

Abstract
Electron detachment dissociation (EDD) Fourier transform mass spectrometry has recently been shown to be a useful method for tandem mass spectrometry analysis of sulfated glycosaminoglycans (GAGs). EDD produces abundant glycosidic and cross-ring fragmentation that is useful for localizing sites of sulfation in GAG oligosaccharides. While EDD fragmentation can be used to characterize GAGs in a single tandem mass spectrometry experiment, SO3 accompanies many peaks, and complicates the resulting mass spectra. In this work we demonstrate the ability to significantly decrease SO3 loss by selection of the proper ionized state of GAG precursor ions. When the degree of ionization is greater than the number of sulfate groups in an oligosaccharide, a significant reduction in SO3 loss is observed in the EDD mass spectra. This data suggested that SO3 loss is reduced when an electron is detached from carboxylate groups instead of sulfate. Electron detachment occurs preferentially from carboxylate versus sulfate for thermodynamic reasons, provided that carboxylate is in its ionized state. Ionization of the carboxylate group is achieved by selecting the appropriate precursor ion charge state, or by the replacement of protons with sodium cations. Increasing the ionization state by sodium cation addition decreases, but does not eliminate, SO3 loss from infrared multiphoton dissociation (IRMPD) of the same GAG precursor ions.
INTRODUCTION
Glycosaminoglycans (GAGs) participate in numerous important biological processes such as cell-cell signaling [1, 2], the regulation of biochemical pathways [3, 4], inhibiting proteolysis [5], and effecting angiogenesis [6]. GAGs also play an important role in pathogenic infections [7–9], and may undergo alteration in certain types of cancer [10]. Both intra- and extracellular GAG proteoglycans can be found in a wide variety of organisms, from bacteria to humans [11]. At the molecular level, GAGs are sulfated, linear carbohydrates consisting of alternating acidic sugar and basic sugar residues. GAG complexity arises through variable sulfation, N-modification of the basic residues, and C5 stereochemistry of the hexuronic acid residues. There is significant interest in developing methods to determine the pattern of modification in GAG oligosaccharides, as these are believed to influence the biological function of the GAG molecule. While 1D and 2D NMR can be used to determine the pattern and type of GAG modification [12], GAGs must be isolated from natural sources and may not often be available in the quantity and purity that is required for NMR analysis. Mass spectrometry is much more sensitive than NMR, and can be used to examine mixtures, particularly when combined with online chromatography. A wide variety of mass spectrometry and tandem mass spectrometry techniques have been developed to analyze this challenging class of molecules [13–20].
Recent work from this laboratory has demonstrated the analytical utility of electron detachment dissociation (EDD) [21] for the structural analysis of negatively-charged GAG oligosaccharides varying in length from tetrasaccharides to decasaccharides [22–24]. EDD produces more abundant glycosidic and cross-ring fragmentation for GAG oligosaccharides than is observed by more conventional dissociation methods such as collisionally activated dissociation (CAD) or infrared multiphoton dissociation (IRMPD). Moreover, it reduces the loss of SO3 from labile sites of sulfation compared to other methods of ion activation. This fragmentation behavior has been proposed to result from a radical anion that is formed from electron detachment, and which yields product ions that are distinctly different from the dissociation products of closed shell anions via low energy or threshold fragmentation methods such as CAD or IRMPD. As in electron ionization mass spectra, the location of the radical site is expected to influence the resulting fragmentation and observed products. Our prior results suggest that electron detachment occurs at a site of negative charge, namely carboxylate and sulfate groups. The charge of the precursor ion determines how many of these sites are ionized, and are available as candidates for electron detachment. Thus the charge state of the precursor ion may have a significant influence on fragmentation, and on the ions produced by EDD.
The charge state of the precursor ion has been shown to influence fragmentation of closed-shell ions. For CAD fragmentation of heparin-like GAGs, Zaia and coworkers demonstrated that glycosidic cleavages increased and SO3 loss decreased as the charge state of the precursor ion increased [25]. The work by Zaia et al. also demonstrated that the addition of Ca2+ to the precursor ion minimized SO3 loss during CAD of closed-shell ions. It is possible that replacing protons with monovalent cations such as sodium may also suppress SO3 loss in the dissociation of sulfated GAG oligosaccharides by EDD. In the present work, we examine the influence of charge state and sodium cationization on the EDD fragmentation of a GAG octasaccharide, and compare these products with those obtained by IRMPD.
MATERIALS AND METHODS
Preparation of DS Oligosaccharides
Dermatan sulfate (DS) octasaccharides were prepared by partial enzymatic depolymerization of porcine intestinal mucosa dermatan sulfate (Celsus Laboratories, Cincinnati, OH). A 20 mg/mL dermatan sulfate solution in 50 mM Tris-HCl/60 mM sodium acetate buffer, pH 8 was incubated at 37°C with chondroitin ABC lyase from Proteus vulgaris, EC 4.2.2.4. (Seikagaku, Japan). The absorbance at 232 nm was monitored to determine when the digestion was 50% completed. The digestion mixture was then heated at 100°C for 3 min. High-molecular-weight oligosaccharides and the glycosidic enzyme were removed by ultra-filtration using a 5000 MWCO membrane. The resulting oligosaccharide mixture was concentrated by rotary evaporation and fractionated by low pressure GPC on a Bio-Gel P10 (Bio-Rad, Richmond, CA) column. The fraction containing DS octasaccharides (dp8) was desalted by GPC on a Bio-Gel P2 column and freeze-dried [26]. Further purification of DS dp8 were carried out using strong anion exchange high-pressure liquid chromatography (SAX-HPLC) on a semi-preparative SAX S5 Spherisorb column (Waters Corp, Milford, MA). The SAX-HPLC fractions containing > 90% of DS dp8 were collected, desalted by GPC, and freeze-dried. The solid was reconstituted in water and purified a second time by SAX-HPLC. Only the top 30% of the chromatographic peak was collected, desalted, and freeze-dried. Concentration of the octasaccharide solution was determined by measuring the absorbance at 232 nm (ε = 3800 M−1 cm−1). The resulting fraction containing DS dp8 was characterized by PAGE, ESI-MS, and high-field nuclear magnetic resonance (NMR) spectroscopy.
Mass Spectrometry Analysis
Experiments were performed with a 9.4 T Bruker Apex Ultra QhFTMS (Billerica, MA) fitted with an Apollo II dual source, a 25 W CO2 laser (Synrad model J48-2, Mukilteo, WA) for IRMPD, and an indirectly heated hollow cathode for generating electrons for ECD and EDD. For studies requiring sodium, the octasaccharide was introduced at a concentration of 150 µM in 50:50 methanol:H2O with 1 mM NaOH (Sigma, St. Louis, MO) by ESI in negative ion mode. To produce abundant [M-3H]3− ions, the octasaccharide was introduced at a concentration of 20 µM in 50:50:1 methanol:H2O:formic acid. All sample solutions were infused at a rate of 120 µL/hour.
For the EDD experiments, precursor ions were isolated in the external quadrupole and accumulated for 1–4 seconds before injection into the FTMS cell. The isolation/cell fill was repeated up to 6 times. The selection of the precursor ion was further refined by using in-cell isolation with a coherent harmonic excitation frequency (CHEF) event [27]. The precursor ions were then irradiated with electrons for 1 second. For electron irradiation the cathode bias was set to −19 V, the extraction lens was set to −18.5 V±0.5 V, and the cathode heater was set to 1.6 A. Ions were excited with an RF frequency chirp that covered the range of 100 – 2000 m/z. 24 acquisitions were signal averaged per mass spectrum. For each mass spectrum, 512K points were acquired at a 2.4 MHz digitization rate, padded with one zero fill, and apodized using a sinebell window. Background spectra were acquired by leaving all parameters the same but setting the cathode bias to 0 V to ensure that no electrons reached the analyzer cell. Background spectra were not subtracted from the EDD spectra, but were used to ensure that very few products were formed during ion accumulation and isolation. IRMPD spectra were acquired using the same experimental setup as EDD, but replacing the electron irradiation event with a laser pulse. For IRMPD, ions were irradiated for 0.01 – 0.2 seconds with beam attenuation set to pass from 40 – 60% of full power. External calibration of IRMPD and EDD mass spectra produced mass accuracy of 5 ppm. Internal calibration was also performed using confidently assigned glycosidic bond cleavage products as internal calibrants, providing mass accuracy of <1 ppm. Due to the larger number of low intensity products formed by EDD, only peaks with S/N > 10 are reported. Product ions were assigned using accurate mass measurement. For the work presented here, fragmentation of the DS octasaccharide is presented using a modification of the Domon and Costello annotation [28] that presents GAG fragmentation with SO3 loss and hydrogen rearrangement that is observed in EDD of GAGs [22].
RESULTS AND DISCUSSION
DS dp8 has eight ionizable acidic sites; four carboxylic acid groups and four sulfate groups. This acidic molecule readily forms negative ions, but the charge state and Na/H heterogeneity depend strongly on solution conditions. The influence of ESI solution conditions is shown in Figures 1A and 1B. The addition of 1% formic acid to the ESI solution results in the mass spectrum shown in Figure 1A, in which abundant [M-4H]4− and [M-3H]3− molecular ions are observed, with only a low level of sodium cationization. The addition of 100 µM NaOH in place of formic acid to the ESI solution produces the more highly charged 5− ion as well as the 4− and 3− ions. The presence of sodium cations in solution also results in the exchange of up to 5 protons by sodium cations, as shown in Figure 1B. Using these two solution conditions the influence of precursor ion charge (3−, 4−, 5−) and sodium cationization (0 – 4) on sulfated GAGs is examined.
Figure 1.
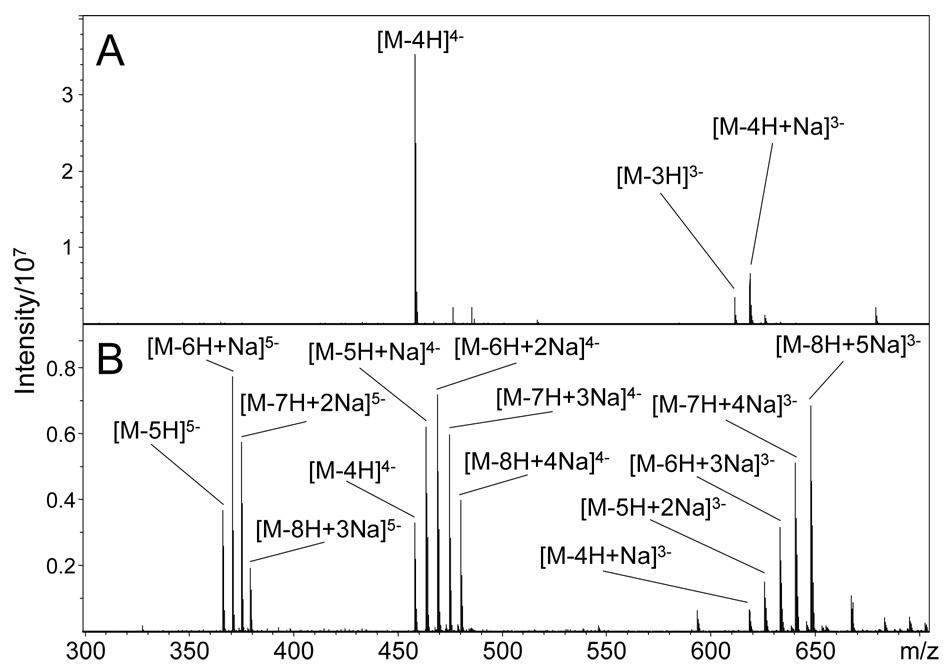
The influence of solution conditions on the observed ESI-FTMS spectra of DS dp8. (A) 15 µM DS dp8 dissolved in 50:50 methanol:H2O with 1% formic acid results in no Na/H heterogeneity and the formation of a [M-3H]3− ion. (B) 150 µM DS dp8 dissolved in 50:50 methanol:H2O with 100 µM NaOH results in abundant Na/H heterogeneity.
Influence of Charge State on EDD Fragmentation of DS dp8
EDD of the [M-4H]4− and [M-3H]3− precursor ions of DS dp8 produce many glycosidic and cross-ring cleavages that are also accompanied by the loss of SO3. No product ions are observed with loss of two equivalents of SO3 for any EDD mass spectrum, regardless of the charge state of the precursor ion. Product ions with SO3 loss generally occur as satellite peaks to similar fragments without SO3 loss, and are easily assigned as the pairs of peaks differ by the exact mass of SO3, 79.957 u. While products ions accompanied by SO3 loss are easily identified, they complicate the EDD mass spectrum by adding additional peaks and do not aid in deciphering the structure of the sulfated GAG. The additional complexity resulting from product ion SO3 is apparent in Figure 2, where many of the product ions accompanied by the loss of SO3 are present in the EDD mass spectrum of DS dp8. The Z3 and Y3 glycosidic cleavages are accompanied by Z3-SO3 and Y3-SO3 product ions, as well as many other products ions accompanied by the loss of SO3 as shown in Figure 2A. However, a much simpler EDD mass spectrum is observed from EDD of the [M-5H]5− precursor ion (Figure 2B) due to the absence of satellite peaks from SO3 loss. As shown in Figure 2B, the Z3 and Y3 glycosidic cleavages are not accompanied by SO3-loss product ions. This behavior is observed throughout the EDD mass spectrum. The number of products accompanied by SO3 loss is similar in EDD of [M-3H]3− and [M-4H]4−, i.e. no decrease in SO3 loss is observed when the precursor ion charge increases from 3− to 4−. In contrast, there is a dramatic decrease in the loss of SO3 for precursor ion charge 5− versus 4− or 3−, as shown in Figures 3A through 3C, respectively. Fragmentation accompanied by loss of SO3 is denoted with an open circle. For EDD of [M-3H]3−, 36 of the 88 observed cleavages are accompanied by SO3 loss, and EDD of[M-4H]4− results in 34 of the 87 products are accompanied by SO3 loss. In contrast to this, 3 of the 56 products from EDD of [M-5H]5− are accompanied by SO3 loss. Most glycosidic cleavages of the 3− or 4− precursor occur with loss of SO3, but are absent for the 5− charge state. Abundant and similar glycosidic and cross-ring fragmentation is observed for EDD of all charge states. For EDD of [M-3H]3−, [M-4H]4−, and [M-5H]5−, cleavage of most glycosidic bonds is observed. EDD of [M-5H]5− and [M-3H]3− produces 26 of the 28 possible products (Figures 3A and 3C, respectively) while EDD of [M-4H]4− precursor produces all 28 possible glycosidic products (Figure 3B). Differences in the number of observed glycosidic cleavages are due to the low abundance of the product ion, or overlap of the product ion with the precursor ion. Glycosidic bond cleavages accompanied by loss of one or two additional hydrogen atoms are observed for EDD of all charge states. Specifically, we observed B-H, C-2H, Y-2H, and Z-2H, which we refer to as B', C", Y", and Z", respectively. We have previously suggested radical mechanisms for the formation of these ions and found them useful for analyzing structural features, such as distinguishing IdoA form GlcA [24].
Figure 2.
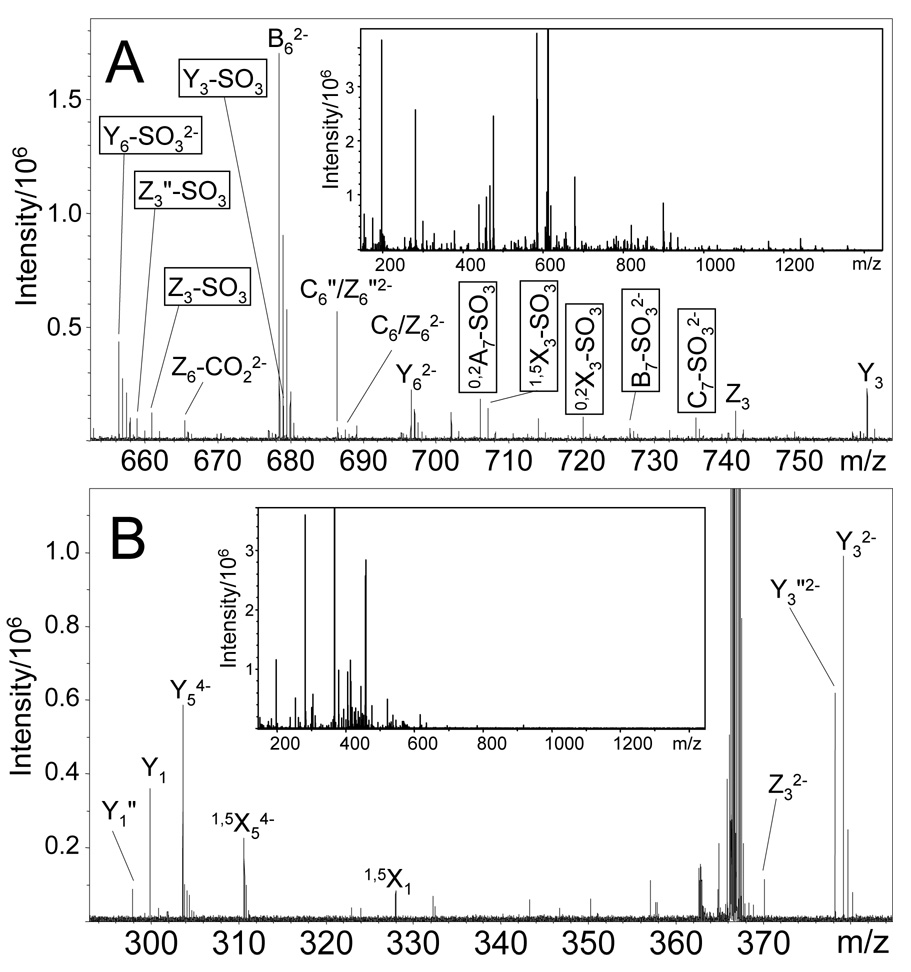
EDD mass spectrum complexity increases when product ions accompanied by SO3 loss are present. (A) EDD of the [M-3H]3− of DS dp8 produces a more complex mass spectrum than (B) EDD of [M-5H]5−.
Figure 3.
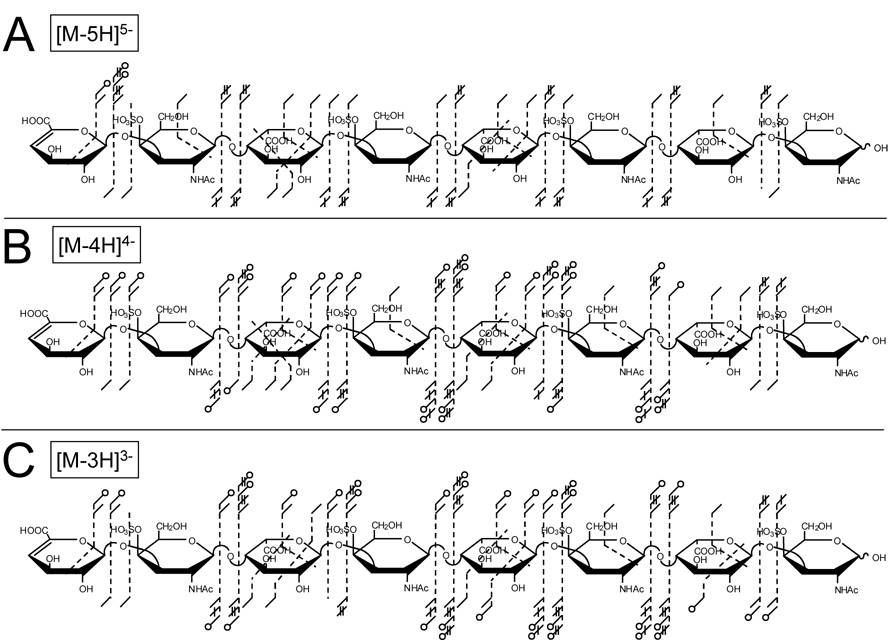
Influence of precursor ion charge on EDD of DS dp8. Product ions observed from EDD of (A) the [M-5H]5− precursor ion, (B) the [M-4H]4− precursor ion, and (C) the [M-3H]3− precursor ion of DS dp8.
Previous results from CAD of sulfated GAGs suggest that the sulfate group readily loses SO3 when it is protonated [25]. Because sulfate is more acidic than carboxylate in solution, sulfate groups will be ionized in preference to carboxyl groups in solution. Products observed from EDD of DS dp8 indicate that these sites of charge are preserved as the molecule moves from solution to the gas phase, and as a result sulfate groups will ionize in preference to carboxyl groups. Therefore it is expected that the [M-5H]5− precursor will have all four sulfate residues charged plus one carboxylate group charged. Electron detachment is expected to occur more readily from the carboxylate anion as it requires ~1.2 eV less energy than from a sulfate anion [23]. For the 3− and 4− precursor ions only sulfates will be ionized, and detachment will occur from the sulfate group. In support of this, EDD of [M-5H]5− produces the charge reduced species minus CO2 ([M-5H-CO2]4−•) and not the charge reduced species minus SO3 ([M-5H-SO3]4−•), indicating a preference for radical site formation at the carboxylate anion. Conversely, for EDD of [M-3H]3− and [M-4H]4−, the charge-reduced species minus SO3 ([M-3H-SO3]2−• and [M-4H-SO3]3−•, respectively) is observed, but not the charge-reduced species minus CO2. These data suggest that electron detachment from a sulfate anion produces a sulfate radical that readily loses SO3 from EDD product ions, while electron detachment from a carboxylate anion does not promote the loss of SO3. Therefore, if the charge on the precursor ion is greater than the number of sulfate groups, at least one ionized carboxyl group is present and available as a radical site after electron detachment, and SO3 loss will greatly reduced in EDD fragmentation.
The pattern of SO3 loss from the charge-reduced species suggests that the initial site of radical formation occurs on a sulfate group of the GalNAc4S residues for the 3− and 4− charge states of DS dp8, and on a carboxylate group of IdoA residues for the 5− charge state of DS dp8. The majority of the cross-ring fragmentation products that are observed, however, originate across IdoA residues for all the charge states of the precursor. The preferential fragmentation of the acidic residues (IdoA) over fragmentation of basic residues (GalNAc4S), regardless of the initial site of electron detachment, can be rationalized by radical site migration from GalNAc4S to IdoA, for example by radical site initiated hydrogen rearrangement. Another explanation for these data is that many of the “EDD” products do not arise from radical site initiated fragmentation, but are a result of ion activation from the interaction of the precursor ions with the 19 eV electrons [29–31]. Clearly, the loss of CO2 or SO3 from the charge-reduced species are radical driven processes, as these products are strongly influenced by the initial site of radical formation. Other products appear to arise both from fragmentation of the odd-electron charge reduced species as well as from activation of the even-electron precursor ion. We have recently examined the electron induced dissociation (EID) of singly-charged GAG tetrasaccharides, for which all products must come from an even-electron precursor (manuscript in preparation). Many cross-ring fragmentation products of the hexuronic acid residues were found in the EID mass spectrum that were not found in the IRMPD mass spectrum of the same precursor ion. Previously, we had proposed the preponderance of cross-ring fragmentation of the hexuronic acid residues over amino sugars in heparan sulfate tetrasaccharides [23] and dermatan sulfate oligosaccharides [22] was related to radical-driven fragmentation processes originating from a carboxy-radical. However, the newer EID data suggests that hexuronic acid residues are more labile, and that fragmentation of the hexuronic acid residues occurs by electron activation of the even-electron precursor ion (EID) as well by as radical-driven fragmentation (EDD).
The gas phase ion conformation of proteins has been shown to influence the products observed by ECD. The combination of IRMPD with ECD, known as activated ion ECD (AI-ECD) [32], have been shown to increase the number of backbone cleavages by ECD, and this effect is also observed by increasing the precursor ion charge state [33]. This increase in fragmentation has been proposed to occur from a change in ion conformation that results from breaking intramolecular noncovalent bonds, producing a more extended structure. To examine the possibility that conformation affects fragmentation for GAG oligomers, activated ion EDD (AI-EDD) was performed on the [M-4H]4− precursor ion using CO2 laser irradiation to warm the ions to the threshold of fragmentation, followed by irradiation with electrons. While the S/N of some product ions increased slightly, AI-EDD does not influence the type or relative abundance of the observed products, specifically products accompanied by SO3 loss (data not shown). Therefore, the difference in SO3 loss in the 3− and 4− versus 5− charge states appears to be a result of electron detachment from a carboxylate anion and not due to a change in gas-phase conformation.
EDD of DS dp8 with Sodium Cations
The loss of SO3 by MS/MS of sulfated GAG oligosaccharides complicates the mass spectrum and reduces its analytical utility. Thus, controlling SO3 loss is important for the analysis of GAG oligosaccharides. The results of the charge state dependence studies show that SO3 loss is reduced when at least one carboxyl group is ionized in the precursor ion. As an alternative to dissociation of higher charge states, which may not have substantial abundance by ESI, we have investigated the replacement of protons by sodium. In such ions, the charge state will be smaller than the number of ionized sites in the precursor. As Figure 1 shows, the [M-5H]5− ion is half as abundant as the [M-5H+Na]4−. Both have five ionized sites and will therefore have an ionized carboxyl group available for electron detachment, providing a way to achieve a higher degree of ionization with precursor ions of a lower overall charge.
Products from EDD of the [M-5H+Na]4− precursor ion of DS dp8 are shown in Figure 4A (mass spectrum provided as supplemental data). When sodium is present in the precursor ion, product ions may occur as protonated or sodium cationized products. Product ions that are observed with sodium are indicated by a number above the fragment notation, with this number indicating the number of sodium cations in the product ion. For the work presented here, products containing sodium are described as the fragment plus the number of sodium cations, such as 0,2X7+Na. EDD of the [M-5H+Na]4− precursor ion of DS dp8 produces abundant glycosidic and cross-ring fragmentation similar to EDD of the non-cationized (i.e. 3−, 4−, and 5−) precursor ions of DS dp8. Glycosidic cleavages accompanied by the loss of 1 or 2 hydrogen atoms are also observed. Compared to EDD of [M-4H]4− (Figure 2B), EDD of [M-5H+Na]4− results in a significant decrease in product ions accompanied by SO3 loss. When compared to the product ions observed from EDD of the 3−, 4−, and 5− precursor ions of DS dp8 (Figure 2), the products observed from EDD of the [M-5H+Na]4− precursor ion resemble more closely the products observed from EDD of the [M-5H]5− precursor ion. For example, cross-ring cleavages occur primarily on IdoA residues. Also, the charge-reduced species minus CO2, [M-5H+Na-CO2]3−•, and not SO3 is observed. SO3 loss accompanying other fragmentation is greatly reduced in EDD of both [M-5H]5− and [M-5H+Na]4−, and the small number of products accompanied by SO3 loss occur near the NRE of the octasaccharide for both precursors. For example, 34 or the 87 observed products from EDD of the [M-4H]4− precursor ion are accompanied by SO3 loss, while 3 of the 56 and 1 of the 70 observed products from EDD of the [M-5H]5− and [M-5H+Na]4− precursor ions, respectively, are accompanied by SO3 loss. Together, these data suggest a total of five ionized sites on DS dp8, with at least one ionized site on a carboxy group. These data are consistent with our proposal that electron detachment preferentially occurs from the carboxylate anion, with EDD of [M-5H+Na]4− exhibiting a significant decrease in SO3 loss and fragmentation similar to EDD of [M-5H]5−. These observations suggest SO3 loss is suppressed when the total number of ionized sites on the GAG is one more than the number of sulfate groups. In further support of this observation, a significant decrease in SO3 loss is observed for EDD of [M-5H+2Na]3− when compared to the EDD data of [M-4H+Na]3− (data not shown).
Figure 4.
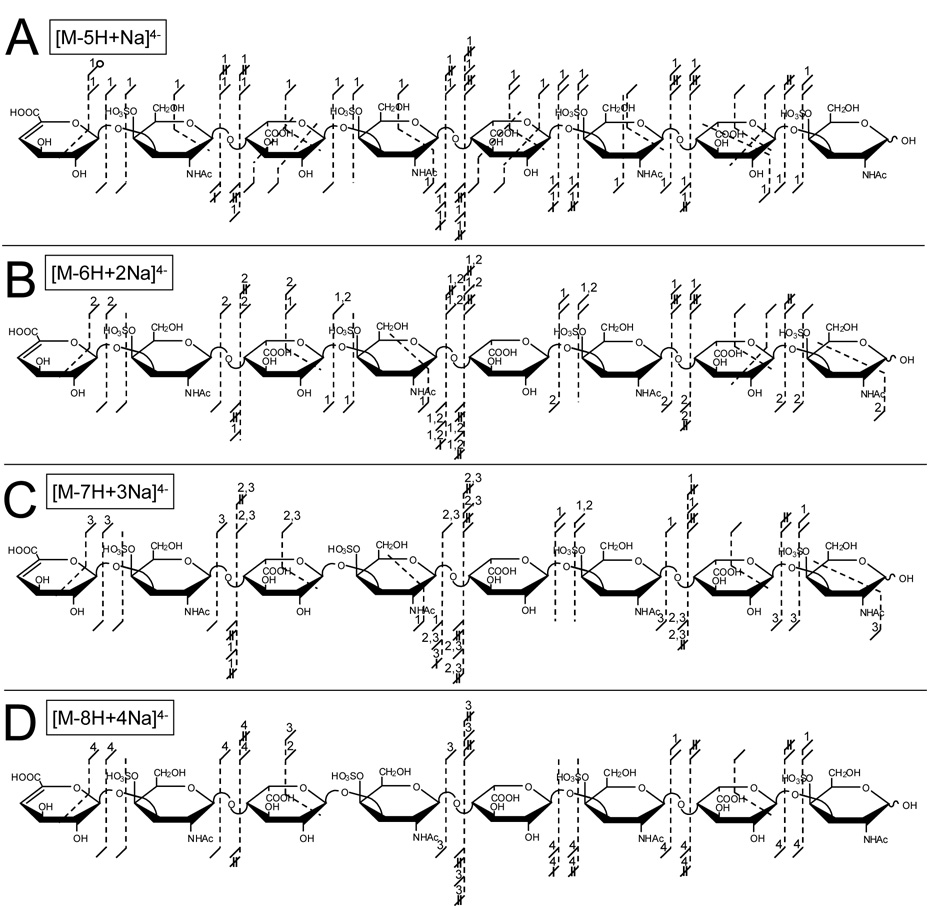
Product ions observed from EDD of the (A) [M-5H+Na]4−, (B) [M-6H+2Na]4−, (C) [M-7H+3Na]4−, and (D) [M-8H+4Na]4− precursor ions of DS dp8.
Additional replacement of protons by sodium cations can be used to increase the number of ionized sites. We have examined the EDD of DS dp8 with up to eight ionized acidic groups. Products from the EDD of the [M-6H+2Na]4−, [M-7H+3Na]4−, and [M-8H+4Na]4− precursor ions are shown in Figure 4B through 4C, respectively (see supplemental data for EDD mass spectra). As shown in Figure 4, increasing the number of sodium cations, and thus the number of ionized sites on the DS dp8 precursor ion decreases the number of observed glycosidic and cross-ring fragmentations. For the 4− ion of DS dp8 with 1 – 4 sodium cations, the charge reduced species minus CO2 is observed for all ions, indicating preference for electron detachment at carboxylate on IdoA residues. While increasing the ionization state of the molecule such that one carboxylate is charged benefits fragmentation by decreasing SO3 loss, further ionization of the GAG with sodium cations reduces EDD fragmentation. The decrease in fragmentation suggests that carboxyl hydrogens may play a role in the mechanism of cross-ring fragmentation, perhaps via hydrogen rearrangement reactions.
IRMPD of DS dp8 with Sodium Cations
SO3 loss during electron activation methods such as EDD of EID differs from that observed during activation of even-electron ions by threshold or low-energy methods. Previous work by Zaia and coworkers suggests that sulfate groups are most labile and readily undergo loss of SO3 during CAD when protonated [25]. While SO3 loss can be minimized and glycosidic cleavages maximized by increasing the charge state so that all sulfate groups are charged, the abundance of precursor ions in which all sulfates are charged may be too small to be useful for MS/MS. The addition of sodium cations to the ESI solution allows IRMPD to be applied to more ionized sulfated GAGs. Products resulting from IRMPD of the [M-4H]4−, [M-5H+Na]4−, [M-6H+2Na]4−, [M-7H+3Na]4−, and [M-8H+4Na]4− precursor ions of DS dp8 are shown in Figures 5A – 5C, respectively. Compared to EDD of the same precursor ions (Figure 2A and Figure 4A – 4C), IRMPD produces fewer glycosidic and cross-ring cleavages as well as more abundant SO3 loss. Unlike EDD, IRMPD produces some product ions which are observed only with SO3 loss. However, product ion SO3 loss decreases as the cationization of the precursor ion increases, for example IRMPD of the [M-4H]4− precursor ion produces 18 products accompanied by SO3 loss, while IRMPD of the [M-8H+4Na]4− produces 6 products accompanied by SO3 loss. These results are similar to the observations by Zaia et al. [25], suggesting that ionization of the sulfate groups prevents loss of SO3 for ion activation via threshold dissociation. The number of glycosidic bond and cross-ring cleavages produced by IRMPD decreases when the ionization of the GAG is increased using sodium cations, similar to the observation for EDD. For example, IRMPD of the [M-4H]4− precursor ion produces a total of 41 glycosidic and cross-ring cleavages, while IRMPD of the [M-8H+4Na]4− produces 23 glycosidic and cross-ring ring cleavages.
Figure 5.
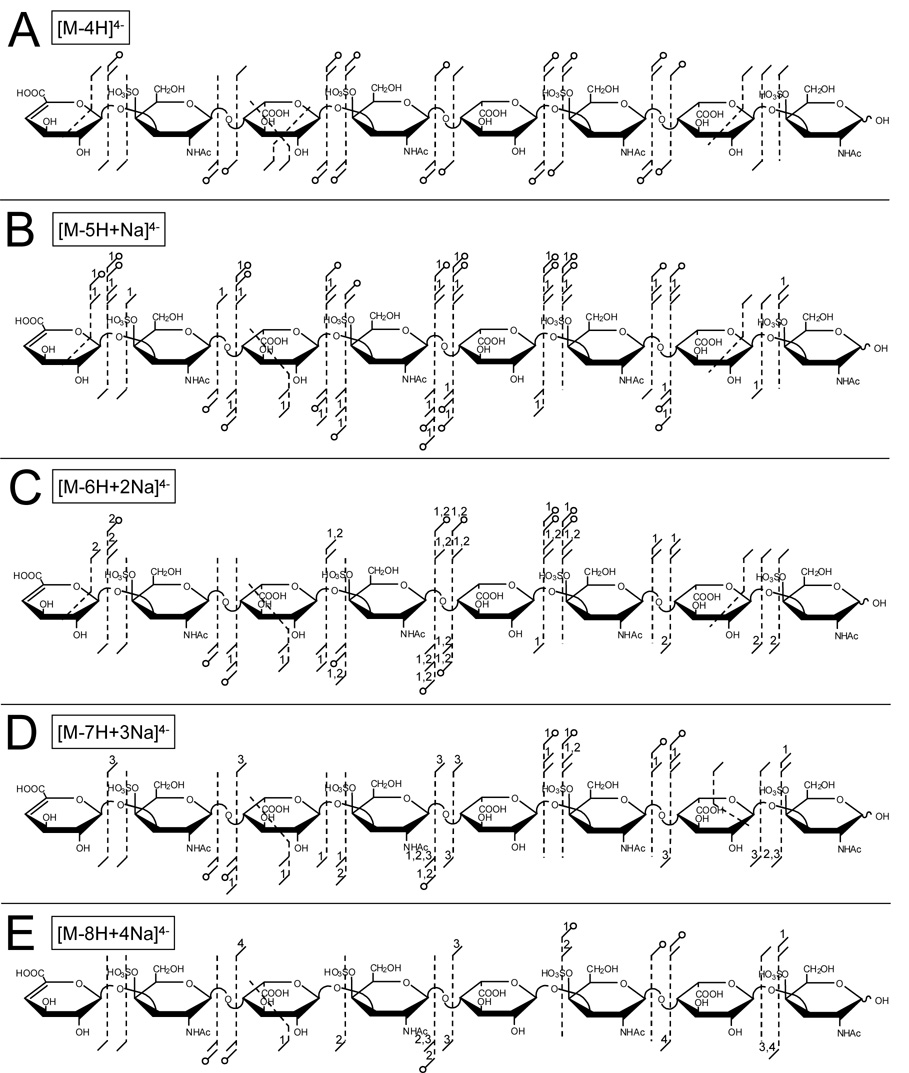
Product ions observed from IRMPD of the (A) [M-4H]4−, (B) [M-5H+Na]4−, (C) [M-6H+2Na]4−, (D) [M-7H+3Na]4−, and (E) [M-8H+4Na]4− precursor ions of DS dp8.
CONCLUSIONS
The ionization state of a sulfated GAG affects the number of ionized sites available for electron detachment. Since electron detachment occurs at a site of negative charge, the pattern of SO3 loss in EDD of sulfated GAGs is dependant on the number of ionized sites on the sulfated GAG. When electron detachment occurs at a sulfate anion, many product ions are accompanied by loss of SO3. When electron detachment occurs at a carboxylate anion there is a significant decrease in the number of product ions that are accompanied by loss of SO3. Increasing the degree of ionization can be achieved by either increasing the precursor ion charge state, or by the replacement of protons with sodium cations. Both methods can be used to ensure ionization of a carboxylate group. For EDD of sulfated GAGs, SO3 loss is minimized if the ionization state of the sulfated GAG is equal to one more than the number of sulfate groups. Further increasing the ionization state of the molecule so that more carboxylate groups are charged eliminates SO3 loss by EDD, but also decreases the number of glycosidic and cross-ring cleavages. For IRMPD of sulfated GAGs, increasing the ionization state of GAG precursor ions by replacement of protons with sodium cations decreases SO3 loss, but also decreases glycosidic and cross-ring fragmentation.
Supplementary Material
ACKNOLWEDGMENTS
The authors wish to acknowledge the seminal contributions of Roman Zubarev to the field of electron-ion interactions, and offer congratulations on his selection as a Biemann awardee. We gratefully acknowledge financial support from the National Institutes of Health grant #2R01-GM038060-16.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Linhardt RJ, Toida T. Role of Glycosaminoglycans in Cellular Communication. Acc. Chem. Res. 2004;37:431–438. doi: 10.1021/ar030138x. [DOI] [PubMed] [Google Scholar]
- 2.Gotte M. Syndecans in Inflammation. FASEB J. 2003;17:575–591. doi: 10.1096/fj.02-0739rev. [DOI] [PubMed] [Google Scholar]
- 3.Fannon M, Forsten KE, Nugent MA. Potentiation and Inhibition of bFGF Binding by Heparin: A Model for Regulation of Cellular Response. Biochemistry. 2000;39:1434–1445. doi: 10.1021/bi991895z. [DOI] [PubMed] [Google Scholar]
- 4.Wu ZL, Zhang L, Yabe T, Kuberan B, Beeler DL, Love A, Rosenberg RD. The Involvement of Heparan Sulfate (HS) in FGF1/HS/FGFR1 Signaling Complex. J. Biol. Chem. 2003;278:17121–17129. doi: 10.1074/jbc.M212590200. [DOI] [PubMed] [Google Scholar]
- 5.Sadir R, Imberty A, Baleux F, Lortat-Jacob H. Heparan Sulfate/Heparin Oligosaccharides Protect Stromal Cell-derived Factor-1 (SDF-1)/CXCL12 against Proteolysis Induced by CD26/Dipeptidyl Peptidase IV. J. Biol. Chem. 2004;279:43854–43860. doi: 10.1074/jbc.M405392200. [DOI] [PubMed] [Google Scholar]
- 6.Iozzo RV, San Antonio JD. Heparan sulfate proteoglycans: heavy hitters in the angiogenesis arena. J. Clin. Invest. 2001;108:349–355. doi: 10.1172/JCI13738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Batinic D, Robey FA. The V3 region of the envelope glycoprotein of human immunodeficiency virus type 1 binds sulfated polysaccharides and CD4-derived synthetic peptides. J. Biol. Chem. 1992;267:6664–6671. [PubMed] [Google Scholar]
- 8.Chen Y, Maguire T, Hileman RE, Fromm JR, Esko JD, Linhardt RJ, Marks RM. Dengue virus infectivity depends on envelope protein binding to target cell heparan sulfate. Nature Med. 1997;3:866–871. doi: 10.1038/nm0897-866. [DOI] [PubMed] [Google Scholar]
- 9.Williams RK, Straus SE. Specificity and affinity of binding of herpes simplex virus type 2 glycoprotein B to glycosaminoglycans. J. Virol. 1997;71:1375–1380. doi: 10.1128/jvi.71.2.1375-1380.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu DF, Shriver Z, Gi YW, Venkataraman G, Sasisekharan R. Dynamic regulation of tumor growth and metastasis by heparan sulfate glycosaminoglycans. Semin. Thromb. Hemost. 2002;28:67–78. doi: 10.1055/s-2002-20565. [DOI] [PubMed] [Google Scholar]
- 11.Perrimon N, Bernfield M. Cellular functions of proteoglycans--an overview. Semin. Cell Dev. Biol. 2001;12:65–67. doi: 10.1006/scdb.2000.0237. [DOI] [PubMed] [Google Scholar]
- 12.Horne A, Gettins P. H-1-Nmr Spectral Assignments for 2 Series of Heparin-Derived Oligosaccharides. Carbohydr. Res. 1992;225:43–57. doi: 10.1016/0008-6215(92)80038-3. [DOI] [PubMed] [Google Scholar]
- 13.Chi LL, Amster J, Linhardt RJ. Mass spectrometry for the analysis of highly charged sulfated carbohydrates. Current Analytical Chemistry. 2005;1:223–240. doi: 10.2174/157341105774573929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dai Y, Whittal RM, Bridges CA, Isogai Y, Hindsgaul O, Li L. Matrix-assisted laser desorption ionization mass spectrometry for the analysis of monosulfated oligosaccharides. Carbohydr. Res. 1997;304:1–9. doi: 10.1016/s0008-6215(97)00195-x. [DOI] [PubMed] [Google Scholar]
- 15.Miller MJC, Costello CE, Malmstrom A, Zaia J. A tandem mass spectrometric approach to determination of chondroitin/dermatan sulfate oligosaccharide glycoforms. Glycobiology. 2006;16:502–513. doi: 10.1093/glycob/cwj093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reinhold VN, Carr SA, Green BN, Petitou M, Choay J, Sinay P. Structural Characterization of Sulfated Glycosaminoglycans by Fast-Atom-Bombardment Mass-Spectrometry - Application to Heparin Fragments Prepared by Chemical Synthesis. Carbohydr. Res. 1987;161:305–313. doi: 10.1016/s0008-6215(00)90088-0. [DOI] [PubMed] [Google Scholar]
- 17.Saad OM, Leary JA. Compositional Analysis and Quantification of Heparin and Heparan Sulfate by Electrospray Ionization Ion Trap Mass Spectrometry. Anal. Chem. 2003;75:2985–2995. doi: 10.1021/ac0340455. [DOI] [PubMed] [Google Scholar]
- 18.Saad OM, Leary JA. Heparin Sequencing Using Enzymatic Digestion and ESI-MS with HOST: A Heparin/HS Oligosaccharide Sequencing Tool. Anal. Chem. 2005;77:5902–5911. doi: 10.1021/ac050793d. [DOI] [PubMed] [Google Scholar]
- 19.Zaia J, Li X-Q, Chan S-Y, Costello CE. Tandem mass spectrometric strategies for determination of sulfation positions and uronic acid epimerization in chondroitin sulfate oligosaccharides. J. Am. Soc. Mass Spectrom. 2003;14:1270–1281. doi: 10.1016/S1044-0305(03)00541-5. [DOI] [PubMed] [Google Scholar]
- 20.Zaia J, McClellan JE, Costello CE. Tandem Mass Spectrometric Determination of the 4S/6S Sulfation Sequence in Chondroitin Sulfate Oligosaccharides. Anal. Chem. 2001;73:6030–6039. doi: 10.1021/ac015577t. [DOI] [PubMed] [Google Scholar]
- 21.Budnik BA, Haselmann KF, Zubarev RA. Electron detachment dissociation of peptide di-anions: an electron-hole recombination phenomenon. Chem. Phys. Lett. 2001;342:299–302. [Google Scholar]
- 22.Wolff JJ, Laremore TN, Busch AM, Linhardt RJ, Amster IJ. Electron Detachment Dissociation of Dermatan Sulfate Oligosaccharides. J. Am. Soc. Mass Spectrom. 2008 doi: 10.1016/j.jasms.2007.10.007. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wolff JJ, Amster IJ, Chi L, Linhardt RJ. Electron Detachment Dissociation of Glycosaminoglycan Tetrasaccharides. J. Am. Soc. Mass Spectrom. 2007;18:234–244. doi: 10.1016/j.jasms.2006.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wolff JJ, Chi LL, Linhardt RJ, Amster IJ. Distinguishing glucuronic from iduronic acid in glycosaminoglycan tetrasaccharides by using electron detachment dissociation. Anal. Chem. 2007;79:2015–2022. doi: 10.1021/ac061636x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zaia J, Costello CE. Tandem Mass Spectrometry of Sulfated Heparin-Like Glycosaminoglycan Oligosaccharides. Anal. Chem. 2003;75:2445–2455. doi: 10.1021/ac0263418. [DOI] [PubMed] [Google Scholar]
- 26.Pervin A, Gallo C, Jandik KA, Han X-J, Linhardt RJ. Preparation and structural characterization of large heparin-derived oligosaccharides. Glycobiology. 1995;5:83–95. doi: 10.1093/glycob/5.1.83. [DOI] [PubMed] [Google Scholar]
- 27.Heck AJR, de Koning LJ, Pinkse FA, Nibbering NMM. Mass-specific selection of ions in Fourier-transform ion cyclotron resonance mass spectrometry. Unintentional off-resonance cyclotron excitation of selected ions. Rapid Commun. Mass Spectrom. 1991;5:406–414. [Google Scholar]
- 28.Domon B, Costello CE. A systematic nomenclature for carbohydrate fragmentations in FAB-MS/MS spectra of glycoconjugates. Glycoconjugate J. 1988;5:397–409. [Google Scholar]
- 29.Budnik BA, Haselmann KF, Elkin YN, Gorbach VI, Zubarev RA. Applications of Electron-Ion Dissociation Reactions for Analysis of Polycationic Chitooligosaccharides in Fourier Transform Mass Spectrometry. Anal. Chem. 2003;75:5994–6001. doi: 10.1021/ac034477f. [DOI] [PubMed] [Google Scholar]
- 30.Cody RB, Freiser BS. Electron impact excitation of ions from organics: an alternative to collision induced dissociation. Anal. Chem. 1979;51:547–551. [Google Scholar]
- 31.Cody RB, Freiser BS. Electron impact excitation of ions in Fourier transform mass spectrometry. Anal. Chem. 1987;59:1054–1056. [Google Scholar]
- 32.Horn DM, Ge Y, McLafferty FW. Activated Ion Electron Capture Dissociation for Mass Spectral Sequencing of Larger (42 kDa) Proteins. Anal. Chem. 2000;72:4778–4784. doi: 10.1021/ac000494i. [DOI] [PubMed] [Google Scholar]
- 33.Sze SK, Ge Y, Oh H, McLafferty FW. Top-down mass spectrometry of a 29-kDa protein for characterization of any posttranslational modification to within one residue. Proc. Natl. Acad. Sci. 2002:251691898. doi: 10.1073/pnas.251691898. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.