

Abstract
West Nile virus (WNV) is a mosquito borne, neurotropic flavivirus that causes a severe central nervous system (CNS) infection in humans and animals. Although commercial vaccines are available for horses, none is currently approved for human use. In this study, we evaluated the efficacy and mechanism of immune protection of two candidate WNV vaccines in mice. A formalin-inactivated WNV vaccine induced higher levels of specific and neutralizing antibodies compared to a DNA plasmid vaccine that produces virus-like particles. Accordingly, partial and almost complete protection against a highly stringent lethal intracranial WNV challenge were observed in mice 60 days after single dose immunization with the DNA plasmid and inactivated virus vaccines, respectively. In mice immunized with a single dose of DNA plasmid or inactivated vaccine, antigen-specific CD8+ T cells were induced and contributed to protective immunity as acquired or genetic deficiencies of CD8+ T cells lowered the survival rates. In contrast, in boosted animals, WNV-specific antibody titers were higher, survival rates after challenge were greater, and an absence of CD8+ T cells did not appreciably affect mortality. Overall, our experiments suggest that in mice, both inactivated WNV and DNA plasmid vaccines are protective after two doses, and the specific contribution of antibody and CD8+ T cells to vaccine immunity against WNV is modulated by the prime-boost strategy.
Keywords: vaccine, immunity, flavivirus, pathogenesis, antibody, CD8+ T cell
1. INTRODUCTION
West Nile virus (WNV) is a mosquito borne, neurotropic, single-stranded RNA Flavivirus, that is closely related to other human pathogens such as Japanese encephalitis, Saint Louis encephalitis, tick-borne encephalitis, yellow fever (YFV) and Dengue viruses. Humans infected with WNV develop a febrile illness that can progress to meningitis or encephalitis or acute flaccid paralysis. The elderly and immunocompromised are at greatest risk for severe encephalitic disease. At present, treatment for WNV infection is supportive and no vaccine is approved for human use [1–3].
Studies in animal models indicate that both innate and adaptive immune responses are required to protect against primary infection by virulent strains of WNV. Type I (α/β) and type II (γ) interferon, γδ T cell activation, an early neutralizing IgM response, and CD4+ and CD8+ T cells all contribute to and orchestrate control and clearance of WNV from peripheral and CNS tissues (reviewed in [4]). The induction of high-titer WNV-specific and neutralizing antibodies after infection has been assumed as a primary mechanism of control during secondary challenge. Indeed, passive transfer of immune serum, monoclonal antibodies or polyclonal antibodies protects rodents from lethal primary WNV infection [5–11].
Since its introduction into United States in 1999, several classes of candidate WNV vaccines have been developed (reviewed in [12]). Although attenuated and inactivated viral vaccines currently protect horses and birds against WNV infection, no vaccine is approved for human use. Formalin-treated WNV protects geese, hamsters, and horses from lethal experimental WNV challenge [13–18], and horses in field trials [16,19,20]. Although inactivated vaccines may be useful for immunocompromised individuals, repeated dosing may be required to induce durable protection. DNA plasmid vaccines also elicit protective immunity in animal models. Administration of plasmid DNA encoding the prM and E proteins prevented both viremia and mortality in horses, mice, and crows [18,21–23]. Plasmid based DNA vaccines induced robust helper T cell immune responses, cytokine production, and humoral immunity [21]. Vaccination with multiple doses of purified recombinant WNV E protein or domain III also elicited neutralizing antibodies and protected mice from WNV challenge [24–26]. However, in some of these studies, mice immunized with recombinant protein succumbed to high dose WNV challenge, suggesting that the efficacy of subunit vaccination may be limited by a failure to stimulate CD8+ T cell immunity. Live-attenuated WNV vaccines derived by serial passage have also been evaluated: vaccination with an attenuated Israeli isolate protected mice and geese from lethal challenge with a virulent WNV stain [27,28]. A plasmid expressing the full-length genome of an attenuated Kunjin strain of WNV was also protective in mice [29]. Chimeric WNV strains have also been developed as candidate vaccines [30,31]. A YFV-WNV chimeric vaccine induced neutralizing antibodies and T cell responses, was completely protective against lethal WNV challenge in monkeys [30], and generated robust immune responses in healthy human volunteers [32].
Many vaccines are believed to protect against infectious agents by inducing humoral and cellular immune responses. A fundamental area of vaccine research is to identify the surrogate markers of humoral and cellular immunity that predict the protective activity of a vaccine in a population that cannot be experimentally challenged. Although most vaccine platforms have been evaluated in terms of production of neutralizing antibodies and induction of T cell responses, their relative importance with respect to protection has not been studied in detail. Herein, we evaluate in detail the development of WNV-specific humoral and CD8+ T cell responses after vaccination with DNA plasmid or formalin-inactivated vaccines. By performing challenge experiments in mice that have been depleted or are genetically deficient in CD8+ T cells, we define the relative roles of antibody and CD8+ T cells in vaccine protection against WNV.
2. METHODS
2.1. Cell culture and virus strains
Baby hamster kidney fibroblast (BHK21) cells were used to determine viral titers as described previously [5]. The lineage I WNV strain (3000.0259) was isolated in New York (2000) and passaged once in C6/36 cells to create a stock virus (2 × 108 PFU/ml) as described previously [5].
2.2. Inactivated virus and DNA plasmid WNV vaccines
A formalin-inactivated whole virion (New York 1999 strain VM2, serial #1666142A) veterinary vaccine, prepared at Fort Dodge Animal Health, was used in the present study. This vaccine is available commercially (West Nile Innovator™) and protects horses when administered as two 1 ml doses, three to four weeks apart [18]. This inactivated virus vaccine is not gradient purified and likely contains additional viral proteins including the non-structural proteins. For experiments in this study, different doses of the inactivated vaccine 100 μl (~106 PFU), 10μl (~105 PFU) or 1μl (~104 PFU) were tested. A DNA plasmid vaccine was prepared at Fort Dodge; it encodes for the premembrane (prM) and the envelope (E) protein of WNV New York 1999 strain resulting in the production of virus-like particles (VLP) [18]: 10 or 100 μg of DNA was used for each immunization per mouse. Both the inactivated and DNA vaccines contained a proprietary oil adjuvant, MetaStim® as well as an excipient. Placebo vaccines contained only the adjuvant and excipient.
2.3. Mice used in experiments
C57BL/6J strain (H-2b) inbred wild type and congenic CD8α-chain−/− (CD8αtm1mak) mice were obtained from Jackson Laboratory (Bar Harbor, Maine). All experiments were performed in the animal facilities with approval and under guidelines of the Washington University Animal Studies Committee.
2.4. Animal immunization and protection studies
Eight week-old mice were immunized with formalin-inactivated virus or DNA plasmid WNV vaccines. In most of the studies with inactivated vaccines, animals received 100 μl of formalin-treated WNV by an intraperitoneal (IP) route. For animals receiving the DNA vaccine, 10 or 100 μg of plasmid was inoculated by an intramuscular (IM) route into the quadriceps muscle through an insulin syringe (100 μl/mouse). A subset of studies was also performed by inoculating varying doses (100 μl, 10 μl and 1 μl) of formalin-inactivated WNV into wild type mice through an IM or IP route. As controls, mice were inoculated with corresponding amounts of placebo vaccine (adjuvant + excipient) by an IM or IP route. For comparison, age-matched mice received 102 PFU of live virulent lineage I WNV (New York 2000: strain 3000.0259 [33]) in 1% heat inactivated fetal bovine serum in Hanks balanced salt solution (HBSS) via an IP route. This infection resulted in a ~60% mortality rate and the remaining survivors were used for analysis of immune responses and subsequent challenge studies. After initial immunization or infection, blood was collected by retro-orbital sampling on days 14 and 60. Serum was recovered after centrifugation and heat inactivation (56ºC for 30 minutes), and stored aliquotted at −80ºC. On day 60, some of the mice were boosted and after 10 days (on day 70), blood was again collected and serum recovered.
In protection studies, mice (on day 60 after primary immunization or day 10 after boosting) were challenged with 101 or 103 PFU of WNV New York by an intracranial (IC) route. After challenge, all mice were monitored for morbidity and mortality for 28 days.
2.5. Detection of WNV-specific antibodies
The levels of WNV specific IgM and IgG were detected by ELISA using purified WNV E protein as described previously [34]. The titer of neutralizing antibody was determined using a previously published plaque reduction neutralization assay protocol with BHK21 cells [34]. Plaques were counted, plotted and the plaque reduction neutralization titer (PRNT50) for 50% inhibition was calculated.
2.6. Passive transfer of immune serum
Serum was collected from the different immunized groups of mice on day 70 (after boost) and heat inactivated. Serum was diluted (to 100 μl) and passively transferred to 5 week-old naïve wild type mice by an IP route. Mice were then infected immediately after with 102 PFU of WNV New York by a subcutaneous (sQ) route. In parallel, the neutralization titer (PRNT50) of the passively transferred pooled serum was measured.
2.7. Depletion of CD8+ T cells
Some of the immunized mice were depleted of CD8+ T cells before challenge with WNV on day 60. As described previously [35,36], 500 μg of rat anti-mouse CD8 or an isotype control antibody (anti-human HLA-DR5) was administered to each mice two days prior to challenge with WNV. This dose of antibody depletes >99% of CD8+ T cells from mice [35].
2.8. Intracellular IFN-γ staining
Intracellular IFN-γ staining was performed on splenocytes at indicated days after infection or vaccination as previously described [37,38]. Briefly, splenocytes from infected or vaccinated mice were stimulated with 1μg/ml of the WNV NS4B peptide (#33, Db-restricted, SSVWNATTAI) or WNV E peptide (#3, Kb-restricted, IALTFLAV) and WNV E peptide (#28, Db-restricted, TVWRNRETL) [38,39] for four hours at 37ºC with the addition of Golgi plug™ (1μl/ml, BD Biosciences), washed and stained with fluorescein isothiocyanate (FITC)-conjugated mAbs to CD8 or a FITC-conjugated isotype control mAb (BD Biosciences) at 4ºC for 30 minutes. After washing and fixing with 1% PFA in PBS, cells were permeabilized with saponin and stained with an allophycocyanin (APC)-conjugated IFN-γ antibody or an isotype control (BD Biosciences) at 4ºC for 30 minutes, washed and analyzed by flow cytometry.
2.9. Data analysis
Data was analyzed using Prism software (Graph Pad, San Diego, CA). Kaplan-Meier survival curves were analyzed by the log-rank test. For antibodies and neutralizing titers, depending on the number of groups, an unpaired T-test or an analysis of variance (ANOVA) was used to determine statistically significant differences.
3. RESULTS
3.1. Humoral immune response induced by candidate vaccines
Although it is straightforward to establish that a candidate vaccine induces specific antibody and T cell responses, it is more challenging to define the relative contribution of each in protection against challenge. For flavivirus vaccines in general, and WNV in particular, such analysis has not been performed in detail. Previous studies have established that a WNV-specific humoral response prevents CNS dissemination during primary infection [5,40]. Moreover, passive administration of immune serum, WNV specific mouse or human monoclonal antibodies (mAbs), or human immune γglobulin completely protects mice from lethal primary WNV infection [5–11,40]. Based on these findings, we expected that the induction of high-titer WNV-specific antibodies by candidate vaccines was essential for protection.
To analyze the humoral response generated by different candidate WNV vaccines, wild type C57BL/6 mice were immunized with a DNA plasmid encoding the prM and E structural genes of WNV New York strain that secretes virus-like particles (VLP) [18], a formalin-inactivated WNV preparation that has been developed as equine vaccine [17], or a placebo vaccine comprised of the adjuvants and excipients used in the DNA and inactivated vaccines. The vaccine-induced humoral responses were further compared to that obtained from the subset of mice (~60%) that survived primary infection with a live virulent WNV New York strain. Serum samples were collected at day 14 and 60 after immunization or primary infection. On day 60, all mice were boosted, and additional serum was collected at day 70 (Fig 1, top). Samples were analyzed for levels of specific antibodies against purified recombinant WNV E protein using a previously validated ELISA [5,40]. With the exception of the placebo-vaccinated mice, all animals showed increased titers of WNV-specific antibodies during the immunization period. Importantly, immunization with a control DNA plasmid that lacked the prM-E genes did not induce
Figure 1.
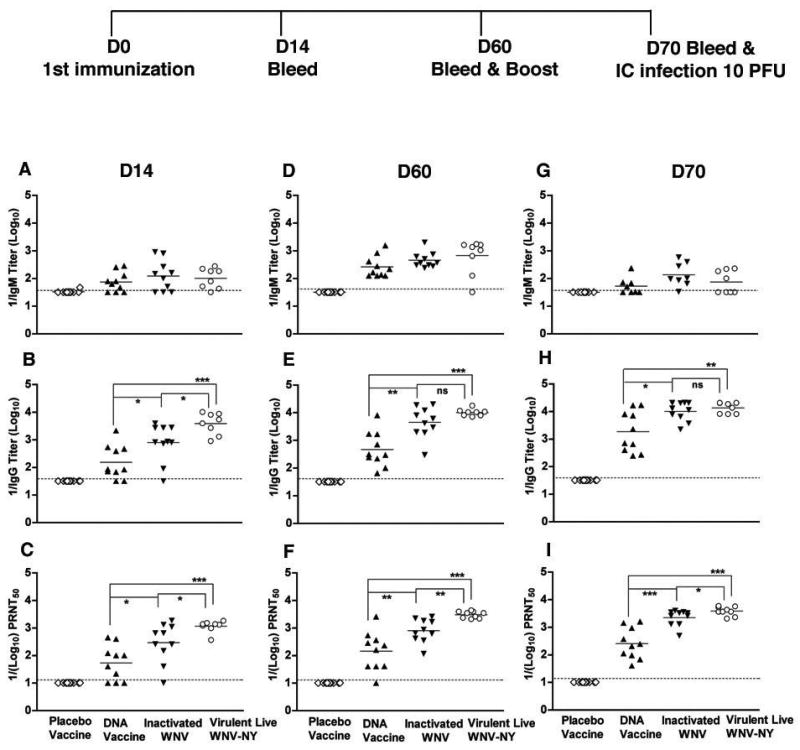
Development of WNV-specific humoral response in wild type mice after immunization at days 60 and 70. Eight week-old wild type C57BL/6 mice were vaccinated with DNA plasmid, formalin-inactivated WNV New York, or placebo. Humoral responses were compared to the subset (60%) of animals surviving primary infection with live virulent WNV New York. Serum was collected on days 14 (A–C) and 60 (D–F). On day 60, mice were boosted and serum was collected 10 days later (G–I). The development of (A, D, and G) IgM or (B, E, and H) IgG anti-WNV E protein was determined by ELISA. C. Neutralizing antibody titers (C, F, and I) were determined by PRNT assay. The data are an average of 10 mice per time point with two independent experiments performed in duplicate. The dotted line represents the limit of sensitivity of the assay. The asterisks denote statistical differences between indicated groups: * P < 0.05, **P < 0.005, ***P < 0.0001. ns indicates no statistical differences were observed between indicated groups.
WNV-specific immune responses in mice ([18] and data not shown). WNV-specific IgM was detected at day 14 in mice receiving the DNA plasmid, formalin inactivated vaccines or the virulent challenge strain. Generally, IgM titers leveled after day 14 and fell by day 70. Among groups, there was no statistical difference in the WNV-specific IgM response at any individual time point (Fig 1A, D, and G, P > 0.4). Although increased levels of WNV-specific IgG were detected at day 14 after immunization in mice receiving the vaccines or virulent WNV strain, significantly higher levels (5 to 25-fold) were detected in mice vaccinated with the formalin-inactivated (P < 0.05) or mice that survived live virulent WNV (P < 0.0001) (Fig 1B) compared to the DNA plasmid vaccine. This higher level of WNV-specific IgG induced by the formalin-inactivated vaccine or in mice that survived virulent WNV infection persisted throughout the time course, and also after boosting (Fig 1E and H). In mice receiving the formalin-inactivated vaccine, the early WNV-specific IgG response was less (~5-fold, P = 0.02) compared to live virulent WNV at day 14. However, by days 60 and 70, statistically significant differences in WNV-specific IgG responses between the inactivated and live virulent WNV were no longer apparent (P > 0.3).
The strong humoral response induced by the formalin-inactivated WNV vaccine could be attributed to the relatively high dose and route (IP) that was used for immunization. To determine how the dose and route of immunization affected the antibody response, several doses (100 μl (~106 PFU), 10 μl (~105 PFU), or 1 μl (~104 PFU)) of the formalin-inactivated WNV vaccine were administered to wild type mice by an IP or IM route. Generally, no major differences in the magnitude of the anti-WNV antibody response were observed at days 14 and 60 when the corresponding dose of the inactivated vaccines was given by the IP and IM routes (Table 1). However, a significant dose effect was observed as IgG titers were ~5 to 90-fold lower at days 14 and 60 (P < 0.01) when smaller amounts of inactivated vaccine was administered by either the IP or IM route.
Table 1.
Anti-WNV antibody production on day 14 and 60 after immunization with different doses and routes of formalin-inactivated WNV in wild type mice
| Vaccination | Day 14 Antibody Titer
|
Day 60 Antibody Titer
|
||
|---|---|---|---|---|
| Dose and Route | IgM | IgG | IgM | IgG |
| Intramuscular route | ||||
| 1 μl (~104 PFU) | < 33 | < 33 | 100 ± 49 | 53 ± 20 |
| 10 μl (~105 PFU) | 55 + 23 | 120 ± 112 | 140 ± 107 | 1082 ± 935 |
| 100 μl (~106 PFU) | 48 ± 22 | 324 ± 165 | 339 ± 229 | 4826 ± 2851 |
| Intraperitoneal route | ||||
| 1 μl (~104 PFU) | < 33 | < 33 | 136 ± 146 | 541 ± 840 |
| 10 μl (~105 PFU) | 55 ± 22 | 66 ± 49 | 147 ± 77 | 412 ± 524 |
| 100 μl (~106 PFU) | 250 ± 321 | 1533 ± 1344 | 569 ± 526 | 7588 ± 7046 |
Eight week-old wild type C57BL/6 mice were vaccinated with different doses (100 μl, 10 μl, or 1μl) of formalin-inactivated WNV vaccines by either an IM or IP route. Serum was collected on days 14 and day 60 after vaccination and WNV-specific IgM or IgG was measured using an ELISA with recombinant, purified WNV E protein. The data are average of 5 to 10 mice per time point and are expressed as geometric mean titers (reciprocal) ± standard deviation. The level of detection of the assay was 1/33.
Previous studies have established that in vitro neutralization activity of anti-WNV antibodies correlates with in vivo protection [7,8,10,11]. To determine how successful the different vaccine candidates were at generating functionally relevant antibodies, we tested serum for neutralizing assay in a plaque reduction assay. Similar to that seen with overall titers, at day 70, the level of neutralizing antibodies was significantly higher (~9 to 15-fold) in mice receiving formalin-inactivated WNV (P < 0.005) or surviving primary infection with live virulent WNV (P < 0.0001) compared to mice vaccinated with the DNA plasmid. In mice surviving infection with live virulent WNV, the neutralizing titers were about 2 to 4 fold higher than (P < 0.05)) those immunized with inactivated WNV (Fig. 1C, 1F and 1I). These experiments suggest that although the DNA plasmid vaccine elicited significant anti-WNV specific IgG and neutralizing responses, they were weaker than that observed with formalin-inactivated or live virulent WNV. To assess whether the decreased humoral response was due to a dosing effect, we also immunized mice with 10-fold higher amounts (100 μg per mouse) of DNA plasmid. Notably, no significant difference in humoral response with a 10 or 100 μg dose of DNA plasmid vaccine was observed (data not shown).
3.2. Challenge experiments with virulent WNV
A successful WNV vaccine should protect against morbidity and mortality when animals are challenged with a virulent strain. Although previous studies [18] suggested that immunization with DNA plasmid or formalin-inactivated vaccine protected mice and horses from lethal WNV challenge, these were performed after iterative boosting. Thus, the survival rate after a limited boosting regimen, which is more likely to be used in humans, was not evaluated.
To determine the efficacy of immunization with DNA plasmid or formalin-inactivated vaccines, mice were initially challenged IP with 102 PFU of WNV after a single immunization on day 60 or a booster immunization on day 70 (Table 2). Control unimmunized mice or those receiving a single dose of placebo vaccine had a 60% survival rate after challenge with a New York strain of WNV. In contrast, all groups of vaccinated mice and those surviving initial virulent strain infection survived IP challenge regardless of boosting. Thus, even though disparities in humoral response were detected among candidate vaccine, the IP challenge model in mice with 102 PFU did not show a difference in phenotypes, as both DNA and inactivated vaccines protected all mice from IP challenge.
Table 2.
Effect of vaccines on IP challenge with WNV in wild type mice
| Vaccination | Challenge Dose WNV PFU | # survived/total | P value | |
|---|---|---|---|---|
| Challenge: Day 60, no boost | ||||
| Mock, no challenge | 0 | 5/5 | ||
| Placebo | 102 | 3/5 | ||
| DNA plasmid | 102 | 5/5 | 0.13 | |
| Inactivated WNV | 102 | 5/5 | 0.13 | |
| Live WNV (survivors) | 102 | 5/5 | 0.13 | |
| Challenge: Day 70, one boost | ||||
| Mock | 102 | 4/5 | ||
| Placebo | 102 | 3/5 | 0.64 | |
| DNA plasmid | 102 | 5/5 | 0.32 | |
| Inactivated WNV | 102 | 5/5 | 0.32 | |
| Live WNV (survivors) | 102 | 5/5 | 0.32 | |
Mice were vaccinated DNA plasmid (10 μg), formalin-inactivated WNV New York (100 μl), or placebo vaccines. Mice were challenge after primary immunization at day 60 or ten days after a boost, at day 70 with 102 PFU of WNV by an IP route and monitored for survival for 28 days. Survival was compared to re-challenge of the subset of animals surviving primary infection with live virulent WNV New York. P values were determined using the log rank test.
To generate a more stringent model, we challenged mice through an intracranial (IC) route. This model was selected because (a) studies with other flaviviruses (e.g., YFV) have used this as a rigorous test of vaccine protection [41]; (b) C57BL/6 adult mice greater than 12 weeks of age have a relatively low mortality rate (20 to 40%) after IP or sQ challenge even with the virulent New York WNV strain (M. Engle and M. Diamond, unpublished results). Thus, very large numbers of mice would be required to discern small statistically significant differences in survival; and (c) IC challenge with a New York strain of WNV is highly lethal in C57BL/6 mice at all ages even at low doses (101–103 PFU) [42]. This model provides a highly stringent, albeit non-physiologic, test of vaccine protection and would at worst, underestimate vaccine efficacy through more conventional challenge routes.
To determine whether the immune responses elicited by the different vaccination strategies could protect against IC infection, immunized mice were challenged with 101 or 103 PFU of WNV New York strain on day 60 (Table 3). As expected, mice that received the placebo vaccine had 100% mortality at either WNV challenge dose. Mice that survived an initial subcutaneous infection with live virulent WNV were almost (90 to 100%, P < 0.0001) completely protected against homologous challenge at either the 101 or 103 PFU dose. In contrast, 30 to 40% (P = 0.1 and P = 0.04, respectively) of mice immunized with the DNA plasmid vaccine and 70 to 80% (P ≤ 0.001) of mice immunized with the formalin-inactivated virus vaccine were protected. To determine whether boosting improved survival rates, mice were re-immunized with DNA plasmid or inactivated virus vaccines on day 60 and then challenged IC with 101 PFU of WNV on day 70. Similar results were observed: inactivated virus preparations provided virtually complete protection whereas the DNA plasmid protected partially (Table 3). These data suggest that the immune response generated with inactivated virus better withstands the stringent challenge of direct intracranial WNV infection.
Table 3.
Effect of vaccines on IC challenge with WNV in wild type mice
| Vaccination | Challenge Dose WNV PFU | # survived/total | P value |
|---|---|---|---|
| Challenge: Day 60, no boost | |||
| Mock, no challenge | 0 | 10/10 | |
| Placebo | 101 | 0/10 | |
| DNA plasmid | 101 | 4/10 | 0.04 |
| Inactivated WNV | 101 | 7/10 | 0.001 |
| Live WNV (survivors) | 101 | 10/10 | < 0.0001 |
| Placebo | 103 | 0/10 | |
| DNA plasmid | 103 | 3/10 | 0.1 |
| Inactivated WNV | 103 | 8/10 | 0.0004 |
| Live WNV (survivors) | 103 | 9/10 | < 0.0001 |
| Challenge: Day 70, one boost | |||
| Mock | 101 | 0/10 | |
| Placebo | 101 | 1/10 | |
| DNA plasmid | 101 | 4/10 | 0.4 |
| Inactivated WNV | 101 | 9/10 | < 0.0001 |
| Live WNV (survivors) | 101 | 10/10 | < 0.0001 |
Wild type mice were vaccinated DNA plasmid (10 μg), formalin-inactivated WNV New York (100 μl), or placebo vaccines. Mice were challenge after primary immunization at day 60 or ten days after a boost, at day 70 with either 101 or 103 PFU of WNV by an IC route and monitored for survival for 28 days. Survival was compared to re-challenge of the subset of animals surviving primary infection with live virulent WNV New York. P values were determined using the log rank test.
To further explore the limits of protection of the formalin-inactivated vaccines, wild type mice were immunized with varying doses (100 μl, 10 μl and 1 μl per mouse) of inactivated vaccines either by IP or IM route. After 60 days, mice were challenged with 101 PFU of WNV New York strain by an IC route. Mice that were immunized with 100 μl of inactivated vaccines via IP or IM routes showed a 70 to 80% survival rate, respectively (Table 4), with no significant difference between the routes of immunization (P = 0.6). In comparison, mice immunized with 100 μl of inactivated vaccines by IM or IP route were completely protected from when IP challenge with 102 PFU WNV (data not shown). Interestingly, although mice immunized with 10 μl of inactivated vaccine by either IM or IP routes were protected completely from challenge with 102 PFU by an IP route (data not shown), only 20 to 30% of mice were protected after the more stringent IC challenge (Table 4). Similarly, when the inactivated vaccine dose was lowered to 1μl per mouse, only 10% of mice were survived IC challenge. Collectively, these experiments show that the protective immune response generated with formalin-inactivated virus is more dependent on dose and less so on the route of vaccination.
Table 4.
Effect of different doses of formalin-inactivated WNV vaccine on IC challenge in wild type mice on day 60
| Vaccination | Challenge Dose WNV PFU | # survived/total | P value |
|---|---|---|---|
| Intramuscular route | |||
| Placebo | 101 | 0/10 | |
| 1 μl (104 PFU) | 101 | 1/10 | 0.1 |
| 10 μl (105 PFU) | 101 | 2/10 | 0.2 |
| 100 μl (106 PFU) | 101 | 8/10 | 0.0003 |
| Intraperitoneal route | |||
| Placebo | 101 | 0/10 | |
| 1 μl (104 PFU) | 101 | 1/10 | 0.1 |
| 10 μl (105 PFU) | 101 | 3/10 | 0.08 |
| 100 μl (106 PFU) | 101 | 7/10 | 0.001 |
Eight week-old wild type mice were vaccinated with different doses of formalin-inactivated WNV vaccines through the indicated routes. Mice were challenged 60 days after immunization with 101 PFU of WNV New York by an IC route and monitored for survival for 28 days. P values were determined using log rank test.
3.3. Passive protection experiments by transfer of immune serum from vaccinated to naïve mice
The antibody analysis and challenge experiments suggested that differences in production of WNV-specific antibody could account for the disparate survival phenotypes. To test directly the relative protective activity of antibodies generated with the different immunization schemes, passive serum transfer experiments were performed. Pooled serum (1 μl diluted to 100 μl) from each group of immunized mice on day 70 was passively transferred to 5 week-old naïve C57BL/6 mice prior to infection with 102 PFU of the New York strain of WNV by the sQ route (Table 5). The dose of transferred serum derived from mice vaccinated with formalin-inactivated WNV vaccine or infected with live virulent WNV had high neutralizing titers (~1/125 and 1/160, respectively) whereas the serum transferred from DNA plasmid vaccinated animals had substantially less neutralizing activity (1/10 titer). Analogous to the challenge results with vaccinated animals, serum from mice surviving live virulent WNV infection or immunized with inactivated WNV protected mice to a greater degree (70 to 80% survival, P < 0.05) than serum from mice vaccinated with placebo or DNA plasmid. When five-fold lower amounts of serum (0.2 μl diluted to 100 μl) were transferred from mice surviving WNV infection or immunized with inactivated virus, ~40% of mice were protected from lethal infection (data not shown). Protection was not observed after transfer of the low dose of serum from placebo or DNA vaccinated mice (data not shown). To reach a similar 70 to 80% protection rate from DNA vaccinated mice, transfer of ten times the amount of serum (neutralizing titer of ~1/135 ) was necessary (Table 5).
Table 5.
Effect of passive transfer of serum from vaccinated or surviving infected mice on sQ challenge of WNV in 5 week-old naïve wild type mice
| Vaccination | Challenge Dose WNV PFU | PRNT50 1/Titer | # Survived/Total | P value |
|---|---|---|---|---|
| A. 1 μl serum transfer | ||||
| No serum | 102 | < 10 | 1/10 | |
| Placebo | 102 | < 10 | 1/10 | 0.8 |
| DNA plasmid | 102 | 10 | 3/10 | 0.2 |
| Inactivated WNV | 102 | 125 | 7/10 | 0.01 |
| Live WNV (survivors) | 102 | 160 | 8/10 | 0.005 |
| B. 10 μl serum transfer | ||||
| Naive serum | 102 | < 10 | 0/10 | |
| DNA plasmid | 102 | 135 | 8/11 | 0.0001 |
Eight week-old wild type mice were immunized with placebo, DNA plasmid (10 μg), or inactivated WNV (100 μl) or infected with live virulent WNV New York and boosted homologously on day 60. Serum was collected on day 70, pooled, heat-inactivated, the PRNT50 was determined (reciprocal titer) and transferred (1 μl or 10 μl diluted to 100 μl) to naïve 5 week-old wild type mice via an IP route. Immediately after, mice were infected with 102 PFU WNV via sQ route and monitored for survival. P values were calculated using the log rank test. The level of detection of the PRNT50 assay was 1/10.
3.4. Vaccine-induced long-term immunity
Our initial analysis focused on the generation of protective antibody responses immediately before or after boosting. Since durable immunity is an important requisite of vaccination, we also analyzed the protective humoral responses at a later time point, three months after boosting. Mice were primed on day 0, boosted on day 60, bled on day 120 and challenged IC with WNV on day 150 (Fig 2, top). Although WNV-specific antibody responses were detected in all groups of mice at day 120, higher levels of IgG (~11-fold, P ≤ 0.0003) and neutralizing antibody (~9 fold, P ≤ 0.0003) were measured in mice receiving inactivated virus vaccine compared to DNA plasmid vaccine (Fig 2A and B). At day 120, no significant difference (P > 0.3) in WNV-specific antibody level or function was observed among mice immunized with inactivated WNV or those surviving infection with live virulent WNV. In challenge experiments at day 150 (Fig 2C), protection correlated with the antibody responses at day 120. Mice that survived infection with live virulent WNV or were immunized with inactivated WNV completely survived infection (P ≤ 0.0001) whereas those receiving placebo vaccine had a 10% survival rate. Mice immunized and boosted with the DNA plasmid vaccine also were significantly protected (P = 0.01 compared to placebo), albeit at a lower rate, as 50% survived IC challenge. Thus, durable responses were generated by all immunization and infection schemes although the DNA plasmid vaccine induced somewhat lower protective antibody titers.
Figure 2.
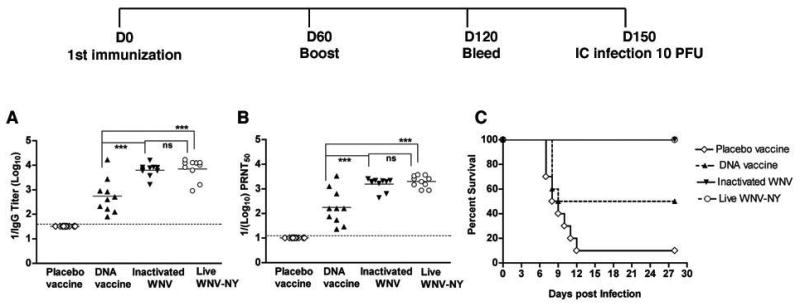
The levels and protective activity of WNV-specific antibody response in wild type mice at 4 to 5 months after vaccination. Eight week-old wild type C57BL/6 mice were vaccinated with DNA plasmid, formalin-inactivated WNV New York, or placebo. Responses were compared to the subset (60%) of animals surviving primary infection with live virulent WNV New York. After boosting on day 60, serum was collected at day 120. The specific titers of (A) IgG were determined by ELISA and the (B) neutralizing titers were determined by PRNT assay. The data are average of 10 mice per time point with two independent experiments performed in duplicate. The dotted line represents the limit of sensitivity of the assay. The asterisks denote statistical differences between indicated groups: ***P < 0.0001. ns indicates no statistical differences were observed between indicated groups. C. Survival analysis of vaccinated or infected mice after WNV challenge. All mice (n = 10 in each group) were challenged with 101 PFU of WNV New York via an IC route on day 150 after initial immunization. Mice were monitored for morbidity and mortality for 28 days and survival curves were constructed using data from two independent experiments. The survival difference after challenge for mice vaccinated with inactivated WNV or surviving initial infection with live virulent WNV was statistically significant (P < 0.0001) compared to placebo.
3.5. Vaccination induces antigen-specific CD8+ T cell responses
CD8+ T cells that are generated during WNV infection protect mice against challenge [38,39,43,44]. Prior immunization studies with live attenuated, DNA plasmid, or protein subunit vaccines against WNV have demonstrated the induction of memory T cell responses [21,25,32], although their role in protection against challenge was not evaluated. To begin to address this question, we first assessed the efficiency of induction of WNV-specific CD8+ T cells after immunzation by analyzing intracellular IFN-γ production ex vivo after antigen-specific re-stimulation with Db and Kb-restricted WNV NS4B and E peptides [37,38]. Splenocytes were harvested from mice on day 14 or 60 after primary immunization or infection or on day 70, 10 days after boosting and the level of CD8+ T cell activation was analyzed by flow cytometry (Fig 3A).
Figure 3.

Detection of IFN-γ producing CD8+ T cells. Eight-week old wild type mice were vaccinated with a placebo vaccine, a DNA plasmid encoding prM and E, inactivated WNV New York, or infected with live virulent WNV New York. Splenocytes were harvested on days 14, 60, and 10 days after boosting on day 70 and stimulated ex vivo with WNV NS4B (peptide 33) and E protein (peptide 3 and 28) specific peptides. The percentage of IFN-γ producing CD8+ T cells was determined by flow cytometry. A. Representative flow cytometry profiles of IFN-γ producing CD8+ T cells on day 60 after vaccination are shown. The percentage of IFN-γ+ CD8+ T cells is indicated in the top right corner. B–D. The percentage of IFN-γ+ CD8+ T after ex vivo restimulation with the indicated peptides on days (B) 14, (C) 60 and (D) 70 after vaccination. The day 70 time point corresponds to 10 days after boosting. Splenocytes from mock-infected mice were used as a negative control. The results are from at least 5 mice per time point, and the bar indicates the mean value.
After primary immunization with the DNA vaccine, inactivated WNV, or infection with live virulent WNV, IFN-γ+ CD8+ T cells were induced after restimulation with peptides from the E protein. The percentage of IFN-γ producing CD8+ T cells was higher (1.4 to 2.0%) on day 14, dropped by day 60 (0.3 to 0.7%), and increased after boosting (1.2 to 1.9%) on day 70 (Fig 3B, C, and D). As a Db-restricted NS4B peptide is immunodominant in C57BL/6 mice during acute WNV infection [39,45], we also assessed the antigen-specific CD8+ T cell response to this peptide. As expected, the placebo or plasmid DNA vaccines, which lack NS4B, showed no CD8+ T cell response to the NS4B peptide (Fig 3D). In contrast, mice receiving inactivated WNV or surviving live virulent WNV infection had NS4B-specific CD8+ T cells that produced IFN-γ on day 14 (1.0 to 2.0%), day 60 (1.0 to 1.8%), and day 70 (0.9 to 1.5%). Collectively, these data suggest that each immunization scheme induced significant numbers of WNV-specific CD8+ T cells. The finding that the inactivated, non-replicating virus vaccine stimulated robust CD8+ T cell responses against NS4B was initially surprising. However, the inactivated virus vaccine preparation is not significantly purified (see Methods) and thus, contains cell substrate debris, including some amount of the viral non-structural proteins. Immunization with the inactivated virus vaccine likely generates CD8+ T cell responses against NS4B through antigen cross-presentation by dendritic cells [46,47].
3.6. The function of CD8+ T cells in the protective vaccine response against WNV
To assess the role of CD8+ T cells in vaccine protection, we immunized mice with DNA plasmid or inactivated WNV or infected with live virulent WNV, and then depleted CD8+ T cells (day 58) immediately prior to IC challenge with 101 PFU of WNV on day 60 (Table 6, top). As observed previously, depletion with an anti-CD8 antibody removed ~99% of circulating CD8+ T cells (data not shown). As expected, after IC challenge with 101 PFU, all mice immunized with placebo vaccine succumbed to infection. In contrast, mice that received live virulent WNV and survived initial infection completely survived re-challenge at 101 PFU regardless of the presence of CD8+ T cells. However, CD8+ T cell depletion appeared to compromise protection by the inactivated virus or DNA plasmid vaccines: an absence of CD8+ T cells increased mortality by ~22 and 50%, respectively, although this difference did not attain statistical significance.
Table 6.
Effect of CD8+ T cell depletion on vaccine protection after IC challenge with WNV in wild type mice.
| Challenge Dose WNV PFU | # Survived/Total | P value | |
|---|---|---|---|
| Placebo vaccine | |||
| Isotype control | 101 | 0/10 | |
| CD8 mAb depletion | 101 | 0/10 | |
| DNA plasmid | |||
| Isotype control | 101 | 8/20 | |
| CD8 mAb depletion | 101 | 4/20 | 0.4 |
| Inactivated WNV | |||
| Isotype control | 101 | 14/15 | |
| CD8 mAb depletion | 101 | 11/15 | 0.2 |
| Live WNV (survivors) | |||
| Isotype control | 101 | 10/10 | |
| CD8 mAb depletion | 101 | 10/10 | 0.7 |
|
| |||
| Placebo vaccine | |||
| Isotype control | 103 | 0/10 | |
| CD8 mAb depletion | 103 | 0/10 | |
| DNA plasmid | |||
| Isotype control | 103 | 6/20 | |
| CD8 mAb depletion | 103 | 2/20 | 0.10 |
| Inactivated WNV | |||
| Isotype control | 103 | 18/20 | |
| CD8 mAb depletion | 103 | 14/20 | 0.08 |
| Live WNV (survivors) | |||
| Isotype control | 103 | 9/10 | |
| CD8 mAb depletion | 103 | 9/10 | 0.6 |
Mice were vaccinated or infected once as described in Table 1 and on day 58 administered 500 μg of rat anti-mouse CD8 or rat anti-human HLA-DR5 (IgG2b isotype control). Two days later mice were challenged IC with 101 or 103 PFU of WNV and monitored for survival. P values were calculated using the log rank test by comparing the isotype control and CD8 mAb-depleted mice for each vaccine.
As a more stringent test of vaccine protection, we repeated the experiments with a 100-fold higher challenge dose, 103 PFU IC. Immunized mice that were depleted of CD8+ T cells generally showed a greater vulnerability to lethal infection (Table 6, bottom). Mice in all groups exhibited a 10 to 25% increase in mortality rate after infection with the higher challenge dose. Again, CD8+ T cell depletion decreased protection in mice immunized with inactivated virus or DNA plasmid vaccines: mortality was increased by ~22 and 67% with a noticeable trend towards statistical significance (P = 0.08 and 0.10, respectively). Taken together, these studies suggest after single dose vaccination with inactivated virus or plasmid DNA, CD8+ T cells have a contributory yet supplemental protective role in the stringent IC challenge model.
To confirm these findings, we repeated immunizations and challenge experiments in CD8−/− mice. Because of the ~90% baseline mortality rate of CD8−/− mice with WNV New York [43] we did not infect mice with live WNV in this cohort. As an initial control, we analyzed the WNV-specific antibody response in CD8−/− mice at days 14 and 60 after vaccination and day 70 after boosting with placebo, DNA plasmid or inactivated virus (Fig 4); previous studies had shown no major defects in anti-WNV antibody response after primary infection in CD8−/− mice [43]. Plasmid DNA and inactivated virus vaccines showed increased levels of WNV-specific antibodies throughout the immunization time course whereas the placebo vaccine did not. Similar to that observed with type mice (see Fig 1), at day 60, the levels of WNV-specific IgG and neutralizing antibodies in CD8−/− mice were higher (4-fold, P ≤ 0.05) in mice immunized with inactivated virus compared to DNA plasmid vaccines. Boosting on day 60 increased the WNV-specific IgG and neutralizing titers 16- and 6-fold, respectively in mice immunized with inactivated virus. Overall, DNA plasmid and inactivated virus vaccines generated similar antibody responses in CD8−/− mice.
Figure 4.
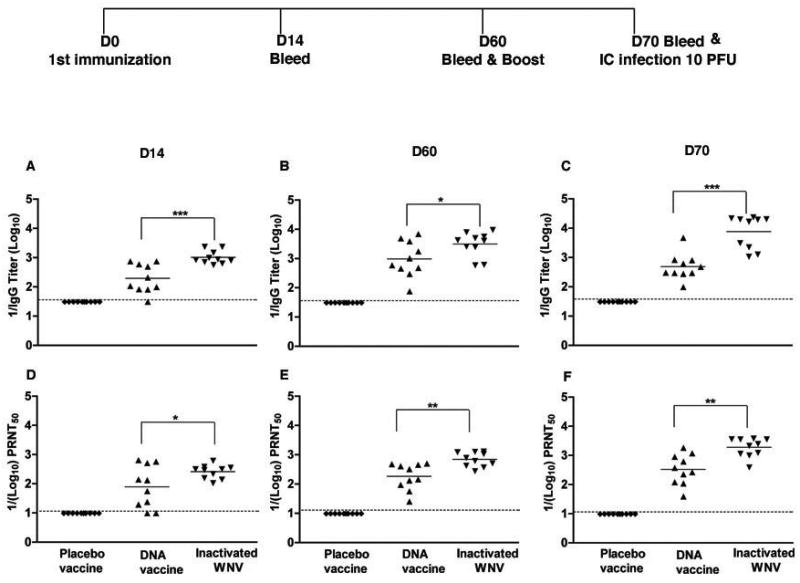
Development of WNV-specific humoral response in CD8−/− mice. Eight week-old CD8−/− mice were vaccinated with DNA plasmid, formalin-inactivated WNV New York, and placebo vaccines. Serum was collected at days 14 (A, D) and 60 (B, E) and after boosting on day 70 (C, F). Serum was analyzed for WNV-specific IgG (A, B and C) and neutralizing titers (D, E and F) as described in Fig 1. The data are average of 10 mice per time point from two independent experiments. The dotted line represents the limit of sensitivity of the assay. The asterisks indicate statistical difference compared to placebo: * P < 0.05, **P < 0.005, ***P < 0.0001.
To further define the contribution of CD8+ T cells to vaccine immunity, CD8−/− mice immunized with placebo, a single dose of DNA plasmid or inactivated virus vaccine were challenged IC on day 60 with 101 PFU of WNV New York strain (Table 7). As anticipated, all mice immunized with placebo vaccines succumbed to infection. However, only 10% and 40% of CD8−/− mice survived challenge after immunization with DNA plasmid or inactivated virus, respectively, values that were significantly lower than the 40% and 70% survival rates seen in age-matched congenic wild type mice (Table 3). In parallel experiments, mice were boosted on day 60 and challenged IC on day 70 with 101 PFU of WNV. Boosting with DNA plasmid or inactivated virus vaccines enhanced the survival rate (DNA, from 10 to 40%; inactivated, from 40 to 80%) after IC challenge. Overall, experiments in CD8−/− mice confirm the depletion results: CD8+ T cells have a role in vaccine protection induced by a single immunization. Boosting, which increases the levels of WNV-specific antibodies, augments protection even in the absence of CD8+ T cells.
Table 7.
Effect of vaccines on IC c hallenge with WNV in CD8−/− mice
| Vaccination | Challenge Dose WNV PFU | # survived/total | P value |
|---|---|---|---|
| Challenge: Day 60, no boost | |||
| Placebo | 101 | 0/10 | |
| DNA plasmid | 101 | 1/10 | 0.2 |
| Inactivated WNV | 101 | 4/10 | < 0.001 |
| Challenge: Day 70, one boost | |||
| Placebo | 101 | 0/10 | |
| DNA plasmid | 101 | 4/10 | 0.005 |
| Inactivated WNV | 101 | 8/10 | < 0.0005 |
CD8−/− mice were vaccinated DNA plasmid, formalin-inactivated WNV New York, and placebo vaccines. Mice were challenge after primary immunization at day 60 or ten days after a boost, at day 70 with either 101 PFU of WNV by an IC route and monitored for survival for 28 days. P values were determined using the log rank test.
4. DISCUSSION
In this study, using both wild type and CD8+ T cell-deficient mice we begin to dissect the mechanism of immune protection by DNA plasmid and inactivated WNV vaccines. Inactivated virus immunization or animals that survived live virulent WNV primary infection induced stronger humoral responses compared to the DNA plasmid vaccine. Although both immunization schemes completely protected mice against IP challenge, differences were observed after challenge by the more stringent IC route: mice that survived initial infection with the virulent WNV strain were most protected, followed by inactivated homologous virus, with the DNA plasmid vaccine providing the least protection. In general, the level of protection correlated with the WNV-specific IgG and neutralizing antibody titers. In mice immunized with a single dose of DNA plasmid or inactivated vaccine, CD8+ T cells also contributed to immunity as acquired or genetic deficiencies lowered the survival rates of immunized mice. In boosted animals, however, WNV antibody titers and protection were greater, and an absence of CD8+ T cells did not affect survival. Our experiments suggest that high doses of inactivated WNV vaccines provide greater protection than the DNA plasmid vaccine, and the relative contribution of antibody and CD8+ T cells to vaccine immunity is modulated by the prime-boost strategy.
Given that the elderly and immunocompromised are targets of severe neuroinvasive WNV infection, and perhaps not suitable candidates for live vaccines, there is a utility for non-infectious WNV vaccine platforms. DNA plasmid vaccines, which propagate within cells, should elicit robust and durable antibody and cellular immunity and carry no risk of infection. DNA plasmid based vaccines have protected animals against challenge with Japanese encephalitis [48,49], dengue [50], Saint Louis encephalitis [51], Murray Valley encephalitis [52], tick-borne encephalitis [53], and WNV [15,18,21]. However, most of these studies did not provide a detailed comparison of potency of immune responses compared to other vaccine modalities. In our study, the WNV-specific protective antibody response generated by the inactivated virus vaccine was significantly higher than that observed with DNA plasmid vaccine. This was not due to inadequate dosing, as a ten-fold higher amount of DNA plasmid did not substantially alter the antibody response. Consistent with our findings, relatively low levels of specific and neutralizing antibodies were generated after immunization of mice and monkeys with DNA plasmid vaccines for dengue and Japanese encephalitis viruses [48,50]. One caveat to our analysis is that the DNA plasmid vaccine, despite having a less robust antibody response, still protected mice against IP challenge with the New York strain of WNV. Similar peripheral protection results with DNA plasmid vaccines were observed in horses with WNV [18] and mice with Japanese encephalitis virus [48].
We observed strong antibody responses and high levels of protection after immunization with the formalin-inactivated WNV vaccine, results that are consistent with published reports on inactivated Dengue virus type-2 vaccines in mice and monkeys [54]. In our single dose immunization scheme, the inactivated virus vaccine induced a WNV-specific antibody response that was greater than the DNA plasmid vaccine yet less than in the subset of animals surviving primary infection with live virulent virus. However, after a single boost, inactivated virus induced a roughly equivalent antibody response and protection against IC challenge compared to mice that had survived live virus infection. An analogously high titer of WNV-specific IgG was achieved after immunization and boost with the same formalin-inactivated virus preparation in baboons [55]. A comparable prime-boost strategy with formalin-inactivated WNV also protected the majority of domestic geese from IC challenge [13,14] and induced neutralizing antibodies in horses [17]. Another caveat of our studies is that lower doses of the formalin-inactivated vaccine induced a less robust antibody response with decreased protection from IC challenge.
Passive transfer of monoclonal or polyclonal antibodies protect mice and hamsters from lethal subcutaneous or intraperitoneal WNV challenge [5–8,10,11,15,40,56]. To address the relative potency of the WNV-specific antibody response generated by the vaccines, we passively transferred serum from immunized to younger naïve mice before lethal peripheral challenge. To our knowledge, this simple experiment has not been performed with WNV vaccinated animals to show directly the utility and potency of the antibody response in vivo. Notably, we observed a direct correlation between absolute ELISA titers, neutralizing titers, and level of protection generated by passive transfer. Ten-fold higher amounts of serum were required from DNA vaccinated mice to achieve a similar level of protection compared to that from mice immunized with inactivated WNV vaccine. In general, more robust WNV-specific antibody responses in vitro directly correlated with increased protection in vivo.
Recent studies have demonstrated the importance of CD8+ T cells in controlling WNV during primary infection [36,43,44]. Although reports have established that CD8+ T cells become activated and transition to memory cells after WNV vaccination [21,57], their role in vaccine protection has remained uncertain. Our study shows that WNV-specific CD8+ T cells are induced by each vaccine platform and their importance for protection depended on the prime-boost strategy. Depletion of CD8+ T cells immediately before IC challenge tended to reduce protection in mice immunized with a single dose of DNA plasmid and inactivated WNV vaccines. These results were supported by experiments with CD8−/− mice. However, an acquired or genetic deficiency of CD8+ T cells had much less effect on protection against IC challenge if mice were boosted, resulting in higher antibody titers and possible sterilizing immunity. Analogous results were observed after three boosts with a DNA vaccine against Japanese encephalitis virus: the humoral immune response was sufficient to protect mice from Japanese encephalitis in the absence of CD8+ T cells [49]. Our results suggests that after boosting the mortality benefit correlates with the magnitude of the neutralizing antibody response, and thus, vaccine optimization should focus on strategies for generating durable long-term protective, antibody responses, perhaps by monitoring the functional activity of the antibodies produced by memory B cells.
5. CONCLUSION
In summary, vaccine induced humoral immunity against WNV E protein plays a critical role in protection against lethal WNV infection. Depending on the vaccine preparation and prime-boost strategy, CD8+ T cells have a supplemental role in protective immunity. Among the non-infectious vaccine candidates, a high dose of the formalin-inactivated WNV provided strong immune responses after a limited prime-boost strategy and significant protection despite a stringent WNV challenge. Nonetheless, further iterative dosing regimens with DNA plasmid vaccines could augment the absolute and neutralizing antibody titer and enhance protection in this and other models.
Acknowledgments
This work was supported by NIH (grants AI061373 (M.S.D.) and U54 AI057160 (Midwest Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research)), and partially by Fort Dodge Animal Health, a division of Wyeth.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Diamond MS. Development of effective therapies against West Nile virus infection. Expert Rev Anti Infect Ther. 2005;3(6):931–944. doi: 10.1586/14787210.3.6.931. [DOI] [PubMed] [Google Scholar]
- 2.Asnis DS, Conetta R, Waldman G, Teixeira AA. The West Nile virus encephalitis outbreak in the United States (1999–2000): from Flushing, New York, to beyond its borders. Ann N Y Acad Sci. 2001;951:161–171. doi: 10.1111/j.1749-6632.2001.tb02694.x. [DOI] [PubMed] [Google Scholar]
- 3.Hubalek Z, Halouzka J. West Nile fever--a reemerging mosquito-borne viral disease in Europe. Emerg Infect Dis. 1999;5(5):643–650. doi: 10.3201/eid0505.990505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Samuel MA, Diamond MS. Pathogenesis of West Nile Virus infection: a balance between virulence, innate and adaptive immunity, and viral evasion. J Virol. 2006;80(19):9349–9360. doi: 10.1128/JVI.01122-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diamond MS, Shrestha B, Marri A, Mahan D, Engle M. B cells and antibody play critical roles in the immediate defense of disseminated infection by West Nile encephalitis virus. J Virol. 2003;77(4):2578–2586. doi: 10.1128/JVI.77.4.2578-2586.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Engle MJ, Diamond MS. Antibody prophylaxis and therapy against West Nile virus infection in wild-type and immunodeficient mice. J Virol. 2003;77(24):12941–12949. doi: 10.1128/JVI.77.24.12941-12949.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oliphant T, Engle M, Nybakken GE, et al. Development of a humanized monoclonal antibody with therapeutic potential against West Nile virus. Nat Med. 2005;11(5):522–530. doi: 10.1038/nm1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Oliphant T, Nybakken GE, Engle M, et al. Antibody recognition and neutralization determinants on domains I and II of West Nile Virus envelope protein. J Virol. 2006;80(24):12149–12159. doi: 10.1128/JVI.01732-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ben-Nathan D, Lustig S, Tam G, Robinzon S, Segal S, Rager-Zisman B. Prophylactic and therapeutic efficacy of human intravenous immunoglobulin in treating West Nile virus infection in mice. J Infect Dis. 2003;188(1):5–12. doi: 10.1086/376870. [DOI] [PubMed] [Google Scholar]
- 10.Throsby M, Geuijen C, Goudsmit J, et al. Isolation and characterization of human monoclonal antibodies from individuals infected with West Nile Virus. J Virol. 2006;80(14):6982–6992. doi: 10.1128/JVI.00551-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gould LH, Sui J, Foellmer H, et al. Protective and therapeutic capacity of human single-chain Fv-Fc fusion proteins against West Nile virus. J Virol. 2005;79(23):14606–14613. doi: 10.1128/JVI.79.23.14606-14613.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chang GJ, Kuno G, Purdy DE, Davis BS. Recent advancement in flavivirus vaccine development. Expert Rev Vaccines. 2004;3(2):199–220. doi: 10.1586/14760584.3.2.199. [DOI] [PubMed] [Google Scholar]
- 13.Malkinson M, Banet C, Khinich Y, Samina I, Pokamunski S, Weisman Y. Use of live and inactivated vaccines in the control of West Nile fever in domestic geese. Ann N Y Acad Sci. 2001;951:255–261. doi: 10.1111/j.1749-6632.2001.tb02701.x. [DOI] [PubMed] [Google Scholar]
- 14.Samina I, Khinich Y, Simanov M, Malkinson M. An inactivated West Nile virus vaccine for domestic geese-efficacy study and a summary of 4 years of field application. Vaccine. 2005;23(41):4955–4958. doi: 10.1016/j.vaccine.2005.03.052. [DOI] [PubMed] [Google Scholar]
- 15.Tesh RB, Siirin M, Guzman H, et al. Persistent West Nile virus infection in the golden hamster: studies on its mechanism and possible implications for other flavivirus infections. J Infect Dis. 2005;192(2):287–295. doi: 10.1086/431153. [DOI] [PubMed] [Google Scholar]
- 16.Gardner IA, Wong SJ, Ferraro GL, et al. Incidence and effects of West Nile virus infection in vaccinated and unvaccinated horses in California. Vet Res. 2007;38(1):109–116. doi: 10.1051/vetres:2006045. [DOI] [PubMed] [Google Scholar]
- 17.Ng T, Hathaway D, Jennings N, Champ D, Chiang YW, Chu HJ. Equine vaccine for West Nile virus. Dev Biol (Basel) 2003;114:221–227. [PubMed] [Google Scholar]
- 18.Davis BS, Chang GJ, Cropp B, et al. West Nile virus recombinant DNA vaccine protects mouse and horse from virus challenge and expresses in vitro a noninfectious recombinant antigen that can be used in enzyme-linked immunosorbent assays. J Virol. 2001;75(9):4040–4047. doi: 10.1128/JVI.75.9.4040-4047.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schuler LA, Khaitsa ML, Dyer NW, Stoltenow CL. Evaluation of an outbreak of West Nile virus infection in horses: 569 cases (2002) J Am Vet Med Assoc. 2004;225(7):1084–1089. doi: 10.2460/javma.2004.225.1084. [DOI] [PubMed] [Google Scholar]
- 20.Siger L, Bowen RA, Karaca K, et al. Assessment of the efficacy of a single dose of a recombinant vaccine against West Nile virus in response to natural challenge with West Nile virus-infected mosquitoes in horses. Am J Vet Res. 2004;65(11):1459–1462. doi: 10.2460/ajvr.2004.65.1459. [DOI] [PubMed] [Google Scholar]
- 21.Yang JS, Kim JJ, Hwang D, et al. Induction of potent Th1-type immune responses from a novel DNA vaccine for West Nile virus New York isolate (WNV-NY1999) J Infect Dis. 2001;184(7):809–816. doi: 10.1086/323395. [DOI] [PubMed] [Google Scholar]
- 22.Chang GJ, Davis BS, Hunt AR, Holmes DA, Kuno G. Flavivirus DNA vaccines: current status and potential. Ann N Y Acad Sci. 2001;951:272–285. [PubMed] [Google Scholar]
- 23.Turell MJ, Bunning M, Ludwig GV, et al. DNA vaccine for West Nile virus infection in fish crows (Corvus ossifragus) Emerg Infect Dis. 2003;9(9):1077–1081. doi: 10.3201/eid0909.030025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang T, Anderson JF, Magnarelli LA, Wong SJ, Koski RA, Fikrig E. Immunization of mice against West Nile virus with recombinant envelope protein. J Immunol. 2001;167(9):5273–5277. doi: 10.4049/jimmunol.167.9.5273. [DOI] [PubMed] [Google Scholar]
- 25.Chu JH, Chiang CC, Ng ML. Immunization of flavivirus West Nile recombinant envelope domain III protein induced specific immune response and protection against West Nile virus infection. J Immunol. 2007;178(5):2699–2705. doi: 10.4049/jimmunol.178.5.2699. [DOI] [PubMed] [Google Scholar]
- 26.Martina BE, Koraka P, van den Doel P, van Amerongen G, Rimmelzwaan GF, Osterhaus AD. Immunization with West Nile virus envelope domain III protects mice against lethal infection with homologous and heterologous virus. Vaccine. 2008;26(2):153–157. doi: 10.1016/j.vaccine.2007.10.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lustig S, Olshevsky U, Ben-Nathan D, et al. A live attenuated West Nile virus strain as a potential veterinary vaccine. Viral Immunol. 2000;13(4):401–410. doi: 10.1089/vim.2000.13.401. [DOI] [PubMed] [Google Scholar]
- 28.Yamshchikov G, Borisevich V, Seregin A, et al. An attenuated West Nile prototype virus is highly immunogenic and protects against the deadly NY99 strain: a candidate for live WN vaccine development. Virology. 2004;330(1):304–312. doi: 10.1016/j.virol.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 29.Hall RA, Nisbet DJ, Pham KB, Pyke AT, Smith GA, Khromykh AA. DNA vaccine coding for the full-length infectious Kunjin virus RNA protects mice against the New York strain of West Nile virus. Proc Natl Acad Sci U S A. 2003;100(18):10460–10464. doi: 10.1073/pnas.1834270100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arroyo J, Miller C, Catalan J, et al. ChimeriVax-West Nile virus live-attenuated vaccine: preclinical evaluation of safety, immunogenicity, and efficacy. J Virol. 2004;78(22):12497–12507. doi: 10.1128/JVI.78.22.12497-12507.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borisevich V, Seregin A, Nistler R, Mutabazi D, Yamshchikov V. Biological properties of chimeric West Nile viruses. Virology. 2006;349(2):371–381. doi: 10.1016/j.virol.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 32.Monath TP, Liu J, Kanesa-Thasan N, et al. A live, attenuated recombinant West Nile virus vaccine. Proc Natl Acad Sci U S A. 2006;103(17):6694–6699. doi: 10.1073/pnas.0601932103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ebel GD, Dupuis AP, 2nd, Ngo K, et al. Partial genetic characterization of West Nile virus strains, New York State, 2000. Emerg Infect Dis. 2001;7(4):650–653. doi: 10.3201/eid0704.010408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shrestha B, Wang T, Samuel MA, et al. Gamma interferon plays a crucial early antiviral role in protection against West Nile virus infection. J Virol. 2006;80(11):5338–5348. doi: 10.1128/JVI.00274-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McClellan JS, Tibbetts SA, Gangappa S, Brett KA, Virgin HWt. Critical role of CD4 T cells in an antibody-independent mechanism of vaccination against gammaherpesvirus latency. J Virol. 2004;78(13):6836–6845. doi: 10.1128/JVI.78.13.6836-6845.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shrestha B, Samuel MA, Diamond MS. CD8+ T cells require perforin to clear West Nile virus from infected neurons. J Virol. 2006;80(1):119–129. doi: 10.1128/JVI.80.1.119-129.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sitati EM, Diamond MS. CD4+ T-cell responses are required for clearance of West Nile virus from the central nervous system. J Virol. 2006;80(24):12060–12069. doi: 10.1128/JVI.01650-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Purtha WE, Myers N, Mitaksov V, et al. Antigen-specific cytotoxic T lymphocytes protect against lethal West Nile virus encephalitis. Eur J Immunol. 2007;37(7):1845–1854. doi: 10.1002/eji.200737192. [DOI] [PubMed] [Google Scholar]
- 39.Brien JD, Uhrlaub JL, Nikolich-Zugich J. Protective capacity and epitope specificity of CD8(+) T cells responding to lethal West Nile virus infection. Eur J Immunol. 2007;37(7):1855–1863. doi: 10.1002/eji.200737196. [DOI] [PubMed] [Google Scholar]
- 40.Diamond MS, Sitati EM, Friend LD, Higgs S, Shrestha B, Engle M. A critical role for induced IgM in the protection against West Nile virus infection. J Exp Med. 2003;198(12):1853–1862. doi: 10.1084/jem.20031223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pincus S, Mason PW, Konishi E, et al. Recombinant vaccinia virus producing the prM and E proteins of yellow fever virus protects mice from lethal yellow fever encephalitis. Virology. 1992;187(1):290–297. doi: 10.1016/0042-6822(92)90317-i. [DOI] [PubMed] [Google Scholar]
- 42.Samuel MA, Diamond MS. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J Virol. 2005;79(21):13350–13361. doi: 10.1128/JVI.79.21.13350-13361.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shrestha B, Diamond MS. Role of CD8+ T cells in control of West Nile virus infection. J Virol. 2004;78(15):8312–8321. doi: 10.1128/JVI.78.15.8312-8321.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Y, Lobigs M, Lee E, Mullbacher A. CD8+ T cells mediate recovery and immunopathology in West Nile virus encephalitis. J Virol. 2003;77(24):13323–13334. doi: 10.1128/JVI.77.24.13323-13334.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Purtha WE, Myers N, Mitaksov V, et al. Antigen-specific cytotoxic T lymphocytes protect against lethal West Nile virus encephalitis. Eur J Immunol. 2007 doi: 10.1002/eji.200737192. In press. [DOI] [PubMed] [Google Scholar]
- 46.Steinman RM, Pope M. Exploiting dendritic cells to improve vaccine efficacy. J Clin Invest. 2002;109(12):1519–1526. doi: 10.1172/JCI15962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sigal LJ, Crotty S, Andino R, Rock KL. Cytotoxic T-cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature. 1999;398(6722):77–80. doi: 10.1038/18038. [DOI] [PubMed] [Google Scholar]
- 48.Chang GJ, Hunt AR, Davis B. A single intramuscular injection of recombinant plasmid DNA induces protective immunity and prevents Japanese encephalitis in mice. J Virol. 2000;74(9):4244–4252. doi: 10.1128/jvi.74.9.4244-4252.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pan CH, Chen HW, Huang HW, Tao MH. Protective mechanisms induced by a Japanese encephalitis virus DNA vaccine: requirement for antibody but not CD8(+) cytotoxic T-cell responses. J Virol. 2001;75(23):11457–11463. doi: 10.1128/JVI.75.23.11457-11463.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simmons M, Porter KR, Hayes CG, Vaughn DW, Putnak R. Characterization of antibody responses to combinations of a dengue virus type 2 DNA vaccine and two dengue virus type 2 protein vaccines in rhesus macaques. J Virol. 2006;80(19):9577–9585. doi: 10.1128/JVI.00284-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Phillpotts RJ, Venugopal K, Brooks T. Immunisation with DNA polynucleotides protects mice against lethal challenge with St. Louis encephalitis virus. Arch Virol. 1996;141(3–4):743–749. doi: 10.1007/BF01718332. [DOI] [PubMed] [Google Scholar]
- 52.Colombage G, Hall R, Pavy M, Lobigs M. DNA-based and alphavirus-vectored immunisation with prM and E proteins elicits long-lived and protective immunity against the flavivirus, Murray Valley encephalitis virus. Virology. 1998;250(1):151–163. doi: 10.1006/viro.1998.9357. [DOI] [PubMed] [Google Scholar]
- 53.Schmaljohn C, Vanderzanden L, Bray M, et al. Naked DNA vaccines expressing the prM and E genes of Russian spring summer encephalitis virus and Central European encephalitis virus protect mice from homologous and heterologous challenge. J Virol. 1997;71(12):9563–9569. doi: 10.1128/jvi.71.12.9563-9569.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Putnak R, Barvir DA, Burrous JM, et al. Development of a purified, inactivated, dengue-2 virus vaccine prototype in Vero cells: immunogenicity and protection in mice and rhesus monkeys. J Infect Dis. 1996;174(6):1176–1184. doi: 10.1093/infdis/174.6.1176. [DOI] [PubMed] [Google Scholar]
- 55.Wolf RF, Papin JF, Hines-Boykin R, et al. Baboon model for West Nile virus infection and vaccine evaluation. Virology. 2006;355(1):44–51. doi: 10.1016/j.virol.2006.06.033. [DOI] [PubMed] [Google Scholar]
- 56.Morrey JD, Siddharthan V, Olsen AL, et al. Humanized monoclonal antibody against West Nile virus envelope protein administered after neuronal infection protects against lethal encephalitis in hamsters. J Infect Dis. 2006;194(9):1300–1308. doi: 10.1086/508293. [DOI] [PubMed] [Google Scholar]
- 57.Monath TP. Prospects for development of a vaccine against the West Nile virus. Ann N Y Acad Sci. 2001;951:1–12. doi: 10.1111/j.1749-6632.2001.tb02680.x. [DOI] [PubMed] [Google Scholar]