

Summary
TRPC3, 6 and 7 channels constitute a subgroup of non-selective, calcium-permeable cation channels within the TRP superfamily that are activated by products of phospholipase C-mediated breakdown of phosphatidylinositol 4,5-bisphosphate (PIP2). A number of ion channels, including other members of the TRP superfamily, are regulated directly by PIP2. However, there is little information on the regulation of the TRPC channel subfamily by PIP2. Pretreatment of TRPC7-expressing cells with a drug that blocks the synthesis of polyphosphoinositides inhibited the ability of the synthetic diacylglycerol, oleyl-acetyl glycerol, to activate TRPC7. In excised patches, TRPC7 channels were robustly activated by application of PIP2 or ATP, but not by inositol 1,4,5-trisphosphate. Similar results were obtained with TRPC6 and TRPC3, although the effects of PIP2 were somewhat less and with TRPC3 there was no significant effect of ATP. In the cell attached configuration, TRPC7 channels could be activated by the synthetic diacylglycerol analog, oleyl-acetyl glycerol. However, this lipid mediator did not activate TRPC7 channels in excised patches. In addition, channel activation by PIP2 in excised patches was significantly greater than that observed with oleyl-acetyl glycerol in the cell attached configuration. These findings reveal complex regulation of TRPC channels by lipid mediators. The results also reveal for the first time direct activation by PIP2 of members of the TRPC ion channel subfamily.
The primary mode of activation of cannonical transient receptor potential (TRPC) channels is believed to be through phospholipase C (PLC) [1–4]. In the case of a subgroup of TRPC channels, specifically TRPC3, TRPC6 and TRPC7, the signal for their activation is thought to be the diacylglycerol formed for phospholipase C-induced degradation of the plasma membrane phospholipid, phosphatidylinositol 4,5-bisphosphate (PIP2) [2;4–6]. A growing number of ion channels have been shown to be regulated in a more direct manner by PIP2 [7;8]. In the vast majority of cases, this regulation involves either activation, or prevention of rundown. A characteristic signature for PIP2 regulation, as well as a putative physiological function for this mode of regulation, is the ability of agonists that activate PLC to inhibit channel activity [8]; however, PLC does not inhibit TRPC channels, rather it causes their activation. We thought it of interest therefore to investigate the role, if any, of PIP2 in regulation of TRPC7, a representative member of this subgroup. Our results indicate that TRPC7, and to some degree also TRPC3 and TRPC6 channels in excised patches are potently activated by PIP2. This is to our knowledge the first direct demonstration of direct PIP2 regulation of this important subfamily of Ca2+-permeable channels.
Methods
Experiments were carried out with HEK293 cells stably expressing either TRPC3, TRPC6 or TRPC7 [9–11]. Ca2+ measurements were carried out with TRPC7-expressing cells loaded with Fura-2-AM and a cell imaging system (Intracellular Imaging, Inc., Cincinnati, OH) as described previously [12]. For Fura-2 measurements, the ratio obtained by dividing the emitted fluorescence from excitation at 340 nm by the emitted fluorescence measured after excitation at 380 nm is reported as in indicator of changes in intracellular Ca2+ concentration. Briefly, cells were loaded with 1 µM of Fura-2/AM for 30 minutes then washed and bathed in Hepes-buffered saline solution (HBSS; in mM, 140 NaCl, 4.7 KCl, 10 CsCl, 2 CaCl2, 1.13 MgCl2, 10 glucose, and 10 Hepes, pH 7.4) for at least 10 min before Ca2+ measurements are made.
Macroscopic membrane ion currents in TRPC7-expressing cells were recorded using the patch-clamp technique in its whole-cell configuration. The currents were acquired using pCLAMP-9.2 (Axon Instruments) and analyzed offline using Origin 6 (Microcal) software. The extracellular solution (osmolarity 310 mosmol/l) contained (in mM): 145 NaCl, 5 KCl, 10 HEPES, 1 MgCl2, 2 CaCl2, pH, 7.3 (adjusted with NaOH). The intracellular pipette solution (osmolarity 290 mosmol/l) contained (in mM): 145 Cs-methanesulfonate, 10 BAPTA, 10 HEPES, 1 MgCl2, and 2.2 CaCl2 (70 nM free Ca2+ determined with Winmaxc 2.05 software, http://www.stanford.edu/~cpatton/maxc.html [13]), pH 7.2 (adjusted with CsOH). Patch pipettes were fabricated from borosilicate glass capillaries (WPI). The resistance of the pipettes varied between 3 and 5 MΩ. Necessary supplements were added directly to the respective solutions, in concentrations that would not significantly change the osmolarity. Changes in the external solutions were carried out using a multibarrel puffing micropipette with common outflow that was positioned in close proximity to the cell under investigation. During the experiment, the cell was continuously superfused with the solution via a puffing pipette to reduce possible artefacts related to the switch from static to moving solution and vice versa.
For the cell attached and inside-out patch-clamp experiments with TRPC3, TRPC6 and TRPC7-expressing cells we used the following intracellular pipette and bath solutions, respectively in mM: 140 KCl, 10 HEPES, 1 CaCl2, 1 MgCl2; and 120 KCl, 20 HEPES, 10 EGTA, 4.1 CaCl2, 1 MgCl2 (80 nM free Ca2+, determined as described above). Both solutions were adjusted to pH 7.3 with KOH. Signals were low-pass filtered at 2 kHz, digitized at 20 kHz and then analyzed using the Clampfit 9.2 software (Axon Instruments).
In experiments with cell-attached or excised inside-out patches, control data were collected at +60 mV (command potential of −60 mV) after sealing or after excision. PIP2, diacylglycerols or MgATP were then added and data collection continued. Therefore statistical evaluations of effects were based on paired t-tests. Detailed methods for collection and analysis of TRPC7 single channel data were described previously [14].
All reagents were purchased from Sigma (St. Louis, MO), except oleyl acetyl glycerol (OAG) (Calbiochem, San Diego, CA) and PIP2 (Echelon Biosciences Inc., Salt Lake City, UT).
Results
As previously demonstrated [15;16], in HEK293 cells stably expressing TRPC7, application of the membrane permeant diacylglycerol analog, oleyl-acetyl glycerol (OAG) activated Ca2+ entry (Figure 1A). Pretreatment of the cells with the phosphoinositide lipid kinase inhibitor, LY294002 [17;18] caused near total inhibition of the [Ca2+]i signal (Figure 1A). This inhibition does not likely result from a direct action on TRPC7 channels, because addition of LY294002 after the Ca2+ signal had developed had little or no effect (Figure 1B); however, removal of OAG from the bathing solution resulted in a decline in Ca2+ entry (data not shown). The increased Ca2+ entry by OAG in TRPC7 expressing cells results from activation of a non-selective TRPC7 cation current [15;16] (Figure 2A, B). This current was also strongly inhibited by pretreatment with LY294002 (Figure 2A, B). This result is consistent with findings for other ion channels known to be activated by, or to require PIP2 [8]. Application of LY294002 at lower concentrations, which would specifically inhibit PI 3-kinase, had no effect on OAG-activated Ca2+ signals (data not shown). However, addition of a water soluble PIP2 to the patch pipette solution did not significantly activate TRPC7 currents, nor did it potentiate the effects of OAG (Figure 2C). This is perhaps not surprising, as the micellar nature of dispersed phospholipids may make their diffusion into cells through patch pipettes difficult. In addition, if PIP2 is a necessary co-factor rather than an activator, there may be sufficient PIP2 present in intact cells such that additional effects of added lipid are not observed. Thus, we next examined the effects of PIP2 more directly by application of the lipid to excised patches containing TRPC7 channels. As shown in Figure 3, excised patches from TRPC7-expressing cells show limited spontaneous activity with low open probability (NPo = 0.011±0.003). Application of 10 µM synthetic di-octanyl PIP2 resulted in rapid and reversible channel activation (NPo = 0.487±0.232). The size of these single channel openings was similar to that previously reported by us in cell attached configuration (69.8 ± 0.4 pS [14;16]). No such channels were ever observed in non-transfected cells (5 patches, and 5 patches in our previous study [16]). To obtain information on the kinetics of single channel behavior, we analyzed the dwell time distributions for apparent open and closed times. We analyzed the kinetics of single channel opinings in excised patches in the presence and absence of PIP2 and in cell-attched patches activated by OAG. In cell attached mode without OAG, openings were too infrequent for reliable analysis. The data were well fitted by the sum of two and three (for cell attached) or four (excised patches) closed exponential components, corresponding to open and closed states, respectively. Table 1 summarizes the time constants and corresponding relative abundance for these states. Open time distributions showed a major, highly populated component with a time constant of ~0.25 ms which is somewhat shorter than we observed for overexpressed TRPC7 in DT40 lymphocytes [14]. Open time distributions did not differ significantly among the three groups, indicating that the basic kinetic behavior of open TRPC7 channels is not altered by excision, or by interaction with OAG or PIP2. Closed time distributions showed a range of components. In excised patches, the increase in NPo due to PIP2 was associated with a decrease in the time constants of the three slower components.
Figure 1. The inositol lipid kinase inhibitor, LY294002, inhibits Ca2+ entry responses of TRPC7-expressing HEK293 cells to OAG.
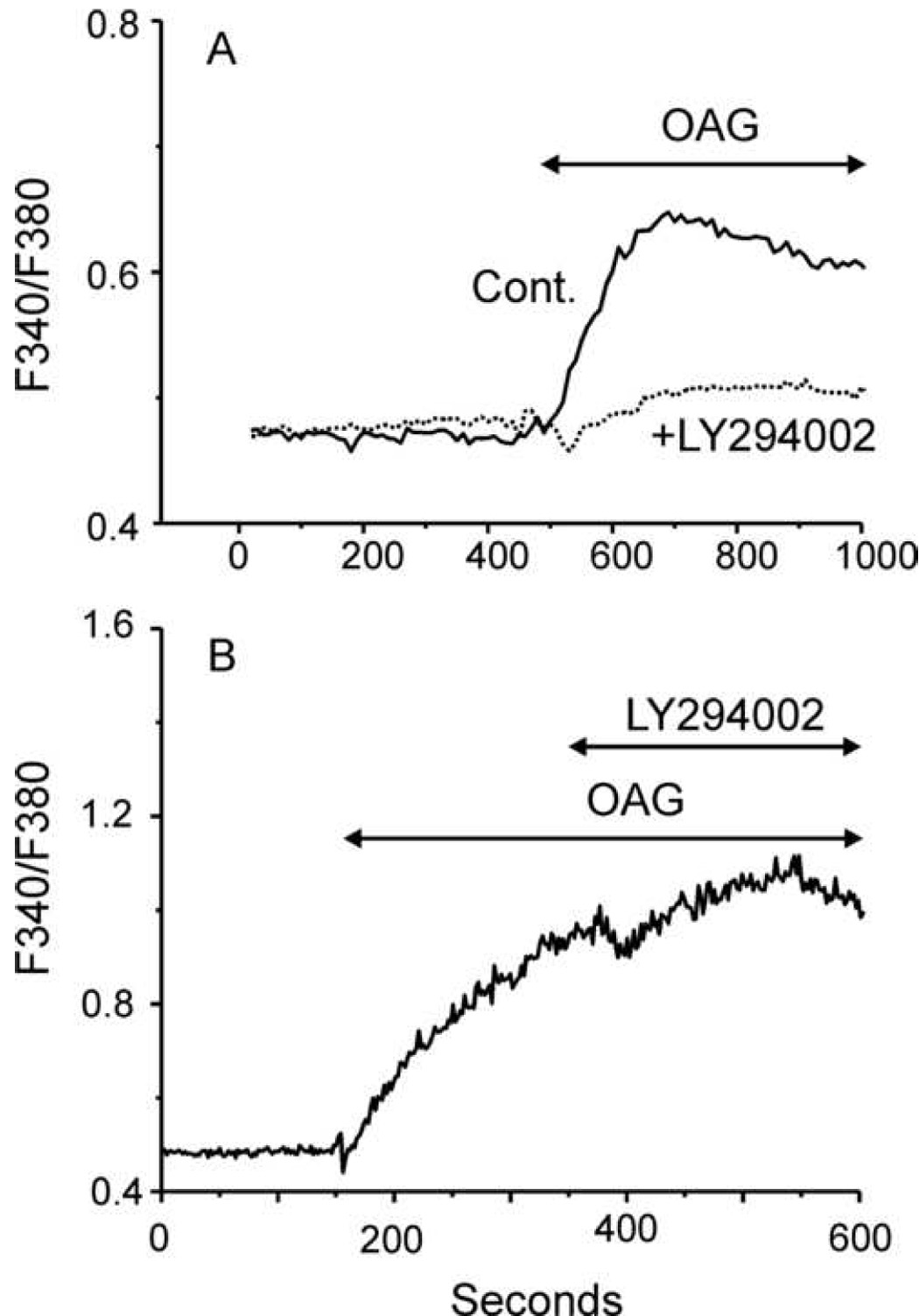
A: Time course of average fluorescence ratio in TRPC7-HEK cells loaded with Fura-2 and pretreated (LY, n=55, 2 independent experiments) or not (Con, n=74, 2 independent experiments) with 100 µM LY294002. LY294002 was added 5 minutes prior to the beginning of data collection and was present throughout. Cells were exposed to 30 µM OAG as indicated by the horizontal bar. Results are presented as mean ± SEM, and are representative of a total of 6 independent experiments. B: The protocol was similar to that for A, except LY294002 was added after activation of Ca2+ entry by OAG, as indicated (n=20, from one experiment, representative of four).
Figure 2. Effects of LY294002 and PIP2 on whole-cell TRPC7 currents.
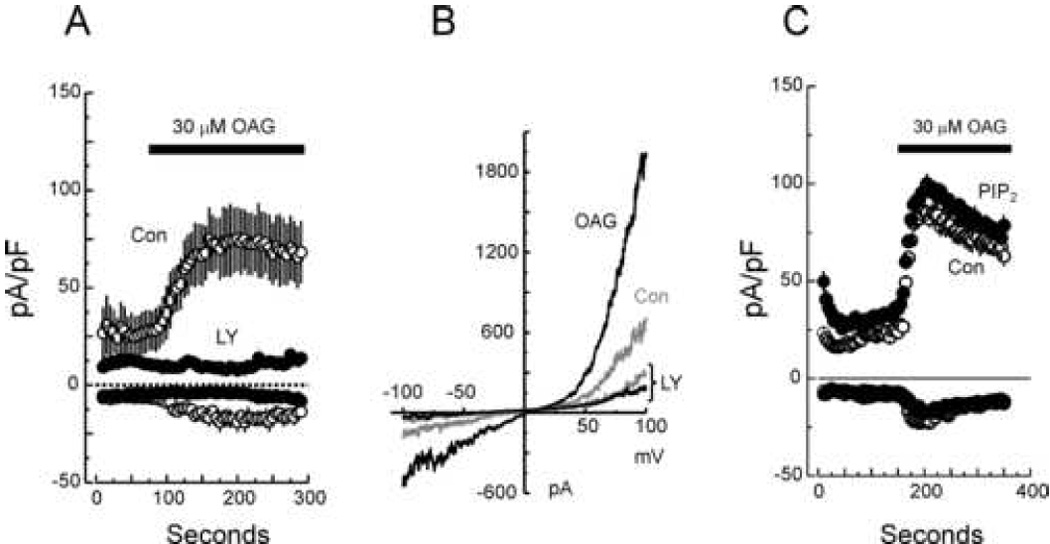
A: Average time courses (mean ± SEM) of whole-cell current development at −100 and +100 mV in TRPC7-expressing HEK293 cells pretreated (LY, solid circle, n=5) or not (Con, open circle, n=5) with 100 µM LY294002 for 5 minutes before break-in, and then exposed to 30 µM OAG as indicated by the horizontal bar. As in A, LY294002 was maintained in the bath during the whole course of the recording. B: Current-voltage relationships for OAG-activated TRPC7 currents. The IV relationships are representative of TRPC7-HEK cells pretreated (LY) or not with 100 µM LY294002 and then sequentially exposed to a control (gray traces) or a 30 µM OAG-containing solution (black traces). C: Average time courses (mean ± SEM) of whole-cell current development at −100 and +100 mV in TRPC7-expressing HEK293 cells activated by 30 µM OAG. Shown are average current from Control experiments (Con, open circles, n=5), and from experiments in which the pipette solution contained 100µM PIP2 (PIP2, solid circles, n=5).
Figure 3. PIP2 activates TRPC7 channels in inside-out patches.
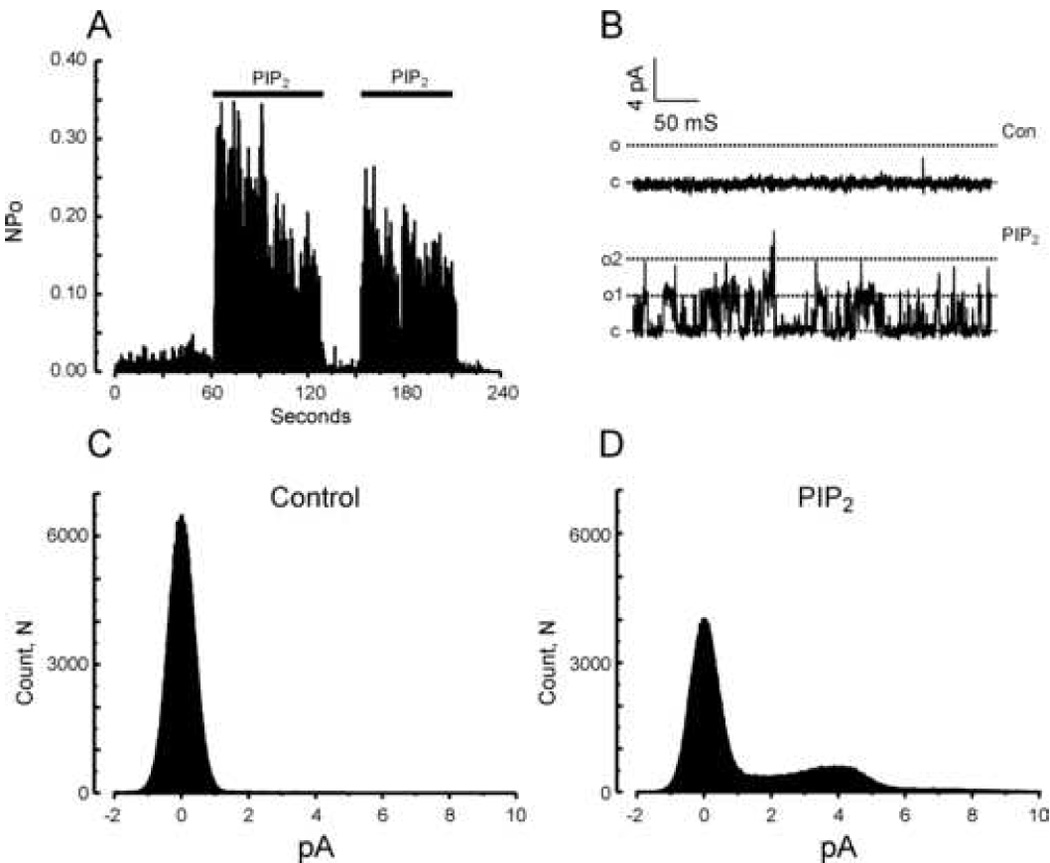
A: Histogram summarizing TRPC7 channel open probability over time at +60 mV during repeated applications of a 10 µM PIP2-containing solution. The figure is representative of results obtained in 12 independent inside-out patches. B: Examples of currents recorded in an inside-out patch clamped at +60 mV and exposed to a standard (Con, top panel) or a 10 µM PIP2-containing solution (bottom panel). “c” and “o” denote respectively the closed and open states. C and D: All-points histograms summarizing single channel amplitudes at +60 mV in the inside-out configuration before (Control) and after addition of 10 µM PIP2.
Table 1.
Time constants (abundance) for open closed states of single TRPC7 channels activated by OAG in cell attached mode, and with or without addition of PIP2 in excised patches.
| TRPC7 (cell attached + OAG) | TRPC7 (excised) | TRPC7 (excised + PIP2) | |
|---|---|---|---|
| τ open (ms) | 0.258 ± 0.099 (69.7 ± 4.1%) | 0.257 ± 0.106 (70.8 ± 4.3%) | 0.284 ± 0.102 (64.4 ± 4.0%) |
| 1.29 ± 0.15 (30.3 ± 4.5%) | 1.46 ± 0.18 (29.2 ± 4.4%) | 1.48 ± 0.13 (35.6 ± 4.3%) | |
| τ closed (ms) | 0.220 ± 0.079 (40.7 ± 2.2%) | 0.213 ± 0.105 (37.2 ± 3.0%) | 0.212 ± 0.082 (45.5 ± 2.4%) |
| 2.19 ± 0.08 (32.9 ± 1.6%) | 2.67 ± 0.11 (26.9 ± 1.8%) | 1.69 ± 0.12 (30.0 ± 2.4%) | |
| 15.8 ± 0.1 (26.52 ± 1.6%) | 24.4 ± 0.15 (20.3 ± 1.9%) | 8.98 ± 0.21 (20.3 ± 2.4%) | |
| 212 ± 0.15 (15.6 ± 1.7%) | 56.2 ± 0.8 (4.2 ± 2.1%) | ||
The activation by PIP2 would likely reflect in part loss of PIP2 in the excised patches due to rapid action of phosphatases. Thus, we next tested the effect of addition of MgATP, which would serve as substrate for endogenous kinases to replenish PIP2 levels. As shown in Figure 4, ATP also robustly activated TRPC7 channel activity, although to a somewhat lesser degree than PIP2 (Control NPo = 0.007±0.003; MgATP, 0.096±0.038). Figure 5 summarizes the effects of both PIP2 and ATP from multiple experiments. Also shown in Figure 5 is the lack of effect of the head group of PIP2, inositol 1,4,5-trisphosphate (IP3) (Control NPo = 0.012±0.006; IP3, 0.0136±0.008). The statistics reflect the high degree of variability in NPo in ATP- and PIP2-activated patches. Nonetheless, ATP increased NPo in 8 of 8 patches, and PIP2 increased NPo in 14 of 14 patches.
Figure 4. MgATP activates TRPC7 channels in inside-out patches.
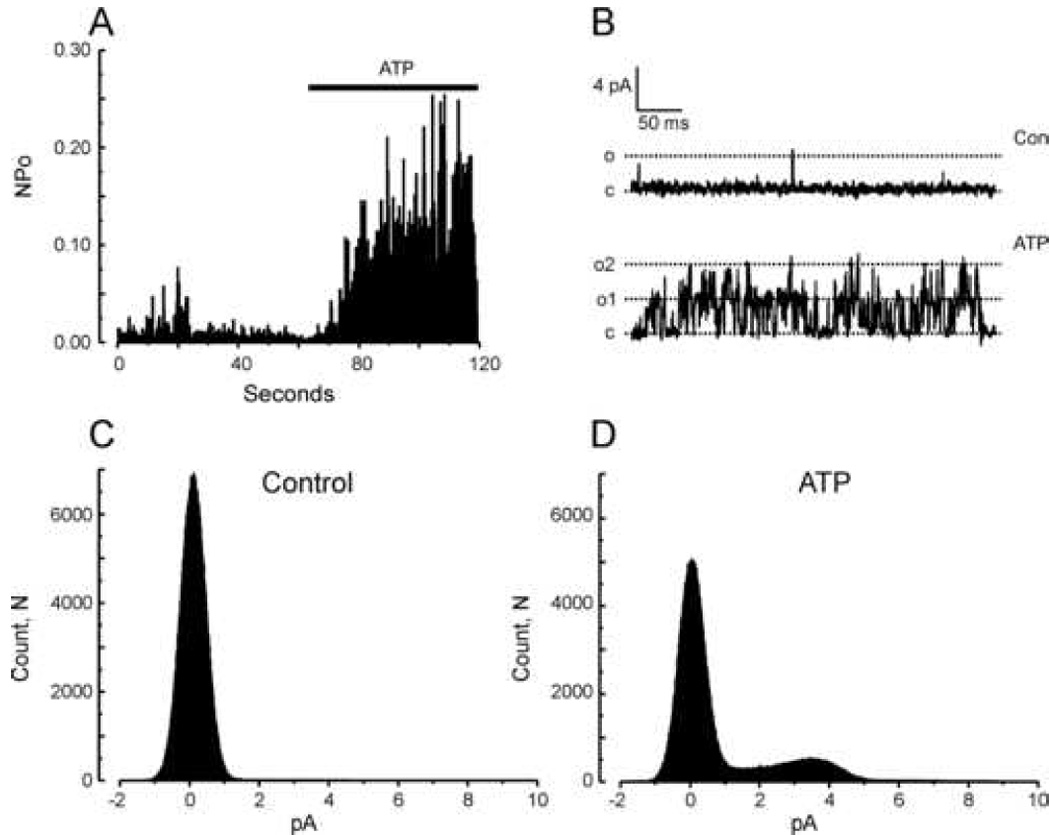
A: Histogram summarizing TRPC7 channel open probability over time at +60 mV when challenged with 1 mM MgATP. The figure is representative of results obtained in 9 independent inside-out patches. B: Representative traces of currents recorded in an inside-out patch clamped at +60 mV and exposed to a control (top panel) or 1 mM MgATP-containing solution (bottom panel). “c” and “o” denote respectively the closed and open states. C: All-points histograms summarizing single channel amplitudes at +60 mV in the inside-out configuration before (Con) and after addition of 1 mM MgATP (ATP).
Figure 5. Summarized properties of TRPC7 channels.
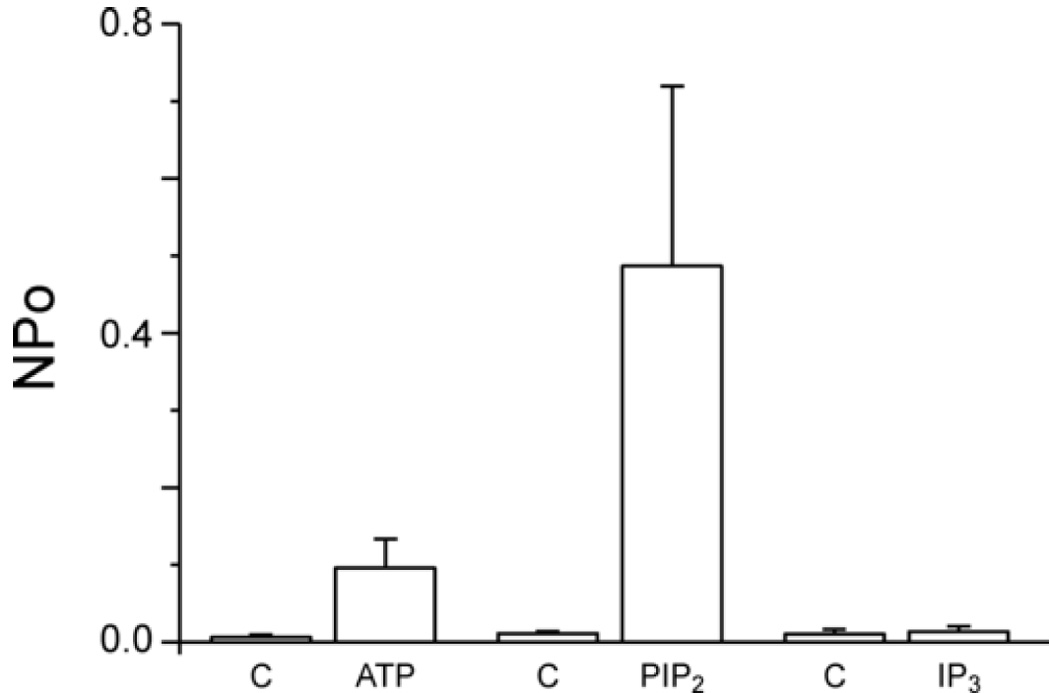
Histograms summarizing TRPC7 single channel activity at +60 mV in the inside-out configuration when challenged with 1 mM MgATP (n=9 patches), 10 µM PIP2 (n=12 patches) or 10 µM IP3 (n=4 patches) (C = Control). Results are presented as mean ± SEM.
We also examined effects of PIP2 on TRPC6 and TRPC3, channels that are structurally similar to TRPC7 and which are also activated by diacylglycerols [2;19]. HEK293 cells stably expressing TRPC6 or TRPC3 showed single channel openings with conductances similar to those reported by others (TRPC6, 60.8 ± 0.1 pS [20]; TRPC3, 70.2 ± 0.3 pS [21]). Figure 6 illustrates effects of PIP2 on TRPC6 single channel activity. TRPC6 channels were reproducibly activated by bath application of either PIP2 (Control NPo = 0.0015±0.0005; PIP2, 0.0197±0.0072) or ATP (Control NPo = 0.0196±0.0103; ATP, 0.0729±0.0440), although to a somewhat lesser extent than TRPC7. However, ATP increased NPo in 10 of 10 patches and PIP2 increased NPo in 10 of 10 patches. In Figure 7 we show results of similar experiments for TRPC3. TRPC3 was activated by PIP2 to a lesser extent than either TRPC7 or TRPC6 (Control NPo = 0.0105±0.0034; PIP2, 0.0232±0.0128), and not in all patches (5 of 9), and was not significantly activated by ATP (Control NPo = 0.0032±0.0008; ATP, 0.0022±0.0010).
Figure 6. TRPC6 channels in inside-out patches are activated by PIP2.
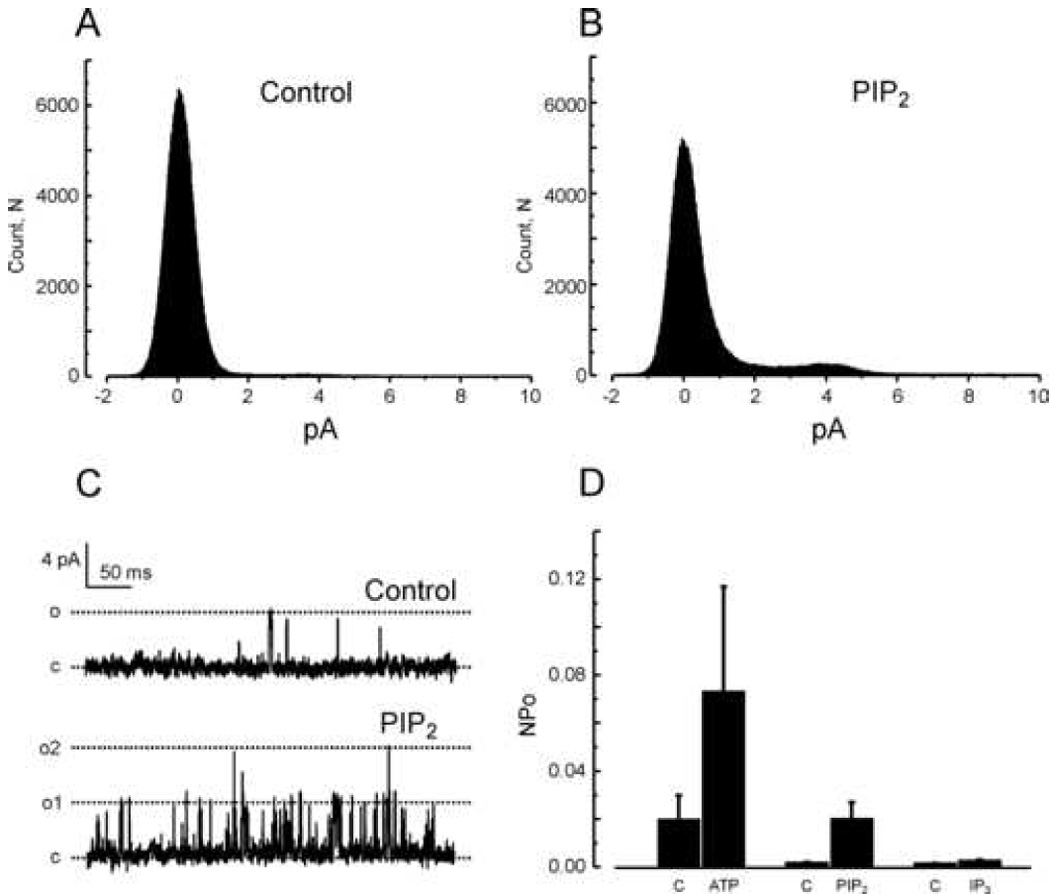
A and B: All-points histograms summarizing single channel amplitudes at +60 mV in the inside-out configuration before (A, Control) and after addition of 10 µM PIP2 (B, PIP2). C: Examples of TRPC6 currents recorded in an inside-out patch clamped at +60 mV and exposed to a standard (top panel) or 10 µM PIP2-containing solution (bottom panel). “c” and “o” denote respectively the closed and open states. D: Histograms summarizing TRPC6 single channel activity at +60 mV in the inside-out configuration when challenged with 1 mM MgATP (n=5 patches), 10 µM PIP2 (n=8 patches) or 10 µM IP3 (n=5 patches). Results are presented as mean ± SEM.
Figure 7. TRPC3 channels in inside-out patches are not significantly affected by PIP2.
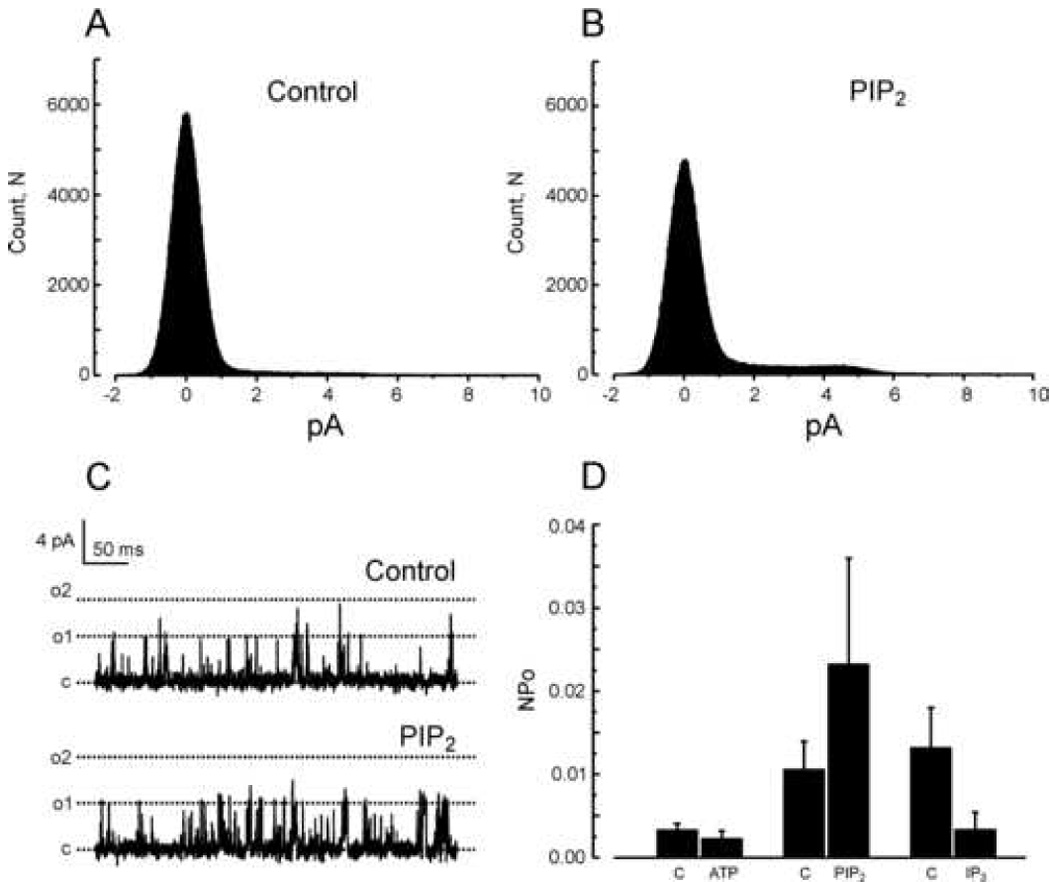
A and B: All-points histograms summarizing single channel amplitudes at +60 mV in the inside-out configuration before (A, Control) and after addition of 10 µM PIP2 (B, PIP2). C: Examples of TRPC3 currents recorded in an inside-out patch clamped at +60 mV and exposed to a regular (top panel) or a 10 µM PIP2-containing solution (bottom panel). “c” and “o” denote respectively the closed and open states. D: Histograms summarizing the variations of TRPC3 single channel activity at +60 mV in the inside-out configuration when challenged with 1 mM MgATP (n=5 patches), 10 µM PIP2 (n=10 patches) or 10 µM IP3 (n=5 patches). Results are presented as mean ± SE.
The robust activation of TRPC7 channels by PIP2 or ATP in excised patches was somewhat surprising given that these regulators are expected to be present constitutively in cells. In fact, as shown in Figure 8, NPo of TRPC7 channels in cell attached mode, when PIP2 and ATP would presumed to be present, was very low. Addition of a maximal concentration of OAG significantly increased NPo (8 of 9 patches), but to a much lower level than with either ATP or PIP2 in excised patches (Control NPo = 0.0003±0.0002; OAG, 0.0122±0.0067). Furthermore, constitutive activity of TRPC7 channels in cell attached mode was considerably less than in excised patches. Finally, we tested the effects of OAG in excised patches; surprisingly, as shown in Figure 8, we observed no TRPC7 channel activation by OAG in this recording configuration (Control NPo = 0.0080±0.0023; OAG, 0.0077±0.0020). Addition of OAG to patches previously activated with PIP2 or ATP also showed no increase in activity (data not shown).
Figure 8. Effects of OAG on TRPC7 channel activity in cell attached and excised patch configurations.
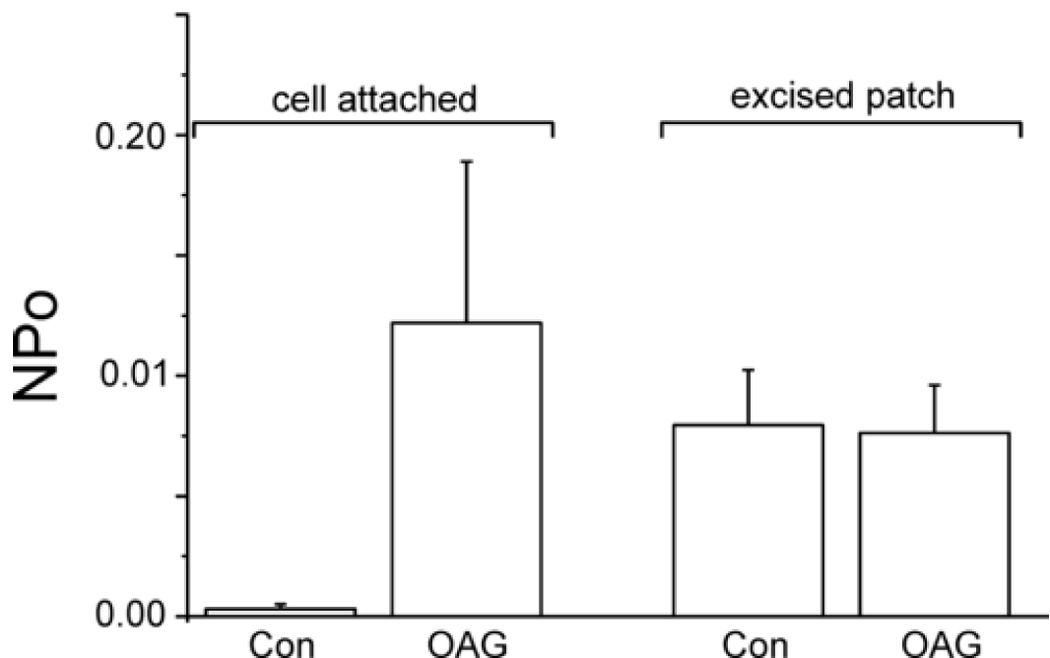
Histograms summarizing effects of 30 µM OAG (Con = controls) on TRPC7 single channel activity (Po = open probability) at +60 mV in the cell attached or excised, inside out patch configuration. Shown are averages ± SEM for 9 experiments for cell attached patch, and 10 experiments for excised patch.
Discussion
A wide variety of ion channels and plasma membrane transporters are known to be regulated by the acidic phospholipid, PIP2 [7;8]. Generally, this modulation is a positive one. Since PIP2 is constitutively present in the membrane of cells and can be decreased by activation of PLC-coupled receptors, this may provide a means for negative regulation of ion channel signaling pathways. That appears to be the case for a number of channels in the TRP superfamily that are activated by PIP2 and negatively regulated by PLC [22–29]. However, until the present study there have been no published findings on PIP2 regulation of TRPC channels, which are known to be activated by PLC [30–33]. Our findings indicate that all three members of the TRPC3,6,7 subgroup of TRPC channels are, to varying degrees, sensitive to activation by PIP2 in excised patches. We focused on the regulation of TRPC7, as a representative of the TRPC3,6,7 subgroup, which was of the three most strongly activated by PIP2.
In a recent report, Kwon et al. [34] investigated binding of various inositol phospholipids to TRPC6, and determined that PIP3 bound with highest affinity to a site in the C-terminal cytoplasmic region. These authors concluded that PIP3 acts by displacing an inhibitory calmodulin from this site, although no experiments involving direct application of phospholipids were reported. It cannot be determined at present if the regulation by PIP3 described by Kwon et al. represents the same mode of regulation we have observed for PIP2. The failure of low concentrations of LY294002 to affect TRPC7 activation argues against a role for PIP3. In addition, the results in Figure 2 show that activation in excised patches by PIP2 is reversible. Thus, if the mechanism of activation involves displacement of calmodulin, that calmodulin must remain available in excised patches for rebinding upon removal of PIP2. There is evidence for regulation of voltage-dependent Ca2+ channels by tethered calmodulin [35], but it is not known whether a similar situation exists for TRPC channels.
The members of this subgroup were originally thought to be activated directly by diacylglycerols, because TRPC6 channel activation was observed by direct application of synthetic diacylglycerols to channels in excised patches [19]. However, this does not necessarily mean that activation is direct, only that if intermediate mediators are involved, they may be retained in the excised patch. A striking finding in the current study was that excision of patches containing TRPC7 channels resulted in loss of regulation by diacylglycerols and the acquisition of a strong activation by PIP2.2
We have previously suggested that the activation of TRPC3 channels by OAG did not likely reflect a direct action of OAG on the channels. This was based on two observations. First, OAG activation of TRPC3 required the tyrosine kinase activity of the proto-oncogene, src, while constitutive activity of the channels did not have this requirement [36]. Second, when TRPC3 channels were translocated to the plasma membrane due to activation of the epidermal growth factor receptor, the newly translocated channels showed normal constitutive activity, but were incapable of responding to OAG [37]. The loss of responsiveness of TRPC7 channels to OAG following excision in the current study is thus consistent with our hypothesis, and may result from the loss from the patch of a factor or factors required for OAG to act. This may also explain in part the somewhat variable rundown of TRPC7 currents we previously observed in whole-cell mode [16]. The loss of such a factor or factors may result in the unusual degree of activation by PIP2, but this idea cannot be tested until these factors are identified. However, consistent with this interpretation, we failed to observe any activation of TRPC7 channels by inclusion of PIP2 in patch pipettes while recording currents in whole-cell mode (Figure 2C). Differences in losses of PIP2 and other regulatory factors may also explain the differences between our findings and those in the earlier study by Hofmann et al. [19] if their patches, taken from CHO-K1 cells, more efficiently retained PIP2 and diacylglycerol-regulated components. Consistent with this idea, Hofmann et al. reported that they saw no effect of PIP2 on TRPC6 channels [19]. The more efficient depletion of PIP2 from the HEK293 cell membranes may thus be fortuitous in revealing the potent activation of TRPC7 channels by PIP2 and the loss of regulation by diacylglycerols.
Our findings thus indicate two potentially opposing roles for PIP2 and PLC for regulation of TRPC channels. In the process of receptor signaling to TRPC7 channels, PLC degrades PIP2 to produce the channel activator, diacylglycerol. Yet in so doing the necessary co-factor, PIP2, may be diminished, possibly resulting in inhibition of the channels. However, we previously reported that in TRPC7-expressing HEK293 cells activation of PLC through muscarinic receptors did not inhibit OAG-activated TRPC7 channels [10]. Thus, either the requirement for PIP2 is satisfied by low levels of PIP2, or possibly the lipid kinases can keep pace with the PLC thereby maintaining PIP2 at necessary levels.
Acknowledgements
Drs. David Armstrong, Jerrel Yakel and Guillermo Vazquez read the manuscript and provided helpful comments. This work was supported by funds from the Intramural Program of the NIEHS and NIH.
Footnotes
Although we failed to see activation of TRPC7 by OAG in excised patches, we have confirmed some limited activation of TRPC3 and TRPC7 by another synthetic diacylglycerol, stearoyl-arachidonoyl glycerol; the maximally activated NPo averaged less than 0.01 (data not shown).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Clapham DE, Runnels LW, Strübing C. The TRP ion channel family. Nature Rev Neurosci. 2002;2:387–396. doi: 10.1038/35077544. [DOI] [PubMed] [Google Scholar]
- 2.Trebak M, Vazquez G, Bird GStJ, Putney JW., Jr The TRPC3/6/7 subfamily of cation channels. Cell Calcium. 2003;33:451–461. doi: 10.1016/s0143-4160(03)00056-3. [DOI] [PubMed] [Google Scholar]
- 3.Hardie RC. Regulation of trp channels via lipid second messengers. Ann Rev Physiol. 2003;65:735–759. doi: 10.1146/annurev.physiol.65.092101.142505. [DOI] [PubMed] [Google Scholar]
- 4.Dietrich A, Schnitzler M, Kalwa H, Storch U, Gudermann T. Functional characterization and physiological relevance of the TRPC3/6/7 subfamily of cation channels. Naunyn Schmiedebergs Arch Pharmacol. 2005;371:257–265. doi: 10.1007/s00210-005-1052-8. [DOI] [PubMed] [Google Scholar]
- 5.Trebak M, Bird GStJ, McKay RR, Birnbaumer L, Putney JW., Jr Signaling mechanism for receptor-activated TRPC3 channels. J Biol Chem. 2003;278:16244–16252. doi: 10.1074/jbc.M300544200. [DOI] [PubMed] [Google Scholar]
- 6.Groschner K, Rosker C. TRPC3: a versatile transducer molecule that serves integration and diversification of cellular signals. Naunyn Schmiedebergs Arch Pharmacol. 2005;371:251–256. doi: 10.1007/s00210-005-1054-6. [DOI] [PubMed] [Google Scholar]
- 7.Hilgemann DW, Feng S, Nasuhoglu C. The complex and intriguing lives of PIP2 with ion channels and transporters. Science's STKE. 2001;2001(111):RE19. doi: 10.1126/stke.2001.111.re19. [DOI] [PubMed] [Google Scholar]
- 8.Suh BC, Hille B. Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr Opin Neurobiol. 2005;15:370–378. doi: 10.1016/j.conb.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 9.McKay RR, Szmeczek-Seay CL, Lièvremont J-P, et al. Cloning and expression of the human transient receptor potential 4 (TRP4) gene: localization and functional expression of human TRP4 and TRP3. Biochem J. 2000;351:735–746. [PMC free article] [PubMed] [Google Scholar]
- 10.Lievremont JP, Bird GS, Putney JW., Jr Canonical transient receptor potential TRPC7 can function as both a receptor- and store-operated channel in HEK-293 cells. Am J Physiol Cell Physiol. 2004;287:C1709–C1716. doi: 10.1152/ajpcell.00350.2004. [DOI] [PubMed] [Google Scholar]
- 11.Lievremont JP, Bird GS, Putney JW., Jr Mechanism of inhibition of TRPC cation channels by 2-aminoethoxydiphenylborane. Mol Pharmacol. 2005;68:758–762. doi: 10.1124/mol.105.012856. [DOI] [PubMed] [Google Scholar]
- 12.Trebak M, Hempel N, Wedel BJ, Smyth JT, Bird GS, Putney JW., Jr Negative regulation of TRPC3 channels by protein kinase C-mediated phosphorylation of serine 712. Mol Pharmacol. 2005;67:558–563. doi: 10.1124/mol.104.007252. [DOI] [PubMed] [Google Scholar]
- 13.Patton C, Thompson S, Epel D. Some precautions in using chelators to buffer metals in biological solutions. Cell Calcium. 2004;35:427–431. doi: 10.1016/j.ceca.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 14.Vazquez G, Bird GS, Mori Y, Putney JW., Jr Native TRPC7 Channel Activation by an Inositol Trisphosphate Receptor-dependent Mechanism. J Biol Chem. 2006;281:25250–25258. doi: 10.1074/jbc.M604994200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okada T, Inoue R, Yamazaki K, et al. Molecular and functional characterization of a novel mouse transient receptor potential protein homologue TRP7. Ca2+-permeable cation channel that is constitutively activated and enhanced by stimulation of G protein-coupled receptor. J Biol Chem. 1999;274:27359–27370. doi: 10.1074/jbc.274.39.27359. [DOI] [PubMed] [Google Scholar]
- 16.Lemonnier L, Trebak M, Lievremont JP, Bird GS, Putney JW., Jr Protection of TRPC7 cation channels from calcium inhibition by closely associated SERCA pumps. FASEB J. 2006;20:503–505. doi: 10.1096/fj.05-4714fje. [DOI] [PubMed] [Google Scholar]
- 17.Linseman DA, McEwen EL, Sorensen SD, Fisher SK. Cytoskeletal and phosphoinositide requirements for muscarinic receptor signaling to focal adhesion kinase and paxillin. J Neurochem. 1998;70:940–950. doi: 10.1046/j.1471-4159.1998.70030940.x. [DOI] [PubMed] [Google Scholar]
- 18.Sorensen SD, Linseman DA, McEwen EL, Heacock AM, Fisher SK. A role for wortmannin-sensitive phosphatidylinositol-4-kinase in the endocytosis of muscarinic cholinergic receptors. Mol Pharmacol. 1998;53:827–836. [PubMed] [Google Scholar]
- 19.Hofmann T, Obukhov AG, Schaefer M, Harteneck C, Gudermann T, Schultz G. Direct activation of human TRPC6 and TRPC3 channels by diacylglycerol. Nature. 1999;397:259–262. doi: 10.1038/16711. [DOI] [PubMed] [Google Scholar]
- 20.Jung S, Mühle A, Schaefer M, Strotmann R, Schultz G, Plant TD. Lanthanides potentiate TRPC5 currents by an action at extracellular sites close to the pore mouth. J Biol Chem. 2003;278:3562–3571. doi: 10.1074/jbc.M211484200. [DOI] [PubMed] [Google Scholar]
- 21.Kiselyov K, Xu X, Mozhayeva G, et al. Functional interaction between InsP3 receptors and store-operated Htrp3 channels. Nature. 1998;396:478–482. doi: 10.1038/24890. [DOI] [PubMed] [Google Scholar]
- 22.Hardie RC, Raghu P, Moore S, Juusola M, Baines RA, Sweeney ST. Calcium influx via TRP channels is reguired to maintain PIP2 levels in Drosophila photoreceptors. Neuron. 2001;30:149–159. doi: 10.1016/s0896-6273(01)00269-0. [DOI] [PubMed] [Google Scholar]
- 23.Runnels LW, Yue L, Clapham DE. The TRPM7 channel is inactivated by PIP2 hydrolysis. Nature Cell Biol. 2002;4:329–336. doi: 10.1038/ncb781. [DOI] [PubMed] [Google Scholar]
- 24.Liu D, Liman ER. Intracellular Ca2+ and the phospholipid PIP2 regulate the taste transduction ion channel TRPM5. Proc Natl Acad Sci U S A. 2003;100:15160–15165. doi: 10.1073/pnas.2334159100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu B, Zhang C, Qin F. Functional recovery from desensitization of vanilloid receptor TRPV1 requires resynthesis of phosphatidylinositol 4,5-bisphosphate. J Neurosci. 2005;25:4835–4843. doi: 10.1523/JNEUROSCI.1296-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu B, Qin F. Functional control of cold- and menthol-sensitive TRPM8 ion channels by phosphatidylinositol 4,5-bisphosphate. J Neurosci. 2005;25:1674–1681. doi: 10.1523/JNEUROSCI.3632-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rohacs T, Lopes CM, Michailidis I, Logothetis DE. PI(4,5)P2 regulates the activation and desensitization of TRPM8 channels through the TRP domain. Nat Neurosci. 2005;8:626–634. doi: 10.1038/nn1451. [DOI] [PubMed] [Google Scholar]
- 28.Lee J, Cha SK, Sun TJ, Huang CL. PIP2 activates TRPV5 and releases its inhibition by intracellular Mg2+ J Gen Physiol. 2005;126:439–451. doi: 10.1085/jgp.200509314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nilius B, Mahieu F, Prenen J, et al. The Ca2+-activated cation channel TRPM4 is regulated by phosphatidylinositol 4,5-biphosphate. EMBO J. 2006;25:467–478. doi: 10.1038/sj.emboj.7600963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vazquez G, Wedel BJ, Aziz O, Trebak M, Putney JW., Jr The mammalian TRPC cation channels. Biochim Biophys Acta. 2004;1742:21–36. doi: 10.1016/j.bbamcr.2004.08.015. [DOI] [PubMed] [Google Scholar]
- 31.Putney JW., Jr Physiological mechanisms of TRPC activation. Pflugers Arch. 2005;451:29–34. doi: 10.1007/s00424-005-1416-4. [DOI] [PubMed] [Google Scholar]
- 32.Dietrich A, Kalwa H, Rost BR, Gudermann T. The diacylgylcerol-sensitive TRPC3/6/7 subfamily of cation channels: functional characterization and physiological relevance. Pflugers Arch. 2005;451:72–80. doi: 10.1007/s00424-005-1460-0. [DOI] [PubMed] [Google Scholar]
- 33.Freichel M, Vennekens R, Olausson J, et al. Functional role of TRPC proteins in native systems: implications from knockout and knock-down studies. J Physiol. 2005;567:59–66. doi: 10.1113/jphysiol.2005.092999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kwon Y, Hofmann T, Montell C. Integration of phosphoinositide- and calmodulin-mediated regulation of TRPC6. Mol Cell. 2007;25:491–503. doi: 10.1016/j.molcel.2007.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Peterson BZ, DeMaria CD, Yue DT. Calmodulin Is the Ca2+ Sensor for Ca2+-Dependent Inactivation of L-Type Calcium Channels. Neuron. 1999;22:549–558. doi: 10.1016/s0896-6273(00)80709-6. [DOI] [PubMed] [Google Scholar]
- 36.Vazquez G, Wedel BJ, Kawasaki BT, Bird GS, Putney JW., Jr Obligatory role of Src kinase in the signaling mechanism for TRPC3 cation channels. J Biol Chem. 2004;279:40521–40528. doi: 10.1074/jbc.M405280200. [DOI] [PubMed] [Google Scholar]
- 37.Smyth JT, Lemonnier L, Vazquez G, Bird GS, Putney JW., Jr Dissociation of regulated trafficking of TRPC3 channels to the plasma membrane from their activation by phospholipase C. J Biol Chem. 2005;281:11712–11720. doi: 10.1074/jbc.M510541200. [DOI] [PubMed] [Google Scholar]