

Abstract
Long-term facilitation (LTF) is a form of respiratory neuroplasticity frequently induced by acute intermittent isocapnic hypoxia (AIH, three 5 min isocapnic hypoxic episodes). Although repetitive apnoeas are a frequent natural occurrence producing brief (< 30 s) episodes of hypoxia and hypercapnia, it is unknown if repetitive apnoeas also elicit LTF. Apnoea-induced LTF may preserve upper airway patency during sleep, thereby limiting further apnoeic events. We tested the hypothesis that repeated, brief ventilator-induced apnoeas are sufficient to induce serotonin-dependent phrenic and hypoglossal (XII) LTF in anaesthetized rats. Anaesthetized, male Sprague–Dawley rats were exposed to three or six 25 s ventilator apnoeas with 5 min intervals, and compared to time control and AIH-treated rats. Three and six ventilator apnoeas induced phrenic and XII LTF with a magnitude similar to AIH. Both apnoea-induced and AIH-induced LTF were associated with a decreased CO2 recruitment threshold for phrenic and XII activity (∼4 mmHg). Spinal methysergide, a serotonin receptor antagonist, blocked apnoea-induced LTF but not changes in the CO2-recruitment threshold. Thus, brief ventilator apnoeas elicit phrenic and XII LTF. Similar to AIH-induced LTF, apnoea-induced LTF is serotonin dependent, and the relevant serotonin receptors for phrenic LTF are located in the cervical spinal cord. Apnoea-induced LTF may have implications for the maintenance of breathing stability, particularly during sleep.
Respiratory long-term facilitation (LTF) is a form of respiratory neuroplasticity that can be elicited by acute intermittent hypoxia (Mitchell et al. 2001; Feldman et al. 2003; Mahamed & Mitchell, 2007). LTF was first described in anaesthetized cats when phrenic nerve activity was assessed following intermittent stimulation of the carotid sinus nerve (originally described as ‘long-term potentiation’; Millhorn et al. 1980a,b; Millhorn & Eldridge, 1986). LTF was expressed as a sustained (> 90 min) increase in phrenic nerve activity following intermittent carotid sinus nerve stimulation. LTF was subsequently demonstrated following intermittent isocapnic or poikilocapnic hypoxia in multiple species (for review see Mitchell et al. 2001), including rats (Hayashi et al. 1993; Bach & Mitchell, 1996; Peng et al. 2003). LTF has been expressed in pulmonary ventilation in sleeping humans with inspiratory flow limitation (Aboubakr et al. 2001; Babcock et al. 2003), and awake humans with modest hypercapnia (Harris et al. 2006). In recent years, considerable progress has been made towards understanding the cellular mechanisms of phrenic LTF, largely based on studies in anaesthetized rats (Feldman et al. 2003; Baker-Herman et al. 2004; Mahamed & Mitchell, 2007). Most studies of LTF to date have utilized acute intermittent isocapnic hypoxia (AIH) as an inducing stimulus, typically with 3–5 min hypoxic exposures, separated each by 5 min of baseline conditions (for review, see Mitchell et al. 2001; Feldman et al. 2003; Mahamed & Mitchell, 2007).
Despite the potential parallels between AIH protocols typically used to elicit LTF in experimental studies and those experienced in sleep apnoea syndromes (SAS), rarely have studies faithfully mimicked the timing and blood gas oscillations typical of these patients. Peng et al. (2003) report that ten 15 s exposures to 12% inspired O2 with 5 min intervals of 100% O2 elicits an LTF of ∼40%. Though the timing of hypoxic episodes in this study is similar in some respects to hypoxaemia expected during apnoeas in human subjects, the magnitude of the swings in O2 tensions are much greater than typically experienced during an apnoea, and concomitant hypercapnia was not experienced by the animals. Since hypercapnia elicits α2-adrenergic receptor-dependent depression of respiratory activity (Bach & Mitchell, 1998; Baker et al. 2001), it is possible that concomitant hypoxia and hypercapnia may mitigate LTF expression.
In a recent study, anaesthetized but spontaneously breathing rats exhibited facilitation of genioglossus, but not diaphragmatic EMGs, following ten 15 s airway occlusions over 15 min (Tadjalli et al. 2006). The failure to induce diaphragm (or phrenic) long-term facilitation in anaesthetized, spontaneously breathing rats is consistent with an earlier report that AIH fails to elicit phrenic LTF, possibly due to specific features characteristic of this experimental preparation (Janssen & Fregosi, 2000).
In the present study, we directly compared the ability of repetitive ventilator apnoeas and standardized AIH protocols to elicit phrenic and XII LTF in an anaesthetized, paralysed, vagotomized and ventilated rat model frequently used to study cellular mechanisms of respiratory LTF (Mitchell et al. 2001; Feldman et al. 2003; Mahamed & Mitchell, 2007). We provide evidence that repeated 25 s ventilator apnoeas that induce only mild hypoxia and hypercapnia elicit robust phrenic and XII LTF. Further, despite major differences in the timing and severity (level of hypoxaemia) of the inducing stimulus (apnoea versus AIH), the magnitude of LTF was comparable. We also provide the first description of changes in the CO2 apnoeic and recruitment thresholds (an important determinant of ventilatory stability) following repetitive apnoeas or AIH. Lastly, we confirm that apnoea-induced LTF is serotonin dependent, suggesting fundamental similarities in the mechanisms of LTF induced by repetitive apnoeas and AIH.
Methods
Animals
All experiments were performed on male Sprague–Dawley rats (Harlan colony 217, n = 47) aged 106 ± 10 days and weighing 378 ± 31 g. All protocols were approved by the Institutional Animal Care and Use Committee of the School of Veterinary Medicine at the University of Wisconsin, Madison.
Surgical preparation
Surgical procedures have been described in detail elsewhere (Baker-Herman & Mitchell, 2002; Baker-Herman et al. 2004) and are summarized here. After anaesthesia induction with isoflurane (2.5–3.5% in 50% O2), the rats were bilaterally vagotomized, tracheotomized and pump ventilated (Rodent Ventilator 683, Harvard Apparatus, Holliston, MA, USA). The tail vein and femoral artery were catheterized for fluid infusions and to allow arterial blood sample analysis, respectively. The left hypoglossal and phrenic nerves were isolated via dorsal approach and prepared for nerve recording (cut distally, desheathed and submerged in mineral oil). Some rats were also prepared for intrathecal injections by performing a laminectomy at C2 and making a small incision in the dura. In those rats, a 2-French silicone catheter (BC-2S, Access Technologies, Skokie, IL, USA) connected to a 50 μl syringe (Hamilton, Gastight no. 1705) was advanced into the intrathecal space until the tip rested over the rostral edge of C4. Rats were converted from isoflurane to urethane anaesthesia (1.6–1.8 g kg−1, i.v.) over a period of 15–25 min and subjected to neuromuscular block with pancuronium bromide (2.5 mg kg−1, i.v.), and an intravenous fluid infusion was initiated (2–4 ml h−1; 11 : 1 lactated Ringer solution with 8.4% sodium bicarbonate). Upon completion of experimental protocols, all animals were killed by urethane overdose.
Nerve recordings
Nerves were prepared for recordings of their activity with bipolar silver electrodes. Their electrical activity was amplified and band-pass filtered (10 000× at 0.1–5 kHz). Raw signals were passed in parallel to a data acquisition system sampling (8 kHz; WinDAQ, DI-720), oscilloscope (Hitachi, VC-6045), audio monitor (AM10, Grass Technologies/Astro-Med Inc., West Warwick, RI, USA) and paynter filter (MA-821RSP, CWE Inc., Ardmore, PA, USA) for rectification and integration (time constant: 50 ms).
Physiological measurements
CO2 was measured (Capnogard, Respironics Novametrix, Wallingford, CT, USA) from the expired gas and used as an indicator of arterial CO2 for the purpose of maintaining isocapnia. These values were confirmed at key times during protocols with blood gas analysis (ABL 500, Radiometer, Copenhagen, Denmark) on 0.2 ml arterial blood samples. Blood pressure was monitored continuously. Data from rats were excluded from analysis if blood pressure dropped more than 30 mmHg over the course of an experiment relative to baseline conditions. Prior to beginning a protocol, the adequacy of anaesthetic depth was assessed by pinching the paw pad, and observing any responses in blood pressure and/or respiratory nerve activity. Supplemental urethane anaesthetic was given as necessary to prevent such responses. Depth of anaesthesia was confirmed at the end of every experiment by the continued absence of any response to pinching of the paw pad.
Pulse oximetry
Four rats underwent apnoea protocols consisting of 15, 25 and 35 s apnoeas for the purpose of estimating the average arterial O2 desaturation. A pulse oximeter (8600V, Nonin Medical Inc., Plymouth, MN, USA) was placed on the hind paw pad of the uncatheterized leg. Each rat was randomly exposed to nine ventilator apnoeas (3 of each duration: 15, 25 and 30 s). These test apnoeas were separated by 5 min intervals.
Intrathecal injections
In six rats, methysergide maleate (Sigma-Aldrich, M137; 250 μg kg−1i.t.) was administered intrathecally. Methysergide was dissolved in artificial cerebrospinal fluid (aCSF) consisting of 120 mm NaCl, 3 mm KCl, 2 mm CaCl, 2 mm MgCl, 23 mm NaHCO3 and 10 mm glucose bubbled with 5% CO2–95% O2 to obtain a pH of 7.4. Ten microlitres of the solution (warmed to 37°C) was injected in 1 μl boluses over a period of 2 min approximately 25–30 min prior to beginning a protocol.
Protocol
Protocols generally lasted ∼2.5 h. Baseline conditions were established approximately 1 h after conversion to urethane anaesthesia while the rat breathed enriched O2 mixtures ( . To establish consistent baseline conditions, the CO2 apnoeic and recruitment thresholds for phrenic nerve activity were determined in each rat. The apnoeic threshold was determined by monitoring
. To establish consistent baseline conditions, the CO2 apnoeic and recruitment thresholds for phrenic nerve activity were determined in each rat. The apnoeic threshold was determined by monitoring 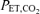 while slowly lowering inspired CO2 or raising ventilation until rhythmic activity was no longer detected in phrenic recordings. The recruitment threshold for rhythmic phrenic activity was then determined while slowly raising inspired CO2 or lowering ventilation. To establish a baseline condition, the
while slowly lowering inspired CO2 or raising ventilation until rhythmic activity was no longer detected in phrenic recordings. The recruitment threshold for rhythmic phrenic activity was then determined while slowly raising inspired CO2 or lowering ventilation. To establish a baseline condition, the 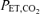 was raised and maintained 2 mmHg above the recruitment threshold. After the amplitude of the nerve signals were steady for at least 15 min (typically 25–40 min post-threshold determination), an arterial blood sample was drawn. The
was raised and maintained 2 mmHg above the recruitment threshold. After the amplitude of the nerve signals were steady for at least 15 min (typically 25–40 min post-threshold determination), an arterial blood sample was drawn. The  at this point was maintained within 1 mmHg throughout the remainder of the protocol (except during apnoeas). Apnoeic and recruitment thresholds were determined again at the end of each protocol to assess any changes due to experimental treatments (see below).
at this point was maintained within 1 mmHg throughout the remainder of the protocol (except during apnoeas). Apnoeic and recruitment thresholds were determined again at the end of each protocol to assess any changes due to experimental treatments (see below).
Five experimental groups were studied: (1) time control (TC, n = 8) rats received neither hypoxia nor apnoeas, but isocapnia was maintained throughout the experiment; (2) acute intermittent hypoxia (AIH; n = 11), consisting of three 5 min episodes of 11% O2 separated by 5 min of baseline conditions (hypoxia and isocapnia were confirmed from an arterial sample drawn during the fourth minute of the first hypoxic exposure); (3) six 25 s ventilator apnoeas (6Apnoea, n = 9) with 5 min intervals; (4) three 25 s apnoeas (3Apnoea, n = 9) with 5 min intervals; and (5) six 25 s apnoeas in rats pretreated with intrathecal methysergide (6Apnoea + methy, n = 6). All apnoeas were induced by stopping the ventilator for 25 s.
In each protocol, arterial blood samples were drawn at 15, 30 and 60 min post-treatment, and adjustments (change in ventilation or inspired CO2) were made as necessary to regulate  within 1 mmHg of the baseline value. After reassessing the apnoeic and recruitment thresholds at the end of each protocol, an additional 5 min hypoxia or 25 s apnoea was imposed, followed by a maximal response to hypercapnia
within 1 mmHg of the baseline value. After reassessing the apnoeic and recruitment thresholds at the end of each protocol, an additional 5 min hypoxia or 25 s apnoea was imposed, followed by a maximal response to hypercapnia  to assure that nerve recordings had been maintained and as an indicator of maximal nerve activity in this experimental preparation. Subsequently, the paw pad was pinched to ensure that adequate anaesthesia had been successfully maintained.
to assure that nerve recordings had been maintained and as an indicator of maximal nerve activity in this experimental preparation. Subsequently, the paw pad was pinched to ensure that adequate anaesthesia had been successfully maintained.
Data analysis
Nerve recordings were analysed using custom software (LabView 6.1, National Instruments, Austin, TX, USA) designed to characterize the integrated phrenic and hypoglossal neurograms. Nerve signals during each apnoea were characterized by the largest 10 s average in peak amplitudes over the duration of the exposure. All other data are averages in 1 min bins sampled immediately before blood samples were drawn to confirm adequate maintenance of isocapnia. Changes are reported as differences from or percentage changes from baseline.
All data are presented as means ±s.e.m. unless stated otherwise. A two-factor ANOVA and repeated measures ANOVA (time) were used to detect significant interactions or effects of treatment and time (SigmaStat 2.03, Systat Software Inc., San Jose, CA, USA). Individual differences were then determined by post hoc Student–Newman–Keuls tests at a significance level of 0.05.
Results
There were no differences between mean age, weight or body temperature among treatment groups. There were no significant within or between rat differences in arterial  in any of the treatment groups at any time point, indicating that strict isocapnia was successfully maintained during experiments. When considering all rats, regardless of treatment group, there were statistically significant decreases in blood pressure over time, similar to many previous studies from our laboratory using this same experimental preparation; the average drop in mean arterial pressure was less than 20 mmHg in all groups with no differences in this decrease between groups.
in any of the treatment groups at any time point, indicating that strict isocapnia was successfully maintained during experiments. When considering all rats, regardless of treatment group, there were statistically significant decreases in blood pressure over time, similar to many previous studies from our laboratory using this same experimental preparation; the average drop in mean arterial pressure was less than 20 mmHg in all groups with no differences in this decrease between groups.
Oximetry
We assessed the magnitude of arterial O2 desaturation to various lengths of ventilator induced apnoeas in order to help draw comparison between this and other means of exposure to hypoxia. In four rats, the average arterial O2 desaturation experienced with 15, 25 and 30 s apnoeas were −6.6 ± 1%, −20.1 ± 1.9% and −34.7 ± 2.5%, respectively, and were all significantly lower than baseline and one another (P < 0.001). The desaturation experienced at progressively longer apnoeas was significantly more severe (P < 0.001). Though not analysed, we observed a slight increase in blood pressure during each apnoea in contrast to a precipitous drop in blood pressure during hypoxic exposures in AIH. This drop prevented accurate measures of O2 saturation (likely through the loss of robust pulsatile blood flow in the hindlimb paw pad). However, since typical rat values of P50 are ∼40 mmHg (Ostojic et al. 2002), and the arterial  during hypoxic episodes was near 40 mmHg, we anticipate a level of desaturation approaching 50%.
during hypoxic episodes was near 40 mmHg, we anticipate a level of desaturation approaching 50%.
Acute nerve responses during hypoxic or apnoeic exposures
We compared acute responses to apnoea and hypoxia to determine if there were any differences in nerve burst amplitude and frequency. Figure 1 shows the typical nerve amplitude and blood pressure response to a single 25 s ventilator apnoea and a single 5 min episode of hypoxia (10% inspired O2). There were no significant differences in peak phrenic amplitudes (last apnoea: 111 ± 13% baseline versus last hypoxia: 127 ± 25% baseline, P = 0.614). The same was true of the hypoglossal nerve amplitude responses (Apnoeas: 230 ± 30% baseline versus Hypoxia: 344 ± 90% baseline, P = 0.380). There were no significant differences in the frequency responses to apnoea versus hypoxia (last apnoea: 21 ± 2 breaths min−1 increase over baseline versus last hypoxia: 17 ± 4 breaths min−1 increase over baseline; P = 0.248). Whereas isocapnic hypoxia, similar to previous studies, decreased mean arterial pressure from 131 ± 6 mmHg to 93 ± 6 mmHg (P < 0.001), there were no such decreases in mean arterial pressure during ventilator apnoeas, most likely due to their limited duration.
Figure 1. Representative traces of blood pressure and integrated phrenic nerve activity.

Top traces show blood pressure responses to 5 min of hypoxia (left) and 25 s of apnoea (right). Note the large ∼40 mmHg drop in pressure during hypoxia. Peak integrated phrenic nerve activity is shown on the bottom traces. Arrows denote the approximate start of an exposure to hypoxia or apnoea. Note that maximum peak amplitudes are similar for hypoxia (left) and 25 s apnoea (right).
LTF in phrenic and hypoglossal burst amplitude
Peak amplitude of the integrated neurograms are well correlated with muscular effort and breathing movements in spontaneously breathing rats (Eldridge, 1971). As such, we used these measures as our indices of increased respiratory motor output. Typical integrated phrenic and hypoglossal neurograms during experimental protocols are shown in Fig. 2. Integrated phrenic burst amplitude significantly increased with time (RMANOVA, P < 0.001). The effect of time depended on protocol (P < 0.001). Therefore the nerve amplitudes expressed as a percentage increase over baseline in time controls differed significantly from AIH, 6Apnoea and 3Apnoea at 60 min (TC = 6 ± 4%versus AIH = 46 ± 11%, P = 0.013; 6Apnoea = 60 ± 7%, P = 0.003; 3Apnoea = 76 ± 21%, P < 0.001) but did not differ from one another at that same 60 min time point (AIH versus 6Apnoea, P = 0.338; AIH versus 3Apnoea, P = 0.102; and 6Apnoea versus 3Apnoea, P = 0.268). Though there was a significant effect of time at 30 min within AIH (29 ± 10%, P = 0.016), 6Apnoea (39 ± 10%, P < 0.001) and 3Apnoea (61 ± 18%, P < 0.001), only the 3Apnoea protocol demonstrated a significant treatment effect at 30 min (P = 0.005). There were no remarkable differences at 15 min within or between protocols. These results are summarized in Fig. 3. Similarly, integrated hypoglossal burst amplitude significantly increased with time (RMANOVA, P < 0.001). The effect of time depended on protocol (P = 0.040). Therefore the nerve amplitudes expressed as a percentage increase over baseline in time controls differed significantly from AIH, 6Apnoea and 3Apnoea at 60 min (TC = 4 ± 8%versus AIH = 46 ± 13%, P = 0.033; 6Apnoea = 63 ± 14%, P = 0.024; 3Apnoea = 60 ± 23%, P = 0.023) but did not differ from one another at that same 60 min time point (AIH versus 6Apnoea, P = 0.650; AIH versus 3Apnoea, P = 0.461; and 6Apnoea versus 3Apnoea, P = 0.914). There was a significant effect of time at 30 min within AIH (34 ± 21%, P = 0.011), 6Apnoea (39 ± 14%, P = 0.026) and 3Apnoea (43 ± 15%, P = 0.027). At 15 min the only remarkable difference was that within AIH a significant effect of time was already evident (36 ± 22%, P = 0.024). These results are summarized in Fig. 4. The facilitation in motor output observed here was qualitatively similar to previous reports in this same experimental preparation (Fuller et al. 2001; Bavis & Mitchell, 2003).
Figure 2. Representative integrated neurogram tracings typical of the overall results.

Sixty minutes post-exposure to AIH, 6 apnoeas or 3 apnoeas, long-term facilitation in integrated nerve burst amplitudes over time is evident compared to time controls. Phrenic traces on left and hypoglossal traces on right are not from the same animals.
Figure 3. Time course of the increase in phrenic nerve burst amplitude.

There is a time-dependent increase in integrated phrenic nerve burst amplitude first evident at 30 min and continuing to 60 min. AIH (n = 8), 6 apnoeas (n = 8) and 3 apnoeas (n = 8) are not different from each other but are all different from time controls (n = 6). All values are expressed as a percentage change from baseline. (*Significant difference from baseline; †difference from equivalent time in control, RMANOVA; P < 0.05.)
Figure 4. Time course of the increase in hypoglossal nerve burst amplitude.

There is a time-dependent increase in integrated hypoglossal nerve burst amplitude first evident at 30 min and continuing to 60 min. AIH (n = 8), 6 apnoeas (n = 8) and 3 apnoeas (n = 8) are not different from each other but are all different from time controls (n = 6). All values are expressed as a percentage change from baseline. (*Significant difference from baseline, †difference from equivalent time in control, Rmanova; P < 0.05.)
Frequency LTF
As in previous reports, frequency LTF in this experimental preparation was small (Baker-Herman et al. 2004). Overall, there was a significant, time-dependent increase in nerve burst frequency within experimental groups following repetitive apnoeas and AIH (Rmanova, P < 0.001). This increase differed significantly among protocols (P = 0.046) with only a slight upward drift of burst frequency in time control rats that was not significant (P = 0.082). There were no statistically significant comparisons (P > 0.05) of time between control and other experimental groups (e.g. repetitive apnoea and AIH versus TC), nor were there significant differences between 6Apnoea, 3Apnoea or AIH protocols. These results are summarized in Fig. 5. Because of its limited magnitude and the observation that no significant changes from control were observed at 60 min, frequency LTF will not be discussed further.
Figure 5. Time course of nerve burst frequency.

There is a time-dependent increase in fictive breathing frequency in AIH (n = 8), 6 apnoeas (n = 8) and 3 apnoeas (n = 8) treated rats but not in time controls (n = 6) when expressed as a change from baseline. This is indicative of a frequency component to LTF. (*Significant difference from baseline; †difference from same time point in time control, Rmanova; P < 0.05.)
Intrathecal methysergide
Since the amount and magnitude of LTF seemed similar after six apnoeas compared to AIH, we next sought to determine if they resulted from similar mechanisms of plasticity. To do so we administered methysergide to six-apnoea treated rats. Methysergide is a broad spectrum serotonin antagonist that has previously been shown to block AIH-induced LTF (Baker-Herman & Mitchell, 2002). Qualitatively, spinal methysergide appeared to increase baseline phrenic burst amplitude without changes in XII burst amplitude. Because of the elevated baseline value, the average burst amplitude response to apnoeas was diminished when expressed as a percentage increase from this new baseline value (last apnoea with methysergide: 44 ± 8% baseline versus without methysergide: 111 ± 13% baseline, P = 0.004). No similar effect was observed in XII burst amplitude during apnoeas (with methysergide: 168 ± 25% baseline versus control: 240 ± 37% baseline, P = 0.124). Intrathecal methysergide blocked phrenic amplitude LTF at 60 min following six apnoeas (1 ± 10% increase over baseline, P = 0.915) while hypoglossal LTF persisted (52 ± 14% increase over baseline, P = 0.029). See Fig. 6 for a summary of this result. As was expected, due to the localized administration of methysergide, the magnitude of hypoglossal LTF at 60 min was not different from that of uninjected rats (intrathecal injection: 52 ± 14% baseline versus no intrathecal injection: 63 ± 14, P = 0.660).
Figure 6. Effect of intrathecal methysergide administration.

Methysergide effectively blocks phrenic (spinal) amplitude LTF (n = 5) while preserving hypoglossal (cranial) amplitude LTF (n = 6). The amount of hypoglossal LTF in drug-administered rats was not different from that observed after AIH, 3 apnoeas or 6 apnoeas (not shown). (*Significant difference from baseline, Rmanova; P < 0.05.)
Changes in apnoeic/recruitment threshold
We assessed the CO2 apnoea and recruitment thresholds before and after each protocol to determine if LTF is associated with changes in these thresholds, or the difference between them. Figure 7 summarizes recruitment thresholds in each protocol (the  at which phrenic activity resumed after hypocapnia-induced quiescence). The threshold decreased from the beginning to the end of all protocols resulting in significant phrenic LTF (3Apnoea: −4.9 ± 1.3 mmHg, P = 0.041; 6Apnoea: −4.2 ± 1.0 mmHg, P = 0.033; AIH: −4.9 ± 0.3 mmHg, P < 0.001) compared to time controls (TC: −1.0 ± 0.5 mmHg). This decrease in recruitment threshold was still evident after blocking phrenic LTF with intrathecal methysergide (6ApnoeaIT: −3.7 ± 0.8 mmHg; P = 0.016). Recruitment thresholds are typically several mmHg higher than the apnoeic CO2 threshold in anaesthetized rats. This difference ([recruitment threshold]–[apnoeic threshold]=[apnoeic-to-recruitment threshold gap]) was estimated by end-tidal CO2 (not arterial) and was larger following protocols leading to significant phrenic LTF. The apnoeic-to-recruitment threshold gap was significantly larger after exposures compared to before (change in the apnoeic to recruitment threshold gap after AIH: 2.1 ± 0.7 mmHg, P = 0.029; 3Apnoea: 2.2 ± 0.6, P = 0.012; and 6Apnoea: 4.2 ± 0.6, P < 0.001; Fig. 8). This difference was not significant in time control experiments (TC: 1.1 ± 0.7 mmHg, P = 0.233; Fig. 8). The increase in the apnoeic to recruitment threshold gap was no longer evident even after blocking phrenic LTF with spinal methysergide (6ApnoeaIT: 2.2 ± 0.8 mmHg, P = 0.057). The post-exposure increase in the apnoeic-to-recruitment threshold gap after 6Apnoea was significantly greater than in any other protocol (P < 0.05). There was no correlation between the apnoea-to-recruitment threshold gap and the magnitude of phrenic or hypoglossal LTF 60 min post-apnoeas or post-AIH (P = 0.756). However, we noted that some differences in thresholds detected using
at which phrenic activity resumed after hypocapnia-induced quiescence). The threshold decreased from the beginning to the end of all protocols resulting in significant phrenic LTF (3Apnoea: −4.9 ± 1.3 mmHg, P = 0.041; 6Apnoea: −4.2 ± 1.0 mmHg, P = 0.033; AIH: −4.9 ± 0.3 mmHg, P < 0.001) compared to time controls (TC: −1.0 ± 0.5 mmHg). This decrease in recruitment threshold was still evident after blocking phrenic LTF with intrathecal methysergide (6ApnoeaIT: −3.7 ± 0.8 mmHg; P = 0.016). Recruitment thresholds are typically several mmHg higher than the apnoeic CO2 threshold in anaesthetized rats. This difference ([recruitment threshold]–[apnoeic threshold]=[apnoeic-to-recruitment threshold gap]) was estimated by end-tidal CO2 (not arterial) and was larger following protocols leading to significant phrenic LTF. The apnoeic-to-recruitment threshold gap was significantly larger after exposures compared to before (change in the apnoeic to recruitment threshold gap after AIH: 2.1 ± 0.7 mmHg, P = 0.029; 3Apnoea: 2.2 ± 0.6, P = 0.012; and 6Apnoea: 4.2 ± 0.6, P < 0.001; Fig. 8). This difference was not significant in time control experiments (TC: 1.1 ± 0.7 mmHg, P = 0.233; Fig. 8). The increase in the apnoeic to recruitment threshold gap was no longer evident even after blocking phrenic LTF with spinal methysergide (6ApnoeaIT: 2.2 ± 0.8 mmHg, P = 0.057). The post-exposure increase in the apnoeic-to-recruitment threshold gap after 6Apnoea was significantly greater than in any other protocol (P < 0.05). There was no correlation between the apnoea-to-recruitment threshold gap and the magnitude of phrenic or hypoglossal LTF 60 min post-apnoeas or post-AIH (P = 0.756). However, we noted that some differences in thresholds detected using  were not discernable when
were not discernable when  was used as an indicator, possibly due to changes in the end-tidal to arterial
was used as an indicator, possibly due to changes in the end-tidal to arterial  difference arising from changes in pulmonary function over time in this preparation.
difference arising from changes in pulmonary function over time in this preparation.
Figure 7. Changes in the arterial CO2 at the recruitment threshold.

All protocols that produce LTF also resulted in a significant decrease in the arterial CO2 at the recruitment threshold compared to time control. (*Significant difference from TC, ANOVA; P < 0.05.)
Figure 8. End-tidal CO2 apnoeic-to-recruitment threshold gap before and after each protocol.

AIH, 3 apnoeas and 6 apnoeas result in a significant increase in the gap between the apnoeic and recruitment threshold as measured by end-tidal CO2. This gap is greater after 6 apnoeas than in any other protocol. (*Significant difference in the threshold gap between before and after; †significant difference between all other groups after, Rmanova; P < 0.05.)
Discussion
This study demonstrates that repetitive apnoeas, with only modest hypoxia and hypercapnia, elicit both phrenic and hypoglossal LTF to an extent nearly identical to previously used protocols of isocapnic acute intermittent hypoxia (AIH; Fuller et al. 2000; Baker et al. 2001; McGuire et al. 2002). Apnoea-induced phrenic LTF requires serotonin receptor activation in the cervical spinal cord, suggesting a common mechanism to AIH-induced phrenic LTF (Baker-Herman & Mitchell, 2002). Both repetitive apnoeas and AIH decrease the CO2-recruitment threshold, and widen the apnoeic to recruitment threshold difference. Thus, in this preparation, phrenic and hypoglossal LTF are relatively insensitive to major differences in the temporal profile, the severity of hypoxaemia (or arterial desaturation) and the simultaneous occurrence of hypoxia and hypercapnia during LTF inducing protocols.
Insensitivity of LTF to duration or severity of intermittent hypoxia
Here we demonstrated that the duration and severity of hypoxaemia are not critical determinants of phrenic or hypoglossal LTF. The 25 s ventilator apnoeas used in this study result in modest hypoxaemia compared to other protocols utilizing 5 min exposures to isocapnic hypoxia. The modest level of hypoxaemia during 25 s apnoeas is in part due to the prevailing hyperoxic inspired gas mixture during baseline conditions (50% O2), as well as to the brevity of the apnoea. For example, during a 25 s apnoea, a desaturation of 20% was observed (expected 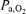 of approximately 70 mmHg) versus an expected desaturation of nearly 50% in rats exposed to 40 mmHg for 5 min. Regardless of these major differences in desaturation, equivalent phrenic and hypoglossal LTF was observed. The insensitivity of LTF to the severity of hypoxaemia is consistent with a previously published meta-analysis of data utilizing this same experimental preparation (Fuller et al. 2000). In this meta-analysis, we concluded that the severity of hypoxia between 28 and 60 mmHg was not a significant predictor of pLTF magnitude (Fuller et al. 2000).
of approximately 70 mmHg) versus an expected desaturation of nearly 50% in rats exposed to 40 mmHg for 5 min. Regardless of these major differences in desaturation, equivalent phrenic and hypoglossal LTF was observed. The insensitivity of LTF to the severity of hypoxaemia is consistent with a previously published meta-analysis of data utilizing this same experimental preparation (Fuller et al. 2000). In this meta-analysis, we concluded that the severity of hypoxia between 28 and 60 mmHg was not a significant predictor of pLTF magnitude (Fuller et al. 2000).
In the meta-analysis of Fuller et al. (2000), one significant predictor of pLTF was the magnitude of the short-term hypoxic phrenic response. Although the apnoea-induced hypoxaemia was considerably less than that encountered during AIH protocols, the magnitude of increase in phrenic nerve activity was similar between the two, possibly due to the coincidence of hypoxia and hypercapnia during an apnoea. Thus, the magnitude of the apnoea versus AIH response may suggest a critical factor in the induction of pLTF, namely the activation of respiratory neurons in the ventral respiratory column that project to, and subsequently activate, raphe, serotonergic neurons; these raphe neurons and their activation are most likely the critical determinant of LTF (Mitchell et al. 2001; Feldman et al. 2003). Thus, in this important respect, repetitive apnoeas and AIH may be similar stimuli to the induction of LTF.
In a recent study, Tadjalli et al. (2007) demonstrate the opposite, that magnitude of facilitation is dependent upon severity of hypoxia. We cannot fully reconcile our results with theirs; they used an immature rat in a perfused working heart brainstem preparation compared to the in vivo adult rats used in our study. Their preparation expresses LTF in frequency, while our preparation expresses predominantly amplitude LTF. This difference may represent a fundamental mechanistic difference in the origins of facilitated motor output: rhythmogenesis versus spinal motor plasticity (Powell et al. 1998). Frequency LTF may be more prominent in the immature respiratory control system or in decerebrate, perfused preparations whereas amplitude LTF may be more prominent in adult or anaesthetized and vagotomized rats. Ventilatory LTF in unanaesthetized, spontaneously breathing rats appears to exhibit both frequency and tidal volume facilitation (Olson et al. 2001; McGuire et al. 2002) after acute intermittent hypoxia, although the tidal volume response is most prominent when hypocapnia is prevented (Olson et al. 2001) or during deep non-REM sleep (Nakamura et al. 2006). While our vagotomized preparation is not a good model to study mechanisms of frequency LTF, it excels as a model to study amplitude.
Intermittent hypercapnia induces a long-lasting depression of respiratory activity lasting many minutes to hours (Bach & Mitchell, 1998; Baker et al. 2001). Thus, one concern had been that repetitive apnoeas may not induce LTF due to hypercapnic depression, offsetting hypoxia-induced LTF during repetitive apnoeas. On the other hand, the effects of intermittent hypercapnia were limited to severe hypercapnia; more moderate hypercapnia did not elicit long-term depression. Thus, one goal of the present study has been to determine if coincident hypoxia/hypercapnia during repetitive apnoeas would mitigate the ability to induce LTF. Since repetitive apnoeas induce respiratory LTF, the evidence suggests that even mild hypoxia prevails over potential inhibitory influences of hypercapnia in this experimental model. Indeed, the repetitive moderate hypercapnia may have actually potentiated the effects of hypoxia by augmenting the phrenic response during apnoeas (see paragraph above).
Although respiratory LTF was insensitive to the duration or severity of hypoxia in this study, the interval between hypoxic episodes must be of major importance since sustained hypoxia is not sufficient to elicit LTF (Baker & Mitchell, 2000) whereas 30 min intervals are too long for LTF expression (Bach et al. 1999). Details concerning the critical range of interhypoxic interval necessary for LTF expression are not known with greater accuracy than this 30 min time-frame.
Although hyperoxic baseline conditions were established in these experiments on repetitive apnoeas, as in most earlier studies using AIH (50% inspired oxygen), Bavis & Mitchell (2003) demonstrated that the hyperoxic background is not in fact necessary for LTF expression. Thus, LTF induction appears to be more sensitive to repetitive oxygen swings versus a return to hyperoxic conditions or the severity of hypoxaemia during hypoxic episodes.
Recent evidence demonstrates that reactive oxygen species (ROS) are necessary for AIH-induced LTF (Wilkerson et al. 2007; MacFarlane & Mitchell, 2008). In one rat pretreatment with a superoxide dismutase mimetic prevented apnoea-induced LTF, suggesting common reliance on ROS formation (authors’ unpublished observation). Repetitive swings in oxygenation have been postulated to generate greater ROS formation than sustained hypoxia (Peng & Prabhakar, 2003; Prabhakar et al. 2007), possibly explaining why swings in oxygenation versus severity of hypoxia are as equally effective at inducing LTF as the longer and more severe hypoxic episodes characteristic of AIH.
Another implication of this study is that large changes in arterial blood pressure during hypoxic episodes are not necessary for respiratory LTF since blood pressure does not change with repetitive apnoeas versus profound hypotension during AIH protocols. This conclusion is consistent with earlier studies demonstrating a disassociation between systemic hypotension and respiratory LTF induced by AIH (Bach & Mitchell, 1996; Neverova et al. 2007), although it extends this finding by demonstrating that LTF is induced in the complete absence of hypotension (and even brief hypertension) during repetitive apnoeas.
Apnoea-induced pLTF requires spinal serotonin receptor activation
AIH-induced pLTF requires cervical spinal serotonin receptor activation for its initiation, but not for its maintenance (Fuller et al. 2001; Baker-Herman & Mitchell, 2002). Similar to these previous studies of AIH-induced pLTF, intrathecal administration of methysergide, a broad spectrum serotonin receptor antagonist, blocked pLTF induced by repetitive apnoeas, suggesting a common mechanism. In this preparation hypoglossal LTF is preserved following intrathecal methysergide, indicating that drug distribution at effective concentrations was restricted to the spinal cord due to the site of delivery and the limited total dose of drug. Although full expression of pLTF following both AIH and repetitive apnoeas requires spinal serotonin receptor activation (Baker-Herman & Mitchell, 2002), this observation does not completely rule out additional mechanisms in the brainstem or periphery. We interpret these data with some caution since baseline phrenic burst amplitude increased slightly following intrathecal methysergide injection, possibly masking LTF. This seems unlikely since a substantial portion (∼50%) of responsiveness to apnoea remained.
Peripheral chemoreceptor plasticity is relatively unlikely since Peng et al. (2003) reported that AIH, applied in a pattern that elicits pLTF, does not elicit long-lasting sensory facilitation in the carotid sinus nerve of normal rats. This conclusion does not imply that carotid body chemoreceptors are not necessary for pLTF, since these receptors are envisioned to activate the raphe serotonergic neurons that initiate pLTF (Fuller et al. 2000; Mitchell et al. 2001; Feldman et al. 2003). In agreement, pLTF is attenuated (although not abolished) by carotid denervation, indicating that peripheral chemoreflex pathway activation is necessary for full expression of pLTF following AIH (Bavis & Mitchell, 2003). On the other hand, since spinal serotonin receptor activation is sufficient to induce pLTF (Lovett-Barr et al. 2006), and intrathecal methysergide completely abolishes LTF in phrenic burst amplitude (Baker-Herman & Mitchell, 2002), mechanisms other than spinal serotonin receptor activation are not necessary to explain AIH-induced pLTF. This conclusion does not rule out additional central mechanisms associated with, but different from, those contributing primarily to pLTF expression.
Apnoea and recruitment CO2 thresholds
There have been no previous studies concerning the effects of AIH or repetitive apnoeas on the apnoeic and/or recruitment CO2 thresholds. Here we report that all protocols producing LTF decrease the apnoeic CO2 threshold (∼4 mmHg), indicating that there is a greater drive to breathe for a given level of CO2. The results also indicate that LTF caused an increase in the gap between the apnoeic and recruitment threshold, another indicator of elevated drive. The neural substrate of this increased respiratory drive is not clear from the present experiments. However, spinal methysergide, which effectively blocks LTF in phrenic burst amplitude, was ineffective at reversing this decrease in the apnoeic CO2 threshold. Thus, the effects of AIH and repetitive apnoeas on the CO2 thresholds are likely to be independent of the spinal, serotonin receptor-dependent component of pLTF. Further, the coincidence of CO2 thresholds for both hypoglossal and phrenic nerve activity under all conditions investigated in this study suggests that changes in the CO2 thresholds are due to mechanisms operating in brainstem respiratory neurons that distribute respiratory activity to both motor pools. Additional evidence for a distinct CNS effect was the increased (although small) frequency LTF, an effect that is unlikely to be attributable to serotonin receptor activation on respiratory motor neurons in this paralysed and vagotomized experimental preparation.
One functional implication of decreased CO2 apnoea/recruitment thresholds is an increase in CO2 reserve (Dempsey, 2005), shifting the system along the isometabolic hyperbola toward an area of lower ‘plant gain’, thus improving ventilatory stability (Khoo, 2000). On the other hand, we are uncertain of the extent of changes in CO2 sensitivity, another determinant of overall breathing stability. Were it increased, one would expect a decrease in breathing stability. We are uncertain what changes in CO2 sensitivity have occurred and thus we cannot report what the effect of repetitive apnoeas or AIH are on the stability of breathing since it depends on the relative contributions of these two potentially opposing effects. However, there are suggestive data for a predominantly stabilizing influence of pLTF since AIH decreases the variability of expiratory time in spontaneously breathing, anaesthetized cats and in normal human subjects (Morris & Gozal, 2004).
The relationship between changes in intercept (threshold) versus sensitivity in establishing pLTF is somewhat unclear in this study since all of pLTF is blocked by intrathecal methysergide, but the change in apnoeic/recruitment threshold is not.
Another factor of potential significance to ventilatory stability, particularly during sleep, is the manifestation of LTF in upper airway motor pools (e.g. hypoglossal). Whereas XII LTF is not expected to contribute to ventilatory stability/instability via feedback loop interactions, it may stabilize the upper airways and thereby contribute to overall ventilatory stability by preventing recurrent upper airway obstructions. The potential impact of phrenic LTF on breathing stability is less clear. On one hand, phrenic LTF may destabilize breathing by increasing chemoreceptor gain (Khoo, 2000). On the other hand, the decrease in apnoeic CO2 threshold may counterbalance this effect. These considerations become even more complicated with a prior history of chronic intermittent hypoxia (versus studies on the naïve rats used in these experiments). Additional experimentation is required to clarify these issues.
Summary
The present study demonstrates that acute clustering of brief asphyxic periods (hypoxia and hypercapnia) results in a lasting increase in upper airway and diaphragm muscle inspiratory nerve motor output. These episodes also result in a significant reduction in the CO2 recruitment threshold and apnoeic threshold. Both phenomena have the potential to alter breathing stability. Furthermore, the former effect requires spinal serotonin receptor activation while the latter effect does not. These results suggest a possible role of apnoeas in shaping the responses to subsequent apnoeas. Given the potential implications of our findings for the expression of unstable breathing, further studies for such a role are warranted.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (HL80209). S. Mahamed was supported by a postdoctoral fellowship from the Canadian Institutes of Health Research.
References
- Aboubakr SE, Taylor A, Ford R, Siddiqi S, Badr MS. Long-term facilitation in obstructive sleep apnea patients during NREM sleep. J Appl Physiol. 2001;91:2751–2757. doi: 10.1152/jappl.2001.91.6.2751. [DOI] [PubMed] [Google Scholar]
- Babcock M, Shkoukani M, Aboubakr SE, Badr MS. Determinants of long-term facilitation in humans during NREM sleep. J Appl Physiol. 2003;94:53–59. doi: 10.1152/japplphysiol.00476.2002. [DOI] [PubMed] [Google Scholar]
- Bach KB, Kinkead R, Mitchell GS. Post-hypoxia frequency decline in rats: sensitivity to repeated hypoxia and α2-adrenoreceptor antagonism. Brain Res. 1999;817:25–33. doi: 10.1016/s0006-8993(98)01181-0. [DOI] [PubMed] [Google Scholar]
- Bach KB, Mitchell GS. Hypoxia-induced long-term facilitation of respiratory activity is serotonin dependent. Respir Physiol. 1996;104:251–260. doi: 10.1016/0034-5687(96)00017-5. [DOI] [PubMed] [Google Scholar]
- Bach KB, Mitchell GS. Hypercapnia-induced long-term depression of respiratory activity requires α2-adrenergic receptors. J Appl Physiol. 1998;84:2099–2105. doi: 10.1152/jappl.1998.84.6.2099. [DOI] [PubMed] [Google Scholar]
- Baker TL, Fuller DD, Zabka AG, Mitchell GS. Respiratory plasticity: differential actions of continuous and episodic hypoxia and hypercapnia. Respir Physiol. 2001;129:25–35. doi: 10.1016/s0034-5687(01)00280-8. [DOI] [PubMed] [Google Scholar]
- Baker TL, Mitchell GS. Episodic but not continuous hypoxia elicits long-term facilitation of phrenic motor output in rats. J Physiol. 2000;529:215–219. doi: 10.1111/j.1469-7793.2000.00215.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Herman TL, Fuller DD, Bavis RW, Zabka AG, Golder FJ, Doperalski NJ, Johnson RA, Watters JJ, Mitchell GS. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nat Neurosci. 2004;7:48–55. doi: 10.1038/nn1166. [DOI] [PubMed] [Google Scholar]
- Baker-Herman TL, Mitchell GS. Phrenic long-term facilitation requires spinal serotonin receptor activation and protein synthesis. J Neurosci. 2002;22:6239–6246. doi: 10.1523/JNEUROSCI.22-14-06239.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bavis RW, Mitchell GS. Intermittent hypoxia induces phrenic long-term facilitation in carotid-denervated rats. J Appl Physiol. 2003;94:399–409. doi: 10.1152/japplphysiol.00374.2002. [DOI] [PubMed] [Google Scholar]
- Dempsey JA. Crossing the apnoeic threshold: causes and consequences. Exp Physiol. 2005;90:13–24. doi: 10.1113/expphysiol.2004.028985. [DOI] [PubMed] [Google Scholar]
- Eldridge FL. Relationship between phrenic nerve activity and ventilation. Am J Physiol. 1971;221:535–543. doi: 10.1152/ajplegacy.1971.221.2.535. [DOI] [PubMed] [Google Scholar]
- Feldman JL, Mitchell GS, Nattie EE. Breathing: rhythmicity, plasticity, chemosensitivity. Annu Rev Neurosci. 2003;26:239–266. doi: 10.1146/annurev.neuro.26.041002.131103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller DD, Bach KB, Baker TL, Kinkead R, Mitchell GS. Long term facilitation of phrenic motor output. Respir Physiol. 2000;121:135–146. doi: 10.1016/s0034-5687(00)00124-9. [DOI] [PubMed] [Google Scholar]
- Fuller DD, Zabka AG, Baker TL, Mitchell GS. Phrenic long-term facilitation requires 5-HT receptor activation during but not following episodic hypoxia. J Appl Physiol. 2001;90:2001–2006. doi: 10.1152/jappl.2001.90.5.2001. discussion 2000. [DOI] [PubMed] [Google Scholar]
- Harris DP, Balasubramaniam A, Badr MS, Mateika JH. Long-term facilitation of ventilation and genioglossus muscle activity is evident in the presence of elevated levels of carbon dioxide in awake humans. Am J Physiol Regul Integr Comp Physiol. 2006;291:R1111–R1119. doi: 10.1152/ajpregu.00896.2005. [DOI] [PubMed] [Google Scholar]
- Hayashi F, Coles SK, Bach KB, Mitchell GS, McCrimmon DR. Time-dependent phrenic nerve responses to carotid afferent activation: intact vs. decerebellate rats. Am J Physiol Regul Integr Comp Physiol. 1993;265:R811–R819. doi: 10.1152/ajpregu.1993.265.4.R811. [DOI] [PubMed] [Google Scholar]
- Janssen PL, Fregosi RF. No evidence for long-term facilitation after episodic hypoxia in spontaneously breathing, anesthetized rats. J Appl Physiol. 2000;89:1345–1351. doi: 10.1152/jappl.2000.89.4.1345. [DOI] [PubMed] [Google Scholar]
- Khoo MC. Determinants of ventilatory instability and variability. Respir Physiol. 2000;122:167–182. doi: 10.1016/s0034-5687(00)00157-2. [DOI] [PubMed] [Google Scholar]
- Lovett-Barr MR, Mitchell GS, Satriotomo I, Johnson SM. Serotonin-induced in vitro long-term facilitation exhibits differential pattern sensitivity in cervical and thoracic inspiratory motor output. Neuroscience. 2006;142:885–892. doi: 10.1016/j.neuroscience.2006.06.036. [DOI] [PubMed] [Google Scholar]
- MacFarlane PM, Mitchell GS. Respiratory long-term facilitation following intermittent hypoxia requires reactive oxygen species formation. Neuroscience. 2008 doi: 10.1016/j.neuroscience.2007.12.003. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuire M, Zhang Y, White DP, Ling L. Effect of hypoxic episode number and severity on ventilatory long-term facilitation in awake rats. J Appl Physiol. 2002;93:2155–2161. doi: 10.1152/japplphysiol.00405.2002. [DOI] [PubMed] [Google Scholar]
- Mahamed S, Mitchell GS. Is there a link between intermittent hypoxia-induced respiratory plasticity and obstructive sleep apnoea? Exp Physiol. 2007;92:27–37. doi: 10.1113/expphysiol.2006.033720. [DOI] [PubMed] [Google Scholar]
- Millhorn DE, Eldridge FL. Role of ventrolateral medulla in regulation of respiratory and cardiovascular systems. J Appl Physiol. 1986;61:1249–1263. doi: 10.1152/jappl.1986.61.4.1249. [DOI] [PubMed] [Google Scholar]
- Millhorn DE, Eldridge FL, Waldrop TG. Prolonged stimulation of respiration by a new central neural mechanism. Respir Physiol. 1980a;41:87–103. doi: 10.1016/0034-5687(80)90025-0. [DOI] [PubMed] [Google Scholar]
- Millhorn DE, Eldridge FL, Waldrop TG. Prolonged stimulation of respiration by endogenous central serotonin. Respir Physiol. 1980b;42:171–188. doi: 10.1016/0034-5687(80)90113-9. [DOI] [PubMed] [Google Scholar]
- Mitchell GS, Powell FL, Hopkins SR, Milsom WK. Time domains of the hypoxic ventilatory response in awake ducks: episodic and continuous hypoxia. Respir Physiol. 2001;124:117–128. doi: 10.1016/s0034-5687(00)00197-3. [DOI] [PubMed] [Google Scholar]
- Morris KF, Gozal D. Persistent respiratory changes following intermittent hypoxic stimulation in cats and human beings. Respir Physiol Neurobiol. 2004;140:1–8. doi: 10.1016/j.resp.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Wenninger JM, Olson EB, Jr, Bisgard GE, Mitchell GS. Ventilatory long-term facilitation following intermittent hypoxia is state-dependent in rats. J Physiol Sci. 2006;56:S75. doi: 10.1152/japplphysiol.90778.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neverova NV, Saywell SA, Nashold LJ, Mitchell GS, Feldman JL. Episodic stimulation of a1-adrenoreceptors induces protein kinase C-dependent persistent changes in motoneuronal excitability. J Neurosci. 2007;27:4435–4442. doi: 10.1523/JNEUROSCI.2803-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson EB, Jr, Bohne CJ, Dwinell MR, Podolsky A, Vidruk EH, Fuller DD, Powell FL, Mitchel GS. Ventilatory long-term facilitation in unanesthetized rats. J Appl Physiol. 2001;91:709–716. doi: 10.1152/jappl.2001.91.2.709. [DOI] [PubMed] [Google Scholar]
- Ostojic H, Cifuentes V, Monge C. Hemoglobin affinity in Andean rodents. Biol Res. 2002;35:27–30. doi: 10.4067/s0716-97602002000100005. [DOI] [PubMed] [Google Scholar]
- Peng YJ, Overholt JL, Kline D, Kumar GK, Prabhakar NR. Induction of sensory long-term facilitation in the carotid body by intermittent hypoxia: implications for recurrent apneas. Proc Natl Acad Sci U S A. 2003;100:10073–10078. doi: 10.1073/pnas.1734109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng YJ, Prabhakar NR. Reactive oxygen species in the plasticity of respiratory behavior elicited by chronic intermittent hypoxia. J Appl Physiol. 2003;94:2342–2349. doi: 10.1152/japplphysiol.00613.2002. [DOI] [PubMed] [Google Scholar]
- Powell FL, Milsom WK, Mitchell GS. Frontiers in Respiration Physiology: Time domains of the hypoxic ventilatory response. Resp Physiol. 1998;112:123–134. doi: 10.1016/s0034-5687(98)00026-7. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR, Kumar GK, Nanduri J, Semenza GL. ROS signaling in systemic and cellular responses to chronic intermittent hypoxia. Antioxid Redox Signal. 2007;9:1397–403. doi: 10.1089/ars.2007.1732. Review. [DOI] [PubMed] [Google Scholar]
- Tadjalli A, Duffin J, Li YM, Hong H, Peever JH. Inspiratory activation is not required for episodic hypoxia-induced respiratory long-term facilitation in postnatal rats. J Physiol. 2007;585:593–606. doi: 10.1113/jphysiol.2007.135798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadjalli A, Duffin J, Peever JH. Clustered obstructive apneas induce long-term facilitation of upper airway motor outlfow in spontaneously breathing rats in-vivo. The Xth Oxford Conference, Lake Louise; 2006. Abstract A56. [Google Scholar]
- Wilkerson JE, Macfarlane PM, Hoffman MS, Mitchell GS. Respiratory plasticity following intermittent hypoxia: roles of protein phosphatases and reactive oxygen species. Biochem Soc Trans. 2007;35:1269–1272. doi: 10.1042/BST0351269. [DOI] [PubMed] [Google Scholar]