (Kwong and Sohal, 2000). Decylubiquinol used in this assay was prepared as previously described (Trumpower and Edwards, 1979). Complex IV activity was measured in n-dodecyl-β-D-maltoside (1 mM) treated mitochondrial isolates by a colorimetric assay kit (Sigma-Aldrich Inc., Saint Louis, MO) following the decrease in absorbance at 550 nm of ferrocytochrome c by cytochrome c oxidase (21.82/mM/cm). Oligomycin-sensitive F1F0-ATPase (Complex V) activity was determined by the decrease in absorbance at 340 nm caused by pyruvate-mediated oxidation of NADH in the absence and presence of 3 µg of oligomycin (Darley-Usmar et al., 1987). An extinction coefficient of 6.22/mM/cm was used to calculate Complex V specific activity.
2.7. Mitochondrial respiration and ATP production
Mitochondrial respiration was determined at 30°C in continuously stirred mitochondria (1 mg/ml) in (in mM) KCl 110, KH2PO4 5, Pyruvate 2.5, Malate 2.5 and MOPS 10 (pH 7.3). Mitochondrial oxygen consumption was determined using an oxygen selective electrode (Jahangir et al., 2001). Data were acquired and processed using the Bioquest software (Holmuhamedov et al., 1998). Mitochondrial respiration was determined in the absence (state 2) or presence (state 3) of 250 µM ADP (Holmuhamedov et al., 1999). The rate of ADP-induced respiration was derived from changes in oxygen concentration continuously monitored in the mitochondrial suspension following addition of ADP (Ozcan et al., 2001). The maximal respiratory capacity of mitochondria was determined following a challenge of 2,4-dinitrophenol (25 µM), a mitochondrial uncoupler (Holmuhamedov et al., 2004). Mitochondrial ATP production was measured in K2CO3-MOPS neutralized HClO4-soluble mitochondrial extracts by high performance liquid chromatography (HPLC, Hewlett Packard, Waldbronn, Germany) as described (Dzeja et al., 1999). Following addition of ADP (1 mM), two aliquots of mitochondrial suspension (200 µl each) were removed at 60 sec interval (1 mM) into 20 µl HClO4 (3.3 M) and precipitated proteins separated by centrifugation (14,000 RPM). After neutralization with 100 µl K2CO3 (2.5 M K2CO3 in 1 M HEPES) and removal of precipitate (centrifugation at 14,000 RPM), the concentration of ATP within extracts was determined and used to define the rate of ATP synthesis (Holmuhamedov et al., 2001).
2.8. Western Blot Analysis
Frozen ventricular tissue (~40 mg) from adult and aged rats (same tissue saved from gene expression analysis) were homogenized in a Urea-modified RIPA buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 1% Triton X-100, 1% SDS, 3 M urea) with protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN). After protein concentration was determined (BIO-RAD Laboratories, Hercules, CA) aliquots of extracted protein (20 µg) solubilized in Laemmli sample buffer containing dithiothretiol (200 mM) were boiled (90°C for 5 min), separated on 15% SDS-PAGE gel and transferred to nitrocellulose membranes (Invitrogen, Carlsbad, CA). Membranes were blocked overnight in Western blocking reagent (Roche Applied Science, Indianapolis, IN) and then incubated for 12 hr in primary antibodies. Primary antibodies for Cytochrome c oxidase subunits Va, Vb, VIc, ATP synthase subunit alpha and beta were diluted to 0.5 mg/ml for succinate dehydrogenase subunit A and Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) to 1:5,000 and citrate synthase diluted to 1:1,000. Membranes were then exposed for 1 hr to secondary antibody (1:30,000). SuperSignal West Pico chemiluminescent substrate (Pierce Biotechnology, Rockford, IL) was used for detection in the AC-1 Autochemi system (UVP, Upland, CA). All primary antibodies, except for citrate synthase (a kind gift from Dr. J.O. Holloszy, Washington University, St. Louis, MO), were purchased from Molecular Probes (Invitrogen Corporation, Carlsbad, CA) and all secondary antibodies purchased from Jackson ImmunoResearch Laboratories (West Grove, PA).
2.9. Electron microscopy imaging and mitochondrial numeric density
Electron microscopy images (Jeol 1200 TEM, Tokyo, Japan) from adult and aged left ventricular free wall were used at 4,000 final magnification, to quantitatively assess mitochondrial numeric density (Frenzel and Feimann, 1984). Using a point counting method, randomly selected areas from micrographs of adult and aged tissue were picked to determine the area density of mitochondria and reported as number of mitochondria/100 µM2.
2.10. Statistical analysis
Data for OxPhos complexes western blots, enzymatic activities, mitochondrial oxygen consumption and ATP production are here reported as mean ± S.E.M. Student's t-test was used for data analysis and differences at p < 0.05 were considered significant.
3. Experimental Results
3.1. Aging alters the expression profile of genes coding for cardiac mitochondrial proteins
Gene expression pattern in adult (6 months, n=5) versus aged (24 months old, n=5) rat ventricles were compared using heat map and hierarchical clustering (Fig. 1A). Heat maps of 614 genes coding for mitochondrial proteins displayed their relative level of expression within each individual sample shown in green (downregulated), red (upregulated) or black (unchanged). Hierarchical clustering revealed a pattern where each individual sample from adult and aged ventricles clustered within the same age group, differentiating the adult from the aged transcriptome (Fig. 1A). The reproducibility of results for level of gene expression of adult and aged hearts are also shown as dot-plots in Figure 1B, highlighting the clustering of altered gene expression within each age group with the statistical significance of age-related changes between the two groups for the selected genes. Furthermore, differences in expression of genes in the aged ventricles plotted as a volcano map defined the degree of significance in expression according to their p-value and fold change (Fig. 1C). Ninety four out of these 614 genes (15%) were differentially expressed in the aging ventricles at a p value < 0.01 (Fig 1B). The majority (95%) of these genes (89 out of 94) were downregulated (Fig. ID, green bar), while 5% (5 out of 94) were upregulated (Fig. 1D, red bar). Age-associated alteration in the expression of genes coding for mitochondrial proteins in the ventricles were confined to those encoded by nuclear DNA (nDNA) with no change in expression of mitochondrial DNA (mtDNA) encoded genes. Distribution of 94 differentially expressed genes according to their molecular function (www.affymetrix.com/analysis/index.affx), demonstrates that 65% percent of altered genes were confined to pathways regulating mitochondrial metabolism and energetics that includes changes in genes coding for OxPhos (41%), substrate metabolism (18%) and tricarboxylic acid (TCA) cycle (6%, Fig. 1D).
Fig. 1.
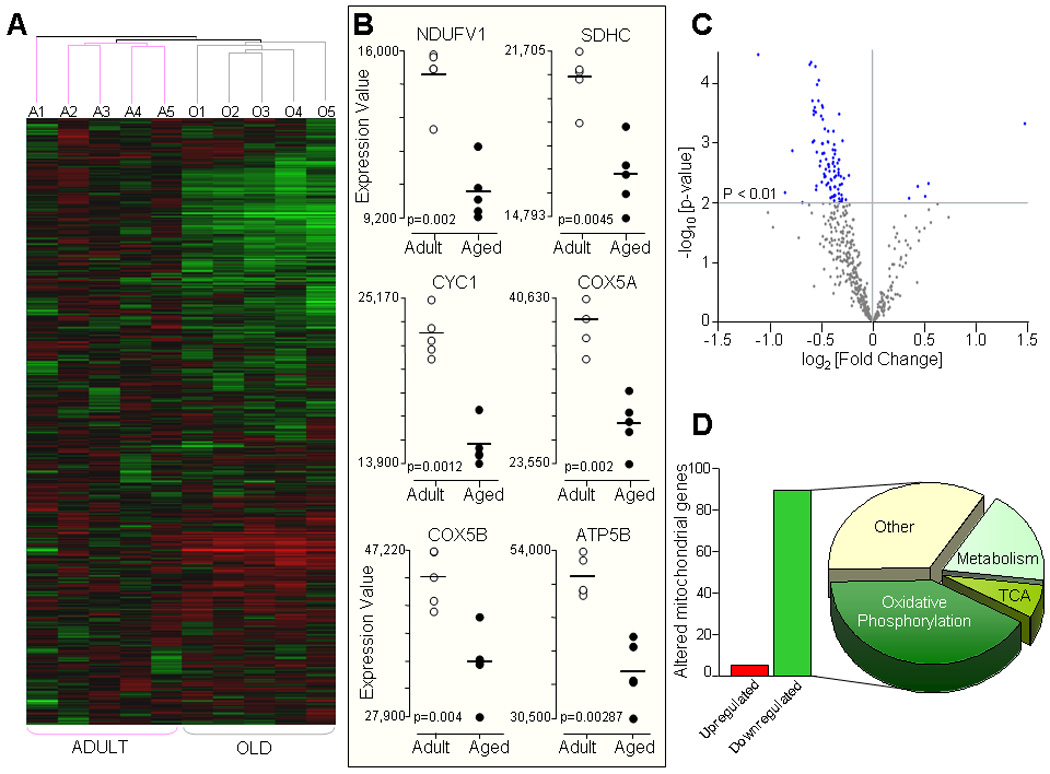
Aging-associated alterations in mitochondrial gene transcripts. A) Hierarchical clustering and heat map of samples from adult (6 month old, n=5) and aged (24 month old, n=5) rat ventricles. Shown are the expression pattern of 614 genes coding for mitochondrial proteins where Green color indicates downregulation, Red color upregulation, and Black color no change in the level of expression of genes. Samples from adult (Pink line) and aged (Gray line) animals clustered together as illustrated by condition tree (A1–A5 adult, O1–O5 aged samples). B) Expression values for selected genes coding for subunits of the mitochondrial OxPhos complexes from adult (n=5, White dots) and aged (n=5, Black dots) ventricles are shown as dot-plots highlighting the clustering of gene expression within each age group with the statistical significance of age-related changes between the two groups shown with their p values. C) Volcano plot illustrating the statistical distribution of 614 genes in aged compared with adult ventricles. There are 94 genes that were significantly different (p < 0.01) in the aged compared to adults (Blue dots). D) Number of significantly downregulated (89 genes, Green bar) and upregulated genes (5 genes, Red bar) in the aged heart and distribution according to their molecular function; 65% changes in genes coding for mitochondrial energetic pathways (41% for oxidative phosphorylation, 18% for substrate metabolism pathways and 6% for TCA cycle) and remaining 35% for other mitochondrial functions.
3.2. Aging decreases the expression of genes regulating oxidative phosphorylation
The expression of genes coding for several subunits of the mitochondrial respiratory chain complexes was downregulated with aging (Fig. 2). Out of 78 genes that code for subunits of Complexes I, II, III and IV, 30 genes (39%) were decreased in ventricles of aged rats (Fig. 2, colored Green). These included 16 genes coding for subunits of Complex I, 2 genes for Complex II, 4 genes for Complex III and 8 genes for Complex IV (Fig. 2 and 3). Also, out of 21 genes known to code for the mitochondrial F0F1ATP synthase complex (www.mitop.de/), 9 genes ATP5A1, ATP5B, ATP5C1, ATP5D, ATP5G1, ATP5G2, ATP5H, ATP5J and ATP5O that respectively code for subunits α, β, γ, and δ of the F1 subcomplex; subunits c isoform 1, c isoform 2, d, and F6 of the F0 subcomplex and subunit O of the stalk that links F1 to F0 complex, were downregulated (Fig. 2, Complex IV, colored Green). Specific genes coding for subunits of the ETC and F0F1ATP synthase complex, along with their description, gene ID, fold change and p-Value are summarized in Fig. 3, collectively indicating a broad downregulation of the components of OxPhos pathway.
Fig. 2.
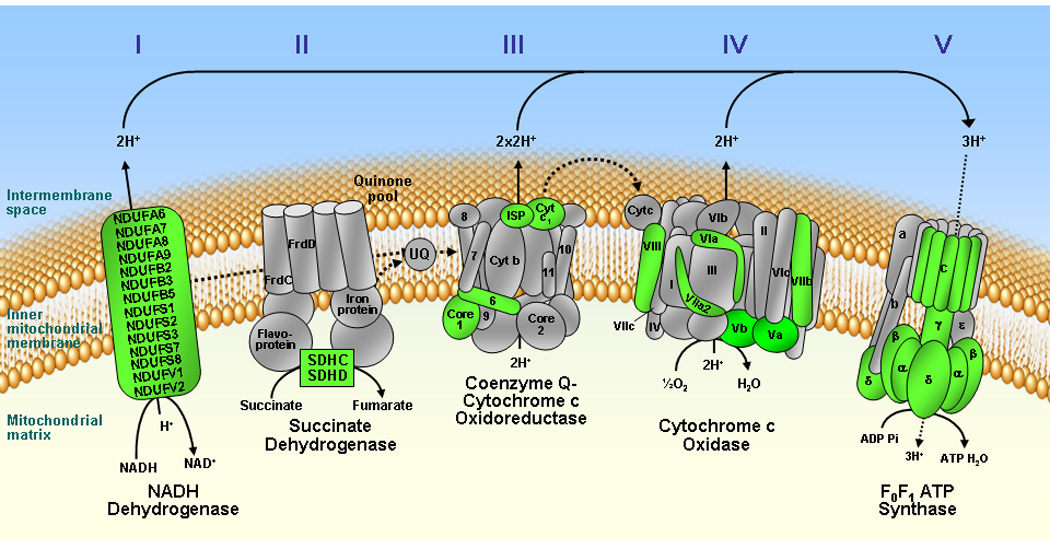
Schematic representation of genes coding for subunits of mitochondrial oxidative phosphorylation complexes. Genes coding for subunits of the electron transport chain (Complexes I – IV) and F0F1 ATP synthase (Complex V) that are significantly downregulated in the aging ventricle are colored Green, while subunits not altered with aging are shown in Gray. None of the genes coding for subunits of the oxidative phosphorylation pathway were upregulated in the aged hearts.
Fig. 3.
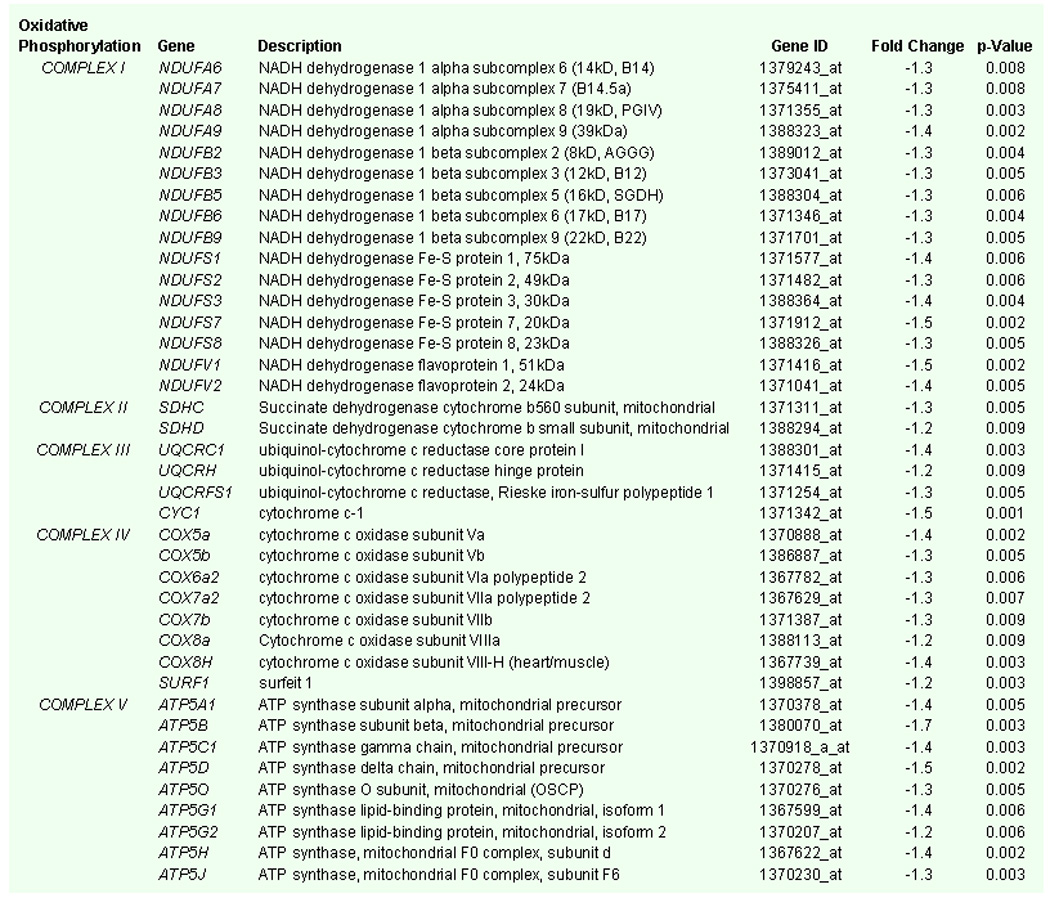
List of genes coding for mitochondrial oxidative phosphorylation pathway that are significantly (p < 0.01) downregulated in the aged rat heart with gene name, description, gene identification code (ID), fold change and p-values.
To verify age-related changes in the transcriptional profile of genes determined by microarray analysis, expression of selected genes in adult and aged ventricles were assessed by real time-polymerase chain reaction (RT-PCR). The expression of CS, COX5a, COX5b and ATP5B encoding four critical subunits of mitochondrial enzymes, citrate synthase, cytochrome c oxidase Va, cytochrome c oxidase Vb and ATP synthase beta respectively, were significantly reduced in the aging ventricles. The mRNA levels of these enzymes determined by RT-PCR were reduced by 1.9, 1.4, 1.5 and 1.9 fold in aged ventricles compared to adults for CS, COX5a, COX5b and ATP5B genes respectively (p < 0.01), a downregulation similar to that detected in the microarray analysis, i.e., 1.4, 1.3, 1.4 and 1.7 fold reduction for each of the tested genes respectively (Fig. 4).
Fig. 4.

Comparison of aging associated reduction in expression of selected genes coding for mitochondrial oxidative phosphorylation subunits determined by microarray and Real Time-Polymerase Chain Reaction (RT-PCR) techniques; CS=Citrate Synthase, COX5a=Cytochrome c oxidase subunit 5a, COX5b=Cytochrome c oxidase subunit 5b and ATP5B=ATP synthase beta.
3.3. Aging selectively decreases protein expression and activities of complexes of the mitochondrial oxidative phosphorylation pathway
The effect of aging-associated changes in the expression of selected genes for subunits of the mitochondrial ETC and ATP synthase complexes was also confirmed at the peptide level using western blots of ventricular tissue from aged (24 month old, n=5) and adult (6 month old, n=5) rats. Protein expression of cytochrome c oxidase subunits Va, Vb, VIc and ATP synthase subunit beta was decreased with aging by 20%, 29%, 40% and 20%, respectively (p<0.01, Fig.5A), while ATP synthase subunit alpha, succinate dehydrogenase subunit A and citrate synthase showed no significant change (10%, 4% and 2% decrease in aged respectively, p=NS). The differences in the activity of mitochondrial ETC and F0F1 ATPase complexes between adult and aged were also determined in mitochondria isolated from ventricles. The activity of ETC Complex I, as determined by the rotenone-sensitive reduction of ubiquinone-1 was 249±10 nmol NADH/min/mg of protein in adult (n=6) compared to 135±10 nmol NADH/min/mg protein in aged (n=6) mitochondria from heart, indicating a 46% reduction in Complex I activity with age (p < 0.01, Fig. 5B). The activity of Complex II of the ETC was however not significantly altered by aging. The rate of reduction of ubiquinone-2 by succinate was 92±11 nmol/min/mg protein in adult (n=6), similar to 77±18 nmol/min/mg protein in aged mitochondria (n=6, p=NS, Fig. 5B). The activity of Complex III, as defined by the rate of reduction of ferricytochrome c by decylubiquinone was 263±9 nmol/min/mg protein for adult (n=6), not significantly different from 243±14 nmol/min/mg protein in aged counterparts (n=6, p=NS, Fig. 5B). Decrease in absorbance of ferrocytochrome c by cytochrome c oxidase, reflective of Complex IV activity, was also not significantly different between the two age groups (498±25 vs 476±75 nmol/min/mg protein in adult and aged respectively, n=6, p=NS, Fig. 5B). Oligomycin-sensitive oxidation of NAD+, reflecting F0F1 ATPase (Complex V) activity was, however significantly reduced from 499±31 nmol NADH/min/mg protein in adults (n=6) to 377±19 nmol NADH/min/mg protein in aged mitochondria (n=6, p < 0.01, Fig. 5B). Overall, the capacity of mitochondria to utilize oxygen and synthesize ATP from ADP was significantly reduced in the aged heart. Specifically, the rate of ADP stimulated oxygen consumption (state 3 respiration) in malate-pyruvate containing media was 230±10 ng-atoms O2/min/mg protein in mitochondria isolated from adult heart (n=6) compared to 140±7 ng-atoms O2/min/mg protein from aged hearts (n=6, p < 0.01, Fig. 5 inset). This was associated with a reduced rate of ATP production measured at 500±55 nmoles ATP/min/mg protein in aged (n=6) compared to 636±40 nmoles ATP/min/mg protein in mitochondria isolated from adult hearts (n=6, p < 0.05, Fig. 5 inset). Thus, with aging the efficiency of mitochondria for OxPhos and the capacity to synthesize ATP are significantly decreased, along with a selective reduction in the activity of Complexes I and V of the mitochondrial OxPhos pathway.
Fig. 5.
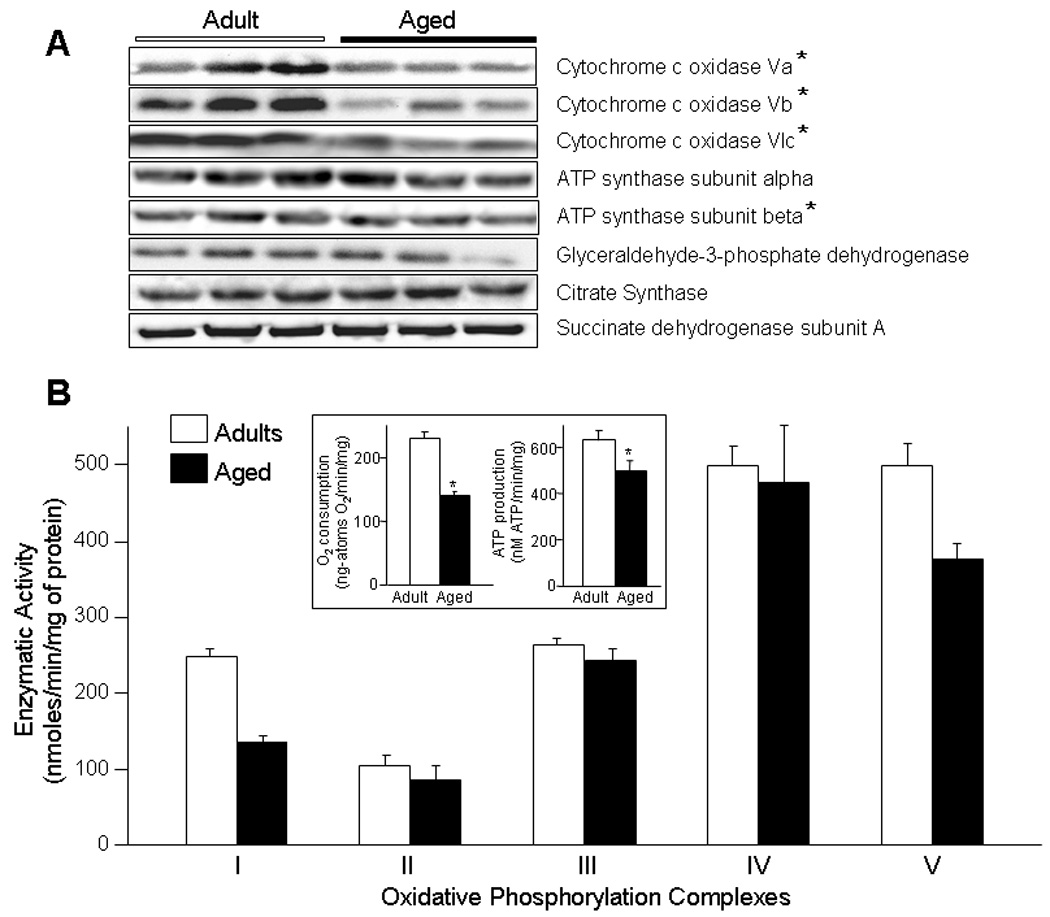
A) Western blots of selected protein subunits of the mitochondrial electron transport chain and ATP synthase complexes from adult (6 month old) and aged (24 month old) rat ventricles. Cytochrome c oxidase subunits Va, Vb, VIc and ATP synthase subunit beta are decreased with aging by 20%, 29%, 40% and 20% respectively (p < 0.01). There was no significant difference between adult and aged in expression of ATP synthase subunit alpha, glyceraldehyde-3-phosphate dehydrogenase, citrate synthase and succinate dehydrogenase subunit A. B) Enzymatic activities of Complexes I to V of the mitochondrial oxidative phosphorylation pathway in adult (n=6) and aged (n=6) ventricles. Compared to adults, functional activities of Complexes I and V are significantly decreased in the aged mitochondria (p < 0.01), while no significant differences were observed in activities of Complexes II, III and IV. All activities are expressed as nmoles/min/mg of mitochondrial protein; Inset, the rate of oxygen consumption and adenosine triphosphate (ATP) production in mitochondria from aged (n=6) ventricle is significantly decreased compared to adult (n=6). White bars = adult, black bars = aged, * p < 0.01.
3.4. Aging decreases gene expression of transcription co-activator factors regulating mitochondrial biogenesis in heart
To determine whether aging alters expression of genes regulating mitochondrial biogenesis, an important adaptive program that remodels cellular energetics in response to metabolic demands (Finck and Kelly, 2006), the transcription profile of genes coding for regulatory transcriptional co-activator factors was also defined. While no change in expression of genes coding for the myocyte enhancer factor 2 (MEF2) transcription factor family was seen with aging, a significant reduction in the expression of MAPK14 (−1.2 fold), PPARGC1A (−2.2 fold), PPARGC1B (−1.5 fold), PPARD (−1.5 fold), ESRRG (−1.4 fold), NRF2L1 (−1.3 fold) and NRF2L2 (1.2 fold) encoding for mitogen-activated protein kinase 14 (p38MAPK), peroxisome proliferator-activated receptor-γ coactivator-1 alpha (PGC-1α), peroxisome proliferator-activated receptor-γ coactivator-1 beta (PGC-1β), peroxisome proliferator-activated receptor delta (PPAR-δ), estrogen receptor gamma (ERR-γ), nuclear factor erythroid-derived 2-like 1 and nuclear factor erythroid-derived 2-like 2 respectively, was observed in ventricles of aged rats (p < 0.01, Fig. 6). To asses whether changes in expression of these transcription co-activator factors results in a decrease in the amount of mitochondria within the ventricles, the total protein content of mitochondrial isolates and a mitochondrial marker enzyme citrate synthase content was determined by western blot. Differences in the mitochondrial numeric density were also evaluated by electron micrography (4,000 magnification) of left ventricular free wall myocardium from adult and aged rats. There was a non-significant increase in protein content of isolated mitochondria in aged (23±3 µg/µl, n=10) compared to adults (25±2 µg/µl, n=10, p=NS) with no increase in citrate synthase content by western blot (Fig. 5A). The number of mitochondria per 100 µm2 was not reduced but rather increased in the aged (77±6/100 µm2) when compared to adult ventricles (41±7/100 µm2, p < 0.01, Fig. 7), suggesting a compensatory remodeling.
Fig. 6.

List of genes coding for transcription co-activator factors that are significantly (p < 0.01) altered with aging with gene name, description, gene identification code (ID), fold change and p-values.
Fig. 7.
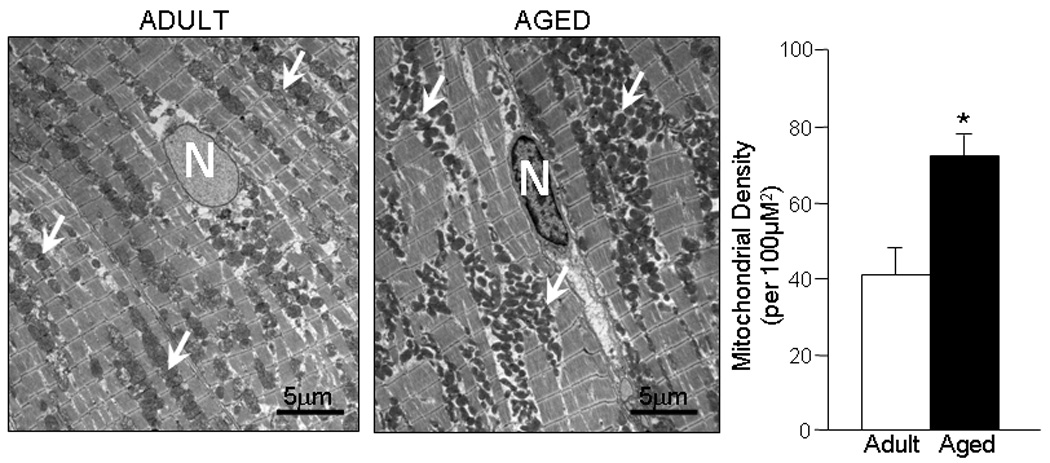
Electron microscopy of ventricular tissue and numeric density of mitochondria in adult (6 month old, n=8) and aged (24 month old, n=8) rat ventricles. N=nucleus, White arrows point to mitochondrial clusters, White bars=adult, Black bars=aged, * p < 0.01.
4. Discussion
Aging is associated with reduced functional reserve and altered responsiveness of the heart to stress but the molecular basis for this deficiency is not entirely clear (Chantler et al., 2006; Goldspink, 2005; Lakatta and Sollott, 2002). Here, we focused on mitochondrial energetic pathways and demonstrated that with aging a widespread suppression of the mitochondrial energy production system and transcription co-activator factors that regulate mitochondrial genes involved in substrate metabolism and OxPhos occurs at the transcriptional level. These changes were associated with reduced activities of Complex I and V of the OxPhos pathway that translated into diminished capacity of cardiac mitochondria to utilize oxygen and to produce ATP from ADP.
Mitochondrial ATP is synthesized through a coordinated transfer of electrons across the inner mitochondrial membrane through complexes I to IV of the electron transport chain. This electron transfer is accompanied by efflux of protons from mitochondrial matrix into the inter-membrane space generating a negative mitochondrial membrane potential (Δψ) and a proton gradient across the inner mitochondrial membrane which is used to phosphorylate ADP to ATP as the protons flow back into the mitochondria through the F0F1ATP synthase complex (Navarro and Boveris, 2007). Electrons delivered to the mitochondrial respiratory chain complexes are derived from NADH and/or FADH2 generated through the TCA cycle from Acetyl Co-A. With aging, a widespread reduction in the expression of genes coding for proteins of various complexes of the mitochondrial OxPhos pathway in aged rat ventricles was here demonstrated. Although, the fold change of most of respiratory transcripts did not exceed 2-fold, the association with a decline in mitochondrial functional capacity for ATP synthesis highlighted the biological significance of these findings. It is becoming clear that the biological outcome of gene expression changes is not necessarily dependent on the level of differential expression (Someya et al., 2007), since highly expressed genes may portray a less than 2-fold difference in microarrays while lowly expressed genes may show a greater than 2-fold change (Klebanov et al., 2007; Mutch et al., 2002). This is why we report age-associated changes in gene expression not only by fold change but also by their statistical significance at p value < 0.01. This is also highlighted by dot plots (Fig. 1B) showing clustering of expression changes within each group with p values < 0.01 indicating reproducibility of expression patterns within each age group. We have not considered any change that had a statistical significance with p > 0.01; however, functional significance of these small changes (p > 0.01) cannot be entirely excluded.
Complex I of the ETC, composed of three subcomplexes (Iα, Iβ and Iλ) with 14 central and 32 accessory subunits (Brandt, 2006), harbors NADH dehydrogenase and oxidoreductase activities, catalyzing the first step of NADH oxidation and transferring electrons from NADH and FMN derived from the TCA cycle to the respiratory chain (Fig. 2). Seven of the subunits of Complex I are encoded by the mitochondrial genome, while the remaining 39 subunits are products of nuclear genes. Mutations identified in genes encoding for complex I subunits have been associated with complex I disassembly, instability and increase in reactive oxygen species production, resulting in degenerative diseases (Benit et al., 2003; Kirby et al., 2004; Loeffen et al., 2001; Mamelak et al., 2005). In the present study no significant alteration in expression of the mtDNA coded genes that are included in the Affymetrix Rat Genome 230 2.0 were found, whereas expression of 16 out of the 37 genes coded by the nDNA was decreased. This observation that changes in expression of nDNA did not alter expression of mtDNA coded genes included in the microarray is of interest and indicates that crosstalk between nDNA and mtDNA may not be tightly coordinated. This is consistent with our previous observation that even with complete depletion of mtDNA, the expression, import and activity of nDNA encoded proteins into mitochondrial scaffolds was not inhibited in mitochondria of rho mutant cells lacking mtDNA, therefore “mismatch” subunits can exist, although the mitochondrial function may be impaired (Holmuhamedov et al., 2003). This was also demonstrated in pathological conditions such as cancer, where an imbalance between nuclear and mitochondrial encoded proteins may occur (Mazzanti and Giulivi, 2006). The alteration in transcript levels of Complex I nDNA encoded genes in the aged hearts (Fig. 3) was associated with functional decline and a 46% reduction in enzymatic activity as determined by the rotenone-sensitive reduction of ubiquinone-1 (Fig. 5B) and decreased state 3 respiration in malate-pyruvate NAD-dependent substrate. These results are consistent with previous reports indicating a reduction in Complex I activity in mitochondria isolated from old brain, liver, kidney and skeletal muscle (Benzi et al., 1992; Kumaran et al., 2005; Lenaz et al., 1997; Nakahara et al., 1998; Navarro and Boveris, 2004; Sugiyama et al., 1993). These changes may contribute to the reduced energetic reserves and increased susceptibility of the aged heart to stress and oxidative injury (Navarro and Boveris, 2007), underscoring the relevance of the present findings.
Complex II, succinate dehydrogenase (SDH), of the ETC is the membrane-bound component of the TCA cycle and is composed of 4 nuclear encoded subunits that are involved in the oxidation of succinate and electron transfer from FADH2 to Coenzyme Q, thus linking the TCA cycle to the mitochondrial respiratory chain promoting electron flow, ATP production and reactive oxygen species generation (Cecchini, 2003). Despite downregulation of SDHC (p=0.002) and SDHD (p=0.007, Fig. 3) that code for subunits c and d of the SDH, we found no significant change in Complex II activity in the aged heart (Fig. 5B) as determined by the reduction of ubiquinone-2 by succinate (Darley-Usmar et al., 1987). These findings are similar to previous reports demonstrating no age-associated change in the activity of SDH in mitochondria isolated from rat skeletal muscle as well as from mice kidneys and liver (Kwong and Sohal, 2000; Sugiyama et al., 1993; Yarian et al., 2006).
Complex III (Coenzyme Q-cytochrome c oxido-reductase) of the ETC is composed of 10 subunits, 9 encoded by the nDNA and 1 by mtDNA and oxidizes ubiquinol and transfer protons from mitochondrial matrix into the inter membrane space reducing cytochrome c. In the aged heart, the expression of UQCRC1, UQCRH, UQCRFS1 and CYC1 encoding for the core protein I, hinge protein, Rieske iron-sulfur polypeptide 1 and cytochrome c-1 of the Coenzyme Q-cytochrome c oxido-reductase complex were downregulated (Fig. 3). However, this was not associated with a significant change in the activity of Complex III as determined by the rate of reduction of ferricytochrome c by decylubiquinol (Fig. 5B). Differences in maximum enzyme activity has been previously described between frozen-thawed mitochondria compared to detergent solubilized and could be due to methodological differences.
Cytochrome c oxidase (COX) is the terminal enzyme of the mitochondrial respiratory chain (Complex IV). It is a multi-subunit enzyme complex that couples the transfer of electrons from cytochrome c to molecular oxygen and contributes to proton efflux into the inter-membrane space (Richter and Ludwig, 2003). The complex consists of 13 subunits encoded by 3 mitochondrial and 17 nuclear genes (www.mitop.de/). The mitochondrial-encoded subunits perform electron transfer of proton pumping activities while the function of nuclear-encoded subunits is unclear but may play a role in the regulation and assembly of the whole complex. In our study no change in the expression of any of the three mtDNA encoded subunits was observed, while 8 of the 17 nDNA encoded genes including COX5A, COX5B, COX6A2, COX7A2, COX7B, COX8A, COX8H and SURF1 genes were all downregulated in the aged heart (Fig. 3). Despite a decrease in the expression of these genes, no significant reduction in cytochrome c oxidase functional activity (Fig. 5B) was observed in aged rat heart mitochondrial fragments, which may reflect preserved expression of functionally active mtDNA encoded subunits. These results are consistent with findings reported in mitochondrial fragments from aged rat hearts (Fannin et al., 1999) but different from those described in brain and liver mitochondria from old rats where a 24–28% reduction in Complex IV activity was demonstrated (Navarro and Boveris, 2004). This discrepancy in the effect of aging on Complex IV activity could be related to tissue specific differences in isoenzymes of Complex IV or other complexes (Capaldi et al., 1988) that may respond differently to aging. In addition, methodological differences such as the use of polarographic determination of cytochrome c oxidase activity demonstrating reduced Complex IV activity in aged cardiac mitochondria may explain some of the reported differences (Navarro and Boveris, 2007). Moreover, differences in the activities of OxPhos complexes in the literature could result from the use of different subpopulations of mitochondria in different studies (Fannin et al., 1999; Palmer et al., 1977). It has been previously demonstrated that interfibrillar mitochondria exhibit a selective decline in Complex III and IV activity which is not observed in the subsarcolemmal population of mitochondria (Fannin et al., 1999; Lesnefsky and Hoppel, 2006; Suh et al., 2003). In our study, mitochondria were not separated and a mixed population was used, therefore any selective decrease in the activity of ETC complexes in one subpopulation could have been masked.
Mitochondrial F0F1 ATP synthase (Complex V) catalyzes ATP synthesis, utilizing the electrochemical gradient generated by proton efflux across the inner membrane during electron transfer (Walker and Dickson, 2006). Complex V is composed of two linked multi-subunit complexes, F0, the integral membrane-spanning component comprising the proton channel and F1, the catalytic portion of mitochondrial ATP synthase linked by a central and peripheral stalk (Carbajo et al., 2005; Walker and Dickson, 2006). We found that 9 of the 21 genes that code for subunits of complex V were downregulated in aged hearts (Fig. 2, Fig. 3). This is consistent with previous reports in mitochondria from rat and mice brain and heart demonstrating downregulation of subunits of F0F1 ATPase (Nicoletti et al., 1998). Subunit ATP6 encoded by mtDNA that was reported to be downregulated by Manczak et.al, (Manczak et al., 2005) were not observed in our analysis. The changes in transcript levels of subunits of the F0F1 ATPase (Fig. 3) in our study were associated with a reduction in functional activity of Complex V demonstrating a 25% reduction in the oligomycin-sensitive F0F1 ATPase activity (Fig. 5B), similar to that reported in mitochondria from rat liver and heart (Davies et al., 2001; Guerrieri et al., 1996). In conditions associated with reduced substrate availability or hypoxia, such as during myocardial ischemia, the enzyme complex switches from an ATP generating to ATP consuming system that utilizes ATP to maintain mitochondrial electrochemical gradient (Das, 2003), thus preserving mitochondrial function and integrity. Reduction in the activity of Complex V in the aging heart may reduce mitochondrial functional capacity and ability to maintain hyperpolarized membrane potential during metabolic stress and thereby increase predisposition to cellular injury (Di Lisa and Bernardi, 2005).
Although expression of specific mitochondrial genes have been previously demonstrated to be altered within the aging heart (LeMoine et al., 2006), the overall gene expression profiling of the myocardium in the present study indicates a more generalized downregulation of the transcriptional regulatory program in the aging heart. This could be explained by downregulation of upstream genes such as transcription co-activator factors that modulate expression of nuclear and/or mitochondrial DNA (Lehman et al., 2000; Molkentin and Dorn, 2001; Nisoli et al., 2003). Indeed, a significant decrease in the expression of transcription co-activator factor genes coding for PGC-1α, PGC-1β, PPAR-δ, ERR-γ, nuclear factor erythroid-derived 2-like 1 and nuclear factor erythroid-derived 2-like 2, which modulates gene expression of enzymes regulating mitochondrial metabolism and mitogenesis (Lin et al., 2005) was observed in the senescent heart (Fig. 6; p < 0.01). The significance of reduction in these factors to mitogenesis in the senescent heart needs to be further explored. However, we did not observe any apparent reduction in the number of mitochondria in cardiomyocytes from senescent heart nor differences in protein content of isolated mitochondria or mitochondrial marker citrate synthase between adult and aged hearts indicating that reduced mitochondrial content was not responsible for the overall reduction in the capacity of mitochondria to synthesize ATP but instead reflects a functional impairment of energy production (Nakahara et al., 1998).
Thus, we here demonstrate aging associated decline in mitochondrial capacity to synthesize ATP with a widespread reduction in expression of genes coding for various subunits of mitochondrial OxPhos complexes. While there is a broad downregulation of the OxPhos complex genes, selective dysfunction of certain complexes were identified opening the possibility of targeted interventions. The absence of non-selective shutdown of OxPhos on the other hand maybe due to adaptive remodeling, which include an increase in the number of mitochondria at least partially compensating for deficiency in Complex I and V allowing the mitochondria to maintain a baseline function, albeit rendering them susceptible to stress.
Acknowledgments
Arshad Jahangir is supported by grants from the National Institute on Aging (AG-21201), National Heart, Lung and Blood Institute (HL089542), the Mayo Clinic Robert and Arlene Kogod Program on Aging and the Marriott Mitochondrial Medicine Award. Current address for Ekhson L. Holmuhamedov is Department of Cell and Developmental Biology, University of North Carolina at Chapel Hill. We are grateful to Dr. Petras Dzeja for technical advice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abete P, Ferrara N, Cioppa A, Ferrara N, Bianco S, Calabrese C, Cacciatore F, Longobardi G, Rengo F. Preconditioning does not prevent post-ischemic dysfunction in aging heart. J Am Coll Cardiol. 1996;27:1777–1786. doi: 10.1016/0735-1097(96)00070-8. [DOI] [PubMed] [Google Scholar]
- Benit P, Beugnot R, Chretien D, Giurgea I, De Lonlay-Debeney P, Issartel JP, Corral-Debrinski M, Kerscher S, Rustin P, Rotig A, Munnich A. Mutant NDUFV2 subunit of mitochondrial complex I causes early onset hypertrophic cardiomyopathy and encephalopathy. Hum Mutat. 2003;21:582–586. doi: 10.1002/humu.10225. [DOI] [PubMed] [Google Scholar]
- Benzi G, Pastoris O, Marzatico F, Villa RF, Dagani F, Curti D. The mitochondrial electron transfer alteration as a factor involved in the brain aging. Neurobiol Aging. 1992;13:361–368. doi: 10.1016/0197-4580(92)90109-b. [DOI] [PubMed] [Google Scholar]
- Brandt U. Energy Converting NADH:Quinone Oxidoreductase (Complex I) Annu Rev Biochem. 2006 doi: 10.1146/annurev.biochem.75.103004.142539. [DOI] [PubMed] [Google Scholar]
- Capaldi RA, Halphen DG, Zhang YZ, Yanamura W. Complexity and tissue specificity of the mitochondrial respiratory chain. J Bioenerg Biomembr. 1988;20:291–311. doi: 10.1007/BF00769634. [DOI] [PubMed] [Google Scholar]
- Carbajo RJ, Kellas FA, Runswick MJ, Montgomery MG, Walker JE, Neuhaus D. Structure of the F1-binding domain of the stator of bovine F1Fo-ATPase and how it binds an alpha-subunit. J Mol Biol. 2005;351:824–838. doi: 10.1016/j.jmb.2005.06.012. [DOI] [PubMed] [Google Scholar]
- Cecchini G. Function and structure of complex II of the respiratory chain. Annu Rev Biochem. 2003;72:77–109. doi: 10.1146/annurev.biochem.72.121801.161700. [DOI] [PubMed] [Google Scholar]
- Chantler PD, Goldspink DF, Clements RE, Sharp L, Schlosshan D, Tan LB. Congestive heart failure: extent of cardiac functional changes caused by aging and organ dysfunction. Heart. 2006;92:686–688. doi: 10.1136/hrt.2005.063578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darley-Usmar VM, Rickwood D, Wilson MT. Mitochondria: a practical approach. Oxford; Washington, DC: IRL Press; 1987. [Google Scholar]
- Das AM. Regulation of the mitochondrial ATP-synthase in health and disease. Mol Genet Metab. 2003;79:71–82. doi: 10.1016/s1096-7192(03)00069-6. [DOI] [PubMed] [Google Scholar]
- Davies SM, Poljak A, Duncan MW, Smythe GA, Murphy MP. Measurements of protein carbonyls, ortho- and meta-tyrosine and oxidative phosphorylation complex activity in mitochondria from young and old rats. Free Radic Biol Med. 2001;31:181–190. doi: 10.1016/s0891-5849(01)00576-7. [DOI] [PubMed] [Google Scholar]
- Di Lisa F, Bernardi P. Mitochondrial function and myocardial aging. A critical analysis of the role of permeability transition. Cardiovasc Res. 2005;66:222–232. doi: 10.1016/j.cardiores.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Dzeja PP, Pucar D, Redfield MM, Burnett JC, Terzic A. Reduced activity of enzymes coupling ATP-generating with ATP-consuming processes in the failing myocardium. Mol Cell Biochem. 1999;201:33–40. doi: 10.1023/a:1007016703229. [DOI] [PubMed] [Google Scholar]
- Fannin SW, Lesnefsky EJ, Slabe TJ, Hassan MO, Hoppel CL. Aging selectively decreases oxidative capacity in rat heart interfibrillar mitochondria. Arch Biochem Biophys. 1999;372:399–407. doi: 10.1006/abbi.1999.1508. [DOI] [PubMed] [Google Scholar]
- Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. 2006;116:615–622. doi: 10.1172/JCI27794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenzel H, Feimann J. Age-dependent structural changes in the myocardium of rats. A quantitative light- and electron-microscopic study on the right and left chamber wall. Mech Ageing Dev. 1984;27:29–41. doi: 10.1016/0047-6374(84)90080-0. [DOI] [PubMed] [Google Scholar]
- Goldspink DF. Ageing and activity: their effects on the functional reserve capacities of the heart and vascular smooth and skeletal muscles. Ergonomics. 2005;48:1334–1351. doi: 10.1080/00140130500101247. [DOI] [PubMed] [Google Scholar]
- Guerrieri F, Vendemiale G, Turturro N, Fratello A, Furio A, Muolo L, Grattagliano I, Papa S. Alteration of mitochondrial F0F1 ATP synthase during aging. Possible involvement of oxygen free radicals. Ann N Y Acad Sci. 1996;786:62–71. doi: 10.1111/j.1749-6632.1996.tb39052.x. [DOI] [PubMed] [Google Scholar]
- Holmuhamedov E, Jahangir A, Bienengraeber M, Lewis LD, Terzic A. Deletion of mtDNA disrupts mitochondrial function and structure, but not biogenesis. Mitochondrion. 2003;3:13–19. doi: 10.1016/S1567-7249(03)00053-9. [DOI] [PubMed] [Google Scholar]
- Holmuhamedov EL, Jahangir A, Oberlin A, Komarov A, Colombini M, Terzic A. Potassium channel openers are uncoupling protonophores: implication in cardioprotection. FEBS Lett. 2004;568:167–170. doi: 10.1016/j.febslet.2004.05.031. [DOI] [PubMed] [Google Scholar]
- Holmuhamedov EL, Jovanovic S, Dzeja PP, Jovanovic A, Terzic A. Mitochondrial ATP-sensitive K+ channels modulate cardiac mitochondrial function. Am J Physiol. 1998;275:H1567–H1576. doi: 10.1152/ajpheart.1998.275.5.H1567. [DOI] [PubMed] [Google Scholar]
- Holmuhamedov EL, Ozcan C, Jahangir A, Terzic A. Restoration of Ca2+-inhibited oxidative phosphorylation in cardiac mitochondria by mitochondrial Ca2+ unloading. Mol Cell Biochem. 2001;220:135–140. doi: 10.1023/a:1010894427373. [DOI] [PubMed] [Google Scholar]
- Holmuhamedov EL, Wang L, Terzic A. ATP-sensitive K+ channel openers prevent Ca2+ overload in rat cardiac mitochondria. J Physiol. 1999;519(Pt 2):347–360. doi: 10.1111/j.1469-7793.1999.0347m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahangir A, Ozcan C, Holmuhamedov EL, Terzic A. Increased calcium vulnerability of senescent cardiac mitochondria: protective role for a mitochondrial potassium channel opener. Mech Ageing Dev. 2001;122:1073–1086. doi: 10.1016/s0047-6374(01)00242-1. [DOI] [PubMed] [Google Scholar]
- Juhaszova M, Rabuel C, Zorov DB, Lakatta EG, Sollott SJ. Protection in the aged heart: preventing the heart-break of old age? Cardiovasc Res. 2005;66:233–244. doi: 10.1016/j.cardiores.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Kirby DM, Salemi R, Sugiana C, Ohtake A, Parry L, Bell KM, Kirk EP, Boneh A, Taylor RW, Dahl HH, Ryan MT, Thorburn DR. NDUFS6 mutations are a novel cause of lethal neonatal mitochondrial complex I deficiency. J Clin Invest. 2004;114:837–845. doi: 10.1172/JCI20683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanov L, Qiu X, Welle S, Yakovlev A. Statistical methods and microarray data. Nat Biotechnol. 2007;25:25–26. doi: 10.1038/nbt0107-25. [DOI] [PubMed] [Google Scholar]
- Kumaran S, Subathra M, Balu M, Panneerselvam C. Supplementation of L-carnitine improves mitochondrial enzymes in heart and skeletal muscle of aged rats. Exp Aging Res. 2005;31:55–67. doi: 10.1080/03610730590882846. [DOI] [PubMed] [Google Scholar]
- Kwong LK, Sohal RS. Age-related changes in activities of mitochondrial electron transport complexes in various tissues of the mouse. Arch Biochem Biophys. 2000;373:16–22. doi: 10.1006/abbi.1999.1495. [DOI] [PubMed] [Google Scholar]
- Lakatta EG, Sollott SJ. Perspectives on mammalian cardiovascular aging: humans to molecules. Comp Biochem Physiol A Mol Integr Physiol. 2002;132:699–721. doi: 10.1016/s1095-6433(02)00124-1. [DOI] [PubMed] [Google Scholar]
- Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeMoine CM, McClelland GB, Lyons CN, Mathieu-Costello O, Moyes CD. Control of mitochondrial gene expression in the aging rat myocardium. Biochem Cell Biol. 2006;84:191–198. doi: 10.1139/o05-169. [DOI] [PubMed] [Google Scholar]
- Lenaz G, Bovina C, Castelluccio C, Fato R, Formiggini G, Genova ML, Marchetti M, Pich MM, Pallotti F, Parenti Castelli G, Biagini G. Mitochondrial complex I defects in aging. Mol Cell Biochem. 1997;174:329–333. [PubMed] [Google Scholar]
- Lesnefsky EJ, Gallo DS, Ye J, Whittingham TS, Lust WD. Aging increases ischemia-reperfusion injury in the isolated, buffer-perfused heart. J Lab Clin Med. 1994;124:843–851. [PubMed] [Google Scholar]
- Lesnefsky EJ, Hoppel CL. Oxidative phosphorylation and aging. Ageing Res Rev. 2006;5:402–433. doi: 10.1016/j.arr.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- Loeffen J, Elpeleg O, Smeitink J, Smeets R, Stockler-Ipsiroglu S, Mandel H, Sengers R, Trijbels F, van den Heuvel L. Mutations in the complex I NDUFS2 gene of patients with cardiomyopathy and encephalomyopathy. Ann Neurol. 2001;49:195–201. doi: 10.1002/1531-8249(20010201)49:2<195::aid-ana39>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Maggioni A, Maseri A, Fresco C, Francosi M, Mauri F, Santoro E, Tognoni G. Age-related increase in mortality among patients with first myocardial infarctions treated with thrombolysis. N Engl J Med. 1993;329:1442–1448. doi: 10.1056/NEJM199311113292002. [DOI] [PubMed] [Google Scholar]
- Mamelak AJ, Kowalski J, Murphy K, Yadava N, Zahurak M, Kouba DJ, Howell BG, Tzu J, Cummins DL, Liegeois NJ, Berg K, Sauder DN. Downregulation of NDUFA1 and other oxidative phosphorylation-related genes is a consistent feature of basal cell carcinoma. Exp Dermatol. 2005;14:336–348. doi: 10.1111/j.0906-6705.2005.00278.x. [DOI] [PubMed] [Google Scholar]
- Manczak M, Jung Y, Park BS, Partovi D, Reddy PH. Time-course of mitochondrial gene expressions in mice brains: implications for mitochondrial dysfunction, oxidative damage, and cytochrome c in aging. J Neurochem. 2005;92:494–504. doi: 10.1111/j.1471-4159.2004.02884.x. [DOI] [PubMed] [Google Scholar]
- Mazzanti R, Giulivi C. Coordination of nuclear- and mitochondrial-DNA encoded proteins in cancer and normal colon tissues. Biochim Biophys Acta. 2006;1757:618–623. doi: 10.1016/j.bbabio.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Molkentin JD, Dorn IG., 2nd Cytoplasmic signaling pathways that regulate cardiac hypertrophy. Annu Rev Physiol. 2001:63. doi: 10.1146/annurev.physiol.63.1.391. [DOI] [PubMed] [Google Scholar]
- Mutch DM, Berger A, Mansourian R, Rytz A, Roberts MA. The limit fold change model: a practical approach for selecting differentially expressed genes from microarray data. BMC Bioinformatics. 2002;3:17. doi: 10.1186/1471-2105-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakahara H, Kanno T, Inai Y, Utsumi K, Hiramatsu M, Mori A, Packer L. Mitochondrial dysfunction in the senescence accelerated mouse (SAM) Free Radic Biol Med. 1998;24:85–92. doi: 10.1016/s0891-5849(97)00164-0. [DOI] [PubMed] [Google Scholar]
- Navarro A, Boveris A. The mitochondrial energy transduction system and the aging process. Am J Physiol Cell Physiol. 2007;292:C670–C686. doi: 10.1152/ajpcell.00213.2006. [DOI] [PubMed] [Google Scholar]
- Navarro A, Boveris A. Rat brain and liver mitochondria develop oxidative stress and lose enzymatic activities on aging. Am J Physiol Regul Integr Comp Physiol. 2004;287:R1244–R1249. doi: 10.1152/ajpregu.00226.2004. [DOI] [PubMed] [Google Scholar]
- Nicoletti VG, Tendi EA, Console A, Privitera A, Villa RF, Ragusa N, Giuffrida-Stella AM. Regulation of cytochrome c oxidase and FoF1-ATPase subunits expression in rat brain during aging. Neurochem Res. 1998;23:55–61. doi: 10.1023/a:1022449403619. [DOI] [PubMed] [Google Scholar]
- Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, Carruba MO. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- Ozcan C, Holmuhamedov EL, Jahangir A, Terzic A. Diazoxide protects mitochondria from anoxic injury: implications for myopreservation. J Thorac Cardiovasc Surg. 2001;121:298–306. doi: 10.1067/mtc.2001.111421. [DOI] [PubMed] [Google Scholar]
- Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem. 1977;252:8731–8739. [PubMed] [Google Scholar]
- Pimentel AE, Gentile CL, Tanaka H, Seals DR, Gates PE. Greater rate of decline in maximal aerobic capacity with age in endurance-trained than in sedentary men. J Appl Physiol. 2003;94:2406–2413. doi: 10.1152/japplphysiol.00774.2002. [DOI] [PubMed] [Google Scholar]
- Richter OM, Ludwig B. Cytochrome c oxidase--structure, function, and physiology of a redox-driven molecular machine. Rev Physiol Biochem Pharmacol. 2003;147:47–74. doi: 10.1007/s10254-003-0006-0. [DOI] [PubMed] [Google Scholar]
- Someya S, Yamasoba T, Prolla TA, Tanokura M. Genes encoding mitochondrial respiratory chain components are profoundly down-regulated with aging in the cochlea of DBA/2J mice. Brain Res. 2007;1182:26–33. doi: 10.1016/j.brainres.2007.08.090. [DOI] [PubMed] [Google Scholar]
- Sreekumar R, Unnikrishnan J, Fu A, Nygren J, Short KR, Schimke J, Barazzoni R, Nair KS. Effects of caloric restriction on mitochondrial function and gene transcripts in rat muscle. Am J Physiol Endocrinol Metab. 2002;283:E38–E43. doi: 10.1152/ajpendo.00387.2001. [DOI] [PubMed] [Google Scholar]
- Sugiyama S, Takasawa M, Hayakawa M, Ozawa T. Changes in skeletal muscle, heart and liver mitochondrial electron transport activities in rats and dogs of various ages. Biochem Mol Biol Int. 1993;30:937–944. [PubMed] [Google Scholar]
- Suh JH, Heath SH, Hagen TM. Two subpopulations of mitochondria in the aging rat heart display heterogenous levels of oxidative stress. Free Radic Biol Med. 2003;35:1064–1072. doi: 10.1016/s0891-5849(03)00468-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumpower BL, Edwards CA. Purification of a reconstitutively active iron-sulfur protein (oxidation factor) from succinate cytochrome c reductase complex of bovine heart mitochondria. J Biol Chem. 1979;254:8697–8706. [PubMed] [Google Scholar]
- Walker JE, Dickson VK. The peripheral stalk of the mitochondrial ATP synthase. Biochim Biophys Acta. 2006;1757:286–296. doi: 10.1016/j.bbabio.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Wallace DC. A mitochondrial paradigm for degenerative diseases and ageing. Novartis Found Symp. 2001;235:247–263. doi: 10.1002/0470868694.ch20. discussion 263–6. [DOI] [PubMed] [Google Scholar]
- Yarian CS, Toroser D, Sohal RS. Aconitase is the main functional target of aging in the citric acid cycle of kidney mitochondria from mice. Mech Ageing Dev. 2006;127:79–84. doi: 10.1016/j.mad.2005.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]