

Abstract
Our prior work showed that brief exposure of pregnant C3H mice to inorganic arsenic-induced hepatocellular carcinoma (HCC) formation in adult male offspring. The current study examined the early hepatic events associated with this oncogenic transformation. Pregnant mice were exposed to a known carcinogenic dose of arsenic (85 ppm) in the drinking water from gestation days 8 to 18. The dams were allowed to give birth and liver samples from newborn males were analyzed for arsenic content, global DNA methylation and aberrant expression of genes relevant to the carcinogenic process. Arsenic content in newborn liver reached 57 ng/g wet weight, indicating arsenic had crossed the placenta, reached the fetal liver and that significant amounts remained after birth. Global methylation status of hepatic DNA was not altered by arsenic in the newborn. However, a significant reduction in methylation occurred globally in GC-rich regions. Microarray and real-time RT-PCR analysis showed that arsenic exposure enhanced expression of genes encoding for glutathione production and caused aberrant expression of genes related to insulin growth factor signaling pathways and cytochrome P450 enzymes. Other expression alterations observed in the arsenic-treated male mouse newborn liver included the overexpression of cdk-inhibitors and stress response genes including increased expression of metallothionein-1 and decreased expression of betaine-homocysteine methyltransferase and thioether S-methyltransferase. Thus, transplacental exposure to arsenic at a hepatocarcinogenic dose induces alterations in DNA methylation and a complex set of aberrant gene expressions in the newborn liver, a target of arsenic carcinogenesis.
Keywords: Arsenic, Transplacental exposure, Newborn mouse liver, DNA methylation, Gene expression
1. Introduction
Inorganic arsenic is a human carcinogen, producing tumors of the skin, lung, liver, urinary bladder, prostate, kidney, and possibly other sites (NRC, 2001; Centeno et al., 2002; IARC, 2004). Inorganic arsenic can readily cross the rodent and human placenta and enters the fetal system (NRC, 2001; Devesa et al., 2006). We have shown that short-term exposure to inorganic arsenic in utero produces a variety of internal tumors (liver and adrenal tumors in males, tumors of the lung, ovary and the urogenital system in females) in the off-spring when mice reached adulthood (Waalkes et al., 2003, 2004a, 2006a,b). Gestation is a period of high sensitivity to chemical carcinogenesis in rodents and probably in humans (Anderson et al., 2000), and the transplacental carcinogenic risks observed in rodents could also occur in humans. To define early events associated with transplacental arsenic carcinogenesis should enhance our understanding of the mechanisms of arsenic carcinogenesis.
Inorganic arsenic is enzymatically methylated to mono- and di-methylated species in most mammals and occurs at a high level in the liver (Aposhian and Aposhian, 2006; Thomas et al., 2007). Arsenite is sequentially methylated to form methylarsonate (MMA5+) and dimethylarsinic acid (DMA5+) by arsenic methyltransferase (AS3MT or Cyt19) using S-adenosylmethionine (SAM) as a methyl group donor (Thomas et al., 2007). SAM is also required for most other cellular methylation reactions, including DNA methylation (Baylin et al., 1998). Chronic exposure of rat liver epithelial cells to inorganic arsenic induces SAM depletion in rodent liver cells, causing a global loss of DNA methylation during malignant transformation (Zhao et al., 1997). Chronic exposure of intact animals to inorganic arsenic also produces hepatic DNA hypomethylation (Chen et al., 2004; Xie et al., 2004). Hypomethylation of DNA is thought to be a nongenotoxic mechanism of carcinogenesis that acts by facilitating aberrant gene expression, and can be a causative factor in hepatocarcinogenesis (Goodman and Watson, 2002), especially during a critical period of genetic programming (Anderson et al., 2000).
The liver is clearly a potential target of arsenic in humans and arsenic exposure has been associated with development of hepatocellular carcinomas as well as other hepatic lesions (Chen et al., 1997; Lu et al., 2001; Centeno et al., 2002; Zhou et al., 2002; Mazumder, 2005). In accord with human data, transplacental exposure to inorganic arsenic induced a marked, dose-related increase in hepatocellular tumors including carcinoma, in adult male mice (Waalkes et al., 2003, 2004a, 2006b). Hypomethylation of the promoter region of the estrogen receptor-α (ER-α) is thought to be responsible for aberrant estrogen signaling and may play a role in the formation of HCC during carcinogenesis induced by in utero arsenic exposure (Waalkes et al., 2004b). Thus, the present study was designed to investigate genome-wide and site-specific DNA methylation in newborn mouse liver following in utero exposure to a carcinogenic dose of inorganic arsenic. In addition, microarray and real-time RT-PCR were used to profile gene expression changes. The results indicate that a brief exposure to inorganic arsenic during gestation induced DNA hypomethylation changes at the GC-rich regions and was associated with aberrant gene expressions in newborn mouse liver. These could be key early events leading to arsenic-induced hepatocarcinogenesis later in life.
2. Materials and methods
2.1. Chemicals
Sodium arsenite (NaAsO2) was obtained from Sigma Chemical Co. (St. Louis, MO) and dissolved in the drinking water at 85 mg arsenic/L (85 ppm). Customer-designed cDNA microarrays (600 genes) were purchased from BD Biosciences Clontech, Inc. (Palo Atlo, CA). [α-32P]-dATP was purchased from Perkin-Elmer, Inc. (Boston, MA), and 3H labeled S-adenosyl-methionine ([3H]-SAM) was from Amersham (Arlington Heights, IL). All other reagents are of reagent grade.
2.2. Animal treatment and sample collection
Timed pregnant C3H mice were given drinking water containing 85 ppm arsenic as sodium arsenite or unaltered water ad libitum from days 8 to 18 of gestation. At day 21 of gestation, dams were allowed to give birth, and the newborn mice were killed by CO2 asphyxiation and livers removed. Animal care was provided in accordance with the US Public Health Policy on the Care and Use of Animals, and the study protocol was approved by the Institutional Animal Care and Use Committee of National Cancer Institute at Frederick.
2.3. Hepatic arsenic levels
A portion of the frozen liver (120–150 mg) was digested in nitric acid. Total arsenic, which would include inorganic and organic forms, was determined using graphic furnace atomic absorption spectrometry (Perkin-Elmer AAanalst100, Norwalk, CT). Results were expressed as μg arsenic/g wet weight liver, as shown in previous publications (Xie et al., 2004).
2.4. Global DNA methylation assay
Genomic DNA was extracted from liver tissue and purified using DNeasy Kits (Qiagen, Valencia, CA). Global DNA methylation status was assessed by methyl acceptance assay (Chen et al., 2004). Briefly, 1 μg DNA was incubated at 37 °C for 2 h in a 30 μl mixture containing 1.25 μM (3 μCi) [3H]-SAM, 4 units of CpG methylase (M. Sss I) (New England Biolabs, Inc., Beverly, MA), 10 mM DDT, Tris-EDTA buffer (100 mM Tris, 10 mM EDTA, pH 8.0) and 100 mM NaCl. The reaction was terminated on ice, and transferred onto a Whatman DE81 filter (Whatman International Ltd., Maidstone, UK). The filter was washed on a vacuum filtration apparatus with 2 ml 0.5 M phosphate buffer (pH 7.0) five times, followed by a wash with 2 ml of 70% ethanol and 2 ml of absolute ethanol. After the filter was dried, the bound radioactivity was measured by scintillation (Beckman LS 6500 Scintillation Counter).
2.5. Methylation analysis of GC-rich regions
Genomic DNA was digested with RsaI alone (methylation-insensitive), and RsaI plus MspI or HpaII, both are methylation sensitive enzymes (Watson et al., 2003). Restriction digests were performed with 1 μg of DNA and 5 units of RsaI in Roche buffer L. After 1 h incubation at 37 °C, two 2.5 unit aliquots of MspI or HpaII were added, 2 h apart. Total incubation time was 18 h. The enzymes were inactivated by 10-min incubation at 65 °C, and the digests were used for PCR using a single primer (5′ -AACCCTCACCCTAACCCCGG-3′ ) that arbitrarily binds within GC-rich regions of DNA (Watson et al., 2003). Routine PCR reactions were performed with 1 μg digested DNA, 20 pmols of primer, 1.5 mM MgCl2, 25 pmol dNTPs, and 1 unit of Taq polymerase. Thermocycle conditions were 5 min at 94 °C, 35 cycles at 94 °C30s,55 °C 15 s, and 72 °C 1 min, and the last cycle at 72 °C was extended to 5 min. PCR products were resolved on 1.8% agarose gel electrophoresis and images were analyzed using Image J software.
2.6. cDNA microarray analysis
Microarray analysis was performed as previously described (Xie et al., 2004). Briefly, total RNA was extracted from liver tissues with Trizol reagent and purified with RNeasy columns (Qiagen, Valencia, CA). Five micrograms of pooled RNA was converted to [α-32P]-dATP-labeled cDNA probe with Atlas specific cDNA synthesis primers (BD Biosciences Clontech Inc., Palo Atlo, CA). The probe was purified with a NucleoSpin column (Clontech), denatured at 100 °C for 2–3 min, and hybridized to the membrane in triplicate with Expresshyb buffer (Clontech) at 68 °C overnight. The membranes were washed at 68 °C four times (30 min each) in 2 × SSC/1% SDS, twice in 0.1 × SSC/0.5% SDS, and exposed to a phosphoimage screen. Images were acquired by PhosphorImager Scanner (Molecular Dynamics Model Storm 860, Sunnyvale, CA) and analyzed densitometrically using AtlasImage software (version 2.01, Clontech).
2.7. Real-time RT-PCR analysis
Total RNA was reverse transcribed with MMLV reverse transcriptase and oligodT primers. The PCR primers were designed with Primer Express software and synthesized by Sigma-Genosys (Woodlands, TX). The SYBR Green DNA PCR kit (Applied Biosystems, Foster City, CA, USA) was used for real-time RT-PCR analysis. Differences in gene expression between groups were calculated using cycle time (Ct) values, which were normalized against β-actin and expressed as relative to controls.
2.8. Statistics
Data are expressed as mean ± S.E.M. For comparisons between two groups, Students’ t-test was used. For comparisons among three or more groups, data were analyzed using a one-way analysis of variance, followed by Duncan's multiple range test. The level of significance was set at p < 0.05 in all cases.
3. Results
3.1. Hepatic arsenic content and global DNA methylation analysis
Arsenic was present in the livers of newborn mice exposed to arsenic in utero, but undetectable in livers of controls. The average arsenic concentration in newborn mouse liver was 57 ± 12 ng/g wet weight, approximately 10% of the concentration detected in maternal liver (750 ± 95 ng/g). These data indicate that arsenic did cross placental barrier, reached fetal liver and significant amounts remained in the newborn mouse liver 3 days after termination of exposure.
Global DNA methylation was assessed by methyl acceptance assay. This assay uses a bacterial DNA methyltransferase that indiscriminately methylates all unmethylated cytosines in DNA using SAM labeled with [3H] at the donated methyl group. Thus, higher methyl incorporation corresponds to a lower degree of global DNA methylation (i.e. hypomethylation). Compared to control global DNA methylation (100 ± 9.3%), DNA methylation in arsenic-treated mice (92.9 ± 10.4%) was not significantly different from control (Fig. 1). This result is different from the global DNA hypomethylation seen in the liver after chronic arsenic exposure in adult mice (Xie et al., 2004; Chen et al., 2004), perhaps due to the brief period of in utero arsenic exposure. In addition, global methylation measures both quiescent and active areas of DNA within the genome.
Fig. 1.

Effect of in utero arsenic exposure on the global methylation status in newborn mouse liver by the methyl acceptance assay. Data are mean ± S.E.M. (n = 6).
3.2. Methylation of the GC-rich regions
Methylation of GC-rich regions was evaluated by amplification with PCR of GC-rich DNA products following restrictive digestion of genomic DNA with methylation sensitive restriction enzymes (Watson et al., 2003). As illustrated in Fig. 2, the major PCR amplification bands (around 600 kDa) were similar following digestion with the methyl insensitive RsaI between control and arsenic groups. However, the intensity of this 600 kDa band in arsenic-treated livers was significantly reduced following the digestion with RsaI plus MspI (80% reduction), or following the digestion with RsaI plus HapII (60% reduction). Both MspI and HpaII are methylation sensitive enzymes, indicating that GC-rich regions from arsenic-treated livers were less methylated as compared to controls.
Fig. 2.
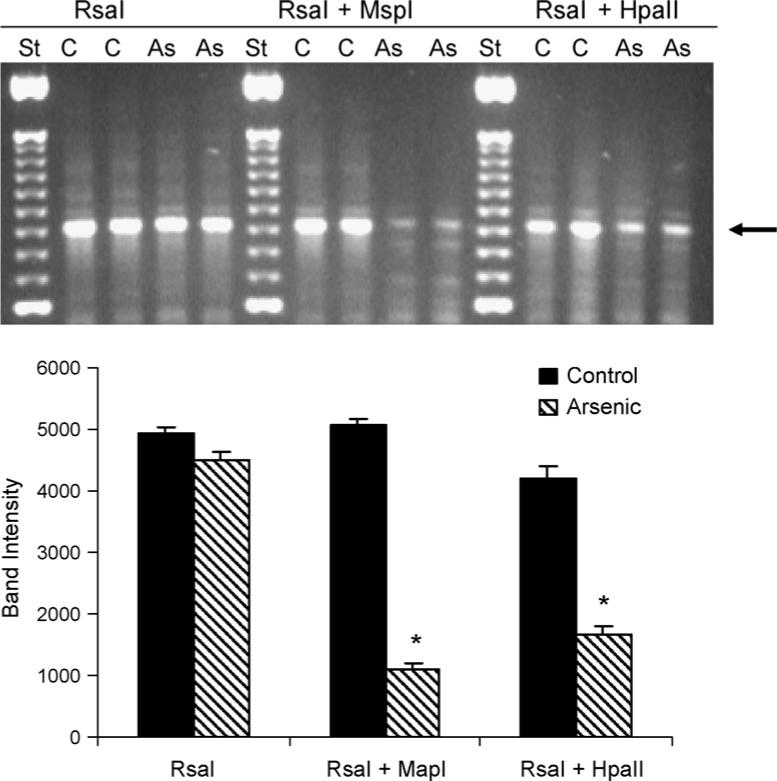
Effect of in utero arsenic exposure on the methylation of the CG-rich regions in newborn mouse liver by restrictive DNA digestion and PCR amplification of CG-rich regions. Upper panel: representative gel electrophoresis images; lower panel: quantification of the indicated band with Image J software. Data are mean ± S.E.M. of three experiments. *Significantly different from controls, p < 0.05. Arrow indicates the bands of interest.
3.3. Gene expression analysis by cDNA microarray and real-time RT-PCR
Among the 600 genes examined via microarray analysis, 40 showed markedly increased or decreased expression in newborn livers following in utero arsenic exposure. The hybrid intensity (ratio to control value) was calculated for comparison. The genes showing the most significant changes by microarray are listed in Table 1. These altered genes in newborn liver included increased expression of genes of the glutathione system, metabolic enzyme genes, stress-related genes, and others.
Table 1.
Microarray analysis of gene expression in livers of newborn male mice exposed in utero to 85 ppm arsenic from the gestation days 8−18
| Gene description | Accession | Control | Arsenic | As/con | p |
|---|---|---|---|---|---|
| Stress-related genes | |||||
| Glutathione S-transferase theta 1 | X98055 | 1791 | 8447 | 4.72 | 0.000 |
| Mu-class glutathione S-transferase | U24428 | 3360 | 5121 | 1.52 | 0.003 |
| Glutathione S-transferase, alpha 4 | NM_009997 | 2103 | 3925 | 1.87 | 0.000 |
| Glutathione S-transferase pi | D30687 | 14986 | 24659 | 1.65 | 0.000 |
| Glucose regulated protein, 78 kDa | D78645 | 10937 | 17619 | 1.61 | 0.007 |
| NF-KB essential modulator | AF069542 | 2486 | 3913 | 1.57 | 0.010 |
| AP endonuclease | U12273 | 1953 | 3061 | 1.57 | 0.002 |
| Bcl-xL apoptosis regulator | L35049 | 2385 | 3727 | 1.56 | 0.018 |
| Genes for metabolic enzymes | |||||
| Cytochrome P450, 2A4 | J03549 | 2791 | 4335 | 1.55 | 0.001 |
| UDP-glucuronosyltransferase 1 | D87866 | 20531 | 13581 | 0.66 | 0.000 |
| Hydroxysteroid (17-β) dehydrogenase 5 | NM_030611 | 13616 | 9569 | 0.63 | 0.001 |
| Cytochrome P450, 2E1, ethanol inducible | L11650 | 6436 | 2682 | 0.42 | 0.001 |
| Betaine-homocysteine methyltransferase | AF033381 | 11619 | 4839 | 0.42 | 0.001 |
| Cytochrome P450 CYP4A10 | AB018421 | 4339 | 1160 | 0.27 | 0.000 |
| Cytochrome P450, 4A14 | NM_007822 | 6572 | 1745 | 0.27 | 0.000 |
| Cytochrome P450 1A2 | X00479 | 2028 | 395 | 0.20 | 0.002 |
| Cytochrome P450 3A25 | Y11995 | 7706 | 1335 | 0.17 | 0.000 |
| Hydroxysteroid sulfotransferase | L02335 | 3390 | 818 | 0.24 | 0.000 |
| Thioether S-methyltransferase | NM_009349 | 1942 | 1393 | 0.72 | 0.002 |
| Glutathione peroxidase 1 | X03920 | 15941 | 22266 | 1.40 | 0.001 |
| Growth factors | |||||
| Insulin-like growth factor 2 | M14951 | 17107 | 13765 | 0.80 | 0.002 |
| Insulin-like growth factor receptor II | U04710 | 4455 | 8517 | 1.91 | 0.001 |
| Insulin growth factor binding protein 1 | X81579 | 11183 | 7861 | 0.70 | 0.017 |
| Insulin-like growth factor 1 | X04480 | 6219 | 4130 | 0.66 | 0.008 |
| Insulin-like growth factor binding protein 5 | X81583 | 3673 | 934 | 0.25 | 0.009 |
| Insulin-like growth factor binding protein 2 | X81580 | 13418 | 3131 | 0.23 | 0.001 |
| p57kip2; cdk-inhibitor kip2 | U20553 | 2160 | 10683 | 4.95 | 0.000 |
| Genes related to hormone metabolism | |||||
| Sex hormone binding globulin | U85644 | 865 | 1360 | 1.57 | 0.014 |
| Thyroid hormone receptor alpha | X51983 | 1873 | 2836 | 1.51 | 0.003 |
| Glucocorticoid receptor 1 | X13358 | 4073 | 6116 | 1.50 | 0.001 |
To confirm and extend microarray results, real-time RT-PCR analysis of selected genes was performed using individual samples from control and arsenic-exposed male newborn livers. In general, real-time RT-PCR confirmed microarray results, and RT-PCR appeared to be more sensitive and reliable based on individual samples. Thus, the data descriptions and discussions henceforth are based on the real-time RT-PCR analysis. For expression of genes in the glutathione system (Fig. 3), including the expression of glutathione S-transferase alpha (GST-alpha), GST-mu, GST-pi, GST-theta, and glutathione peroxidase-1 (GPX1) were all significantly increased between 1.5 and 3.5 fold in response to arsenic. The expression of the stress gene, metallothionein 1 (MT-1), was doubled. The expression of genes of methyl metabolism, such as thioether S-methyltransferase (TEMT) and betaine-homocysteine methyltransferase (BHMT), were decreased by 50%. The expression of p57kip2 (Table 1), were increased with arsenic, while the expression of p16 (a tumor suppressor gene) was depressed.
Fig. 3.

Effect of in utero arsenic exposure on glutathione and stress-related gene expression in newborn mouse liver. Data are mean ± S.E.M. (n = 6). *Significantly different from controls, p < 0.05.
Fig. 4 illustrates arsenic-induced alterations in expression of genes encoding metabolism and insulin-like growth factor signaling system. The expression of CYP2A4 was increased, but in general, the expression of cytochrome P450 enzyme genes was decreased, including decreased expression of CYP3A25, CYP2F2, CYP2J5, CYP7B1, and hydroxysteroid 17β dehydrogenase-5. Arsenic also altered several genes in the insulin-like growth factor signaling system. In particular, there was a significant increase in IGF receptor 2, but the expression of IGF-1, IGFBP1 and IGFBP3 were decreased with arsenic.
Fig. 4.

Effect of in utero arsenic exposure on expression of genes related to cytochrome P450 enzymes and the IGF signaling systems. Data are mean ± S.E.M. (n = 6). *Significantly different from controls p < 0.05.
4. Discussion
The present study clearly demonstrated that in utero arsenic exposure can induce a loss of DNA methylation in the GC-rich regions in the newborn mouse liver. In utero arsenic exposure also produced various aberrant gene expressions related to the glutathione utilization system, insulin-like growth factor signaling, and stress responses including potentially the response to overutilization of SAM in arsenic metabolism (i.e. methyl stress). Thus, in utero exposure to a carcinogenic dose of arsenic produces complex set of molecular alterations, which could play an early role in hepatic carcinogenesis. This gestational arsenic exposure may act through disruption of early genetic programming leading to tumor formation much later in life in a fashion seen with estrogenic carcinogens (Cook et al., 2005).
DNA hypomethylation can occur after chronic in vitro exposure of rat liver cells to inorganic arsenic (Zhao et al., 1997), or in intact animals chronically exposed to arsenic (Okoji et al., 2002; Chen et al., 2004; Xie et al., 2004). Alterations in methylation (such as reduced methylation of the promoter of the ER-α gene) were also evident in adult mice following transplacental arsenic exposure (Waalkes et al., 2004b). Thus, arsenic-induced alterations of DNA methylation could be an important epigenetic mechanism for arsenic carcinogenesis, at least in the liver. In the present study, global DNA methyaltion was unaltered, possibly due to the short arsenic exposure period. However, the methylation within GC-rich regions was clearly reduced. Thus, altered DNA methylation occurred in the newborn mouse liver after in utero arsenic exposure. In our recent studies, arsenic-induced hypomethylation of the ER-α gene is associated with aberrant hepatic gene expression, hepatocellular proliferation and carcinogenesis (Chen et al., 2004; Waalkes et al., 2004b). On the other hand, arsenic-induced hypermethylation of the tumor suppressor genes p53 and p16 has also been observed in an arsenic-exposed human population (Chanda et al., 2006). It is not uncommon for both hypomethylation and hypermethylation of DNA to occur during the carcinogenic process. Efforts are currently underway to examine the methylation status of selected individual genes following in utero arsenic exposure in connection with their expression.
DNA hypomethylation is an important mechanism involved in aberrant gene expression and carcinogenesis (Baylin et al., 1998; Goodman and Watson, 2002). In particular, it is thought that aberrant DNA methylation is important in the development of liver cancers (Goodman and Watson, 2002), and is a significant epigenetic mechanism that underlines the aberrant expression of genes involved in mouse liver carcinogenesis (Counts et al., 1997). In the present study, arsenic-exposed newborn mouse liver showed various gene expression changes, accounting for approximately 5% of genes on the array. These included glutathione-related genes, stress-related genes, genes encoding for metabolic enzymes and genes involved in insulin-like growth factor signaling. Previous work has shown these categories to be potentially related to aberrant cell growth and neoplasia (Xie et al., 2004).
Glutathione (GSH) systems play important roles in arsenic toxicity, adaptation, and perhaps carcinogenesis (NRC, 2001; Xie et al., 2004; Liu et al., 2006). In the present study, the expression of glutathione S-transferase mu (GST-mu), GST-pi, GST-alpha and GST-theta were all increased by in utero arsenic exposure in newborn mouse liver. The largest increases occurred in GST-theta (>350%). GSTs are a large group of enzymes and some can catalyze the conjugation of GSH with arsenic (Liu et al., 2001; Leslie et al., 2004; Xie et al., 2004). For instance, increases in GST expression and activity, particularly for GST-pi, play important roles in arsenic adaptation through increased cellular efflux of arsenic-GSH conjugates (Liu et al., 2001; Leslie et al., 2004). GST expression and activity in humans also are associated with altered arsenic metabolism (Chiou et al., 1997; Marnell et al., 2003) and GST polymorphisms may be a susceptibility factor for human arsenic intoxication (Marnell et al., 2003). Taken together, these data indicate that disruption of GST gene expressions and/or functions are consistent events associated with arsenic carcinogenicity and toxicity.
Oxidative stress is proposed to play an important role in arsenic toxicity and carcinogenesis (NRC, 2001; Xie et al., 2004). In addition to GSTs, other biomarkers for arsenic-induced oxidative stress such as metallothionein-1 (Liu et al., 2006) and methyl stress are evident. The expression of betainehomocysteine methyltransferase (BHMT) and thioether S-methyltransferase (TEMT), were decreased in arsenic-exposed fetal livers in prior (Liu et al., 2007) as well as in neonatal livers in the present study. The aberrant expression of the genes encoding for these enzymes would potentially contribute to abnormal production or utilization of SAM, a key donor for DNA methylation. In arsenic-induced hepatocellular carcinoma, decreased expression of BHMT is also evident (Liu et al., 2004, 2006). Most importantly, recent studies have indicated that altered methyl group metabolism could provide a potential mechanism for inducing epigenetic expression changes in the preimplanation embryo including genes such as BHMT and TEMT (Steele et al., 2005), which could disrupt the DNA methylation process, an important gene imprinting mechanism in prenatal development.
The expression of various metabolic enzyme genes was also altered. The increased expression of the female-predominant CYP2A4 in newborn liver is consistent with our prior observations in arsenic transplacental carcinogenesis studies in adult liver (Waalkes et al., 2004b; Liu et al., 2006) as well as in fetal liver (Liu et al., 2007). CYP2A4 encodes for hepatic microsomal androstenedione 15α-hydroxylase and its overexpression would imply altered steroid metabolism. On the other hand, the expression of male-predominant CYP genes, including CYP3A25, CYP2F2, CYP2J5 and CYP7B1, were decreased. The expression of HSD17β5 dehydrogenase, an enzyme catalyzing the transformation of 4-androstenedione (4-dione) into testosterone, was also decreased. These changes are consistent with male liver feminization and a role of aberrant estrogen signaling seen in our series of transplacental arsenic carcinogenesis studies (Waalkes et al., 2004b, 2006a,b; Liu et al., 2006, 2007; Shen et al., 2007).
We observed altered transcript levels of genes encoding for the insulin-like growth factor (IGF) signaling system, which can be influenced by ER and have been implicated in the lung oncogenic process (Gielen et al., 2005). The aberrant expression of insulin-like growth factors, such as IGFR2, is observed in the fetal mouse lung exposed to arsenic in utero (Shen et al., 2007) and in arsenic-exposed neonatal liver. Although the expression of IGFBP1 was increased in arsenic-exposed fetal lung (Shen et al., 2007), it was decreased in arsenic-exposed neonatal livers. Such differences could be due to different tissues and different time of sampling. In any event, the disruption of the IGF signaling systems could be an early event in arsenic hepatocarcinogenesis following in utero arsenic exposure. Aberrant promoter hypermethylation of 5′ CpG islands of key tumor suppressor genes, including p16 and p53, could also be important events in arsenic-induced tumorigenesis (Chanda et al., 2006).
Although the applied dose in these arsenic-treated mice (85 ppm) is much higher than those seen in environmentally exposed humans, they are non-toxic with respect to maternal or newborn body weights or pregnancy outcome (Waalkes et al., 2007). Furthermore, toxicokinetic differences exist between mice and humans. This exposure level in mice actually results in fetal blood levels of inorganic arsenic (Devesa et al., 2006) that are similar to blood levels in humans chronically exposed to much lower, but commonly occurring environmental levels of arsenic in the drinking water (∼0.4 ppm; Pi et al., 2002).
In summary, the present study demonstrated that in utero exposure to inorganic arsenic in the drinking water resulted in the accumulation of arsenic in the newborn liver, the alteration of DNA methylation in GC-rich regions, and alterations in gene expression in newborn mouse livers potentially related to arsenic adaptation or carcinogenesis. These findings indicate that in utero arsenic exposure can trigger early molecular changes, including potentially aberrant DNA methylation, which could alter gene expression and possibly precipitate genetic reprogramming. All of these factors could potentially lead to hepatocarcinogenesis much later in life. A recent epidemiology study in Chile found increased lung cancer mortality in young adults was related to in utero or early life exposure to arsenic (Smith et al., 2006). Thus, protection of pregnant women from excessive arsenic exposure could help prevent arsenic carcinogenesis in humans.
Acknowledgements
The authors thank Drs. Erik Tokar, Jean-Francosis Coppin and Larry Keefer for their critical review on this manuscript. Research was supported in part by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, The National Center for Toxicogenomics and National Cancer Institute under contract NO1-CO-12400. The authors have no competing financial interest and the content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services.
References
- Anderson LM, Diwan BA, Fear NT, Roman E. Critical windows of exposure for children's health: cancer in human epidemiological studies and neoplasms in experimental animal models. Environ. Health Perspect. 2000;108(Suppl 3):573–594. doi: 10.1289/ehp.00108s3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aposhian HV, Aposhian MM. Arsenic toxicology: five questions. Chem. Res. Toxicol. 2006;19:1–15. doi: 10.1021/tx050106d. [DOI] [PubMed] [Google Scholar]
- Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv. Cancer Res. 1998;72:141–196. [PubMed] [Google Scholar]
- Centeno JA, Mullick FG, Martinez L, Page NP, Gibb H, Longfellow D, Thompson C, Ladich ER. Pathology related to chronic arsenic exposure. Environ. Health Perspect. 2002;110(Suppl 5):883–886. doi: 10.1289/ehp.02110s5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chanda S, Dasgupta UB, Guhamazumder D, Gupta M, Chaudhuri U, Lahiri S, Das S, Ghosh N, Chatterjee D. DNA hypermethylation of promoter of gene p53 and p16 in arsenic-exposed people with and without malignancy. Toxicol. Sci. 2006;89:431–437. doi: 10.1093/toxsci/kfj030. [DOI] [PubMed] [Google Scholar]
- Chen CJ, Yu MW, Liaw YF. Epidemiological characteristics and risk factors of hepatocellular carcinoma. J. Gastroenterol. Hepatol. 1997;12:S294–S308. doi: 10.1111/j.1440-1746.1997.tb00513.x. [DOI] [PubMed] [Google Scholar]
- Chen H, Li S, Liu J, Diwan BA, Barrett JC, Waalkes MP. Chronic inorganic arsenic exposure induces hepatic global and individual gene hypomethylation: implications for arsenic hepatocarcinogenesis. Carcinogenesis. 2004;25:1779–1786. doi: 10.1093/carcin/bgh161. [DOI] [PubMed] [Google Scholar]
- Chiou HY, Hsueh YM, Hsieh LL, Hsu LI, Hsu YH, Hsieh FI, et al. Arsenic methylation capacity, body retention, and null genotypes of glutathione S-transferase M1 and T1 among current arsenic-exposed residents in Taiwan. Mutat. Res. 1997;386:197–207. doi: 10.1016/s1383-5742(97)00005-7. [DOI] [PubMed] [Google Scholar]
- Cook JD, Davis BJ, Cai SL, Barrett JC, Conti CJ, Walker CL. Interaction between genetic susceptibility and early life environmental exposure determines tumor suppressor gene penetrance. Proc. Natl. Acad. Sci. U.S.A. 2005;102:8644–8649. doi: 10.1073/pnas.0503218102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Counts JL, McClain RM, Goodman JI. Comparison of effect of tumor promoter treatments on DNA methylation status and gene expression in B6C3F1 and C57BL/6 mouse liver and in B6C3F1 mouse liver tumors. Mol. Carcinog. 1997;18:97–106. doi: 10.1002/(sici)1098-2744(199702)18:2<97::aid-mc5>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Devesa V, Adair BM, Liu J, Waalkes MP, Diwan BA, Styblo M, Thomas DJ. Speciation of arsenic in the maternal and fetal mouse tissues following gestational exposure to arsenite. Toxicology. 2006;224:147–155. doi: 10.1016/j.tox.2006.04.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gielen SC, Hanekamp EE, Blok LJ, Huikeshoven FJ, Burger CW. Steroid-modulated proliferation of human endometrial carcinoma cell lines: any role for insulin-like growth factor signaling? J. Soc. Gynecol. Investig. 2005;12:58–64. doi: 10.1016/j.jsgi.2004.08.003. [DOI] [PubMed] [Google Scholar]
- Goodman JI, Watson RE. Altered DNA methylation: a secondary mechanism involved in carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 2002;42:501–525. doi: 10.1146/annurev.pharmtox.42.092001.141143. [DOI] [PubMed] [Google Scholar]
- IARC (International Agency for Research on Cancer) Some Drinking Water Disinfectants and Contaminants, Including Arsenic. IARC; Lyon, France: 2004. IARC monographs on the evaluation of carcinogenic risks to humans, vol. 84: arsenic in drinking water. pp. 269–477. [PMC free article] [PubMed] [Google Scholar]
- Leslie EM, Haimeur A, Waalkes MP. Arsenic transport by the human multidrug resistance protein 1 (MRP1/ABCC1). Evidence that a tri-glutathione conjugate is required. J. Biol. Chem. 2004;279:32700–32708. doi: 10.1074/jbc.M404912200. [DOI] [PubMed] [Google Scholar]
- Liu J, Chen H, Miller DS, Saavedra JE, Keefer LK, Johnson DR, Klaassen CD, Waalkes MP. Overexpression of glutathione S-transferase II and multidrug resistance transport proteins is associated with acquired tolerance to inorganic arsenic. Mol. Pharmacol. 2001;60:302–309. doi: 10.1124/mol.60.2.302. [DOI] [PubMed] [Google Scholar]
- Liu J, Xie Y, Ward JM, Diwan BA, Waalkes MP. Toxicogenomic analysis of aberrant gene expression in liver tumors and nontumorous livers of adult mice exposed in utero to inorganic arsenic. Toxicol. Sci. 2004;77:249–257. doi: 10.1093/toxsci/kfh055. [DOI] [PubMed] [Google Scholar]
- Liu J, Xie Y, Ducharme DMK, Shen J, Diwan BA, Merrick BA, Grissom SF, Tucker CJ, Paules PS, Tennant R, Waalkes MP. Global gene expression associated with hepatocarcinogenesis in adult male mice induced by in utero arsenic exposure. Environ. Health Perspect. 2006;114:404–411. doi: 10.1289/ehp.8534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Xie Y, Cooper R, Ducharme DMK, Tennant R, Diwan BA, Waalkes MP. Transplacental exposure to inorganic arsenic at a hepatocarcinogenic dose induces fetal gene expression changes in mice indicative of aberrant estrogen signaling and disrupted steroid metabolism. Toxicol. Appl. Pharmacol. 2007 doi: 10.1016/j.taap.2007.01.018. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T, Liu J, LeCluyse EL, Zhou YS, Cheng ML, Waalkes MP. Application of cDNA microarray to the study of arsenic-induced liver diseases in the population of Guizhou, China. Toxicol. Sci. 2001;59:185–192. doi: 10.1093/toxsci/59.1.185. [DOI] [PubMed] [Google Scholar]
- Marnell LL, Garcia-Vargas GG, Chowdhury UK, Zakharyan RA, Walsh B, Avram MD, Kopplin MJ, Cebrian ME, Silbergeld EK, Aposhian HV. Polymorphisms in the human monomethylarsonic acid (MMA(V)) reductase/hGSTO1 gene and changes in urinary arsenic profiles. Chem. Res. Toxicol. 2003;16:1507–1513. doi: 10.1021/tx034149a. [DOI] [PubMed] [Google Scholar]
- Mazumder DN. Effect of chronic intake of arsenic-contaminated water on liver. Toxicol. Appl. Pharmacol. 2005;206:169–175. doi: 10.1016/j.taap.2004.08.025. [DOI] [PubMed] [Google Scholar]
- NRC (National Research Council) Arsenic in the Drinking Water (Update) NRC, National Academy; Washington, DC: 2001. pp. 1–225. [Google Scholar]
- Okoji RS, Yu RC, Maronpot RR, Froines JR. Sodium arsenite administration via drinking water increases genome-wide and Ha-ras DNA hypomethylation in methyl-deficient C57BL/6J mice. Carcinogenesis. 2002;23:777–785. doi: 10.1093/carcin/23.5.777. [DOI] [PubMed] [Google Scholar]
- Pi J, Yamauchi H, Kumagai Y, Sun G, Yoshida T, Aikawa H, Hopenhayn-Rich C, Shimojo N. Evidence for induction of oxidative stress caused by chronic exposure of Chinese residents to arsenic contained in drinking water. Environ. Health Perspect. 2002;110:331–336. doi: 10.1289/ehp.02110331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Liu J, Xie Y, Diwan BA, Waalkes MP. Fetal onset of aberrant gene expression relevant to pulmonary carcinogenesis in lung adenocarcinoma development induced by in utero arsenic exposure. Toxicol. Sci. 2007;95:313–320. doi: 10.1093/toxsci/kfl151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AH, Marshall G, Yuan Y, Ferreccio C, Liaw J, von Ehrenstein O, Steinmaus C, Bates MN, Selvin S. Increased mortality from lung cancer and bronchiectasis in young adults after exposure to arsenic in utero and in early childhood. Environ. Health Perspect. 2006;114:1293–1296. doi: 10.1289/ehp.8832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele W, Allegrucci C, Singh R, Lucas E, Priddle H, Denning C, Sinclair K, Young L. Human embryonic stem cell methyl cycle enzyme expression: modelling epigenetic programming in assisted reproduction? Reprod. Biomed. Online. 2005;10:755–766. doi: 10.1016/s1472-6483(10)61120-0. [DOI] [PubMed] [Google Scholar]
- Thomas DJ, Li J, Waters SB, Styblo M. Arsenic (+3 oxidation state) methyltransferase and the methylation of arsenicals. Exp. Biol. Med. 2007 in press. [PMC free article] [PubMed] [Google Scholar]
- Waalkes MP, Ward JM, Liu J, Diwan BA. Transplacental carcinogenicity of inorganic arsenic in the drinking water: induction of hepatic, ovarian, pulmonary, and adrenal tumors in mice. Toxicol. Appl. Pharmacol. 2003;186:7–17. doi: 10.1016/s0041-008x(02)00022-4. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Ward JM, Diwan BA. Induction of tumors of the liver, lung, ovary and adrenal in adult mice after brief maternal gestational exposure to inorganic arsenic: promotional effects of postnatal phorbol ester exposure on hepatic and pulmonary, but not dermal cancers. Carcinogenesis. 2004a;25:133–141. doi: 10.1093/carcin/bgg181. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Liu J, Chen H, Xie Y, Achanzar WE, Zhou YS, Cheng ML, Diwan BA. Estrogen signaling in livers of male mice with hepatocellular carcinoma induced by exposure to arsenic in utero. J. Natl. Cancer Inst. 2004b;96:466–474. doi: 10.1093/jnci/djh070. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Liu J, Ward JM, Powell DA, Diwan BA. Urogenital system cancers in female CD1 mice induced by in utero arsenic exposure are exacerbated by postnatal diethylstilbestrol treatment. Cancer Res. 2006a;66:1337–1445. doi: 10.1158/0008-5472.CAN-05-3530. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Liu J, Ward JM, Diwan BA. Enhanced urinary bladder and liver carcinogenesis in male CD1 mice exposed to transplacental inorganic arsenic and postnatal diethylstilbestrol or tamoxifen. Toxicol. Appl. Pharmacol. 2006b;215:295–305. doi: 10.1016/j.taap.2006.03.010. [DOI] [PubMed] [Google Scholar]
- Waalkes MP, Liu J, Diwan BA. Transplacental arsenic carcinogenesis in mice. Toxicol. Appl. Pharmacol. 2007 2007 January 12; doi: 10.1016/j.taap.2006.12.034. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson RE, Curtin GM, Doolittle DJ, Goodman JI. Progressive alterations in global and GC-rich DNA methylation during tumorigenesis. Toxicol. Sci. 2003;75:289–299. doi: 10.1093/toxsci/kfg190. [DOI] [PubMed] [Google Scholar]
- Xie Y, Trouba KJ, Liu J, Waalkes MP, Germolec DR. Biokinetics and subchronic toxic effects of oral arsenite, arsenate, monomethylarsonic acid, and dimethylarsinic acid in v-Ha-ras transgenic (Tg. AC) mice. Environ. Health Perspect. 2004;112:1255–1263. doi: 10.1289/txg.7152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao CQ, Young MR, Diwan BA, Coogan TP, Waalkes MP. Association of arsenic-induced malignant transformation with DNA hypomethylation and aberrant gene expression. Proc. Natl. Acad. Sci. U.S.A. 1997;94:10907–10912. doi: 10.1073/pnas.94.20.10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YS, Du H, Cheng M-L, Liu J, Zhang XJ, Xu L. The investigation of death from diseases caused by coal-burning type of arsenic poisoning. Chin. J. Endemiol. 2002;21:448–484. [Google Scholar]