

Abstract
Inclusion body myopathy with Paget disease of the bone (PDB) and/or frontotemporal dementia (IBMPFD, OMIM 167320), is a progressive autosomal dominant disorder caused by mutations in the Valousin-containing protein (VCP, p97 or CDC48) gene. IBMPFD can be difficult to diagnose. We assembled data on a large set of families to illustrate the number and type of misdiagnoses that occurred. Clinical analysis of 49 affected individuals in nine families indicated that 42 (87%) of individuals had muscle disease. The majority were erroneously diagnosed with limb girdle muscular dystrophy (LGMD), facioscapular muscular dystrophy, peroneal muscular dystrophy, late adult onset distal myopathy, spinal muscular atrophy, scapuloperoneal muscular dystrophy, or amyotrophic lateral sclerosis (ALS) among others. Muscle biopsies showed rimmed vacuoles characteristic of an inclusion body myopathy in 7 of 18 patients (39%), however, inclusion body myopathy was correctly diagnosed among individuals in only families 5 and 15. Frontotemporal dementia (FTD) was diagnosed in 13 individuals (27%) at a mean age of 57 years (range 48.9–60.2 years); however, several individuals had been diagnosed with Alzheimer disease. Histopathological examination of brains of three affected individuals revealed a pattern of ubiquitin positive neuronal intranuclear inclusions and dystrophic neurites. These families expand the clinical phenotype in IBMPFD, a complex disorder caused by mutations in VCP. The presence of PDB in 28 (57%) individuals suggests that measuring serum alkaline phosphatase (ALP) activity may be a useful screen for IBMPFD in patients with myopathy.
Keywords: autosomal dominant, hereditary inclusion body myopathy, limb-girdle muscular dystrophy, Paget disease of bone, frontotemporal dementia, chromosome 9p13.3-12, VCP (valosin-containing protein)
INTRODUCTION
Hereditary inclusion body myopathy (HIBM) with early-onset Paget disease of bone (PDB) and frontotemporal dementia (FTD), or IBMPFD, is an autosomal dominant disorder (MIM 167320) associated with a variable expressivity. Kimonis et al. [2000] described a family with 11 affected members with autosomal dominant limb-girdle muscular dystrophy (LGMD) associated with early-onset PDB. The association of inclusion body myopathy and FTD was established by Kovach et al. [2001] among 49 affected individuals from the original family and three other unrelated families. This disorder is now called IBMPFD: HIBM, PDB, and FTD and it is caused by mutations in the valosin-containing protein (VCP; 601023) [Watts et al., 2004]. In this report, we review the clinical variability in 49 individuals among nine families, where the diagnosis was confirmed by the presence of VCP mutations and illustrate the difficulty in making an accurate diagnosis.
PATIENTS AND METHODS
Clinical Evaluation and Diagnosis
The study was reviewed and approved by Children’s Hospital Boston IRB. This is a cross-section study of individuals referred from North American clinical centers including those self-referred. VCP mutations were confirmed in 49 individuals of nine families, and major clinical characteristics derived from medical records and family histories.
We present the details of the three principal physical findings: myopathy, PDB, and dementia. The diagnosis of myopathy was based on the presence of muscular weakness, elevated creatine kinase measurements, and in several patients by EMG and/or muscle biopsy findings. Physical examination findings used to make a diagnosis of myopathy included various combinations of an abnormal gait, lumbar lordosis from the proximal weakness, difficulty raising the arms or climbing stairs, and mild weakness of the hands. Tendon reflexes were reduced or absent. Sections of unfixed muscle were stained by standard procedures. Indirect immunofluorescence analysis and immunoblotting of skeletal muscle were performed and electron microscopy was done on unfixed frozen sections [Askanas et al., 1993; Mirabella et al., 1996; Askanas and Engel, 1998].
Clinical symptoms suggestive of PDB included spine or hip pain, pathologic fractures, or long bone or cranial bone deformity. Whenever possible, PDB was confirmed by skeletal radiographic surveys. Typical findings of PDB include coarse trabeculation, cortical thickening, and spotty sclerosis. Radio-nuclide scans were used to demonstrate increased bony uptake and are more sensitive indicators of PDB than plain survey films. The diagnosis of PDB was made by nuclide scans in 13 patients and radiographs in 21.
FTD was diagnosed by neuropsychological assessments and imaging studies when available, together with typical clinical features of behavioral alteration (e.g., personal/social unawareness, perseveration, abulia, disinhibition), early expressive or receptive language dysfunction, and relative preservation of memory, orientation, or praxis [Miller et al., 1997]. Neuropsychological evaluations included the administration of a combination of the following tests: Neurobehavioral Cognitive Status Examination, Boston Naming Test, Wechsler Adult Intelligence Scale, Controlled Oral Word Association Test, Categories Verbal Fluency Test, Eisenson Reading Passages, Hooper Visual Organization Test, Draw a Clock by Command, Rey Osterreith Complex Figure Drawing, Bender Visual-Motor Gestalt Test, Benton Visual Retention Test, Memory for Unrelated Sentences Test, Memory Assessment Scale, Trailmaking Tests A & B, Stroop Neuropsychological Screening Test, Grooved Pegboard, McCarthy Verbal Fluency Test, Electronic Finger Tapping Test, and the Multi-score Depression Inventory. In several deceased individuals, medical history was abstracted from their medical records.
Antibody Studies
We present a summary of the neuropathology studies previously described in detail [Forman et al., 2006].
Statistical Analysis
Tables of clinical variables for statistical analysis were generated by cross-tabulation of the available clinical and laboratory data for the affected members of the nine families. Data are presented as means ± SE unless otherwise specified.
Clinical Reports (Grouped by Family)
Family 5
The proband (IV:3) of this four-generation family (Fig. 1A) of German extraction was diagnosed with limb girdle muscular dystrophy (LGMD). She noticed weakness of the left index finger followed by weakness of both hands and reduced wrist mobility, and toe weakness, and pain associated with tripping in her 20’s. The distal weakness was followed by proximal weakness in the arms and legs resulting in difficulty climbing stairs in her late 30s. She has shortness of breath and has sleep apnea. Examination revealed proximal and distal weakness of hand and foot extensors, and decreased deep tendon reflexes. Muscle biopsy revealed non-specific myopathic changes with fiber size variation, necrosis, centrally located nuclei, focal endomysial fibrosis, and rimmed vacuoles within the muscle fibers consistent with inclusion body myopathy. PDB was not apparent on bone scans. Her brother, individual (IV:4) had a 10–15 years history of progressive distal and proximal muscle weakness and back and hip pain from PDB and was using an automated wheelchair since the age of 39 years. Her younger brother (IV:5) had chronic low back pain and was diagnosed with PDB after finding elevated alkaline phosphatase (ALP) levels at age 39 years, following enrollment in our study. Individual IV:15, a 33-year-old male cousin of the proband, developed proximal muscle weakness, pain, spasms, and tremors in the arms and legs and severe back pain at 23 years, the latter attributed to PDB. A muscle biopsy of the left deltoid muscle revealed significant variation of muscle fiber size with fiber splitting, fragmentation, and atrophic and hypertrophic fibers and rimmed vacuoles within muscle fibers, consistent with inclusion body myopathy.
Fig. 1.

Pedigrees of IBMPFD families. The filled in top right corner of a symbol represents myopathy, the bottom right corner of a symbol represents Paget disease of the bone (PDB) and the bottom left corner of a symbol represents frontotemporal dementia (FTD).
Family 6
This Canadian family of English origin includes two affected siblings and their mother (II:2) (Fig. 1B), the latter of whom developed progressive weakness in the legs, arms, trunk, and neck in her 40’s and was diagnosed with motor neuron disease. At 56 years, she was confined to a wheelchair and a year later she was fitted with a collar for severe neck muscle weakness. X-ray revealed PDB of the spine, pelvis, skull, and legs, and osteoporosis. She also had bilateral sensorineural hearing loss. One of her daughters (III:1) had onset of progressive proximal muscle weakness and fatigability in the limbs at 21 years and was diagnosed with LGMD at 37 years. She has weakness in her hands and neck but is mobile with an automated wheelchair. Nerve conduction studies were suggestive of axonal sensory neuropathy in her legs. She has restrictive lung disease. She has a history of fractures, and was diagnosed with PDB at 35 years. Her brother (III:4) was diagnosed with PDB at 30 years after sustaining a right humeral fracture. He had a subsequent history of fractures of the forearm, ribs, and left clavicle. EMG studies in both siblings showed neuropathic and myopathic features. Muscle biopsy revealed variation in muscle fiber size and atrophic fibers with <1% rimmed vacuolated fibers.
Family 7
This three-generation family includes eight affected, three of whom are deceased (Fig. 1C). The proband (III:6) had leg muscle cramps in her 30’s and was diagnosed with LGMD at age 47 years. She developed diaphragmatic involvement and required positive pressure oxygen at night. A muscle biopsy revealed non-specific myopathic findings. Her father (II:10) was diagnosed with amyotrophic lateral sclerosis (ALS) and died at the age of 61 years. Her grandmother (I:2) had myopathy and was diagnosed with Parkinson’s disease. Two paternal aunts (II:3, II:5), two paternal uncles (II:6, II:8), and two cousins (III:1, III:2) were diagnosed with either LGMD or ALS, and several with PDB. One paternal aunt (II:3), who was diagnosed with scapuloperoneal muscular dystrophy (SPMD), became wheelchair-bound and developed respiratory distress at age 70 years. An EMG at age 63 years revealed severe myopathy with myotonia. Another aunt (II:5), with similar features had difficulty breathing since age 61 years. and used a motorized scooter since age 66 years. Her son (III:1) had leg weakness since age 34 years, reporting difficulty climbing stairs and lifting his arms overhead. Individual III:3, diagnosed with PDB, had back problems and pain in her hips since her 20s, and had onset of muscle weakness at age 42 years.
Family 9
The female proband (V:2) of this five-generation family was diagnosed at 35 years with PDB at a routine examination based on an elevated ALP of 859 IU/L (Fig. 1D). Bone scan revealed abnormal sclerosis and mild enlargement in the right clavicle, proximal left humerus, distal left femur, left hemipelvis, and thoracic spine. Nerve conduction studies and EMG of the left arm were normal. Her mother (IV:2) was considered unaffected, however had difficulty walking up stairs, and demonstrates a mild peripheral myopathy. A maternal aunt (IV:4) became bed-bound from myopathy at 44 years. She was diagnosed with dementia at age 61 years. Physical exam revealed bilateral foot drop and clawing of the left hand. CT of the brain revealed mild generalized atrophy involving the bitemporal lobes and perisylvian areas. Her son (V:3) was diagnosed at age 40 years. with SPMD.
Family 10
The proband of this four-generation family (pedigree not shown) was diagnosed with LGMD at age 46 years. He has progressive proximal weakness, was unable to lift his arms above his head, had problems climbing stairs, and developed nocturnal hypoventilation at 39 years. He had onset of back pain for 10 years and was diagnosed with PDB in his right hip and L3 vertebrae by X-rays and bone scan at age 46 years. His mother died from myopathy at age 66 years. His brother was diagnosed with myopathy at age 50 years, but chose not to participate in this study.
Family 11
Clinical data in nine individuals from this family were previously reported by Tucker et al. [1982] (Fig. 1E). They were previously diagnosed with autosomal dominant lower motor neuron disease and PDB. Medical information on several of these deceased individuals is included in Table I. We have established contact with individuals from successive generations of the original family. The proband for our study (V:4) was diagnosed with SPMD although his predominant illness was PDB. He developed back pain at age 24 years, and sustained a non-displaced fracture of the distal left fibula, which was slow to heal after a fall at work at age 31 years. He has suffered repetitive leg fractures. Laboratory testing revealed an elevated ALP level (>1,000 IU/L), X-rays revealed increased trabecular pattern of PDB of the right tibia, and bone scan revealed multiple areas of increased uptake throughout the skeleton. Because of concern for metastatic bone disease, he had a biopsy of the right pelvis at age 31 years, which revealed disruption of the normal pattern of cortical and cancellous bone, with a proliferation of fibrous connective tissue in the medullary spaces, hypertrophic trabeculi with mosaic patterns, and focal new bone formation consistent with early PDB. In the areas where the medullary fibrous proliferation was obvious, the pattern suggested fibrous dysplasia. He has had difficulty lifting his arms since the age of 40 years, and difficulty walking since the age of 46 years.
TABLE I.
Clinical and Laboratory Data of Affected Individuals
| Case | Age (years) |
Sex | PDB | Myopathy | Dementia | Age of onset PDB (years) |
Age of onset myopathy (years) |
Age of onset dementia (years) |
ALP U/L |
CPK U/L |
EMG changes |
Muscle biopsy |
Paget Dx. method |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Family 5 463 C >T, R155C | |||||||||||||
| 1 IV:3 | 55 | F | − | +LGMD >HIBM | − | – | 34 | – | 71/80 | 271 | Myopathic | Inclusion body myopathy, rimmed vacuoles | X-ray, bone scan |
| 2 IV:4 | 41 | M | + | +SMA >LGMD | − | 33 | 31 | – | 68 | 564 | Myopathic | Inclusion body myopathy | X-ray |
| 3 IV:5 | 39 | M | + | − | − | 40 | – | – | 1388 | 471 | – | – | X-ray, bone scan |
| 4 IV:15 | 33 | M | + | +Inclusion body myositis | − | 23 | 24 | – | 73 | 251 | Myopathic | Inclusion body myopathy | X-ray, bone scan |
| 5 III:4 | a58 | M | + | + | + | 32 | 32 | 57 | NA | NA | NA | NA | NA |
| 6 III:6 | a60 | M | + | + | + | 34 | NA | NA | NA | NA | NA | ||
| 7 III:9 | a58 | F | + | + | + | 56 | NA | NA | NA | NA | NA | ||
| 8 III:11 | a62 | M | + | + | − | 32 | 32 | – | NA | NA | NA | NA | NA |
| 9 II:4 | a47 | F | + | + | + | 32 | 30 | 46 | NA | NA | NA | NA | NA |
| Mean | 42/a57 | 32 | 31 | 53 | 402 | 389 | |||||||
| Total | 3F/6M | 8 | 8 | 4 | |||||||||
| Family 6 695 C >A, A232E | |||||||||||||
| 1 II:2 | a63 | F | + | +ALS, SNHL | − | 50 | – | NA | NA | Myopathic | NA | X-ray | |
| 2 III:2 | 55 | F | + | +LGMD | − | 37 | – | 1204 | 200 | Myopathic/neurogenic | Myopathic neuropathic | X-ray, total body bone imaging | |
| 3 III:5 | 49 | M | + | +LGMD | − | 30 | 39 | – | 3006 | 124 | Myopathic | Myopathic, neuropathic | X-ray, bone scan |
| Mean | 52/a63 | 30 | 42 | – | 2105 | 162 | |||||||
| Total | 2F/1M | 3 | 3 | 0 | |||||||||
| Family 7 464 G >A, R155H | |||||||||||||
| 1 III:6 | 54 | F | + | +LGMD/SPMD | − | 51 | 47 | – | 265 | 75 | Diffuse denervation | Non-specific myopathic | X-ray, bone scan |
| 2 II:5 | 67 | F | + | +LGMD | − | 65 | 60 | – | 149 | 76 | NA | Non-specific myopathic | X-ray, bone scan |
| 3 III:1 | 43 | M | + | +LGMD/Diabetic neuropathy | − | 40 | 39 | – | 261 | 210 | Active denervation | Inclusion body myopathy | X-ray |
| 4 II:6 | a65 | M | − | + | − | – | 35 | – | NA | NA | NA | NA | NA |
| 5 II:8 | a55 | M | − | + | − | – | 50 | – | NA | NA | NA | NA | NA |
| 6 II:10 | a61 | M | − | +ALS | − | – | 61 | – | NA | NA | NA | NA | NA |
| 7 II:3 | 71 | F | − | +LGMD/SPMD | − | – | 56 | – | 66 | 70 | Severe myopathy, myotonia | Non-specific, neurogenic | NA |
| 8 III:2 | 45 | F | + | −Diabetic neuropathy | − | 45 | – | – | 354 | 113 | Myopathic | Non-specific | X-ray, bone scan |
| Mean | 56/a60 | 50 | 50 | 219 | 109 | ||||||||
| Total | 4F/4M | 4 | 7 | 0 | |||||||||
| Family 9 283 C >G, R95G | |||||||||||||
| 1 V:3 | 42 | M | − | +SPMD | − | – | 40 | – | 157 | 163 | NA | NA | NA |
| 2 V:2 | 36 | F | + | − | − | 35 | – | – | 1200 | 93 | NA | NA | X-ray, bone scan |
| 3 IV:5 | 60 | M | − | +ALS | − | – | 50 | – | 339 | 74 | Non-specific | NA | NA |
| 4 IV:4 | 61 | F | − | +SPMD | + | – | 40 | 58 | 109 | 52 | Non-specific | Neurogenic | NA |
| Mean | 50 | 35 | 43 | 58 | 451 | 96 | |||||||
| Total | 2F/2M | 1 | 3 | 1 | |||||||||
| Family 10 464 G >A, R155H | |||||||||||||
| 1 | 52 | M | + | +LGMD | − | 40 | 46 | – | 493 | – | Non-specific | NA | X-ray, bone scan |
| Mean | 52 | 40 | 46 | 0 | 493 | NA | |||||||
| Total | 1M | 1 | 1 | 0 | |||||||||
| Family 11 464 G >C, R155Q | |||||||||||||
| 1 V:4 | 48 | M | + | +SPMD | − | 31 | 42 | – | 950 | 157 | NT | NT | X-ray, MRI, bone scan, EM |
| 2 V:13 | 35 | F | + | −Early weakness | − | 30 | – | – | 190 | 103 | NT | NT | X-ray, EM |
| V3 IV:3 | a48 | M | + | + | − | 40 | 40 | – | NA | NA | _ | _ | X-ray, EM |
| 4 IV:10 | a49 | M | + | + | − | 35 | 35 | – | NA | NA | _ | _ | X-ray, EM |
| 5 IV:5 | a53 | M | + | − | − | 52 | – | – | NA | NA | _ | _ | NA |
| 6 III:4 | a57 | F | − | + | + | – | 44 | 52 | NA | NA | _ | _ | NA |
| 7 IV:12 | a55 | M | + | +Kugelberg Welander | − | 43 | 45 | – | NA | NA | Neuropathic | _ | NA |
| Mean | 42/a52 | 39 | 41 | 52 | 570 | 130 | |||||||
| Total | 2F/5M | 6 | 5 | 1 | |||||||||
| Family 13 572 G >A, R191Q | |||||||||||||
| 1 III:8 | 53 | F | − | +Polymyositis | − | – | 43 | – | 58 | 119 | Myopathic | Myopathic, neurogenic | MRI, X-ray |
| 2 II:5 | a60 | F | − | +MS | − | – | 40 | – | NA | NA | NA | NA | NA |
| 3 III:7 | a58 | M | − | +LGMD >HIBM | − | – | 48 | – | NA | NA | Myopathic, L5 radiculopathy. | Inclusion body myopathy. | X-ray |
| 4 III:2 | a68 | F | + | + | + | 42 | 45 | 60 | NA | NA | Neuropathic | Myopathic | X-ray, CT scan |
| Mean | 53/a62 | 42 | 44 | 60 | 58 | 119 | |||||||
| Total | 3F/1M | 1 | 4 | 1 | |||||||||
| Family 15 464 G >A, R155H | |||||||||||||
| 1 III:1 | 60 | M | + | +FSH >LGMD | − | 50 | 38 | – | 75 | 34 | Myopathic, neuropathic | Non-specific | X-ray |
| 7 III:2 | a47 | F | − | + | + | – | 38 | – | NA | NA | NA | NA | |
| 2 III:3 | 55 | M | − | +Distal myopathy/OPMD/myofibrillar myopathy | − | – | 33 | – | 92 | 164 | Neuropathic | Rimmed vacuoles | NA |
| 3 III:4 | 53 | F | − | +IBM | − | – | 37 | – | 294 | 82 | Severe myopathic | Rimmed Vacuoles | NA |
| 4 III:7 | 47 | M | − | +IBM | − | – | 37 | – | 357 | 180 | Neuropathic, denervation | Rimmed vacuoles | X-ray, CT |
| 5 II:2 | a81 | F | − | + | + | – | 50 | 79 | NA | NA | NA | NA | NA |
| 6 II:4 | a52 | F | − | + | − | – | 35 | – | NA | NA | NA | NA | NA |
| Mean | 54/a60 | 50 | 38 | 79 | 205 | 115 | |||||||
| Total | 4F/3M | 1 | 7 | 2 | |||||||||
| Family 16 464 G >A, R155H | |||||||||||||
| 1 II:6 | a59 | F | − | + | + | – | 42 | – | NA | NA | NA | NA | NA |
| 2 III:3 | a52 | M | + | + | − | 50 | 44 | – | 47 | 55 | NA | NA | X-ray |
| 3 III:6 | a56 | F | − | +SPMD | + | – | 49 | 51 | 171 | 98 | NA | NA | NA |
| 4 III:8 | a61 | F | + | +SPMD | + | 42 | 48 | 54 | 1160 | 224 | Neuronopathy axonopathy | NA | X-ray |
| 5 III:10 | 58 | F | − | +SPMD | + | – | 50 | 58 | 122 | 32 | NA | NA | NA |
| 6 IV:8 | 43 | M | + | − | – | 38 | – | – | 84 | 114 | NA | NA | X-ray, MRI, bone scan |
| Mean | 51/a57 | 43 | 47 | 54 | 317 | 105 | |||||||
| Total | 4F/2M | 3 | 5 | 4 | |||||||||
| TOTAL | 50/58a | 24F/25M | 28/57% | 43/87% | 13/27% | 40 | 42 | 57 | 457 | 154 | |||
Age individuals deceased, NA, not available; NT, not tested; LGMD, limb girdle muscular dystrophy; ALS, amyotropic lateral sclerosis; OPMD, oculopharyngeal muscular dystrophy; SPMD, scapuloperoneal muscular dystrophy; MS, multiple sclerosis; HIBM/IBM, hereditary/inclusion body myopathy; CPK, creatine phosphokinase; normal range 4–150 U/L; ALK, alkaline phosphatase normal range 30–120 U/L; EM, electron microscopy.
Ultrastructural studies of osteoclasts of bone in four members of this family revealed nuclear inclusions, identical to those of patients with PDB (Fig. 2A,B). The nuclear inclusions consisted of straight, tubular structures of ~15 nm diameter. The nuclear inclusions seen in muscle (Fig. 2C) and osteoclasts (Fig. 2A,B) were structurally very similar.
Fig. 2.
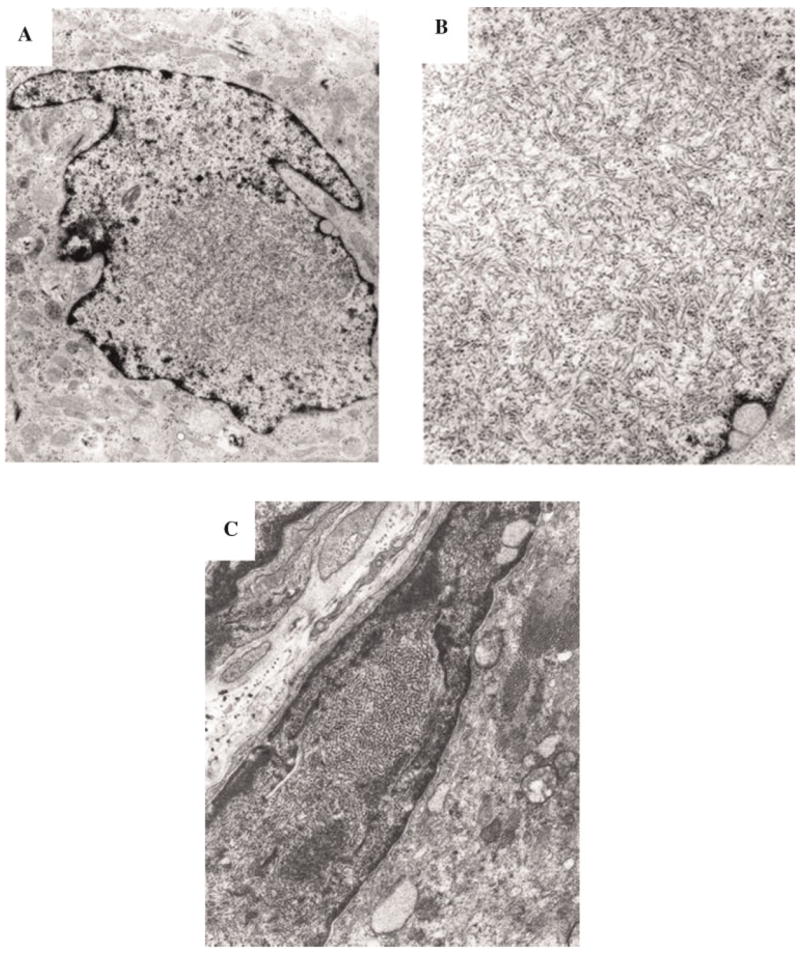
Electron microscopy of typical inclusions in IBMPFD muscle, compared with inclusions in Pagetic nuclei. A: Low magnification of inclusions in a nucleus of a Pagetic osteoclast (×20,000). B: High magnification of PHF in a nucleus of a Pagetic osteoclast (×40,000). C: Nuclei almost completely filled with collections of PHF (paired helical filaments), which appear as inclusions by light microscopy (×16,000).
Family 13
Individual III:8 in this four-generation family of German extraction, an avid sportswoman, developed climbing stairs and back pain at age 43 years (Fig. 1F). A trial of an immunosuppressant for her diagnosis of polymyositis was unsuccessful. EMG studies revealed myopathic changes (legs >arms) and median nerve entrapment at the wrist and ulnar entrapment at the elbow. Two muscle biopsies from the quadriceps showed both neurogenic and myopathic changes with normal staining for dystrophin, alpha and delta sarcoglycans, caveolin, and spectrin, with normal electron microscopy. Individual III:7, diagnosed with LGMD at age 42 years, showed signs of fast progression of his myopathy until his demise at the age of 58 years. In his final year, he became paranoid and depressed, however, he was not diagnosed with dementia. An autopsy was not obtained. His mother (II:5) was diagnosed with PDB at the age of 42 years. She had several limb fractures and had a hip replacement. Non-specific myopathy was diagnosed in her mid 40’s, progression of which led to transitioning to a wheelchair over several years. She developed bizarre, inappropriate behavior in her 50’s, was diagnosed with Alzheimer disease at age 60 years, and died at age 68 years.
Family 15
The proband of Scottish ancestry (III:I) first noted distal and proximal muscle weakness at age 38 years (Fig. 1G). He was initially diagnosed with facioscapulohumeral muscular dystrophy (FSH), this diagnosis being later revised to LGMD. He developed PDB in his 50’s. His mother became wheelchair-bound because of myopathy and later developed dementia. His sister (III:2) also developed myopathy, as did three maternal cousins and their mother prior to her demise. Individual III:4 developed progressive weakness at age 39 years, and had an elevated ALP. Muscle biopsy of the patient’s right deltoid showed small, scattered foci of interstitial chronic inflammation, including focally prominent lymphocytes, indicative of a mild inflammatory component as well as foci of fiber degeneration/regeneration without prominently increased fibrous tissue. Rimmed vacuoles were focally present, though relatively inconspicuous. Staining was negative for amyloid, neurofilament, and tau; however, immunohistochemical stain for ubiquitin showed unusually prominent staining and moderate but variable desmin immunoreactivity and increased cytoplasmic vimentin staining. Ultrastructural inclusions showing features characteristic of inclusion body myopathy or inclusion body myositis were relatively prominent within nuclei. X-rays have been unremarkable. Individual III:7 developed weakness of his left shoulder and atrophy of his deltoid muscle in his 20s and was initially diagnosed with FSH and spinal muscular atrophy. EMG at 36 years showed active denervation in both arms in a C5-7 distribution and no evidence of a myopathic process. Muscle biopsy showed moderately severe chronic-active myopathic changes with numerous fibers showing one or multiple rimmed vacuoles (Fig. 3A,B). He had a left C5-6 laminectomy, however, he later developed weakness of the right arm and leg. Although he has had back pain for several years, and elevated ALP, he was not previously diagnosed with PDB.
Fig. 3.
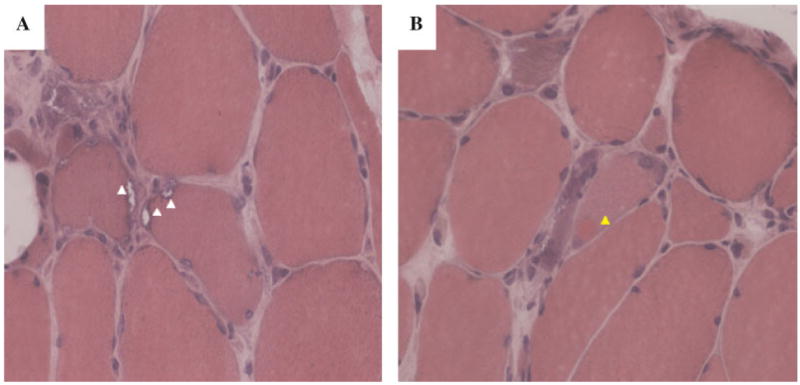
A: Muscle biopsy from individual III:7, age 47 years from family 15 showed moderately severe chronic-active myopathic changes with numerous fibers showing one or multiple rimmed vacuoles (white arrows). B: A single fiber showing a hyaline sarcoplasmic inclusion (yellow arrow) (H and E staining). [Color figure can be viewed in the online issue, which is available at www.interscience.wiley.com.]
Family 16
This family was evaluated extensively by CDS for a history of autosomal dominant early-onset Alzheimer’s disease, later established as FTD (Fig. 1H). Individual II:6 had bilateral foot drop in her 40s and died at the age of 59 years. Brain autopsy showed gross atrophy in the frontal and dorsolateral temporal lobes. Microscopically, neurofibrillary tangles were absent in the medial temporal lobe and elsewhere, and there was cortical neuronal loss with associated lamina II vacuolation in the atrophied areas. There were numerous diffuse senile plaques and no Pick bodies. Her two deceased sisters (II:1 and II:3) were both affected by weakness, bone pain and dementia, but details are lacking and neither underwent autopsy.
The first daughter of II:6 (III:6) died at the age of 56 years. She was diagnosed with SPMD at age 49 years and had a history of progressive decompensation in cognitive, memory, and behavioral functioning from the age of 51 years and lost ambulation by age 55 years. She had no evidence of PDB by ALP or by bone scan at age 48 years, however, testing at age 54 years revealed elevated ALP. She developed personality changes, becoming irritable, suspicious, anxious, agitated, and argumentative. A formal neuropsychological assessment noted impaired verbal skills, visuospatial skills, and measures of attention, concentration, and visual scanning. During her final year, she sat mute in a wheelchair with severe neck dystonia and rigidity. At autopsy, brain neuropathologic examination showed changes of VCP-associated FTD (described below). One of her two daughters has developed mild proximal limb girdle weakness at age 36 years.
Individual III:8, the second daughter of II:6, was diagnosed with PDB after X-rays showed compression fractures of L2,3 and L5 vertebrae at the age of 42 years. PDB also involved the right hemipelvis left humerus and proximal femur. She developed bilateral mixed proximal and distal myopathy associated with a typical steppage gait with a pelvic waddle and was later diagnosed with ‘‘scapuloperoneal dystrophy’’ at the age of 48 years. Subsequently, she began having nocturnal respiratory difficulties. Beginning at age 54 years, she had a rapidly progressive dementia, becoming severely bradykinetic and rigid with no expressive speech in her final year. Brain neuropathologic examination showed changes of VCP-type FTD. Immunohistochemical analysis of the patient’s port-mortem brain tissue demonstrated extensive tau pathology including accumulation of tau-positive inclusions in both neurons and glia. Autopsy showed moderate density of neocortical neuritic senile plaques as well as neurofibrillary tangle pathology in the mesial temporal lobe. One son (IV:3) of her three children developed mild proximal limb girdle type of weakness and PDB.
The third daughter of II:6 (individual III:10) was diagnosed with SPMD with facial involvement. She was on ventilatory support in the last few months of life because of myogenic respiratory failure. Autopsy was not granted. One of her sons (IV:8) was diagnosed with PDB at age 38 years.
Individual II:3, had dementia, muscular dystrophy, and PDB. An affected son (III:3) was confined to a wheelchair due to proximal limb weakness and bilateral foot drop. He had PDB, but no dementia when examined at age 51 years. He died unexpectedly at age 52 years. His sister (III:1) had severe FTD at 49 years with severe expressive dysphasia, frontal lobe release signs, proximal weakness, neck dystonia, rigidity, and PDB.
Neuropsychological and neuropathological testing:
Three affected patients in family 16 (II:6, III:6, III:8) evaluated were in advanced stages of FTD when first seen and the diagnosis was made by a dementia-specialist neurologist on the basis of typical history and neurologic findings of mute unresponsiveness and Parkinsonism. The history in these three individuals was not consistent with Alzheimer’s disease because early sociobehavioral and language symptoms dominated any component of short-term episodic memory impairment. Detailed neurocognitive testing was not possible in these three individuals. In a fourth patient (III:10) where early cognitive change might have been detected, the patient was too ill from respiratory complications to undergo detailed testing; this patient subsequently died without autopsy. Dementia was not suspected in this patient.
Detailed neuropathologic examination by MSF in a 56 years female (III: 6) from family 16 typifies findings in IBMPFD. Post-mortem examination revealed a 940-gram brain with mild frontal lobe atrophy. Microscopic examination revealed mild superficial microvacuolation, neuronal loss, and gliosis throughout the neocortex including the frontal, temporal, and parietal lobes. The limbic system was also affected with mild neuron loss and gliosis. Thioflavin S staining revealed a low density of neuritic plaques in the neocortex and a low density of neurofibrillary tangles in the entorhinal cortex and amygdala. Immunohistochemistry with antibodies to β-amyloid, phosphorylated tau, α-synuclein, and ubiquitin revealed a moderate density of predominantly diffuse senile plaques in the neocortex (Fig. 4A–D) and a low density of tau pathology in the hippocampus and entorhinal cortex. Tau pathology was not identified in the neocortex (Fig. 4B) and there were no Lewy bodies (Fig. 4C). Immunohistochemistry for ubiquitin revealed a moderate to high density of ubiquitinated inclusions throughout the neocortex consisting of abundant nuclear inclusion and dystrophic neurites with only a low density of ubiquitin positive cytoplasmic inclusions (Fig. 4D). Similar, but less robust, ubiquitin pathology was also detected in the hippocampal formation, subcortical nuclei, and brainstem. Moreover, the ubiquitin staining detected the low density of neuritic plaques observed with the Thioflavin S stain.
Fig. 4.
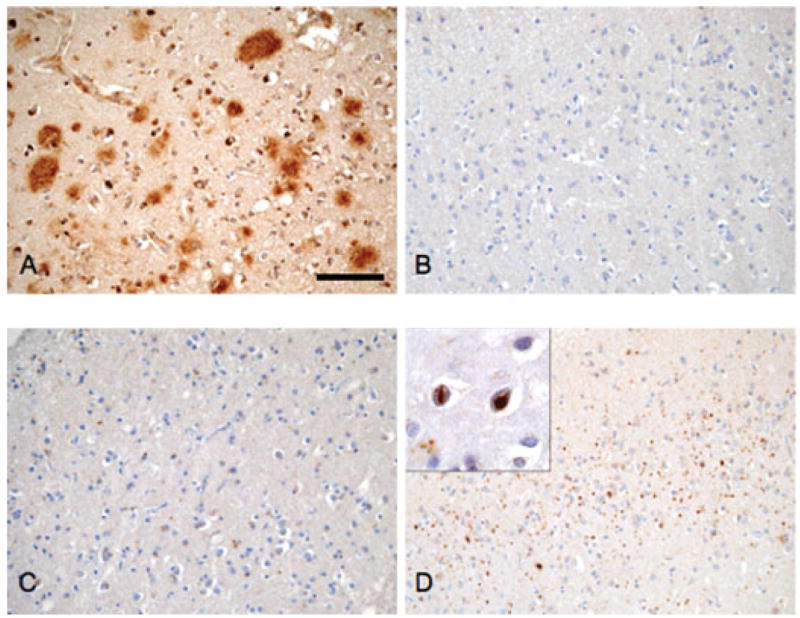
Neuropathology of IBMPFD. Frontal neocortex from patient III:6, family 16 was immunostained with antibodies to B-amyloid (A, 4G8), phosphorylated tau (B, PHF1), alpha synuclein (C, Syn303), and ubiquitin (D, Chemicon). A moderate density of predominantly diffuse senile plaques was present throughout the neocortex (A) and there was no tau (B) or Lewy body (C) pathology identified. In contrast, there were abundant ubiquitin positive inclusions (D) that consisted of nuclear inclusions (inset, panel D) and dystrophic neurites with only a low density of cytoplasmic inclusion. Scale bar = 200 μm.
SUMMARY OF CLINICAL DATA IN NINE FAMILIES
On reviewing the specific diagnoses among individuals from these families in whom records could be obtained, LGMD was the most common diagnosis made in 11 (38%) of 29 individuals, SPMD in eight (28%), ALS in three (10%), SMA in two (7%), diabetic neuropathy in two (7%), and inclusion body myositis, multiple sclerosis, polymyositis, FSH, and distal myopathy/oculopharyngeal muscular dystrophy/myofibrillar myopathy in one individual each (3%), the remaining individuals being diagnosed with a non-specific myopathy. Several individuals had more than one diagnosis made over the course of their illness. Review of the clinical features in the nine families indicate that 42/49 (86%) affected individuals with VCP mutations had progressive proximal muscle weakness and occasionally distal muscle involvement. The onset of weakness was at a mean age of 42 years (range 24–61 years), however, the pattern varied widely, even within families. Mean CPK levels in affected individuals was 154.4 ± 121.4 IU/L compared to 113.6 ± 29.3 in the unaffected VCP mutation negative relatives (data not shown). Muscle biopsy specimens revealed typical myopathic features including variation in muscle fiber size and mildly increased endomysial connective tissue in patchy regions of the biopsy. Large focal regions of ‘‘myopathic grouping’’ were observed in some biopsies. Rimmed vacuoles and cytoplasmic inclusions characteristic of HIBM but also seen in idiopathic inclusion body myositis and in a number of other hereditary distal myopathy types were present in 39% of muscles studied. Two individual from families 5 and 15 were correctly diagnosed with HIBM and in an additional, two individuals the diagnosis was revised from the initial diagnosis of LGMD to HIBM (Table I).
PDB seen in 28/49 (57%) patients occurred early (mean age 40 years, with a range of 23–65 years) with typical distribution in the spine, pelvis, and skull and later progression involved other bones. Elevated ALP levels were seen in 16 individuals diagnosed with PDB (mean 359.3 IU/L, range 58–1,508 IU/L; unaffected relatives mean 86.8 IU/L, normal population 30–130 IU/L). Ultrastructural studies of bone biopsy osteoclasts in four members of family number 6 revealed nuclear and cytoplasmic inclusions morphologically identical to that seen in classic PDB.
The onset of dementia seen in 27% of individuals studied occurred at a mean age of 57 years (range 48.9–60.2 years). It was characterized by personality change and language dysfunction, usually an expressive dysphasia, with early relative sparing of memory, dystonia (particularly neck dystonia), rigidity without tremor and severe gait instability. These features together with findings of early relative sparing of memory, the impairment of executive skills, absence of cortical sensory or visuospatial abnormalities, absence of vertical gaze abnormalities, tremor or other involuntary movements, onset in the fifth and sixth decade, and rapid progression, made the clinical classification of FTD secure. Two of 13 patients had significant visual or auditory hallucinations. The end stage is a wheelchair or bed-confined, mute, rigid individual with no ability to interact meaningfully with the environment. In the case of family 16, the three affected patients were in advanced stages of FTD when first seen, and the diagnosis was made by a dementia-specialist neurologist on the basis of typical history and neurological findings of mute unresponsiveness and Parkinsonism. The history in these three was not consistent with Alzheimer’s disease. Neuropathological findings from three individuals from family 16 who were diagnosed with dementia showed characteristic findings including ubiquitin-positive neuronal intranuclear inclusions, dystrophic neurites, and only rare intracytoplasmic VCP positive inclusions.
DISCUSSION
Clinical, biochemical, and histopathological data of 49 individuals from nine families with a recently described disorder autosomal dominant proximal myopathy associated with PDB and FTD was analyzed for their usefulness in establishing diagnostic criteria. The CPK levels in affected individuals were only mildly elevated, EMG and muscle biopsies revealed non-specific myopathic changes, and only 39% of muscle biopsies showed rimmed vacuoles and cytoplasmic inclusions characteristic of HIBM, the vast majority of individuals revealing non-specific changes.
In contrast to classic PDB seen in 1–2% of the >50 years population [Klein and Norman, 1995], the PDB seen in 57% of patients with IBMPFD presented earlier (mean age of 40 years) with typical distribution in the spine, pelvis, and skull and later progression to involve other bones. Elevation of serum ALP was found to be a good indicator of PDB. Fractures were not typically seen except among affected individuals in family 6 with the severe VCP A232E mutation. The association of familial PDB and neuromuscular disorders is a rare combination, having been reported in a few unrelated families with distinct muscular phenotypes. We have identified the descendants of one family with the familial disorder of combined lower motor neuron degeneration and skeletal disorganization reported by Tucker et al. [1982]. This family has the VCP R155Q mutation. At least two of Tucker’s patients with lower motor neuron degeneration and PDB developed dementia as a terminal event. Evaluation of an affected descendant of this family indicates resemblance to members of the other families.
The VCP protein is widely expressed and is a member of the AAA-ATPase super-family [Beyer, 1997]. It has been implicated in many cellular functions [Wang et al., 2003; Woodman, 2003], and is required for the proteasomal degradation of phosphorylated IκB-α [Dai et al., 1998; Asai et al., 2002], an essential step in NF-κB activation. Interestingly, the causative mutations in VCP all affect the highly conserved CDC 48 domain, which is involved in ubiquitin-binding [Dai and Li, 2001; Rape et al., 2001]. The PDB-causing mutations in the SQSTM1 gene also affect the ubiquitin-binding domain of the gene product, p62 [Layfield and Hocking, 2004], suggesting that the disease processes in PDB associated with SQSTM1 and IBMPFD may be related [Daroszewska and Ralston, 2006]. We believe that IBMPFD is currently underdiagnosed among the patients with myopathy and/or dementia. Using an elevated ALP as a screen, with confirmation by radiography or bone scan, should identify more cases of syndromic PDB.
Dementia, mainly of the frontotemporal type, was diagnosed in 27% individuals (mean age of onset 57 years (range 48.9–60.2 years)). The novel pattern of brain histopathology from three individuals from family 16 who were diagnosed with dementia is different from that previously reported for sporadic and familial FTD with ubiquitin inclusions also known as FTLD with motor neuron type inclusions or motor neuron disease inclusion dementia [McKhann et al., 2001], autosomal-dominant FTD linked to the tau gene on chromosome 17 [Spillantini et al., 1998], to an unknown gene on chromosome 3 [Brown, 1998] and the familial corticobasal syndrome associated with mutations in the progranulin gene [Masellis et al., 2006]. Forman et al. [2006] analyzed neuropathologic changes in eight patients with VCP mutations (six with dementia including these three cases from family 16). Characteristic findings include ubiquitin-positive neuronal intranuclear inclusions and dystrophic neurites. There was no biochemical alteration in the VCP protein and only rare intracytoplasmic inclusions were detected with antibodies to VCP in contrast to the reported findings in a 55-year German patient with VCP gene mutations [Schröder et al., 2005]. The overlap between the histology and composition of the brain protein accumulations in FTD, for example, ubiquitin epitopes, and those found in the muscle fibers of individuals with IBM [Askanas and Engel, 2001] suggests that cellular deterioration may result from a common cascade of events. Mehta et al. [2007] studied Apolipoprotein-E (APOE) as a risk factor for the dementia seen in approximately one-third of patients with IBMPFD based on its known modifier effect in Alzheimer’s disease. They identified a possible association with the presence of one or more alleles of the APOE 4 genotype and FTD (P = 0.002), but not with IBM (P = 0.9), or PDB (P = 0.9). They did not observe an association with FTD and the H2 MAPT haplotype.
The variable presentation of IBMPFD has been noted by other authors. Haubenberger et al. [2005] discovered a novel missense mutation in VCP (R159H, 688G>A) in four Austrian siblings, ages ranging from 60 to 70 years, who presented with both progressive proximal myopathy and PDB in whom dementia was excluded in all members by neuropsychological assessment. In contrast, the majority of patients in two French kindreds [Guyant-Marechal et al., 2006] have been diagnosed with dementia in 100 and 90% of the individuals with a novel R93C missense mutation and a common R155C mutation, respectively. Neuropsychological examination of six affected individuals from the first family identified with dementia was characterized by aggressive behavior, impaired judgment, and paranoia. Fifty percent presented with selectively severe atrophy and symmetric weakness of the proximal and distal muscles of the upper limbs and distal legs (mean age of onset 57 years) and 50% of the patients suffered from PDB at a mean age of onset of 55 years associated with foraminal lumbar stenosis or hearing loss in individuals. In a second family of 30 individuals, previously mistakenly diagnosed with myotonic dystrophy and reported to be linked to chromosome 15q21-24 by Le Ber et al. [2004], 10 presented with proximal and axial muscle weakness with clinical and electrical myotonia and associated FTD. Histological analyses confirmed PDB in six individuals. Hepatitis and variable diagnoses of non-specific hepatic fibrosis, cytolysis, and cholestasis was reported.
We hope to increase awareness of this under-diagnosed disorder which shares features with LGMD, FSH, SPMD, late adult onset distal myopathy, spinal muscular atrophy, ALS, and non-specific dementias, among others, suggesting possible underlying common pathogenic mechanisms. An elevated blood ALP may be a useful screen for PDB, which is seen in over half the patients. Testing for VCP mutations should be considered in any individual with two or more associated features of IBMPFD, or, with one or more features and a suspicious family history. Understanding the basic molecular pathways of VCP disease will hopefully lead to specific therapies as in the case of Paget disease of bone [Deftos, 2005]. Studying this relatively rare disease has important implications for common disorders such as the muscular dystrophies, myopathies, PDB and related bone disorders and the dementias.
ELECTRONIC-DATABASE INFORMATION
Online Mendelian Inheritance in Man (OMIM) http://www3.ncbi.nlm.nih.gov/omim (for LGMD with PDB [MIM 605382], IBM1 [MIM 147420], IBM2 [MIM 600737], IBM3 [MIM 605637], PDB2 [MIM 602080], DMRV, [MIM 605820], FTD presenting with Parkinsonism [MIM 600274], FTD presenting with ALS [MIM 105550]), Pagetoid amyotrophic lateral sclerosis [MIM 167320].
Acknowledgments
We thank the families and their health care providers for their enthusiastic participation and contribution in our research studies in particular Dr. Zachary Simmons, Dr. Javed Towfighi, Dr. Rabi Tawil, Dr. Fred Singer and other colleages and collaborators who referred patients for this study.
Grant sponsor: NIAMS; Grant sponsor: National Institutes of Health; Grant numbers: RO1 AR050236-01A1, R03 AR 46869; Grant sponsor: Muscular Dystrophy Association; Paget Foundation.
References
- Asai T, Tomita Y, Nakatsuka S, Hoshida Y, Myoui A, Yoshikawa H, Aozasa K. VCP (p97) regulates NF kappaB signaling pathway, which is important for metastasis of osteosarcoma cell line. Jpn J Cancer Res. 2002;93:296–304. doi: 10.1111/j.1349-7006.2002.tb02172.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askanas V, Engel WK. Sporadic inclusion-body myositis and its similarities to Alzheimer disease brain. Recent approaches to diagnosis and pathogenesis, and relation to aging. Scand J Rheumatol. 1998;27:389–405. doi: 10.1080/030097498442208. [DOI] [PubMed] [Google Scholar]
- Askanas V, Engel WK. Inclusion-body myositis: Newest concepts of pathogenesis and relation to aging and Alzheimer disease. J Neuropathol Exp Neurol. 2001;60:1–14. doi: 10.1093/jnen/60.1.1. [DOI] [PubMed] [Google Scholar]
- Askanas V, Alvarez RB, Engel WK. Beta-amyloid precursor epitopes in muscle fibers of inclusion body myositis. Ann Neurol. 1993;34:551–560. doi: 10.1002/ana.410340408. [DOI] [PubMed] [Google Scholar]
- Beyer A. Sequence analysis of the AAA protein family. Protein Sci. 1997;6:2043–2058. doi: 10.1002/pro.5560061001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown J. Chromosome 3-linked frontotemporal dementia. Cell Mol Life Sci. 1998;54:925–927. doi: 10.1007/s000180050222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai RM, Li CC. Valosin-containing protein is a multi-ubiquitin chain-targeting factor required in ubiquitin-proteasome degradation. Nat Cell Biol. 2001;3:740–744. doi: 10.1038/35087056. [DOI] [PubMed] [Google Scholar]
- Dai RM, Chen E, Longo DL, Gorbea CM, Li CC. Involvement of valosin-containing protein, an ATPase Co-purified with IkappaBalpha and 26 S proteasome, in ubiquitin-proteasome-mediated degradation of IkappaBalpha. J Biol Chem. 1998;273:3562–3573. doi: 10.1074/jbc.273.6.3562. [DOI] [PubMed] [Google Scholar]
- Daroszewska A, Ralston SH. Mechanisms of disease: Genetics of Paget’s disease of bone and related disorders. Nat Clin Pract Rheumatol. 2006;2:270–277. doi: 10.1038/ncprheum0172. [DOI] [PubMed] [Google Scholar]
- Deftos LJ. Treatment of Paget’s disease-taming the wild osteoclast. NEJM. 2005;353:872–875. doi: 10.1056/NEJMp058184. [DOI] [PubMed] [Google Scholar]
- Forman MS, Mackenzie IR, Markesbery WR, Swanson E, Cairns NJ, Boyer PJ, Jhaveri BS, Karlawish JH, McKeel DW, Watts GD, Markesbery WR, Smith CD, Kimonis VE. Novel ubiquitin brain pathology in frontotemporal dementia with inclusion body myopathy and Paget’s disease. J Neuropathol Exp Neurol. 2006;65:571–581. doi: 10.1097/00005072-200606000-00005. [DOI] [PubMed] [Google Scholar]
- Guyant-Marechal L, Duyckaerts C, Bou J, Dugny F, Le Ber I, Frebourg T, Hannequin D, Campion D. VCP mutations in two kindreds with prominent frontotemporal dementia: Clinical and neuropathological features. Neurology. 2006;67:644–651. doi: 10.1212/01.wnl.0000225184.14578.d3. [DOI] [PubMed] [Google Scholar]
- Haubenberger D, Bittner RE, Rauch-Shorny S, Zimprich F, Mannhalter C, Wagner L, Mineva I, Vass K, Auff E, Zimprich A. Inclusion body myopathy and Paget disease is linked to a novel mutation in the VCP gene. Neurology. 2005;65:1304–1305. doi: 10.1212/01.wnl.0000180407.15369.92. [DOI] [PubMed] [Google Scholar]
- Kimonis VE, Kovach MJ, Waggoner B, Leal S, Salam A, Rimer L, Davis K, Khardori R, Gelber D. Clinical and molecular studies in a unique family with autosomal dominant limb-girdle muscular dystrophy and Paget disease of bone. Genet Med. 2000;2:232–241. doi: 10.1097/00125817-200007000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein RM, Norman A. Diagnostic procedures for Paget’s disease. Radiologic, pathologic, and laboratory testing. Endocrinol Metab Clin North Am. 1995;24:437–450. [PubMed] [Google Scholar]
- Kovach MJ, Waggoner B, Leal SM, Gelber D, Khardori R, Levenstien MA, Shanks CA, Gregg G, Al-Lozi MT, Miller T, Rakowicz W, Lopate G, Florence J, Glosser G, Simmons Z, Morris JC, Whyte MP, Pestronk A, Kimonis VE. Clinical delineation and localization to chromosome 9p13.3-p12 of a unique dominant disorder in four families: Hereditary inclusion body myopathy, Paget disease of bone, and frontotemporal dementia. Mol Genet Metab. 2001;74:458–475. doi: 10.1006/mgme.2001.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layfield R, Hocking LJ. SQSTM1 and Paget’s disease of bone. Calcif Tissue Int. 2004;75:347–357. doi: 10.1007/s00223-004-0041-0. [DOI] [PubMed] [Google Scholar]
- Le Ber I, Martinez M, Campion D, Laquerriere A, Betard C, Bassez G, Girard C, Saugier-Veber P, Raux G, Sergeant N, Magnier P, Maisonobe T, Eymard B, Duyckaerts C, Delacourte A, Frebourg T, Hannequin D. A non-DM1, non-DM2 multisystem myotonic disorder with frontotemporal dementia: Phenotype and suggestive mapping of the DM3 locus to chromosome 15q 21-24. Brain. 2004;127:1979–1992. doi: 10.1093/brain/awh216. [DOI] [PubMed] [Google Scholar]
- Masellis M, Momeni P, Meschino W, Heffner R, Jr, Elder J, Sato C, Liang Y, St George-Hyslop P, Hardy J, Bilbao J, Black S, Rogaeva E. Novel splicing mutation in the progranulin gene causing familial corticobasal syndrome. Brain. 2006;129:3115–3123. doi: 10.1093/brain/awl276. [DOI] [PubMed] [Google Scholar]
- McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ. Clinical and pathological diagnosis of frontotemporal dementia: Report of the work group on frontotemporal dementia and Pick’s disease. Arch Neurol. 2001;58:1803–1809. doi: 10.1001/archneur.58.11.1803. [DOI] [PubMed] [Google Scholar]
- Mehta S, Watts GD, Adamson JL, Hutton M, Umberger G, Xiong S, Ramdeen S, Lovell MA, Kimonis VE, Smith C. APOE is a potential modifier gene in an autosomal dominant form of frontotemporal dementia (IBMPFD) Genet Med. 2007;9:9–13. doi: 10.1097/gim.0b013e31802d830d. [DOI] [PubMed] [Google Scholar]
- Miller BL, Ikonte C, Ponton M, Levy M, Boone K, Darby A, Berman N, Mena I, Cummings JL. A study of the Lund-Manchester research criteria for frontotemporal dementia: Clinical and single-photon emission CT correlations. Neurology. 1997;48:937–942. doi: 10.1212/wnl.48.4.937. [DOI] [PubMed] [Google Scholar]
- Mirabella M, Alvarez RB, Bilak M, Engel WK, Askanas V. Difference in expression of phosphorylated tau epitopes between sporadic inclusion-body myositis and hereditary inclusion-body myopathies. J Neuropathol Exp Neurol. 1996;55:774–786. doi: 10.1097/00005072-199607000-00003. [DOI] [PubMed] [Google Scholar]
- Rape M, Hoppe T, Gorr I, Kalocay M, Richly H, Jentsch S. Mobilization of processed, membrane-tethered SPT23 transcription factor by CDC48 (UFD1/NPL4), a ubiquitin-selective chaperone. Cell. 2001;107:667–677. doi: 10.1016/s0092-8674(01)00595-5. [DOI] [PubMed] [Google Scholar]
- Schröder R, Watts GD, Mehta SG. Mutant valosin-containing protein causes a novel type of frontotemporal dementia. Ann Neurol. 2005;57:457–461. doi: 10.1002/ana.20407. [DOI] [PubMed] [Google Scholar]
- Spillantini MG, Murrell JR, Goedert M, Farlow MR, Klug A, Ghetti B. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA. 1998;95:7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker WSJ, Hubbard WH, Stryker TD, Morgan SW, Evans OB, Freemon FB, Theil GB. A new familial disorder of combined lower motor neuron degeneration and skeletal disorganization. Trans Assoc Am Phys. 1982;95:126–134. [PubMed] [Google Scholar]
- Wang Q, Song C, Li CC. Hexamerization of p97-VCP is promoted by ATP binding to the D1 domain and required for ATPase and biological activities. Biochem Biophys Res Commun. 2003;300:253–260. doi: 10.1016/s0006-291x(02)02840-1. [DOI] [PubMed] [Google Scholar]
- Watts G, Wymer J, Kovach M, Mehta S, Mumm S, Darvish D, Pestronk A, Whyte M, Kimonis V. Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat Genet. 2004;36:377–381. doi: 10.1038/ng1332. [DOI] [PubMed] [Google Scholar]
- Woodman PG. p97, a protein coping with multiple identities. J Cell Sci. 2003;116:4283–4290. doi: 10.1242/jcs.00817. [DOI] [PubMed] [Google Scholar]