

Abstract
Lung maturation at the terminal sac stage of lung development is characterized by a coordinated increase in terminal sac formation and vascular development in conjunction with the differentiation of alveolar type I and type II epithelial cells. The Cited2-Tcfap2a/c complex has been shown to activate transcription of Erbb3 and Pitx2c during mouse development. In this study, we show that E17.5 to E18.5 Cited2-null lungs had significantly reduced terminal sac space due to an altered differentiation of type I and type II alveolar epithelial cells. In addition, E17 Cited2-null lungs exhibited a decrease in the number of apoptotic cells, contributing to the loss in airspace. Consistent with the phenotype, genes associated with alveolar cell differentiation and survival were differentially expressed in Cited2-null fetal lungs compared to those of wild-type littermates. Moreover, expression of Cebpa, a key regulator of airway epithelial maturation, was significantly decreased in Cited2-null fetal lungs. Cited2 and Tcfap2c were present on the Cebpa promoter in E18.5 lungs to activate Cebpa transcription. We propose that the Cited2-Tcfap2c complex controls lung maturation by regulating Cebpa expression. Understanding the function of this complex may provide novel therapeutic strategies for patients with respiratory distress syndromes.
Keywords: Cited2, Tcfap2c, Cebpa, Lung maturation
Introduction
During early lung development, the foregut endoderm-derived epithelial cells migrate into the surrounding splanchnic mesoderm. Through branching morphogenesis, an epithelial tubular structure forms and differentiates to produce respiratory bronchioles and terminal alveolar sacs, which integrate with the endothelial capillary bed (Minoo and King, 1994; Cardoso and Lu, 2006). At birth, respiratory transition from aqueous to air breathing requires a mature lung. The prenatal process of lung maturation is characterized by a coordinated increase in terminal sac formation and vascular development in conjunction with the differentiation of alveolar type I and type II epithelial cells. Cuboidal alveolar type II epithelial cells undergo marked ultrastructural and biochemical changes, including depletion of glycogen, increased synthesis of surfactant proteins and lipids, increased numbers of lamellar bodies, and secretion of surfactant proteins into the air space. Surfactant proteins reduce surface tension at the air-liquid interface and are required for pulmonary homeostasis (Williams and Mason, 1977; Weaver and Conkright, 2001; Boggaram, 2003). Alveolar type II cells terminally differentiate into alveolar type I cells, which are elongated, flattened cells and are capable of fluid reabsorption.
Cebps are basic leucine zipper transcription factors that are expressed in several organs and control the expression of genes important for cell differentiation. In the lung, Cebpa, -b, and -d are expressed in the type II alveolar epithelial cells and bronchiolar epithelial Clara cells and regulate gene expression of surfactant proteins, Abca3 (a lamellar body associated protein), Cyp2b1 (P450 enzyme), and Clara cell secretory protein (Cassel and Nord, 2003; Basseres et al., 2006; Martis et al., 2006). Analyses of the embryonic lung show that the expression of Cebpa, -b, and -c increases during late gestation (Berg et al., 2006; Breed et al., 1997; Rosenberg et al., 2002), but no lung phenotype has been reported in the Cebpb−/−, Cebpd−/−, or Cebpb/Cebpd double knockout mice (Screpanti et al., 1995; Sterneck et al., 1998; Tanaka et al., 1997). In contrast, the phenotype of Cebpa conditional knockouts suggests an important role of Cebpa in the differentiation of respiratory alveolar epithelium such that immature glycogen-rich pre-type II cells become mature surfactant-secreting type II cells in the last few days of gestation (Basseres et al., 2006; Martis et al., 2006). Loss of Cebpa in the respiratory epithelium in embryos leads to respiratory failure at birth due to an arrest in type II alveolar cell differentiation, resulting in lack of both surfactant-secreting type II and differentiated type I alveolar cells.
Cited2 [CBP/p300-interacting transactivators with glutamic acid (E) and aspartic acid (D)-rich tail 2] is one of the founding members of a new family of transcriptional activators. Expression of Cited2 can be induced by hypoxia, cytokines, lipopolysaccharide and shear stress (Bhattacharya et al., 1999; Sun et al., 1998; Yokota et al., 2003). It functions as a negative regulator of Hif1 in vitro and in vivo by competing with Hif1a in binding to the CH1 region of CBP/p300 to inhibit HIF-1-mediated signaling (Bhattacharya et al., 1999; Freedman et al., 2003; Xu et al., 2007). Cited2 also acts as a transcriptional modulator in vitro by interacting with DNA binding proteins, such as Tcfap2, Smad2/3, Lhx2, PPARα, and PPARγ, to regulate transcription of their corresponding target genes (Bhattacharya et al., 2001; Chou et al., 2006, Glenn and Maurer, 1999; Tien et al., 2004).
Cited2 is widely expressed in both embryonic and extra-embryonic tissues during early mouse development (Dunwoodie et al., 1998; Weninger et al., 2005). It is essential for embryonic and extra-embryonic tissue development. Mice lacking Cited2 die at late gestation with numerous developmental defects, including cardiac malformations, adrenal agenesis, exencephaly, neural crest, left-right patterning, and placental defects (Bamforth et al., 2001, 2004; Martinez Barbera et al., 2002; Yin et al., 2002; Weninger et al., 2005; Withington et al., 2006; Xu et al., 2007). Defects in the establishment of left-right axis result in abnormal heart looping and right atrial and right pulmonary isomerism (Bamforth et al., 2004; Weninger et al., 2005). Some of these abnormalities in Cited2−/− embryos are hypothesized to be due to reduced transactivation by Tcfap2 or over-expression of HIF-1-responsive genes (Bamforth et al., 2001, 2004; Yin et al., 2002; Weninger et al., 2005; Xu et al., 2007).
Here we report that Cited2-null fetal lungs displayed defects in terminal sac formation due to altered differentiation and a lower frequency of apoptotic alveolar cells, which likely resulted from significantly decreased expression of Cebpa at late gestation. Furthermore, Cited2 and Tcfap2c interacted with the Cebpa gene promoter in the alveolar epithelium to activate Cebpa transcription. These results strongly suggest that Cited2 plays a novel role in pulmonary maturation through its coactivation function with Tcfap2c to regulate Cebpa expression.
Materials and methods
Mouse strains and generation
Cited2 heterozygous mice were genotyped by Southern blot analysis (Yin et al., 2002) and backcrossed at least thirteen generations into the C57BL/6J genetic background. Twenty-five Cited2-null lungs between E15.5 and E18.5 from the intercrosses of Cited2 heterozygotes on the C57BL/6J genetic background (Table 1) and 28 wild-type lungs from their littermates were dissected. One half of each embryonic lung (right or left lobe) was fixed in 4% paraformaldehyde for histology, immunohistochemistry, in situ hybridization, and/or the terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL) assay, and the other half was frozen for RNA isolation.
Table 1.
Phenotype of Cited2−/− lungs
| Stage | No. of Cited2−/− lungs (RPI / PI) |
|---|---|
| E15.5 | 3 (2 / NA) |
| E16.5 | 5 (3 / NA) |
| E17 | 3 (2 / NA) |
| E17.5 | 8 (6 / 8) |
| E18 | 4 (2 / 4) |
| E18.5 | 2 (1 / 2) |
RPI: right pulmonary isomerism
PI: pulmonary immaturity
NA: not applicable for PI analysis
Morphological analysis
Hematoxylin and eosin (HE) stained sections were used for histological examination. Immunohistochemistry was carried out by using rabbit polyclonal anti-prosurfactant protein B (Chemicon International, Temecula, CA), mouse monoclonal anti-vimentin (40E-C, Developmental Studies Hybridoma Bank, Iowa City, IA), mouse monoclonal anti-cytokeratin (TROMA-I, Developmental Studies Hybridoma Bank), rabbit monoclonal anti-Cebpa (C-18, Santa Cruz Biotechnology, Santa Cruz, CA), rabbit polyclonal anti-Tcfap2c (Santa Cruz Biotechnology), mouse monoclonal anti-Titf1 (8G7G3/1, DakoCytomation, Carpinteria, CA), mouse monoclonal anti-Ki-67 (B56, BD PharMingen, San Diego, CA), and rat monoclonal anti-CD31 (MEC13.3, BD PharMingen) antibodies on cryosections. The reactions were detected by the Labeled Streptavidin Biotin Kit (DakoCytomation). Electron microscopy was performed on lung tissues obtained from E18.5 Cited2-null embryos and littermate controls after fixation in 2.5% glutaraldehyde and 2% paraformaldehyde solution, postfixation in 1.5% potassium ferrocyanide and 1% osmium tetroxide solution, dehydration in ascending alcohols, cleared with propylene oxide, and infiltration with a mixture of Epon resin and propylene oxide. 70-nm sections stained for contrast with uranyl acetate and lead citrate were viewed and photographed on a JEOL 1200CX electron microscope.
X-gal staining and TUNEL assay
X-gal staining was performed on lung cryosections using standard procedures (Martinez Barbera et al., 2002). TUNEL assay was performed on 3 sections from all of the collected E16.5, E17, and E17.5 Citedd2-null lungs (see Table. 1 for actual sample numbers) and three of the matching E16.5, E17, and E17.5 wild-type lungs using ApopTag Peroxidase In Situ Apoptosis Detection Kit (Chemicon International) according to the manufacturer’s instructions. The percentages of TUNEL positive cells were then calculated from 5 random selected fields of each section. On average, approximately 500 alveolar cells were counted in each field.
RNA isolation, northern blot analysis, and real time RT-PCR
Total RNA was isolated from the lung tissue using Trizol reagent following the manufacturer’s protocol (Invitrogen, Carlsbad, CA). Ten-microgram total RNA was loaded per lane for formaldehyde agarose gel electrophoresis and transferred to a nylon membrane. Equal loading was demonstrated by 18S RNA levels. Five-microgram total RNA was used for cDNA synthesis using the SuperScript™ First Strand Synthesis System (Invitrogen). Real time PCR reactions were carried out with diluted RT reaction products using iQ™ SYBR Green Supermix PCR kit and iCycler machine (Bio-Rad). Transcript of β-actin and HPRT was used as an internal control for normalization.
Plasmids, DNA transfection, luciferase reporter assay and siRNA
A segment of the Cebpa gene from nucleotides −805 to +117 containing the entire 5’-UTR was amplified from mouse genomic DNA and subcloned into pGL3 (C/EBPα-805-luc). Mouse Cited2 cDNA was subcloned into pcDNA3.1(−)B and the deletion of C-terminal residues (ΔCR2) was introduced into this Cited2 expression vector (Chou et al., 2006). Tcfap2c cDNA subcloned into pcDNA3.1 was kindly provided by Dr. Frederick E. Domann (Li at al., 2006). CMV-CBP and CMV-CBPΔCH1 were gifts from Dr. Paul K. Brindle (Kasper et al., 2005). HeLa cells were transfected using FuGENE HD Transfection Reagent (Roche Diagnostics) following the manufacturer’s instructions. Luciferase activity in cell lysates was detected by Dual-luciferase Reporter Assay System (Promega, Madison, WI) and normalized according to renilla luciferase activity of co-transfected pRL-CMV. For siRNA knockdown experiment, HeLa cells were transfected with Cited2 siRNA or siRNA control (Chou et al., 2006) using Effectene transfection reagent (Qiagen, Hilden, Germany) for 24 h following the manufacturer’s instruction.
Chromatin immunoprecipitation (ChIP) assay
Lung tissues from 4 each of E18.5 wild-type and Cited2−/− embryos were combined for the ChIP assay using the ChIP Assay Kit (Upstate Cell Signaling Solutions, Lake Placid, NY) according to the manufacturer’s protocol. Chromatin was immunoprecipitated with 1µg rabbit anti-Cited2, -Tcfap2c, -Cebpa (Santa Cruz Biotechnology) or -acetylated-Histone H3 (Upstate Cell Signaling Solutions) antibody. Immunoprecipitated samples were analyzed by PCR with primers specific for the overlapping Sp1/Sp3 and Tcfap2a/c binding sites in the Cebpa promoter (for amplification of −805 to −661 region: sense, 5’-tggagacgcaatgaaaaaga-3’, antisense, 5’-gaactacagggtcccacgg-3’; for −332 to −159: sense, 5’-ctggaagtgggtgacttagagg-3’, antisense, 5’-gggagcatagtgctagtggaga-3’; for −54 to +123: sense, 5’-gatgcccgaccctctataaaa-3’, antisense, 5’-actccatgggggagttagagtt-3’).
Statistical analysis
Statistical difference between two independent groups was assessed by Independent-samples t test; p ≤ 0.05 was considered statistically significant.
Results
Expression of Cited2 in mouse embryonic and fetal lungs
Northern analysis showed that Cited2 was expressed in the E13.5–18.5 lung tissues examined, with higher expression in the E17.5 and E18.5 lungs (Fig. 1a), suggesting its potential involvement in lung maturation. To further explore the developmental expression pattern of Cited2 in mouse embryonic lung, Cited2 expression was assayed on lung cryosections by detecting β-galactosidase activity from the Cited2-lacZ allele in E17.5 heterozygotes, followed by immunostaining with the epithelial marker, cytokeratin, or the mesenchymal marker, vimentin. As shown in Fig. 1b, X-gal positive cells were present in both peripheral epithelial cells that were immunopositive for cytokeratin (Fig. 1b: B and C) and mesenchymal cells that were immunopositive for vimentin (Fig. 1b: E and F). Only a few X-gal positive cells were seen in the proximal airways (Fig. 1b: B).
Fig. 1.
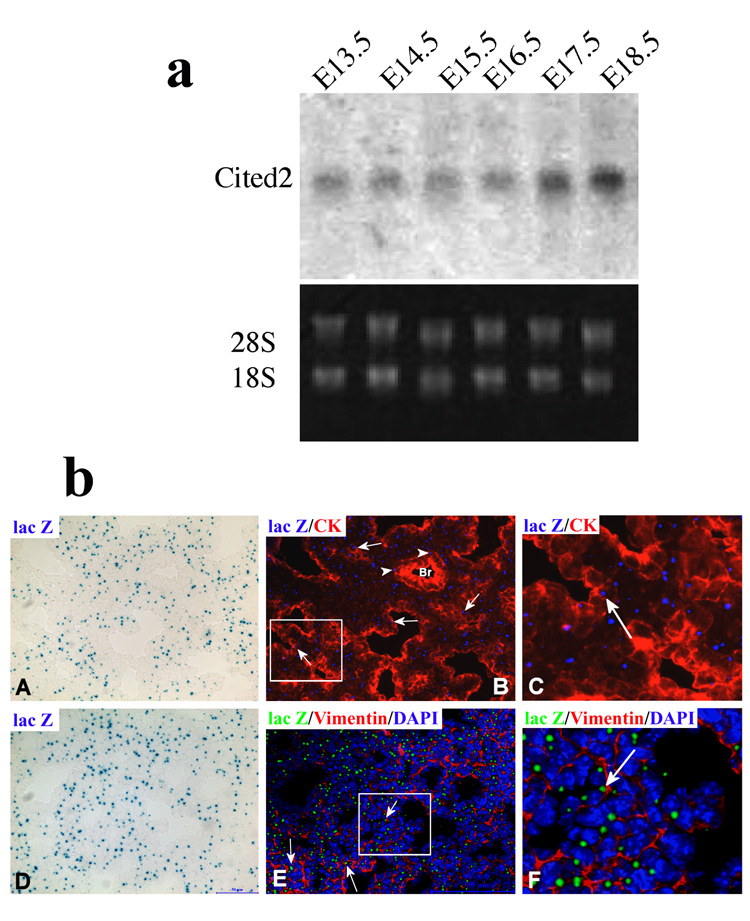
Cited2 expression in developing lungs. (a) Ten-microgram total RNA isolated from E13.5–18.5 lung tissue was subjected to Northern analysis with a specific probe for Cited2. The 18S RNA levels were used as the loading control. (b) Cited2 expression was examined on lung cryosections by detecting β-galactosidase activity from the Cited2-lacZ allele in E17.5 heterozygotes (A, D), followed by immunostaining of cytokeratin (B, C) or vimentin (E, F), showing that X-gal positive cells were present in both cytokeratin-positive cells (arrows in B and C) and vimentin-positive cells (arrows in E and F). Panels C and F are digitally enlarged from portions of panel B and E (shown as rectangles) for visualization purpose. Only a few X-gal positive cells (arrow heads in B) were seen in the proximal airways (B). The X-gal positive signals (blue in A, D) was pseudo-colorized to blue (B, C) and green (E, F) for the ease in visualization. Br: bronchus.
Right pulmonary isomerism and pulmonary immaturity in Cited2-null lungs
Previous studies showed that approximately two thirds of Cited2-null embryos had right pulmonary isomerism: 4 lung lobes on both sides, as opposed to the normal arrangement with 1 lung lobe on the left and 4 lung lobes on the right (Bamforth et al., 2004; Weninger et al., 2005). This overall gross phenotype of Cited2-null lungs was also noted in this study. As shown in Table 1, 16 out of 25 (64%) Cited2-null embryos between E15.5 and E18.5 had right pulmonary isomerism. Furthermore, the lobe sizes of Cited2-null lungs were much smaller in appearance than those of wild-type lungs (Supplemental Fig. 1).
All the lung tissues in Table 1 were examined by HE-stained sections. Essentially, no apparent phenotypic differences could be detected between wild-type (Fig. 2a: A) and Cited2-null (Fig. 2a: B) lungs at E16.5. At E17.5 and E18.5, the dilated terminal sacs of wild-type distal lungs were lined with squamous type I epithelial cells and cuboidal type II cells. The mesenchyme and epithelium were thin with pulmonary capillaries extensively invaded in close proximity to type I epithelial cells (Fig. 2a: C, E), indicating structural maturation at these stages. In contrast, all 14 Cited2-null distal lungs at E17.5 to E18.5 exhibited remarkably abnormal histopathology, characterized by reduced terminal sac space with completely obliterated alveolar structure and the terminal sacs were surrounded by cuboidal epithelial cells and lacked squamous type I cells (Fig. 2a: D, F), consistent with pulmonary immaturity. Moreover, all 14 E17.5 to E18.5 Cited2-null embryos had pulmonary immaturity, but only 9 out of 14 (64%) had right pulmonary isomerism, suggesting that these two are independent phenotypes.
Fig. 2.
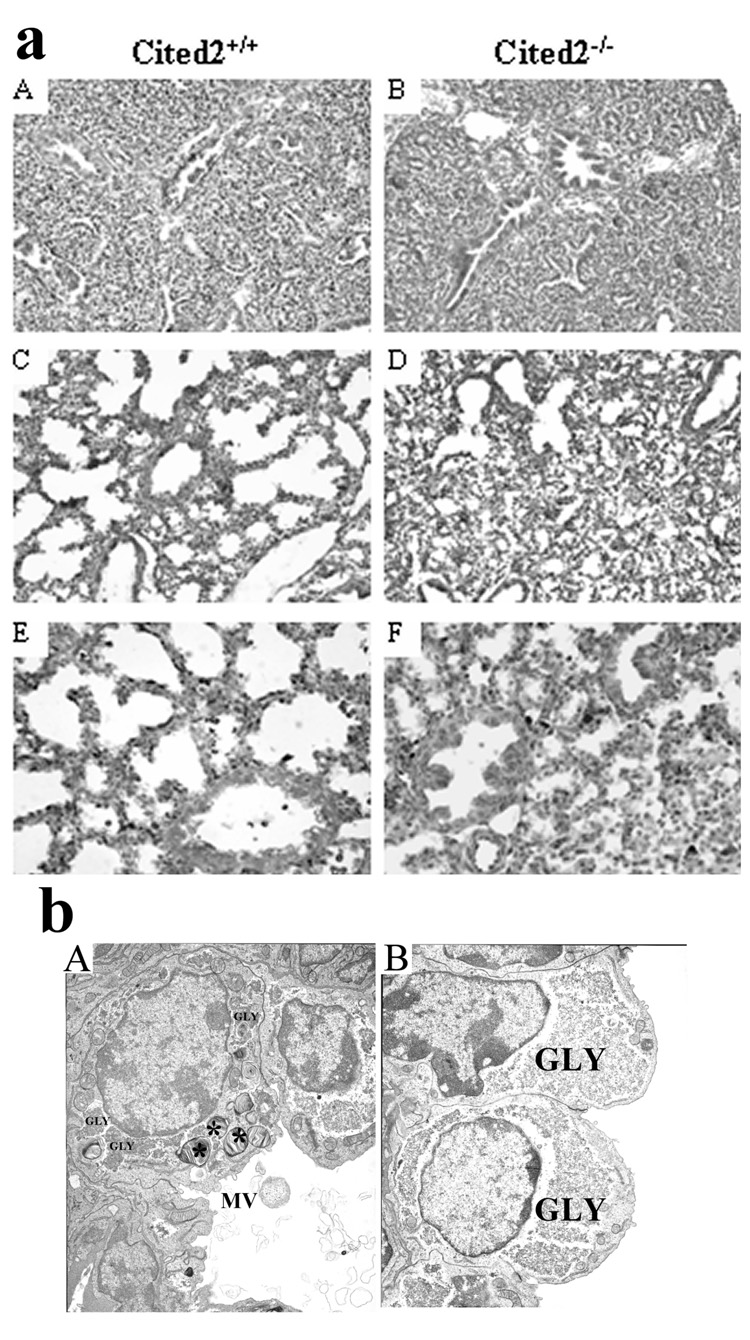
Pulmonary immaturity in Cited2−/− embryonic lungs. (a) HE-stained sections from wild-type (A, C, E) and Cited2−/− (B, D, F) lungs at E16.5 (A, B), E17.5 (C, D), and E18.5 (E, F). Representative sections revealed no apparent phenotypic differences between wild-type (A) and Cited2−/− (B) lungs at E16.5. E17.5 (C) and E18.5 (E) wild-type lungs had dilated terminal sacs lined by both squamous type I epithelial cells (arrow) and cuboidal type II epithelial cells and a thinned mesenchyme and epithelium. In contrast, E17.5 (D) and E18.5 (F) Cited2−/− lungs had reduced terminal sacs that were surrounded by cuboidal type II cells and lacked squamous type I cells. (b) Electron microscopy was performed on lungs from E18.5 Cited2−/− embryos and littermate controls. In controls, cuboidal type II cells contained highly organized rosette glycogen (GLY), apical microvilli (MV), and numerous lamellar bodies (*). Lamellar bodies and secreted surfactant proteins were observed in the lumen of the peripheral airspace (A). In contrast, cytoplasmic glycogen was dispersed, apical microvilli were smaller, and lamellar bodies were not observed in type II epithelial cells of Cited2−/− embryos. The number of squamous type I cells and secreted surfactant proteins were markedly decreased in the airspace of Cited2−/− embryos (B)
Compared with wild-type littermates (Fig. 2b: A), E18.5 Cited2-null lungs (Fig. 2b: B) appeared immature at the ultrastructural level. In controls, cuboidal type II cells contained highly organized rosette glycogen, apical microvilli, and numerous lamellar bodies, which are the intracellular storage forms of surfactant proteins. Lamellar bodies and secreted surfactant proteins were observed in the lumen of peripheral airspaces. In contrast, cytoplasmic glycogen was dispersed, apical microvilli were smaller, and lamellar bodies were markedly decreased in type II epithelial cells of Cited2-null embryos. Squamous type I cells were lacked and secreted surfactant proteins were markedly decreased in the airspace of Cited2-null embryos.
Altered differentiation of alveolar epithelial cells in Cited2-null embryonic lungs
Cellular differentiation is accompanied by the expression of markers that reflects the degree of maturation of the lung epithelium. As the histological analysis indicated that distal epithelial differentiation in Cited2-null fetal lungs was impaired, we examined the expression of lung epithelial markers in wild-type and Cited2-null lungs at E17.5 and E18. Expression of surfactant proteins, SP-A, SP-B, SP-C, and SP-D, in type II epithelial cells increases prior to birth (Boggaram, 2003), while SP-B and SP-C play critical roles in enhancing the spreading and adsorption of phospolipids to an air-liquid interface and promoting the reduction of surface tension (Boggaram, 2003; Weaver and Conkright, 2001). The mRNA levels of SP-A, SP-B, SP-C, and SP-D examined by real-time RT-PCR were significantly down-regulated in the Cited2-null lungs at E17.5 (data not shown) and E18 (Fig. 3a). In addition, Cited2-null fetal lungs showed decreased expression of proSP-B by immunostaining in the peripheral epithelial cells (Fig. 3b). These results suggest that expression of surfactant proteins is decreased in alveolar epithelial cells of Cited2-null fetal lungs.
Fig. 3.
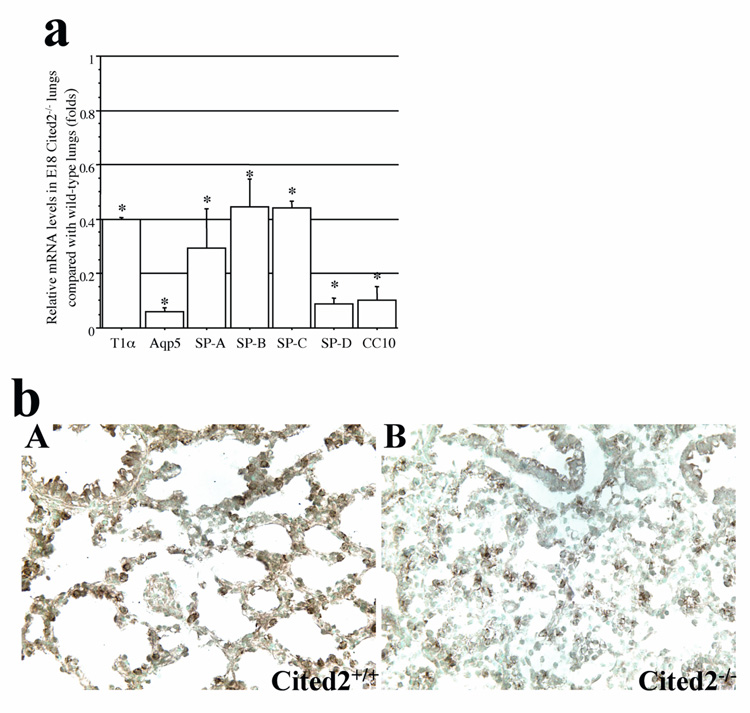
Decreased expression of alveolar and Clara epithelial marker genes in Cited2−/− embryonic lungs. (a) Real time RT-PCR was performed to analyze relative T1α, Aqp5, SP-A, SP-B, SP-C, SP-D, and CC10 mRNA levels in E18 Cited2−/− lungs, compared with those in wild-type littermates. *p < 0.01. (b) Immunohistochemistry for proSP-B confirmed that E18 Cited2−/− lungs (B) had decreased proSP-B expression compared to wild-type littermates (A).
Consistent with the observation that the terminal sacs lacked squamous type I cells in the absence of Cited2 (Fig. 2a: F, 2b: B), the mRNA levels of alveolar type I cell markers, such as T1-α and aquaporin 5, were significantly decreased in Cited2-null fetal lungs (Fig. 3a). These findings support the altered differentiation of alveolar epithelial cells in Cited2-null fetal lungs. In contrast to the immaturity of Cited2-null distal lungs, no obvious histological abnormalities in conducting airways were observed. However, the mRNA level of CC10 (Clara cell 10 kDa protein), a marker for Clara epithelial cells, was decreased in Cited2-null fetal lungs (Fig. 3a).
Proliferation and apoptosis of alveolar cells in Cited2-null fetal lungs
Pulmonary immaturity is dependent on the balance between the rates of cell proliferation, differentiation and programmed cell death (apoptosis). To test whether the cellular accumulation in Cited2-null fetal lungs, which may explain the loss of the airspace (Fig. 2a: D, F), is due to increased cell proliferation, immunostaining of Ki-67, a nuclear antigen present in proliferating cells, was performed on lung sections between E16.5 and E18.5. No obvious difference in the proportion of Ki-67-positive cells was observed in E16.5 to E18.5 Cited2-null lungs compared with that of wild-type lungs (data not shown), suggesting that the cellular accumulation in Cited2-null fetal lungs could not be explained by an increase in cell proliferation alone.
We further investigated alveolar cell apoptosis by the TUNEL assay. Compared with wild-type lungs (Fig. 4a: A), apoptotic alveolar cells in Cited2-null lungs (Fig. 4a: B) at E17 were significantly decreased (p < 0.001), which only had few apoptotic cells. Moreover, the mRNA level of Gli1, which has been shown to be a positive regulator of Bcl2 (Bigelow et al., 2004), was 2 fold higher in E17 Cited2-null lungs than that in wild-type littermates (Fig. 4b). These data suggest that alveolar cell apoptosis induced by Cited2 is a critical mechanism for normal terminal sac formation during lung development.
Fig. 4.
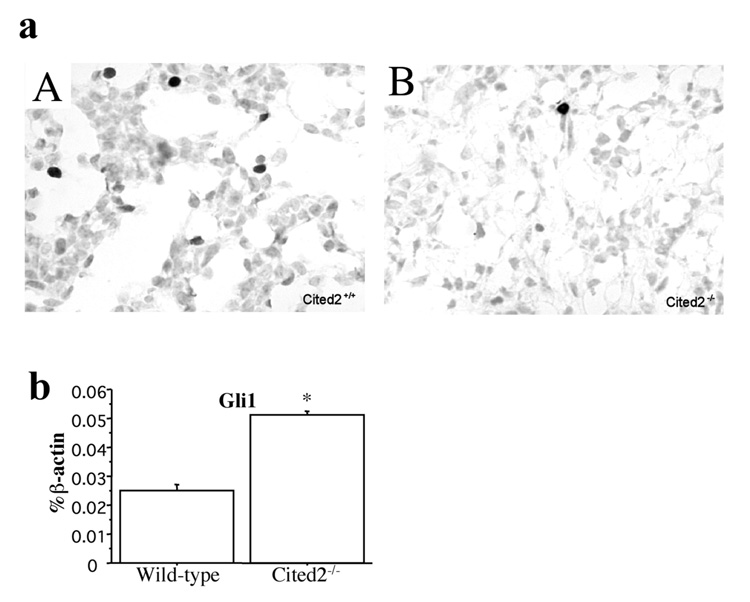
Decreased number of apoptotic alveolar cells and increased expression of Gli1 in Cited2−/− embryonic lungs. (a) Representative pictures of TUNEL assay showed that fewer TUNEL-positive cells were present in E17 Cited2−/− lungs (B) compared to wild-type littermate lungs (A). (b) Real-time RT-PCR showed increased expression of Gli1 in E17 Cited2−/− lungs compared to wild-type littermate lungs. Expression levels are presented as a percentage of β-actin expression. *p < 0.01.
Expression of transcription factors for alveolar epithelial cell differentiation in Cited2-null fetal lungs
We then examined the expression of transcription factors involved in lung branching morphogenesis and alveolar cell differentiation. Cebpa regulates the expression of genes that mediate cell differentiation and proliferation, lipid biosynthesis and metabolism, and host defense (Cassel and Nord, 2003). Titf1, Foxa1, and Foxa2 are expressed in respiratory epithelial cells lining both the conducting and peripheral airways and play distinct roles in lung formation and function. Titf1 and Foxa2 are required for Cebpa gene expression and Cebpa also regulates Foxa2 gene expression (Martis et al., 2006). In this study, there was no obvious difference in Titf1, Foxa1, and Foxa2 mRNA levels between wild-type and Cited2-null lungs at E17.5 (data not shown) and E18 (Fig. 5a). By immunostaining, Titf1 was expressed in the alveolar epithelial cells of wild-type peripheral lungs at E17.5 (data not shown) and E18.5 (Fig. 5b: A), as previously reported (Stahlman et al., 1996). In Cited2-null lungs at E17.5 (data not shown) and E18.5 (Fig. 5b: B), the cuboidal epithelial cells of the terminal sacs expressed Titf1 protein in a relatively homogenous pattern. Interestingly, the mRNA level of Cebpa was significantly decreased in Cited2-null lungs at E17.5 (data not shown) and E18 (Fig. 5a), compared with that in the wild-type lungs. While 31% of the total number of respiratory cells in wild-type lungs at E17.5 (data not shown) and E18.5 (Fig. 5c: A) were immunopositive for Cebpa, only 15% were Cebpa-immunopositive cells in Cited2-null lungs at E17.5 (data not shown) and E18.5 (Fig. 5c: B). Thus, we propose that decreased expression of Cebpa could be responsible for pulmonary immaturity in Cited2-null fetal lungs.
Fig. 5.
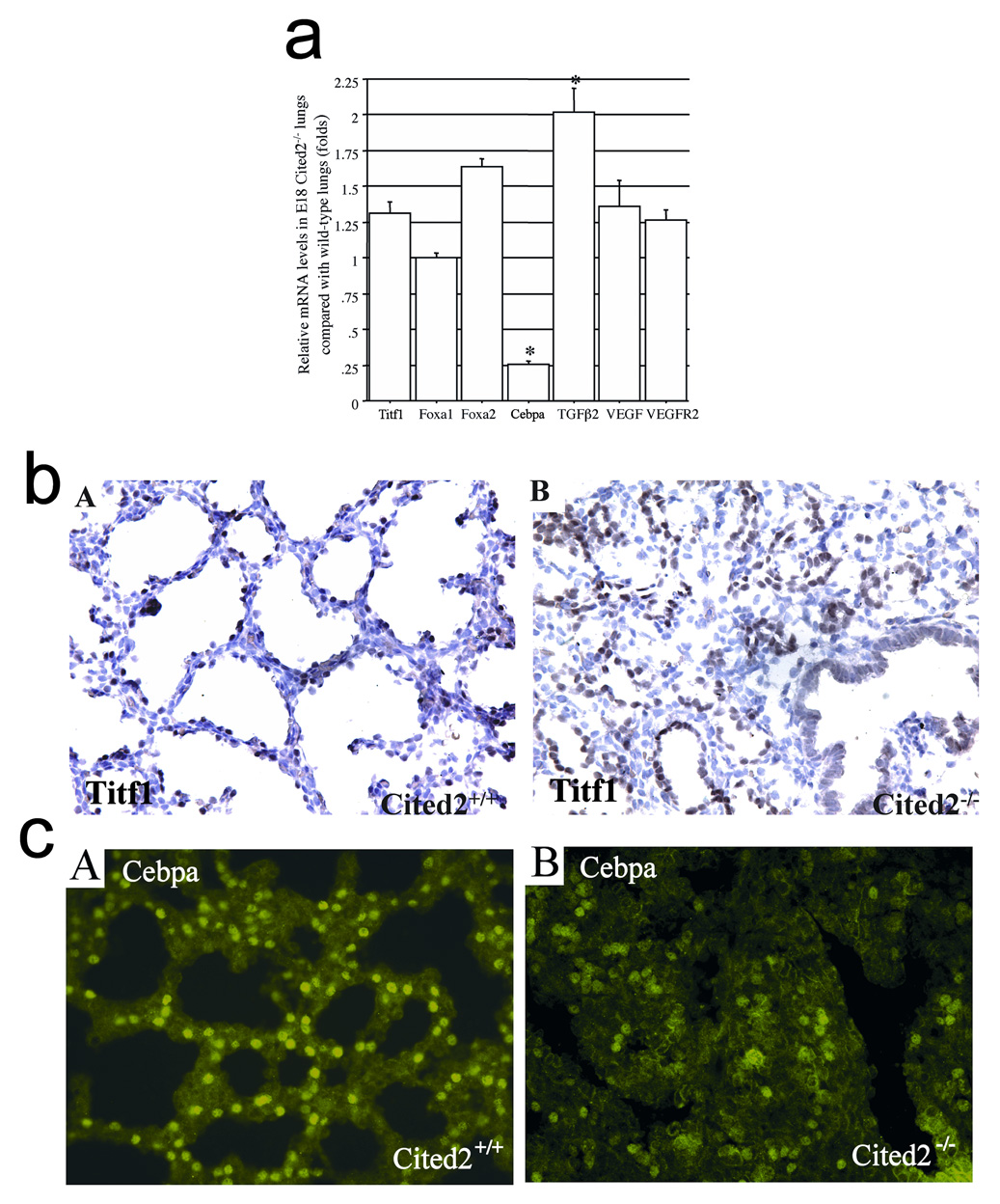
Expression of genes relevant to lung development in Cited2−/− embryonic lungs. (a) Real time RT-PCR was used to analyze relative Titf1, Foxa1, Foxa2, Cebpa, TGFβ2, VEGF, and VEGFR2 mRNA levels in E18 Cited2−/− lungs, compared with those in wild-type littermates. *p < 0.01. (b) Immunohistochemistry for Titf1 showed that alveolar epithelial cells of both wild-type (A) and Cited2−/− (B) peripheral lungs homogeneously expressed Titf1. (c) Compared to the wild-type littermate (A), the proportion of Cebpa-positive cells detected by immunostaining was markedly decreased in E18.5 Cited2−/− lungs where the terminal sac structure was completely obliterated (B).
Cited2 and Tcfap2c interact with the Cebpa promoter
Sp1 and Sp3 are major transcription factors controlling the expression of Cebpa, a pleiotropic transcriptional activator responsible for the expression of various genes during adipogenesis. Sp1/Sp3 mediated transcription of Cebpa is repressed by a weaker transcriptional activator, Tcfap2a, in 3T3-L1 preadipocytes and Schneider cells by competitive binding to overlapping Sp1/Sp3 and Tcfap2a binding sites at nucleotides −252 to −234 (A1 site, Fig. 6a) and +83 to +96 (A2 site, Fig. 6a) of the Cebpa gene (Jiang and Lane, 2002). Tcfap2 represents a family of three closely related and evolutionarily conserved transcription factors, Tcfap2a, -b, and -c, which have regulatory functions in control of apoptosis, cell cycle, and gene expression (Hilger-Eversheim et al., 2000). A previous study showed that Tcfap2c activates Esr1 gene transcription through its high-affinity binding to the imperfect palindrome sequence, CCCTGCGGGG, in the Esr1 promoter (McPherson et al., 1997). Overlapping Sp1/Sp3 and Tcfap2c binding sites are present in the conserved regions among mouse (C site, Fig. 6a), rat, and human Cebpa genes (Fig. 6b), suggesting that they may function as potential DNA binding sites important for Cebpa expression.
Fig. 6.
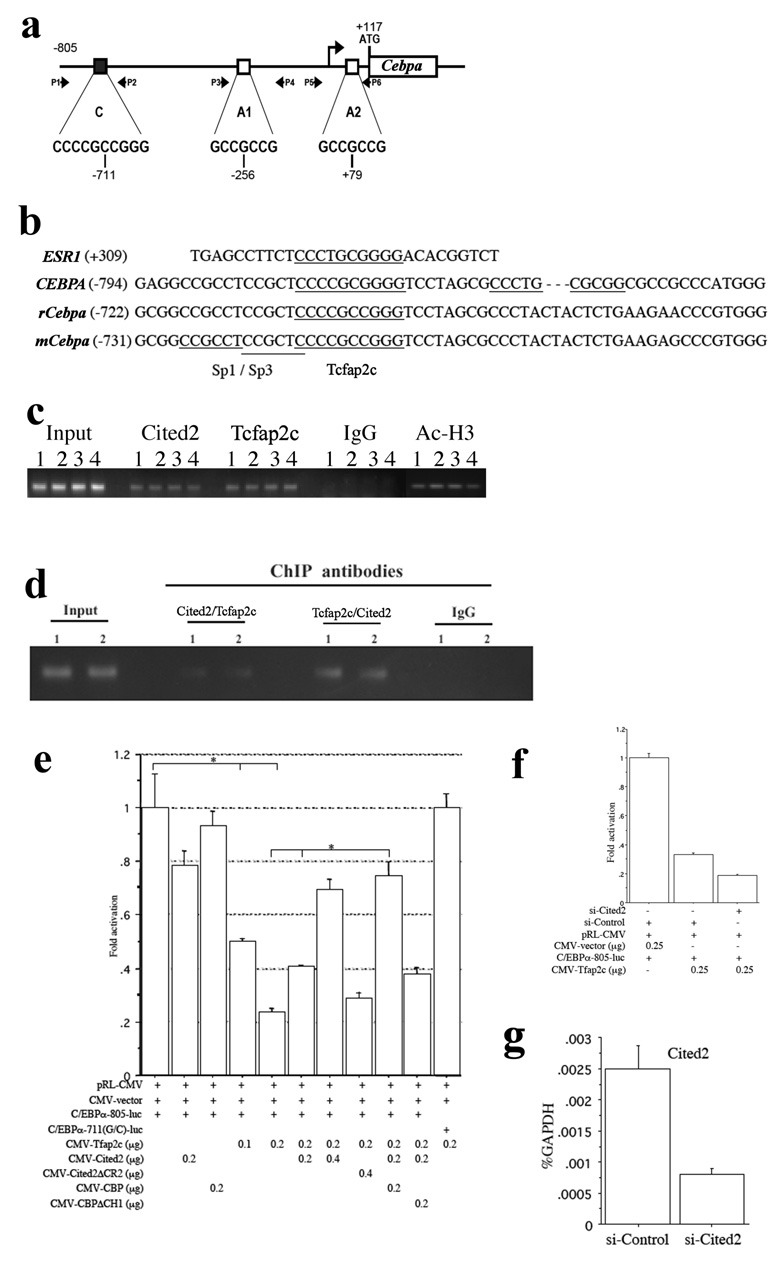
Regulation of the Cebpa promoter by Tcfap2c and Cited2. (a) Overlapping Sp1/Sp3 and Tcfap2a/c binding sites in the Cebpa promoter. C represents overlapping Sp1/Sp3 and Tcfap2c binding site. A1 and A2 represent overlapping Sp1/Sp3 and Tcfap2a binding sites. The positions of PCR primers used for chromatin immunoprecipitation (P1 to P6) are indicated. (b) Comparison of the Tcfap2c binding sequence in the Esr1 promoter with those in the Cebpa promoters from mouse, rat, and human. (c) ChIP assays using E18.5 wild-type lungs with antibodies to Cited2, Tcfap2c, acetylated histone H3, and mouse IgG. Input lane: 1% of total chromatin was used as the PCR template. 1, 2, 3, and 4 are separate samples. (d) Re-ChIP assays by immunoprecipitating with first antibodies to Cited2 or Tcfap2c, then re-immunoprecipitating with second antibodies to Tcfap2c or Cited2. (e) Co-transfection of HeLa cells with C/EBPα-805-luc or C/EBPα-711(G/C)-luc plus Cited2, Cited2ΔCR2, Tcfap2c, CBP, and/or CBPΔCH1 in different combinations, followed by luciferase assays 24 hours after transfection. *p < 0.01. (f) Transfection of HeLa cells with si-Cited2 or si-Control for 24 hours, followed by co-transfection with C/EBPα-805-luc and Tfap2c in different combinations for 24 hours and luciferase assays. (g) Transfection of HeLa cells with si-Cited2 or si-Control for 24 hours, followed by real time RT-PCR with specific primers for human Cited2 and GAPDH.
Since the Cited2-Tcfap2a/c complex activates transcription of Erbb3 and Pitx2c during mouse development (Bamforth et al., 2004; Weninger et al., 2005), we tested whether endogenous Cited2 and Tcfap2 physically interact with overlapping Sp1/Sp3 and Tcfap2a/c binding sites of the Cebpa promoter by the ChIP assay. Using E18.5 wild-type lungs, specific ChIP signals with antibodies to Cited2 were reproducibly detected with primers spanning the C site (Fig. 6c), but not with primers spanning A1 and A2 sites or using Cited2-null lungs (data not shown). In addition, specific and reproducible ChIP signals with antibodies to Tcfap2c and acetylated histone H3 were detected using primers spanning the C site (Fig. 6c). To further examine whether endogenous Cited2 and Tcfap2 simultaneously interact with the C site, re-ChIP assays were performed by immunoprecipitating with first antibodies to Cited2 or Tcfap2c, then reimmunoprecipitating with second antibodies to Tcfap2c or Cited2. As shown in Fig. 6d, specific re-ChIP signals were reproducibly detected with primers spanning the C site. Previous studies have shown that by RT-PCR, Tcfap2c (Oulad-Abdelghani, et al., 1996), but not Tcfap2a and Tcfap2b (Moser et al., 1995), was specifically expressed in adult mouse lung, ovary, and testis. These data therefore suggest that Cited2 and Tcfap2c may specifically interact with the C site of the Cebpa promoter in the respiratory epithelium.
Cited2, Tcfap2c, and CBP activate the Cebpa promoter
To determine whether Cited2 and Tcfap2c could activate the Cebpa promoter, the Cebpa promoter/reporter plasmid (C/EBPα-805-luc) was transiently transfected into HeLa cells that express high levels of Sp1 and Sp3 but low levels of Tcfap2 and Cebpa. As shown in Fig. 6e, C/EBPα-805-luc activity was highly activated when the promoter/reporter alone was transfected. This strong promoter activity, most likely activated by endogenous Sp1 or Sp3, was not affected by the cotransfection of Cited2 or CBP alone, but was inhibited by Tcfap2c (by a factor of 0.24). This inhibition was totally abolished when the Tcfap2c binding site was mutated from C to G at nucleotide −711, suggesting that Tcfap2c competitively binds to the C site with higher affinity. The weak C/EBPα-805-luc activity in the presence of Tcfap2c was further decreased (by a factor of 0.57, Fig. 6f) by 67% knockdown of endogenous expression of Cited2 using siRNA specific for Cited2 (Fig. 6g) and activated by Cited2 (by a factor of 2.92) and Cited2 plus CBP (by a factor of 3.14) but not by Cited2ΔCR2 or CBPΔCH1 (Fig. 6e), which is consistent with previous findings that the C-terminus of Cited2 interacts with the first helix of the Tcfap2c dimerization motif and the CH1 region of CBP to activate Tcfap2 mediated transcription (Braganca et al., 2003). These data suggest that Tcfap2c, Cited2, and CBP can synergistically activate the Cebpa promoter.
Discussion
In this study, we describe pulmonary immaturity in Cited2-null embryos displaying defects in terminal sac formation due to an accumulation of undifferentiated and less apoptotic alveolar cells. Consistent with the phenotype, Cited2-null fetal lungs differed in the expression of genes associated with alveolar cell differentiation and survival. The expression of Cebpa, a key regulator in airway epithelial maturation, was significantly decreased in Cited2-null fetal lungs. Endogenous Cited2 and Tcfap2c physically interacted with the Cebpa gene promoter in the respiratory epithelium at E18.5 and activated Cebpa transcription. These results indicate that we have identified a novel role for Cited2 in pulmonary maturation that explains the pulmonary immaturity in mice lacking Cited2.
Mice lacking Cited2 die at late gestation with numerous developmental defects. Our previous study showed that pulmonary immaturity is independent of cardiac malformations because HIF-1α heterozygosity rescued outflow tract and interventricular septum defects, but did not rescue pulmonary immaturity in Cited2-null embryos (Xu et al., 2007). The present study suggests that pulmonary immaturity is also independent of left-right patterning defects. Only one third of Cited2 knockouts had neural tube defects (Xu et al., 2007), but all Cited2 knockouts had pulmonary immaturity, suggesting that pulmonary immaturity is also independent of neural tube defects. Thus, pulmonary immaturity in Cited2 knockouts is likely a primary defect. Lung epithelium specific knockout of Cited2 is required to address this issue ultimately.
Previous studies have shown that Cebpa expressed in type II pneumocytes regulates the expression of (a) SP-B, Abca3, Scd1, Fas, and Fabp5 that mediate lipid synthesis, (b) SP-B and SP-C that reduce alveolar surface tension, (c) SP-A, SP-D, Lys, and Hc that contribute to the anti-inflammatory activities, (d) Aqp5 and TGF-β2 that mediate alveolar epithelial cell differentiation, and (e) Gli1 with anti-apoptotic function (Cassel and Nord, 2003; Basseres et al., 2006; Martis et al., 2006). Loss of Cebpa in the respiratory epithelium in embryos leads to respiratory failure at birth due to pulmonary immaturity with an arrest in the type II alveolar cell differentiation and reduced peripheral saccules (Basseres et al., 2006; Martis et al., 2006). In this study, histopathological abnormalities consistent with pulmonary immaturity observed in Cited2-null fetal lungs, including reduced alveolar spaces, lack of squamous type I cells in the terminal sacs, and immature type II cells with dispersed cytoplasmic glycogen, smaller apical microvilli, and markedly decreased cytoplasmic lamellar bodies (Fig. 2), are prominent features observed in the lung-specific and conditional Cebpa knockouts (Basseres et al., 2006; Martis et al., 2006). Moreover, significantly decreased expression of Cebpa (Fig. 5) and differentially expressed Cebpa target genes in Cebpa-null lungs including SP-A, -B, -C, -D, Aqp5, TGF-β2, and Gli1 (Fig. 3, 4b, 5a) were demonstrated in Cited2-null fetal lungs. These results strongly suggest that Cited2 functions genetically upstream of Cebpa and the down-regulated expression of Cebpa is most likely responsible for the pulmonary immaturity in Cited2-null fetal lung.
In addition to Cebpa knockouts, lung-specific Cebpa transgenic animals are embryonic lethal with defects in lung branching morphogenesis (Berg et al., 2006). These animal studies indicate that an appropriate level of Cebpa is critical for normal lung development. During adipocyte differentiation, Cebpa transcription is repressed by Tcfap2a through its competitive binding to the A1 and A2 sites with Sp1/Sp3 (Jiang and Lane, 2002). In the present study, the transfection assay showed that strong C/EBPα-805-luc activity, most likely activated by endogenous Sp1/Sp3, was inhibited by Tcfap2c through its competitive binding to the C site on the Cebpa promoter (Fig. 6e). This C site is highly conserved among mouse, rat, and human Cebpa gene (Fig. 6b). In addition, the weak C/EBPα-805-luc activity, through the binding of Tcfap2c, was further activated by Cited2 and CBP (Fig. 6e), suggesting that Cited2 and CBP, transcriptional co-activators for Tcfap2c, may regulate Cebpa transcription to maintain its appropriate level in cells. Several lines of evidence support this idea: (a) endogenous Cited2 and Tcfap2c both occupied the C site of the Cebpa promoter in fetal lungs (Fig. 6c, 6d); (b) acetylated histone H3 can be detected in this region, indicating that it is a transcriptionally active site (Fig. 6c); (c) mutant mice bearing deletions in the CH1 domain of CBP have similar pulmonary defects, including thickened interstitial septa and decreased alveolar airspace, as those observed in Cited2 and Cebpa knockouts (Kasper et al., 2005).
It is also known that Cited2-null embryos develop adrenal agenesis (Bamforth et al., 2001; Val et al., 2007), likely resulting in fetal glucocorticoid insufficiency. Previous study has shown that corticotropin-releasing hormone-deficient mice, which as a consequence are also glucocorticoid insufficient, exhibit morphologic alterations in the lungs at E17.5 including defects of septal thinning and air-space formation (Muglia et al., 1999), which are similar to those observed in Cited2 and Cebpa knockouts. However, these histological alterations in corticotropin-releasing hormone-deficient lungs are considered to be the results of increased alveolar cell proliferation and delayed epithelial cell maturation while there was no obvious difference in the alveolar cell proliferation between wild-type and Cited2-null lungs. In addition, SP-A, B, C, and D mRNA levels were all significantly down-regulated in E17.5 (data not shown) and E18.5 (Fig. 3a) Cited2-null lungs, but only SP-A and SP-B mRNA levels were significantly decreased in E17.5 corticotropin-releasing hormone-deficient lungs. These suggest that fetal glucocorticoid insufficiency alone cannot explain all the phenotypes of the pulmonary immaturity in Cited2-null mice. Whether glucocorticoid insufficiency plays a role in Cited2-null lung phenotype can be directly answered by the analysis of tissue-specific Cited2 knockout mice.
Cited2 is a negative regulator for HIF-1 in vitro and in vivo and in its absence VEGF, a target of HIF-1, is over-expressed in the embryonic heart leading to abnormal coronary vasculature with higher permeability (Bhattacharya et al., 1999; Xu et al., 2007). In the lung, overexpression of VEGF under the control of the SP-C promoter in a transgenic mouse model induces gross abnormalities in lung morphogenesis and an increase in peritubular vascularity, with a concomitant decrease in both epithelial acinar tubules and mesenchyme (Zeng at al., 1998). In this study, there was no obvious difference in VEGF and VEGFR2 mRNA levels between wild-type and Cited2-null lungs at E17.5 (data not shown) and E18 (Fig. 5a). CD31 immunostaining revealed that most pulmonary capillaries closely abutted alveolar spaces in both wild-type and Cited2-null lungs at E18.5 (data not shown). Only a few capillaries embedded in the accumulated cells of E18.5 Cited2-null lungs were observed. These data suggest that the function of Cited2 as a negative regulator for HIF-1 may not be responsible for the pulmonary immaturity in Cited2-null fetal lungs, which is consistent with our previous finding that HIF-1α heterozygosity did not rescue lung defects in Cited2-null embryos (Xu et al., 2007).
Normal lung development is dependent upon reciprocal interactions between epithelial cells and mesenchymal cells during embryonic growth and differentiation, which are mediated by growth and differentiation factors including members of the fibroblast growth factor (FGF) family, Shh, and members of the TGF-β family (Warburton et al., 2003; Mendelson, 2000). Through the coordination with FGF9, FGF10 plays crucial roles in lung branching morphogenesis during early embryonic lung development (Min et al., 1998; Sekine et al., 1998; Colvin et al., 2001). The E18.5 FGF18-null lung is immature, exhibiting reduced alveolar space, thicker interstitial mesenchymal compartments, and embedded capillaries (Usui ey al., 2004), which are prominent features of Cebpa and Cited2 knockout lungs. Cited2 can be induced by hypoxia, cytokines, lipopolysaccharide and shear stress (Bhattacharya et al., 1999; Sun et al., 1998; Yokota et al., 2003). Recently, Haase et al. showed that Cited2 promoter activity and mRNA and protein expression can be induced by basic FGF in NCI-H295R cells (Haase et al., 2007). We therefore propose that upon binding of mesenchyme-expressed FGF18, FGF receptors of the respiratory epithelial cells undergo dimerization and auto-phosphorylation (Szebenyi and Fallon, 1999), leading to activation of Cited2 and Cebpa pathways and subsequent pulmonary maturation. This possibility could be directly addressed by analyzing Cited2 and Cebpa signaling pathways in FGF18-null lungs.
TGF-β plays important roles in lung epithelial cell proliferation and differentiation. Increased expression of TGF-β inhibited lung maturation in late gestation (Zhou et al., 1996; Bartram and Speer, 2004) and the expression of surfactant proteins (Whitsett et al., 1992; Jaskoll et al., 1996). Deletion of Cebpa enhanced expression of TGF-β2 and Smad3, a major transcription factor in the TGF-β signaling, in the respiratory epithelium (Martis et al., 2006), suggesting that the effects of Cebpa deletion may be mediated in part by the activation of TGF-β2 signaling. Expression of TGF-β2 was also increased in Cited2-null lungs, likely through the down-regulated expression of Cebpa in the absence of Cited2 (Fig. 5a). On the other hand, loss of Cited2 may attenuate TGF-β, since Cited2 has been shown to be a transcriptional co-activator for Smad2 and Smad3 to enhance TGF-β-mediated transcription (Chou et al., 2006). Whether increased expression of TGF-β2 or loss of co-activation function of Cited2 for Smad2/Smad3 plays any role in pulmonary immaturity in Cited2-null fetal lungs requires further investigation.
In conclusion, the present study establishes a new role for Cited2 in lung maturation by modulating gene expression of Cebpa, which is one of the key transcription factors for regulating surfactant and Abca3 transcription. Deficiency of surfactant lipids and proteins in the lung is associated with respiratory distress syndrome in preterm infants, which is a common cause of neonatal mortality and morbidity and is associated with environmental risks and mutations in SP-A, SP-B, SP-C, and Abca3 (Ramet et al., 2000; Whitsett et al., 2004; Shulenin et al., 2004). Thus, the knockout mice of the transcription factors and co-activators, including Cited2, which are critical for surfactant and Abca3 transcription, are useful models with which to investigate the etiology and potential therapeutic strategies to reverse or prevent respiratory distress syndrome in humans.
Supplementary Material
Supplemental Fig. 1. Right pulmonary isomerism in Cited2−/− embryonic lungs. Lungs (C, D) were dissected from a wild-type embryo (A) and a Cited2−/− littermate with exencephaly (B) at E18.5. In contrast to the wild-type lung that had 4 lobes on the right side (E, G, I, K) and 1 lobe on the left (M), the Cited2−/− lung had 4 lobes on the right (F, H, J, L) and 4 lobes on the left (N). Right cranial lobe (E, F), right median lobe (G, H), right caudual lobe (I, J), accessory lobe (K, L), and left lobe (M, N) are individually shown.
Acknowledments
We thank Drs. Jeffrey Kern and Balazs Halmos for many helpful discussions. This work was supported by grants from HL075436 (Y.C.Y and M.W.) and NHMRC404805 (S.L.D.). B.X. is supported by American Heart Association Ohio Valley Postdoctoral Fellowship. S.L.D. is a Pfizer Foundation Australia Senior Research Fellow.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bamforth SD, Braganca J, Eloranta JJ, Murdoch JN, Marques FI, Kranc KR, Farza H, Henderson DJ, Hurst HC, Bhattacharya S. Cardiac malformations, adrenal agenesis, neural crest defects and exencephaly in mice lacking Cited2, a new Tfap2 co-activator. Nat. Genet. 2001;29:469–474. doi: 10.1038/ng768. [DOI] [PubMed] [Google Scholar]
- Bamforth SD, Braganca J, Farthing CR, Schneider JE, Broadbent C, Michell AC, Clarke K, Neubauer S, Norris D, Brown NA, Anderson RH, Bhattacharya S. Cited2 controls left-right patterning and heart development through a Nodal-Pitx2c pathway. Nat. Genet. 2004;36:1189–1196. doi: 10.1038/ng1446. [DOI] [PubMed] [Google Scholar]
- Bartram U, Speer CP. The role of transforming growth factor beta in lung development and disease. Chest. 2004;125:754–765. doi: 10.1378/chest.125.2.754. [DOI] [PubMed] [Google Scholar]
- Basseres DS, Levantini E, Ji H, Monti S, Elf S, Dayaram T, Fenyus M, Kocher O, Golub T, Wong KK, Halmos B, Tenen DG. Respiratory failure due to differentiation arrest and expansion of alveolar cells following lung-specific loss of the transcription factor C/EBPalpha in mice. Mol Cell Biol. 2006;26:1109–1123. doi: 10.1128/MCB.26.3.1109-1123.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg T, Didon L, Nord M. Ectopic expression of C/EBPalpha in the lung epithelium disrupts late lung development. Am. J. Physiol. Lung Cell Mol. Physiol. 2006;291:L683–L693. doi: 10.1152/ajplung.00497.2005. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Michels CL, Leung MK, Arany ZP, Kung AL, Livingston DM. Functional role of p35srj, a novel p300/CBP binding protein, during transactivation by HIF-1. Genes Dev. 1999;13:64–75. doi: 10.1101/gad.13.1.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigelow RL, Chari NS, Unden AB, Spurgers KB, Lee S, Roop DR, Toftgard R, McDonnell TJ. Transcriptional regulation of bcl-2 mediated by the sonic hedgehog signaling pathway through gli-1. J. Biol. Chem. 2004;279:1197–1205. doi: 10.1074/jbc.M310589200. [DOI] [PubMed] [Google Scholar]
- Boggaram V. Regulation of lung surfactant protein gene expression. Front. Biosci. 2003;8:d751–d764. doi: 10.2741/1062. [DOI] [PubMed] [Google Scholar]
- Braganca J, Eloranta JJ, Bamforth SD, Ibbitt JC, Hurst HC, Bhattacharya S. Physical and functional interactions among AP-2 transcription factors, p300/CREB-binding protein, and CITED2. J. Biol. Chem. 2003;278:16021–16029. doi: 10.1074/jbc.M208144200. [DOI] [PubMed] [Google Scholar]
- Breed DR, Margraf LR, Alcorn JL, Mendelson CR. Transcription factor C/EBPdelta in fetal lung: developmental regulation and effects of cyclic adenosine 3′,5′-monophosphate and glucocorticoids. Endocrinology. 1997;138:5527–5534. doi: 10.1210/endo.138.12.5637. [DOI] [PubMed] [Google Scholar]
- Cardoso WV, Lu J. Regulation of early lung morphogenesis: questions, facts and controversies. Development. 2006;133:1611–1624. doi: 10.1242/dev.02310. [DOI] [PubMed] [Google Scholar]
- Cassel TN, Nord M. C/EBP transcription factors in the lung epithelium. Am. J. Physiol. Lung Cell Mol. Physiol. 2003;285:L773–L781. doi: 10.1152/ajplung.00023.2003. [DOI] [PubMed] [Google Scholar]
- Chou YT, Wang H, Chen Y, Danielpour D, Yang YC. Cited2 modulates TGF-beta-mediated upregulation of MMP9. Oncogene. 2006;25:5547–5560. doi: 10.1038/sj.onc.1209552. [DOI] [PubMed] [Google Scholar]
- Colvin JS, White AC, Pratt SJ, Ornitz DM. Lung hypoplasia and neonatal death in Fgf9-null mice identify this gene as an essential regulator of lung mesenchyme. Development. 2001;128:2095–2106. doi: 10.1242/dev.128.11.2095. [DOI] [PubMed] [Google Scholar]
- Dunwoodie SL, Rodriguez TA, Beddington RS. Msg1 and Mrg1, founding members of a gene family, show distinct patterns of gene expression during mouse embryogenesis. Mech. Dev. 1998;72:27–40. doi: 10.1016/s0925-4773(98)00011-2. [DOI] [PubMed] [Google Scholar]
- Freedman SJ, Sun ZY, Kung AL, France DS, Wagner G, Eck MJ. Structural basis for negative regulation of hypoxia-inducible factor-1alpha by CITED2. Nat. Struct. Biol. 2003;10:504–512. doi: 10.1038/nsb936. [DOI] [PubMed] [Google Scholar]
- Glenn DJ, Maurer RA. MRG1 binds to the LIM domain of Lhx2 and may function as a coactivator to stimulate glycoprotein hormone alpha-subunit gene expression. J. Biol. Chem. 1999;274:36159–36167. doi: 10.1074/jbc.274.51.36159. [DOI] [PubMed] [Google Scholar]
- Haase M, Schott M, Bornstein SR, Malendowicz LK, Scherbaum WA, Willenberg HS. CITED2 is expressed in human adrenocortical cells and regulated by basic fibroblast growth factor. J. Endocrinol. 2007;192:459–465. doi: 10.1677/JOE-06-0083. [DOI] [PubMed] [Google Scholar]
- Hilger-Eversheim K, Moser M, Schorle H, Buettner R. Regulatory roles of AP-2 transcription factors in vertebrate development, apoptosis and cell-cycle control. Gene. 2000;260:1–12. doi: 10.1016/s0378-1119(00)00454-6. [DOI] [PubMed] [Google Scholar]
- Jaskoll T, Choy HA, Melnick M. The glucocorticoid-glucocorticoid receptor signal transduction pathway, transforming growth factor-beta, and embryonic mouse lung development in vivo. Pediatr. Res. 1996;39:749–759. doi: 10.1203/00006450-199605000-00002. [DOI] [PubMed] [Google Scholar]
- Jiang MS, Lane MD. Sequential repression and activation of the CCAAT enhancer-binding protein-alpha (C/EBPalpha) gene during adipogenesis. Proc. Natl. Acad. Sci. U. S. A. 2000;97:12519–12523. doi: 10.1073/pnas.220426097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper LH, Boussouar F, Boyd K, Xu W, Biesen M, Rehg J, Baudino TA, Cleveland JL, Brindle PK. Two transactivation mechanisms cooperate for the bulk of HIF-1-responsive gene expression. EMBO J. 2005;24:3846–3858. doi: 10.1038/sj.emboj.7600846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Watts GS, Oshiro MM, Futscher BW, Domann FE. AP-2alpha and AP-2gamma are transcriptional targets of p53 in human breast carcinoma cells. Oncogene. 2006;25:5405–5415. doi: 10.1038/sj.onc.1209534. [DOI] [PubMed] [Google Scholar]
- Martinez Barbera JP, Rodriguez TA, Greene ND, Weninger WJ, Simeone A, Copp AJ, Beddington RS, Dunwoodie S. Folic acid prevents exencephaly in Cited2 deficient mice. Hum. Mol. Genet. 2002;11:283–293. doi: 10.1093/hmg/11.3.283. [DOI] [PubMed] [Google Scholar]
- Martis PC, Whitsett JA, Xu Y, Perl AK, Wan H, Ikegami M. C/EBPalpha is required for lung maturation at birth. Development. 2006;133:1155–1164. doi: 10.1242/dev.02273. [DOI] [PubMed] [Google Scholar]
- Mendelson CR. Role of transcription factors in fetal lung development and surfactant protein gene expression. Annu. Rev. Physiol. 2000;62:875–915. doi: 10.1146/annurev.physiol.62.1.875. [DOI] [PubMed] [Google Scholar]
- McPherson LA, Baichwal VR, Weigel RJ. Identification of ERF-1 as a member of the AP2 transcription factor family. Proc. Natl. Acad. Sci. U. S. A. 1997;94:4342–4347. doi: 10.1073/pnas.94.9.4342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min H, Danilenko DM, Scully SA, Bolon B, Ring BD, Tarpley JE, DeRose M, Simonet WS. Fgf-10 is required for both limb and lung development and exhibits striking functional similarity to Drosophila branchless. Genes Dev. 1998;12:3156–3161. doi: 10.1101/gad.12.20.3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minoo P, King RJ. Epithelial-mesenchymal interactions in lung development. Annu. Rev. Physiol. 1994;56:13–45. doi: 10.1146/annurev.ph.56.030194.000305. [DOI] [PubMed] [Google Scholar]
- Moser M, Imhof A, Pscherer A, Bauer R, Amselgruber W, Sinowatz F, Hofstadter F, Schule R, Buettner R. Cloning and characterization of a second AP-2 transcription factor: AP-2 beta. Development. 1995;121:2779–2788. doi: 10.1242/dev.121.9.2779. [DOI] [PubMed] [Google Scholar]
- Muglia LJ, Bae DS, Brown TT, Vogt SK, Alvarez JG, Sunday ME, Majzoub JA. Proliferation and differentiation defects during lung development in corticotropin-releasing hormone-deficient mice. Am. J. Respir. Cell Mol. Biol. 1999;20:181–188. doi: 10.1165/ajrcmb.20.2.3381. [DOI] [PubMed] [Google Scholar]
- Oulad-Abdelghani M, Bouillet P, Chazaud C, Dolle P, Chambon P. AP-2.2: a novel AP-2-related transcription factor induced by retinoic acid during differentiation of P19 embryonal carcinoma cells. Exp. Cell Res. 1996;225:338–347. doi: 10.1006/excr.1996.0184. [DOI] [PubMed] [Google Scholar]
- Ramet M, Haataja R, Marttila R, Floros J, Hallman M. Association between the surfactant protein A (SP-A) gene locus and respiratory-distress syndrome in the Finnish population. Am. J. Hum. Genet. 2000;66:1569–1579. doi: 10.1086/302906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg E, Li F, Reisher SR, Wang M, Gonzales LW, Ewing JR, Malek S, Ballard PL, Notarfrancesco K, Shuman H, Feinstein SI. Members of the C/EBP transcription factor family stimulate expression of the human and rat surfactant protein A (SP-A) genes. Biochim. Biophys. Acta. 2002;1575:82–90. doi: 10.1016/s0167-4781(02)00287-7. [DOI] [PubMed] [Google Scholar]
- Screpanti I, Romani L, Musiani P, Modesti A, Fattori E, Lazzaro D, Sellitto C, Scarpa S, Bellavia D, Lattanzio G, Bistoni F, Frati L, Cortese R, Gulino A, Ciliberto G, Costantini F, Poli V. Lymphoproliferative disorder and imbalanced T-helper response in C/EBP beta-deficient mice. EMBO J. 1995;14:1932–1941. doi: 10.1002/j.1460-2075.1995.tb07185.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekine K, Ohuchi H, Fujiwara M, Yamasaki M, Yoshizawa T, Sato T, Yagishita N, Matsui D, Koga Y, Itoh N, Kato S. Fgf10 is essential for limb and lung formation. Nat. Genet. 1999;21:138–141. doi: 10.1038/5096. [DOI] [PubMed] [Google Scholar]
- Shulenin S, Nogee LM, Annilo T, Wert SE, Whitsett JA, Dean M. ABCA3 gene mutations in newborns with fatal surfactant deficiency. N. Engl. J. Med. 2004;350:1296–1303. doi: 10.1056/NEJMoa032178. [DOI] [PubMed] [Google Scholar]
- Stahlman MT, Gray ME, Whitsett JA. Expression of thyroid transcription factor-1 (TTF-1) in fetal and neonatal human lung. J. Histochem. Cytochem. 1996;44:673–678. doi: 10.1177/44.7.8675988. [DOI] [PubMed] [Google Scholar]
- Sterneck E, Paylor R, Jackson-Lewis V, Libbey M, Przedborski S, Tessarollo L, Crawley JN, Johnson PF. Selectively enhanced contextual fear conditioning in mice lacking the transcriptional regulator CCAAT/enhancer binding protein delta. Proc. Natl. Acad. Sci. U. S. A. 1998;95:10908–10913. doi: 10.1073/pnas.95.18.10908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun HB, Zhu YX, Yin T, Sledge G, Yang YC. MRG1, the product of a melanocyte-specific gene related gene, is a cytokine-inducible transcription factor with transformation activity. Proc. Natl. Acad. Sci. U. S. A. 1998;95:13555–13560. doi: 10.1073/pnas.95.23.13555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland LM, Edwards YS, Murray AW. Alveolar type II cell apoptosis. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2001;129:267–285. doi: 10.1016/s1095-6433(01)00323-3. [DOI] [PubMed] [Google Scholar]
- Szebenyi G, Fallon JF. Fibroblast growth factors as multifunctional signaling factors. Int. Rev. Cytol. 1999;185:45–106. doi: 10.1016/s0074-7696(08)60149-7. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Yoshida N, Kishimoto T, Akira S. Defective adipocyte differentiation in mice lacking the C/EBPbeta and/or C/EBPdelta gene. EMBO J. 1997;16:7432–7443. doi: 10.1093/emboj/16.24.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tien ES, Davis JW, Vanden Heuvel JP. Identification of the CREB-binding protein/p300-interacting protein CITED2 as a peroxisome proliferator-activated receptor alpha coregulator. J. Biol. Chem. 2004;279:24053–24063. doi: 10.1074/jbc.M401489200. [DOI] [PubMed] [Google Scholar]
- Usui H, Shibayama M, Ohbayashi N, Konishi M, Takada S, Itoh N. Fgf18 is required for embryonic lung alveolar development. Biochem. Biophys. Res. Commun. 2004;322:887–892. doi: 10.1016/j.bbrc.2004.07.198. [DOI] [PubMed] [Google Scholar]
- Val P, Martinez-Barbera JP, Swain A. Adrenal development is initiated by Cited2 and Wt1 through modulation of Sf-1 dosage. Development. 2007;134(12):2349–2358. doi: 10.1242/dev.004390. [DOI] [PubMed] [Google Scholar]
- Warburton D, Bellusci S, Del Moral PM, Kaartinen V, Lee M, Tefft D, Shi W. Growth factor signaling in lung morphogenetic centers: automaticity, stereotypy and symmetry. Respir. Res. 2003;4:5. doi: 10.1186/1465-9921-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver TE, Conkright JJ. Function of surfactant proteins B and C. Annu. Rev. Physiol. 2001;63:555–578. doi: 10.1146/annurev.physiol.63.1.555. [DOI] [PubMed] [Google Scholar]
- Weninger WJ, Floro KL, Bennett MB, Withington SL, Preis JI, Barbera JP, Mohun TJ, Dunwoodie SL. Cited2 is required both for heart morphogenesis and establishment of the left-right axis in mouse development. Development. 2005;132:1337–1348. doi: 10.1242/dev.01696. [DOI] [PubMed] [Google Scholar]
- Whitsett JA, Budden A, Hull WM, Clark JC, O'Reilly MA. Transforming growth factor-beta inhibits surfactant protein A expression in vitro. Biochim. Biophys. Acta. 1992;1123:257–262. doi: 10.1016/0005-2760(92)90004-f. [DOI] [PubMed] [Google Scholar]
- Whitsett JA, Wert SE, Trapnell BC. Genetic disorders influencing lung formation and function at birth. Hum. Mol. Genet. 2004;13:R207–R215. doi: 10.1093/hmg/ddh252. [DOI] [PubMed] [Google Scholar]
- Williams MC, Mason RJ. Development of the type II cell in the fetal rat lung. Am. Rev. Respir. Dis. 1977;115:37–47. doi: 10.1164/arrd.1977.115.S.37. [DOI] [PubMed] [Google Scholar]
- Withington SL, Scott AN, Saunders DN, Lopes Floro K, Preis JI, Michalicek J, Maclean K, Sparrow DB, Barbera JP, Dunwoodie SL. Loss of Cited2 affects trophoblast formation and vascularization of the mouse placenta. Dev. Biol. 2006;294:67–82. doi: 10.1016/j.ydbio.2006.02.025. [DOI] [PubMed] [Google Scholar]
- Xu B, Doughman YQ, Turakhia M, Jiang WH, Landsettle CE, Agani FH, Semenza GL, Watanabe M, Yang YC. Partial rescue of defects in Cited2-deficient embryos by HIF-1α heterozygosity. Dev. Biol. 2007;301:130–140. doi: 10.1016/j.ydbio.2006.08.072. [DOI] [PubMed] [Google Scholar]
- Yin Z, Haynie J, Yang X, Han B, Kiatchoosakun S, Restivo J, Yuan S, Prabhakar NR, Herrup K, Conlon RA, Hoit BD, Watanabe M, Yang YC. The essential role of Cited2, a negative regulator for HIF-1alpha, in heart development and neurulation. Proc. Natl. Acad. Sci. U. S. A. 2002;99:10488–10493. doi: 10.1073/pnas.162371799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokota H, Goldring MB, Sun HB. CITED2-mediated regulation of MMP-1 and MMP-13 in human chondrocytes under flow shear. J. Biol. Chem. 2003;278:47275–47280. doi: 10.1074/jbc.M304652200. [DOI] [PubMed] [Google Scholar]
- Zeng X, Wert SE, Federici R, Peters KG, Whitsett JA. VEGF enhances pulmonary vasculogenesis and disrupts lung morphogenesis in vivo. Dev Dyn. 1998;211:215–227. doi: 10.1002/(SICI)1097-0177(199803)211:3<215::AID-AJA3>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Zhou L, Dey CR, Wert SE, Whitsett JA. Arrested lung morphogenesis in transgenic mice bearing an SP-C-TGF-beta 1 chimeric gene. Dev. Biol. 1996;175:227–238. doi: 10.1006/dbio.1996.0110. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Fig. 1. Right pulmonary isomerism in Cited2−/− embryonic lungs. Lungs (C, D) were dissected from a wild-type embryo (A) and a Cited2−/− littermate with exencephaly (B) at E18.5. In contrast to the wild-type lung that had 4 lobes on the right side (E, G, I, K) and 1 lobe on the left (M), the Cited2−/− lung had 4 lobes on the right (F, H, J, L) and 4 lobes on the left (N). Right cranial lobe (E, F), right median lobe (G, H), right caudual lobe (I, J), accessory lobe (K, L), and left lobe (M, N) are individually shown.