

Abstract
The mechanism of the epoxidation of 2-cyclohexen-1-one with tert-butyl hydroperoxide mediated by DBU was studied by a combination of experimental kinetic isotope effects and theoretical calculations. A large 12C/13C (k12C/k13C) isotope effect of ≈1.032 was observed at the C3 (β) position of cyclohexenone, while a much smaller 12C/13C isotope effect of 1.010 was observed at the C2 (α) position. Qualitatively, these results are indicative of nucleophilic addition to the enone being the rate-limiting step. Theoretical calculations support this interpretation. Transition structures for the addition step lead to predicted isotope effects that approximate the experimental values, while the predicted isotope effects for the ring-closure step are not consistent with experiment. The calculations correctly favor a rate-limiting addition step, but suggest that the barriers for the addition and ring-closure steps are crudely similar in energy. The stereochemistry of these epoxidations is predicted to be governed by a preference for an initial axial addition, and the role of this preference in experimental diastereoselectivity observations is discussed.
Introduction
The epoxidation of α,β-unsaturated carbonyls with hydroperoxides under basic conditions, the Weitz-Scheffer reaction, is a valuable transformation in organic synthesis.1,2 The mildness of the reaction conditions and the high selectivity for the oxidation of electron-poor alkenes has allowed the application of these reactions to diverse complex structures.3 The oldest and simplest reaction conditions utilize alkaline hydrogen peroxide, but reactions using alkyl hydroperoxides have advantages in many circumstances.4,5 Diverse approaches to enantioselective epoxidations have been developed,6 including catalysis by polyamino acids (the Juliá-Colonna epoxidation)7 and chiral phase-transfer catalysts,8 reactions employing chiral alkyl hydroperoxides,9 and reactions using zinc reagents and O2 in the presence of a chiral alcohol.10
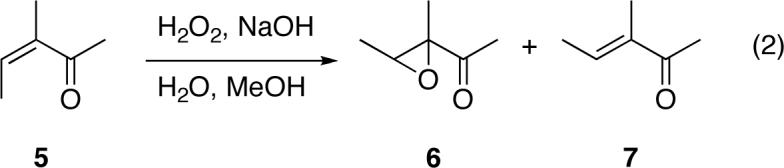
The accepted two-step mechanism of these reactions (eq 1), first proposed by Bunton and Minkoff,2a involves the conjugate addition of a hydroperoxy or alkylperoxy anion 2 to afford a peroxyenolate 3. The peroxyenolate then undergoes a ring-closing intramolecular nucleophilic substitution of C2 on the O-O bond to afford the epoxide 4. Strong evidence for an intermediate includes the nonstereospecificity of these reactions; unlike epoxidations with peracids,11 the stereochemistry of the starting alkene is not necessarily retained in the epoxide. For example, the epoxidations of E- and Z-3-methyl-3-penten-2-one with basic H2O2 in methanol both afford predominantly the E epoxide product.12

While the overall mechanism is well-established, a consistent picture of the rate-limiting step has been elusive. In the reaction of Z-3-methyl-3-penten-2-one (5) with alkaline hydrogen peroxide, House found that Z-E isomerization occurred at rates comparable to epoxidation (eq 2).12 This suggests significant reversibility of the initial addition under these conditions. On the other hand, substituent effect studies of the epoxidation of 4-aryl-3-buten-2-ones have been interpreted as favoring a rate-limiting addition step. 13 In a careful study of the epoxidation of β-deuterium labeled phenyl vinyl ketone with hydrogen peroxide, Kelly and Roberts found that Z-E isomerization was much faster than epoxidation under Juliá-Colonna conditions using polyleucine as catalyst.14 This strongly supports rate-limiting ring closure. In contrast, isomerization and epoxidation were comparable in the absence of the polyleucine. This suggests that the rate-limiting step may vary depending on the detailed reaction conditions. In addition, the rates of additions on nucleophiles to enones varies substantially with substitution, and the rate of the ring closure step should vary with conformational freedom in the intermediate, so enone structure may affect the rate-limiting step.
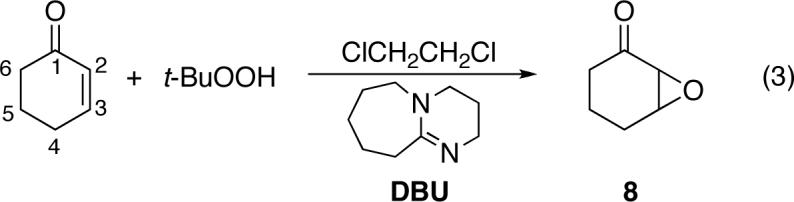
This uncertainty in the rate-limiting step and a lack of knowledge of the structural characteristics of the rate-limiting transition state thwarts a detailed understanding of diastereoselectivity or enantioselectivity in these reactions. This is particularly true for epoxidations with alkyl hydroperoxides under typical synthetic conditions and for epoxidations of cyclic enones, where little mechanistic information is available. We describe here a study of the epoxidation of cyclohexenone with tert-butyl hydroperoxide (t-BuOOH) / DBU (eq 3) using a combination of experimental kinetic isotope effects (KIEs) and theoretical calculations. The results provide insight into the stereoselectivity of these reactions and should aid the further development of stereocontrolled epoxidations.
Results
Experimental Isotope Effects
The epoxidation of an enone using a combination of an alkyl hydroperoxide and DBU in an aprotic solvent was first reported by Schlessinger and Poss.3b Yadav demonstrated the utility of these conditions with a range of α,β-unsaturated carbonyls,5 and they have been commonly applied in enantioselective reactions using chiral alkyl hydroperoxides.9 Under the prototypical epoxidation conditions employed here (stoichiometric DBU and t-BuOOH in dichloroethane at 22 °C), cyclohexenone is converted to 8 cleanly and essentially quantitatively.
The 13C KIEs (k12C/k13C) for the epoxidation of cyclohexenone were determined combinatorially by NMR methodology at natural abundance.15 Two reactions of cyclohexenone were taken to 83±2% and 89±2% conversion, and the unreacted cyclohexenone was recovered by an extractive workup followed by flash chromatography and fractional distillation. The samples of recovered cyclohexenone were analyzed by 13C NMR, along with standard samples that had not been subjected to the reaction conditions. The change in isotopic composition in each position was determined relative to the α-methylene carbon in cyclohexenone,16 with the assumption that isotopic fractionation in this position was negligible. From the percentage conversions and the changes in isotopic composition, the KIEs were calculated as previously described.15
Table 1 shows the results of two separate KIE determinations (each based on six sets of spectra) for each of the two reactions. The independent sets of 13C KIEs agree within the standard deviation of the measurements, though the KIEs here are more variable than in previous KIE determinations on cyclohexenone.17 Despite the variability, the qualitative features of the KIEs are apparent. Only the C2 and C3 KIEs differ significantly from unity, with a relatively large C3 isotope effect and a smaller KIE at C2. The qualitative interpretation of the large C3 KIE is that the rate-limiting step involves a substantial bonding change at C3, as would be expected for a rate-limiting addition of t-BuOO− to cyclohexenone. The medium-sized C2 KIE is less readily interpreted. A more detailed interpretation of these KIEs will be possible with the aid of theoretical calculations.
Table 1.
Experimental and predicted 13C kinetic isotope effects (k12C/k13C) for the epoxidation of cyclohexenone with t-BuOOH / DBU.a
| C1 | C2 | C3 | C4 | C5 | |
|---|---|---|---|---|---|
| Experimental KIEs | |||||
| Expt 1 | 0.998 | 1.010 | 1.033 | 1.005 | 0.996 |
| Expt 2 | 1.003 | 1.009 | 1.027 | 1.002 | 0.999 |
| Expt 1 | b | 1.010 | 1.037 | 1.002 | 0.995 |
| Expt 2 |
b |
1.010 |
1.029 |
1.008 |
1.003 |
| average | 1.001 | 1.010 | 1.032 | 1.004 | 0.998 |
| std dev | 0.000 | 0.004 | 0.003 | 0.004 | |
| Predicted KIEs | |||||
| axial addition (10) | 1.004 | 1.008 | 1.027 | 0.997 | 0.999 |
| equatorial addition (13) | 1.004 | 1.007 | 1.031 | 1.001 | 1.001 |
| axial ring closure (12) | 1.006 | 1.012 | 0.997 | 0.996 | 0.999 |
| equatorial ring closure (15) | 1.005 | 1.012 | 1.000 | 0.999 | 1.001 |
The experimental isotope effects were measured at 22 °C. The predicted isotope effects were calculated for 25 °C.
Due to practical limitations, the NMR data was collected with a time between pulses inadequate for relaxation of the carbonyl carbon, precluding an accurate integration.
Theoretical Mechanisms
The epoxidation of cyclohexenone with t-BuOOH mediated by the simplified DBU model 9 was studied in B3LYP calculations using 6−31G* and 6−31+G** basis sets and using a PCM solvent model for dichloroethane with full geometry optimization in all cases. The involvement of charged species limits the reliability of a calculational mechanistic study of this reaction by itself, but consideration of the experimental isotope effects allows assessment of the calculational predictions. In turn, the prediction of isotope effects from the theoretical models facilitates a detailed interpretation of the experimental isotope effects.
A series of conformationally varying pathways for the epoxidation were explored. The diastereotopic faces of a half-chair cyclohexenone may undergo addition of the tert-butylperoxy anion in either an “axial” or an “equatorial” orientation. (The discussion here will consistently use the axial / equatorial notation to denote epoxidation pathways and products, always basing the description on the orientation of the initial addition.) The pathways explored allowed for 1) axial versus equatorial attack on the cyclohexenone, 2) two possible orientations of DBU model 9, and 3) an anti (O–O--C=C dihedral angle of approximately 180°) versus gauche (O–O--C=C approximately 75°) orientation of the attacking oxygen atoms versus the carbon-carbon double bond. Only the lowest-energy axial and equatorial pathways from the B3LYP/6−31+G**/PCM calculations are presented here (Figure 1); structures and energies associated with the full set of pathways as well as gas-phase and 6−31G* calculations are given in the Supporting Information.
Figure 1.

Lowest energy axial (top) and equatorial (bottom) pathways for the epoxidation of cyclohexenone with t-BuOOH mediated by DBU model 9 in B3LYP/6−31+G**/PCM calculations. Energies (B3LYP/6−31+G**/PCM + zpe) are in kcal/mol relative to separate starting materials.
The lowest-energy calculated pathway involves an axial addition of the peroxide with oxygens oriented anti versus the carbon-carbon double bond, as in transition structure 10. This transition structure leads, by a minimum-energy path, to the zwitterionic enolate/iminium intermediate 11. As a side process, 11 could readily undergo proton transfer to afford neutrals 9 and 3-t-butylperoxycyclohexanone. However, 3-t-butylperoxycyclohexanone is predicted to be 9.6 kcal/mol uphill in free energy (B3LYP/6−31+G**/PCM + zpe + harmonic entropy and enthalpy estimates at 25 °C) from separate starting materials. From this, 3-t-butylperoxycyclohexanone should not build up under the reaction conditions and its formation must be reversible. Alternative side processes such as proton transfer to the enolate oxygen to afford a 3-t-butylperoxycyclohexenol can also be envisioned but are subject to the same limitation.
A low barrier is predicted for 11 to complete the epoxidation by ring-closure via transition structure 12. The alternative equatorial process via 13, 14, and 15 is similar but 2−3 kcal/mol higher in energy. For the axial pathway, the addition step is predicted to be rate limiting, since 10 is 0.7 kcal/mol higher in energy than 12. In notable contrast, the equatorial pathway would have the ring-closure step as mainly rate limiting. In either case, these relative energies should not be considered as highly reliable due to the limitations of the calculational model, the solvent model, and the calculational method itself. A more reliable conclusion from the calculational results is that the addition and ring-closure steps should be crudely comparable in barrier. This is consistent with the idea that the rate-limiting step may depend on the detailed reaction conditions.
The formation of termolecular transition structures such as 10 from separate reactants should involve a substantial entropic penalty. Including an entropy estimate based on the harmonic frequencies at 25 °C, the free-energy barrier (at standard state) for reaction via 10 would be 37.1 kcal/mol. This is too high by about 12−15 kcal/mol for a reaction that proceeds in less than a day at 25 °C. However, the consideration of two issues may ameliorate this problem. The first issue is that the gas phase entropy of compounds is decreased on dissolution in organic solvents, typically by 15 e.u.18 From this, the entropy loss for forming a termolecular transition state might be expected to be decreased compared to a gas-phase calculation by roughly 30 e.u., decreasing the free-energy barrier by 9 kcal/mol. The second issue is that t-BuOOH may form a complex with DBU. The formation of a complex between t-BuOOH and 9 is predicted to be downhill by 4.2 kcal/mol in dichloroethane (B3LYP/6−31+G**/PCM + zpe) but is predicted to be endergonic by 5.6 kcal/mol after inclusion of the gas-phase entropy estimate. However, to roughly evaluate the energetics of complex formation under the solution reaction conditions, one equivalent of DBU was added to a 1 M solution of t-BuOOH in dichloroethane at 22 °C. This leads to a temperature rise of 4 °C. Estimating the heat capacity of the solution as that of dichloroethane (129 J mol−1 K−1),19 this temperature rise translates into a heat of mixing at 1 M of 1.5 kcal/mol, suggesting that a substantial portion of the t-BuOOH / DBU mixture forms a complex. (Complete proton transfer is unlikely – t-BuOOH and DBUH+ have similar pKas in water but proton transfer will be disfavored in less polar solvents.) The predicted free-energy barrier for reaction of a t-BuOOH / 9 complex with cyclohexenone is only 31.5 kcal/mol. Considering these issues along with the limitations of the calculational and solvent models, the calculated barrier is reasonably consistent with the facility of the experimental reaction.
Predicted Isotope Effects
The 13C KIEs associated with the transition structures in Figure 1 were predicted from the scaled theoretical vibrational frequencies20 using conventional transition state theory by the method of Bigeleisen and Mayer.21 Tunneling corrections were applied using the one-dimensional infinite parabolic barrier model.22 Such KIE predictions have proven highly accurate in reactions not involving hydrogen transfer, so long as the calculation accurately depicts the mechanism and transition state geometry.23
The results are summarized in Table 1. The addition transition structures 10 and 13 lead to predicted KIEs of about 1.030 at C3 and 1.008 at C2, with small isotope effects predicted for the remaining carbons. In contrast, ring-closure transition structures 12 and 15 lead to predicted KIEs at C3 near unity and 1.012 at C2. These predictions fit well with conventional expectations. In the addition step, C3 is undergoing a major σ-bonding change and is expected to exhibit a substantial normal isotope effect, while the bonding change at C2 is more minor and the predicted KIE, while still significantly greater than unity, is low. In the ring-closure step, little change is occurring at C3 so the predicted isotope effect is near unity. The predicted isotope effect for C2 at 1.012 is near the low end of primary carbon KIEs (KIEs associated with a σ-bonding change in the rate-limiting step) but is consistent with the relatively low KIEs observed in other epoxidation reactions.23d,24
Stereoselectivity with 4-Methylcyclohexenone
The predicted preference for the axial addition pathway should have stereochemical consequences. With cyclohexenones containing a substituent at C4, C5, or C6, the preferred epoxide product should arise from adding the substituent to 10 in its most favorable position, and this would normally be assumed to have the group in a pseudo-equatorial orientation. However, an equatorial substituent at C4 in 10 could sterically interact with the incoming tert-butylperoxy anion, so that the preferred transition state geometry is not obvious.
To explore this issue, the epoxidation of 4-methylcyclohexenone was investigated. The three lowest-energy transition structures for the addition step, 16 – 18, are shown in Figure 2. The axial addition transition structures 16 and 17, analogues of 10, differ by having pseudo-equatorial and pseudo-axial orientations of the methyl group, respectively, while 18, an analogue of 13, is the most favored transition structure for equatorial addition. Notably, the lowest-energy transition structure 17 places the methyl group in a pseudo-axial position. When the methyl group is pseudo-equatorial, as in 16, the preferred anti orientation of the attacking oxygen atoms versus the carbon-carbon double bond is distorted to avoid interaction of the distal oxygen with the methyl group, so that the O–O--C=C dihedral angle is decreased from 175° in 10 to 157° in 16. With a pseudo-axial methyl group as in 17, the anti orientation of the peroxide is unhindered (the O–O--C=C angle in 17 is 169°), and 17 is favored despite the concomitant strain of the pseudo-axial methyl group. The energy cost of proceeding through an equatorial addition as in 18 remains higher, in part due to a C3-C4 eclipsing interaction present in 18 (and 13) that is not present in 16 and 17 (or 10).
Figure 2.

Lowest-energy transition structures for the addition step of the epoxidation of 4-methylcyclohexenone with t-BuOOH mediated by DBU model 9 in B3LYP/6−31+G**/PCM (for dichloroethane) calculations. Energies (B3LYP/6−31+G**/PCM + zpe) are in kcal/mol relative to separate starting materials. Many hydrogens have been removed for clarity.
Discussion
The natural-abundance 13C KIE determination did not work as well as normal for the current reaction. Aside from the relatively high run-to-run variability in the 13C KIE, particularly at C3 where the standard deviation is 0.004, it is bothersome that the C4 and C5 KIEs were not consistently close to their expected values of near unity. Nonetheless, the overall pattern in the KIEs was sufficiently reproducible for the purpose at hand of distinguishing the rate-limiting step in the epoxidation reaction.
Some care must be taken in the qualitative interpretation of these isotope effects. The observed C3 KIE fits well with conventional expectations and with the β 13C KIEs observed in other additions to α,β-unsaturated carbonyl compounds (Figure 3).25,26 However, as we have previously reported, the presence of weak bonds in reactive intermediates can result in secondary 13C KIEs (13C KIEs that are not the result of a σ-bonding change in the rate-limiting step) that are so large as to mimic primary 13C KIEs.27 Because the C3–O bond in intermediates resembling 3 should be weak, the KIE to be expected at C3 if ring closure were the rate-limiting step was not clear. In addition, the observed C2 KIE of 1.010 is of ambiguous diagnostic value. As noted above, epoxidation reactions tend to exhibit small primary 13C KIEs, in part because the equilibrium isotope effect for an epoxidation is significantly inverse.23d Because of this, the 1.010 is qualitatively consistent with rate-limiting ring closure. However, the α carbon in other additions to α,β-unsaturated carbonyl compounds can exhibit a significantly normal 13C KIE (Figure 3), such as 1.007 in free-radical polymerization.
Figure 3.
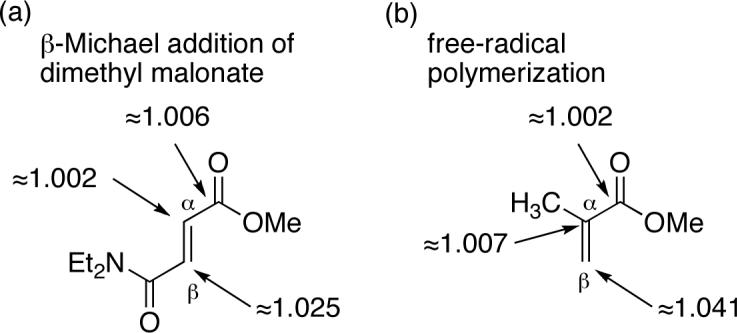
13C KIEs for other additions to α,β-unsaturated carbonyl compounds. (a) Base-catalyzed β-addition of dimethyl malonate in methanol at 64 °C. See ref 25. (b) Free-radical polymerization at 60 °C. See ref 26.
The theoretically predicted isotope effects serve to pin down the rate-limiting step. Despite the concerns expressed above, the predicted C3 13C KIEs based on 12 and 15 for rate-limiting ring closure are near unity. The large observed 13C KIE of approximately 1.030 is thus inconsistent with rate-limiting closure of the epoxide ring. In comparing the experimental KIEs with the predicted KIEs based on 10 and 13 for rate-limiting addition, the match-up is not as good as typically observed in other examples.23 However, considering the difficulty of accurately modeling this reaction due to the involvement of charged intermediates, along with the spread of the experimental KIEs, the correspondence of experimental and predicted KIEs is quite reasonable, particularly for the key C3 and C2 positions. Overall, the KIEs very strongly support rate-limiting addition to the enone.
The choice of rate-limiting step and the factors affecting this choice may be best understood by considering the relative facility of forward versus backward reactions for the enolate intermediates such as 11 or 14. The ring closure of 11 faces a very low barrier because of the weakness of the O–O bond and because its geometry is well positioned for the SN2 displacement by C2. A slight repositioning of the 9•H+ counter-ion to hydrogen bond with the tert-butoxy oxygen appears to aid in catalyzing the displacement. On the other hand, the cleavage of 11 back to starting materials is also understandably facile. From the isotope effect analysis above, we know that the barrier for the backward cleavage of the enolate in reality is higher than that for ring closure. If the ring-closure step were 20% rate-limiting, as would be expected from the calculated relative energies of 10 and 12, the predicted KIE for C3 would be 1.022, at the limit of consistency with experiment. From this, it appears that the difference in energy between addition and ring-closure transition states in reality is at least as high as the calculated difference between 10 and 12.
The higher energy of 14 along the equatorial pathway versus 11 on the axial pathway may be ascribed to a near eclipsing interaction of C3 and C4 in 14 versus the nearly perfectly staggered 11. The barrier for backward cleavage of 14 via 13 is slightly lower than ring closure via 15, so that ring closure in this case would be predicted to be mainly rate limiting. The difference in predicted rate limiting steps for the axial and equatorial pathways may be thought of as resulting from the ease of backward cleavage of 14 via 13, since this loses the strain of the eclipsing interaction.
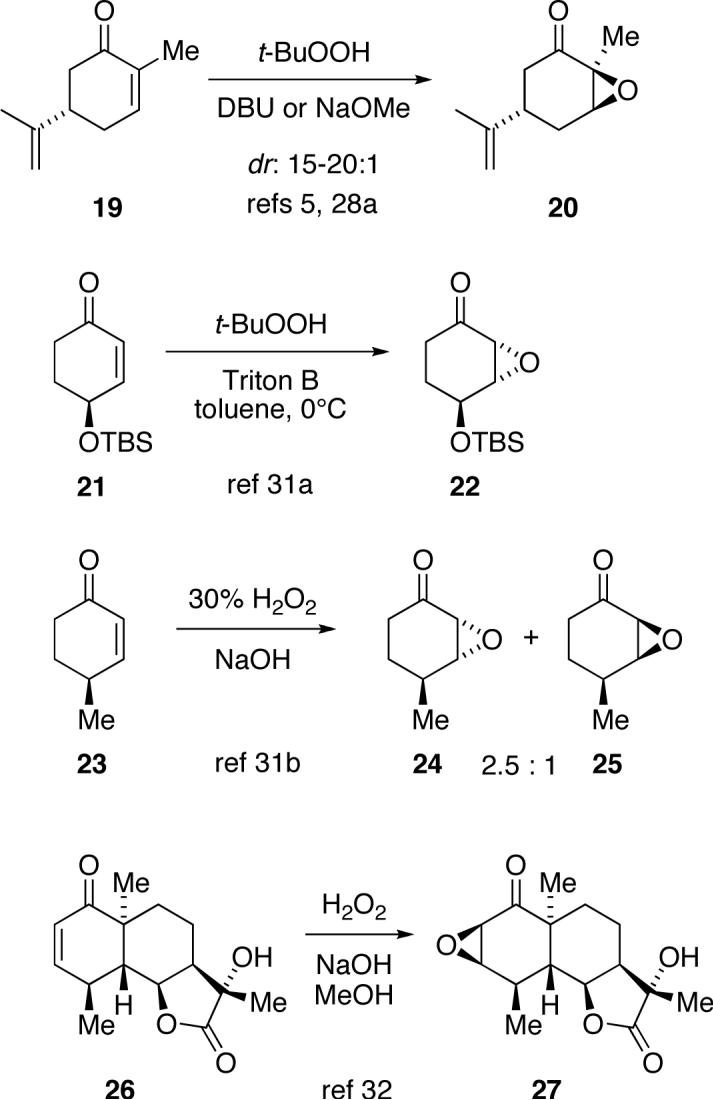
With the knowledge that the addition step is rate limiting, consideration of the calculated transition structures provides insight into the stereochemistry of these epoxidations with substituted cyclohexenones. For 5-substituted cyclohexenones, there is a substantial preference for both hydrogen peroxide- and alkyl hydroperoxide-mediated reactions to afford axial epoxidation.28,29 (We are again defining “axial” epoxidation based on the orientation of the initial addition, assuming that substituents on the cyclohexenone have been placed in a preferred pseudo-equatorial position.) This is illustrated by the widely-used epoxidation of carvone (19) to afford 20.5,296-Substituted cyclohexenones also tend to be epoxidized axially,30 though there has been one report of an exception.31 The observation of axial epoxidation in these cases is in agreement with the calculated preference for axial transition structure 10 over 13 / 15 in the equatorial pathway.
The epoxidation of some other substituted cyclohexenones has until now been stereochemically more enigmatic. With 4-monosubstituted cyclohexenones, such as 21 and 23, there is a tendency for epoxidation to occur on the face opposite the 4-substituent, as in 22 and 24.32 This would be considered equatorial epoxidation if the 4-substituent is pseudo-equatorial during the reaction. However, when the 4-substituent is held rigidly pseudo-equatorial, as in 26, axial epoxidation predominates.33 This strange reversal of stereochemistry between 23 and 26 is difficult to explain if the methyl group in 23 remains pseudo-equatorial. However, the complete set of observations is understandable from the theoretical prediction that the preferred transition state has the 4-substituent flipped to axial, as in 17. When the 4-substituent is equatorial, there is a substantial steric interaction with the distal oxygen of an incoming hydroperoxy or tert-butylhydroperoxy anion. This hindered attack would lead to the minor product 25 from 23. The major product 24 could potentially be formed in two ways, either by placing the methyl group pseudo-axial in an axial transition state analogous to 10, as in 17, or by placing the methyl group pseudo-equatorial in an equatorial pathway analogous to 13 / 15, as in 18. The energy cost of placing the methyl group pseudo-axial, while significant,34 is still less than the substantial cost of the equatorial pathway, and the axial epoxidation pathway remains preferred. In 26, no ring-flip is possible, and the general preference for axial epoxidation affording 27 is likely augmented by a steric effect of the axial bridgehead methyl group inhibiting the rate-limiting ring closure step for the non-observed equatorial epoxidation.
Conclusions
In epoxidations of electron-deficient alkenes mediated by hydroperoxides under basic conditions, reactivity and stereoselectivity will be controlled by the energy of the transition state for the rate-limiting step. Either the addition step or the ring-closure step may in principle be rate limiting, with no obvious qualitative reason to favor one over the other. In the work of Kelly and Roberts on the epoxidation of phenyl vinyl ketone under Juliá-Colonna conditions, it appears clear that the ring-closure step is rate limiting.14 Here, in the first detailed mechanistic study with a cyclic enone, the experimental kinetic isotope effects supplemented by theoretically predicted isotope effects strongly support the addition step as being rate limiting.
The theoretical calculations here correctly predict that the addition step should be rate-limiting, and it is notable that the literature stereochemical observations are generally consistent with a preferred rate-limiting axial-addition step as stereochemically controlling. However, the similarity of barriers for the addition and ring-closure steps should be recognized. Because of this, it is reasonable to suppose that the rate-limiting step may change depending on the detailed reaction conditions and substrate structure. Indeed, the rate-limiting step for the equatorial epoxidation pathway is predicted to be ring closure instead of addition. This overall mechanistic uncertainty complicates the understanding and control of diastereoselectivity or enantioselectivity in these reactions, as it may be uncertain what combination of transition states along major and minor pathways is deciding the selectivity. However, knowledge of the rate-limiting step provides insight into the stereochemistry of epoxidations of interest, and this information is readily available from kinetic isotope effects.
Experimental Section
Epoxidation of 2-Cyclohexenone with tert-Butyl Hydroperoxide and DBU
A mixture of 19.2 g (0.20 mol) of 2-cyclohexenone, 3.4 g of dodecane (internal standard). 27.6 g (0.16 mol) of DBU, and 50 mL (0.21 mol) of 4.1 M tert-BuOOH in dichloroethane35 in 600 mL of dichloroethane was prepared at 0 °C and allowed to warm slowly to 22 °C. Aliquots were periodically removed and the conversion was checked by GC. After 18 h at 22 °C, the conversion was determined to be 83.4%. The reaction mixture was diluted with 300 mL of chloroform and 300 mL of water and stirred for 30 min. The organic layer was separated, washed with H2O, dried (anhydrous MgSO4), and concentrated under vacuum below 30 °C. The residue was then chromatographed on a 5 cm × 45 cm flash silica gel column using 10% ethyl acetate / 30−60° petroleum ether as eluent to afford 9.5 g of a mixture of 2-cyclohexenone and 2,3-epoxy-2-cyclohexenone in the approximate ratio of 17: 83. Vacuum transfer of the volatiles from this mixture on a water aspirator followed by fractional distillation using a 10-cm Vigreaux column afforded a 5.2 g fraction (bp 90−95 °C) of an approximately 45 : 55 mixture of 2-cyclohexenone and 2,3-epoxy-2-cyclohexenone. This fraction was chromatographed on a 4 cm × 30 cm. flash silica gel column using 1:1 chloroform / hexanes as eluent to afford 0.5 g of 2-cyclohexenone ( >99% purity by GC) along with mixtures of 2-cyclohexenone and 2,3-epoxy-2-cyclohexenone in various ratios. A second reaction performed by an analogous procedure proceeded to 89.1% conversion.
NMR Measurements
NMR samples were prepared using 200 mg of cyclohexenone in a 5-mm NMR tube filled to a 5-cm sample height with CDCl3. The 13C spectra were recorded at 100.5 MHz using inverse gated decoupling, 60 s delays, and a 5.0 s acquisition time to collect 400,000 points. Integrations were determined numerically using a constant equal integration region for peaks compared. A zeroth-order baseline correction is generally applied, but in no case was a first-order (tilt) correction applied. Six spectra were obtained for each of two independent samples of cyclohexenone. The raw integration results are shown in the Supporting Information.
Acknowledgements
We thank NIH grant No. GM-45617 and The Robert A. Welch Foundation for support of this research.
Footnotes
Publisher's Disclaimer: This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2−3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
Supporting Information Available. Energies and full geometries of all calculated structures, and NMR integration results for all reactions. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References and Footnotes
- 1.Weitz E, Scheffer A. Chem. Ber. 1921;54:2327–2344. [Google Scholar]
- 2.a Bunton CA, Minkoff GJ. J. Chem.Soc. 1949:665–670. [Google Scholar]; b Emmons WD, Pagano AS. J. Am. Chem. Soc. 1955;77:89–92. [Google Scholar]; c Payne GB, Williams PH. J. Org. Chem. 1961;26:651–659. [Google Scholar]; d Yang NC, Finnegan RA. J. Am. Chem. Soc. 1958;80:5845–5848. [Google Scholar]
- 3.a Still WC. J. Am. Chem. Soc. 1979;101:2493–2495. [Google Scholar]; b Schlessinger GR, Bebernitz GR, Lin P, Poss AJ. J. Am. Chem. Soc. 1985;107:1777–1778. [Google Scholar]; c Danishefsky S, Hirama M, Gombatz K, Harayama T, Berman E, Schuda PF. J. Am. Chem. Soc. 1979;101:7020–7031. [Google Scholar]; d Mehta G, Pan SC. Tetrahedron Lett. 2005;46:5219–5223. [Google Scholar]; e Wada S. –i., Tanaka R. Bioorg. Med. Chem. Lett. 2005;15:2966–2969. doi: 10.1016/j.bmcl.2005.04.052. [DOI] [PubMed] [Google Scholar]; f Rodeschini V, Boiteau J–G, Van de Weghe P, Tarnus C, Eustache J. J. Org. Chem. 2004;69:357–373. doi: 10.1021/jo035065+. [DOI] [PubMed] [Google Scholar]
- 4.a Yang NC, Finnegan RA. 1958;80:5845–5848. [Google Scholar]; b Payne GB. J. Org. Chem. 1960;25:275–276. [Google Scholar]
- 5.Yadav VK, Kapoor KK. Tetrahedron. 1995;51:8573–8584. [Google Scholar]
- 6.Porter MJ, Skidmore J. Chem. Commun. 2000:1215–1225. For a review, see. [Google Scholar]
- 7.Juliá S, Masana J, Vega JC. Angew. Chem. Int. Ed. 1980;19:929–931. [Google Scholar]; Colonna S, Molinari H, Banfi S, Juliá S, Masana J, Alvarez A. Tetrahedron. 1983;39:1635–1641. [Google Scholar]; Banfi S, Colonna S, Molinari H, Juliá S, Guixer J. Tetrahedron. 1984;40:5207–5211. [Google Scholar]; Geller T, Roberts SM. J. Chem. Soc., Perkin Trans. 1. 1999:1397–1398. [Google Scholar]; Kelly DR, Roberts SM. Biopolymers. 2006;84:74–89. doi: 10.1002/bip.20373. [DOI] [PubMed] [Google Scholar]
- 8.Helder R, Hummelen JC, Laane RWPM, Wiering JS, Wynberg H. Tetrahedron Lett. 1976:1831–1834. [Google Scholar]; Wynberg H, Greijdanus B. Chem. Commun. 1978:427–428. [Google Scholar]; Wynberg H, Marsman B. J. Org. Chem. 1980;45:158–161. [Google Scholar]
- 9.a Adam W, Bheema Rao P, Degen H–G, Saha-Möller CR. J. Am. Chem. Soc. 2000;122:5654–5655. [Google Scholar]; b Adam W, Bheema Rao P, Degen H–G, Saha-Möller CR. Eur. J. Org. Chem. 2002:630–639. doi: 10.1021/jo0162078. [DOI] [PubMed] [Google Scholar]; c Aoki M, Seebach D. Helv. Chim. Acta. 2001;84:187–207. [Google Scholar]; d Lattanzi A, Cocilova M, Iannece P, Scettri A. Tetrahedron: Asymmetry. 2004;15:3751–3755. [Google Scholar]
- 10.Enders D, Zhu J, Raabe G. Angew. Chem. Int. Ed. 1996;35:1725. [Google Scholar]; Yu H–B, Zheng X-F, Lin Z-M, Hu Q–S, Huang W-S, Pu L. J. Org. Chem. 1999;64:8149–8155. doi: 10.1021/jo990749w. [DOI] [PubMed] [Google Scholar]
- 11.Bartlett PA, Chouinard PM. J. Org. Chem. 1983;48:3854–3855. For an example of stereospecificity with an electron-poor alkene, see. [Google Scholar]
- 12.House HO, Ro RS. J. Am. Chem. Soc. 1958;80:2428–2433. [Google Scholar]
- 13.Temple RD. J. Org. Chem. 1970;35:1275–1280. [Google Scholar]
- 14.Kelly DR, Caroff E, Flood RW, Heal W, Roberts SM. Chem. Commun. 2004:2016–2017. doi: 10.1039/b404389h. [DOI] [PubMed] [Google Scholar]; Carrea G, Colonna S, Meek AD, Ottolina G, Roberts SM. Tetrahedron: Asymmetry. 2004;15:2945–2949. See also. [Google Scholar]; Kelly DR, Roberts SM. Chem. Commun. 2004:2018–2020. doi: 10.1039/b404390c. [DOI] [PubMed] [Google Scholar]; Mathew SP, Gunathilagan S, Roberts SM, Blackmond DG. Org. Lett. 2005;7:4847–4850. doi: 10.1021/ol051708r. [DOI] [PubMed] [Google Scholar]
- 15.Singleton DA, Thomas AA. J. Am. Chem. Soc. 1995;117:9357–9358. [Google Scholar]
- 16.
- 17.Frantz DE, Singleton DA, Snyder JP. J. Am. Chem. Soc. 1997;119:3383–4. [Google Scholar]; Frantz DE, Singleton DA. J. Am. Chem. Soc. 2000;122:3288–3295. [Google Scholar]
- 18.Wilhelm E, Battino R. Chem. Rev. 1973;73:1–9. [Google Scholar]
- 19.Rastorguev YL, Ganiev YA. Izv. Vyssh. Uchebn. Zaved. Neft Gaz. 1967;10:79–82. [Google Scholar]
- 20.Saunders M, Laidig KE, Wolfsberg M. J. Am. Chem. Soc. 1989;111:8989–8994. The calculations used the program QUIVER. [Google Scholar]; Scott AP, Radom L. J. Phys. Chem. 1996;100:16502–16513. B3LYP frequencies were scaled by 0.9614. The exact choice of scaling factor makes little difference in the calculated KIE – varying the scaling factor from 0.93 to 0.97 changes the 13C KIEs by less than 0.001. [Google Scholar]
- 21.a Bigeleisen J, Mayer MG. J. Chem. Phys. 1947;15:261–267. [Google Scholar]; b Wolfsberg M. Acc. Chem. Res. 1972;5:225–233. [Google Scholar]; c Bigeleisen J. J. Chem. Phys. 1949;17:675–678. [Google Scholar]
- 22.Bell RP. The Tunnel Effect in Chemistry. Chapman & Hall; London: 1980. pp. 60–63. [Google Scholar]
- 23.a Beno BR, Houk KN, Singleton DA. J. Am. Chem. Soc. 1996;118:9984–9985. [Google Scholar]; b Meyer MP, DelMonte AJ, Singleton DA. J. Am. Chem. Soc. 1999;121:10865–10874. [Google Scholar]; c DelMonte AJ, Haller J, Houk KN, Sharpless KB, Singleton DA, Strassner T, Thomas AA. J. Am. Chem. Soc. 1997;119:9907–9908. [Google Scholar]; d Singleton DA, Merrigan SR, Liu J, Houk KN. J. Am. Chem. Soc. 1997;119:3385–3386. [Google Scholar]
- 24.Singleton DA, Merrigan SR. J. Am. Chem. Soc. 2000;122:11035–11036. [Google Scholar]
- 25.Singleton DA, Schulmeier BE. J. Am. Chem. Soc. 1999;121:9313–9317. [Google Scholar]
- 26.Singleton DA, Nowlan DT, III, Jahed N, Matyjaszewski K. Macromolecules. 2003;36:8609–8616. [Google Scholar]
- 27.Singleton DA, Wang Z. J. Am. Chem. Soc. 2005;127:6679–6685. doi: 10.1021/ja0435788. [DOI] [PubMed] [Google Scholar]
- 28.a Gravel D, Bordeleau J. Tetrahedron Lett. 1998;39:8035–8038. [Google Scholar]; b Kreiser W, Körner F. Helv. Chim. Acta. 1999;82:1610–1629. [Google Scholar]; c Hanazawa T, Wada T, Masuda T, Okamoto S, Sato F. Org. Lett. 2001;3:3975–3977. doi: 10.1021/ol016908r. [DOI] [PubMed] [Google Scholar]; d Hatcher MA, Peleg S, Dolan P, Kensler TW, Sarjeant A, Posner GH. 2005;13:3964–3976. doi: 10.1016/j.bmc.2005.04.010. [DOI] [PubMed] [Google Scholar]
- 29.a Daniewski AR, Garofalo LM, Hutchings SD, Kabat MM, Liu W, Okabe M, Radinov R, Yiannikouros GP. J. Org. Chem. 2002;67:1580–1587. doi: 10.1021/jo0161577. [DOI] [PubMed] [Google Scholar]; b Okamura WH, Aurrecoechea JM, Gibbs RA, Norman AW. J. Org. Chem. 1989;54:4072–4083. [Google Scholar]; c Pogrebnoi S, Saraber FCE, Jansen BJM, de Groot A. Tetrahedron. 2006;62:1743–1748. [Google Scholar]; d Zhao XZ, Tu YQ, Peng L, Li XQ, Jia YX. Tetrahedron Lett. 2004;45:3713–3716. [Google Scholar]; e Mehta G, Kumaran RS. Tetrahedron Lett. 2003;44:7055–7059. [Google Scholar]
- 30.Solladie G, Hutt J. J. Org. Chem. 1987;52:3560–3566. [Google Scholar]
- 31.Barrett AGM, Capps NK. Tetrahedron Lett. 1986;27:5571–5574. [Google Scholar]
- 32.a Rodeschini V, Van de Weghe P, Salomon E, Tarnus C, Eustache J. J. Org. Chem. 2005;70:2409–2412. doi: 10.1021/jo047858h. [DOI] [PubMed] [Google Scholar]; b Hoke ME, Brescia M–R, Bogaczyk S, DeShong P, King BW, Crimmins MT. J. Org. Chem. 2002;67:327–335. doi: 10.1021/jo010672n. [DOI] [PubMed] [Google Scholar]
- 33.Moreno-Dorado FJ, Guerra FM, Aladro FJ, Bustamante JM, Jorge ZD, Massanet GM. Tetrahedron Lett. 1999;55:6997–7010. [Google Scholar]
- 34.Rickborn B, Lwo S–Y. J. Org. Chem. 1965;30:2212–2216. [Google Scholar]
- 35.Sharpless KB, Verhoeven TR. Aldrichchimica Acta. 1979;12:63–74. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.