

Abstract
Classical late-infantile neuronal ceroid lipofuscinosis (LINCL) is a hereditary neurodegenerative disease of childhood that is caused by mutations in the gene (CLN2) encoding the lysosomal protease tripeptidyl-peptidase I (TPPI). LINCL is fatal and there is no treatment of demonstrated efficacy in affected children but preclinical studies with AAV-mediated gene therapy have demonstrated promise in a mouse model. Here, we have generated mouse CLN2 mutants that express different amounts of TPPI activity to benchmark levels required for therapeutic benefits. Approximately 3% of normal TPPI activity in brain delayed disease onset and doubled lifespan to a median of ~9 months compared to mice expressing ~0.2% of normal levels. Expression of 6% of normal TPPI activity dramatically attenuated disease, with a median lifespan of ~20 months which approaches that of unaffected mice. While the life-span of this hypomorph is shortened, disease is late-onset, less severe and progresses slowly compared to mice expressing lower TPPI levels. For gene therapy and other approaches that restore enzyme activity, these results suggest that 6% of normal TPPI activity throughout the CNS of affected individuals will provide a significant therapeutic benefit but higher levels will be required to cure this disease.
Keywords: neuronal ceroid lipofuscinosis, mouse model, hypomorph, lysosomal storage disease
INTRODUCTION
The neuronal ceroid lipofuscinoses (NCLs) are a group of genetically distinct but clinically related diseases of which the classical late-infantile form (LINCL) is one of the most frequently encountered [1]. LINCL is a fatal neurodegenerative lysosomal storage disease that results from mutations in the gene CLN2 [2] that encodes the lysosomal protease tripeptidyl-peptidase I (TPPI) [3]. This disease is characterized by seizures, progressive mental decline, loss of vision and locomotor function, and shortened lifespan. Mutations in most LINCL patients are null alleles [4] and typically result in diagnosis at around 4 years and survival to between 7 and 15 years of age. However, like many other lysosomal storage diseases [5], some LINCL cases exhibit a less severe clinical phenotype. Here, compound heterozygosity for a null allele and a presumed hypomorphic Arg447His missense mutation [4] results in a later onset (~8 years) and protracted disease with survival into the third or fourth decade of life.
There is currently no treatment of demonstrated efficacy for LINCL patients but there is promising progress, particularly with respect to adeno-associated virus (AAV)-mediated gene therapy (reviewed in [6]). Initial studies examining the expression of TPPI by recombinant AAV and other viral vectors demonstrated high levels and widespread expression of the protein throughout the central nervous system (CNS) of healthy rodents [7, 8]. More recently, AAV-mediated gene therapy has been evaluated in a mouse model for LINCL. This gene-targeted mouse was created by the disruption of CLN2 with an Arg446His missense mutation (which is equivalent to the late-onset human allele) and insertion of a neomycin selection cassette within an adjacent intron, resulting in a disruption of normal splicing [9]. The synergistic effect of the missense mutation and the splicing defect results in levels of TPPI that are below the threshold of detection (~1% of normal levels) and this mouse recapitulates many of the clinical features of the human disease [9]. Mice appear to be healthy at birth but develop tremors by about 7 weeks. In symptomatic mice, there is pervasive neuronal pathology with a cytoplasmic accumulation of autofluorescent storage material, selective loss of some Purkinje cells and widespread axonal degeneration. Lifespan is greatly shortened with a median survival of ~ 20 weeks.
Treatment of the CLN2-targeted mice with an AAV vector expressing TPPI initially showed expression of the recombinant protein and a slowing of the cellular pathology that is associated with disease [10]. More recently, it has been demonstrated that AAV-mediated gene therapy, in addition to attenuating cellular pathology, can also slow or halt the decline in locomotor function and significantly increase survival of the mutant mice [11, 12]. An important observation to emerge from these studies is that while AAV treatment of symptomatic mice has some positive effects, treatment must be conducted before onset of disease for the greatest therapeutic benefits [11].
Levels of TPPI activity achievable by AAV-mediated gene therapy vary from study to study, reflecting, in addition to other variables, viral serotype and titer as well as the number and location of injection sites. Passini et al [10] achieved levels that approached or exceeded wild-type activities throughout most of the brain. More recently, CNS levels of TPPI have been achieved in AAV-treated mutant mice that were 10 to 100-fold [11] or up to 27-fold [12] higher than found in wild-type mice.
While the induction of such levels is clearly beneficial in the mouse model, it seems unlikely that similar levels of TPPI activity will be attainable throughout the brain of human LINCL patients by gene therapy or enzyme replacement. However, the minimum level of TPPI activity that will be required to successfully treat LINCL remains unknown. In this study, we have taken a direct approach to this fundamental question by investigating the disease phenotype of a series of CLN2-targeted mouse mutants that express different amounts of residual TPPI activity.
MATERIALS AND METHODS
Generation of CLN2 mouse hypomorphs
All experiments and procedures involving live animals were conducted in compliance with approved Institutional Animal Care and Use Committee protocols. The CLN2-targeted mouse model for LINCL (CLN2−/−) has been described previously [9] and was created by the synergistic effect of an Arg446His missense mutation and a splicing defect resulting from the insertion of a floxed neo selection marker (Fig. 1, Panel A).
Figure 1. Schematic of the generation of the CLN2 hypomorph.
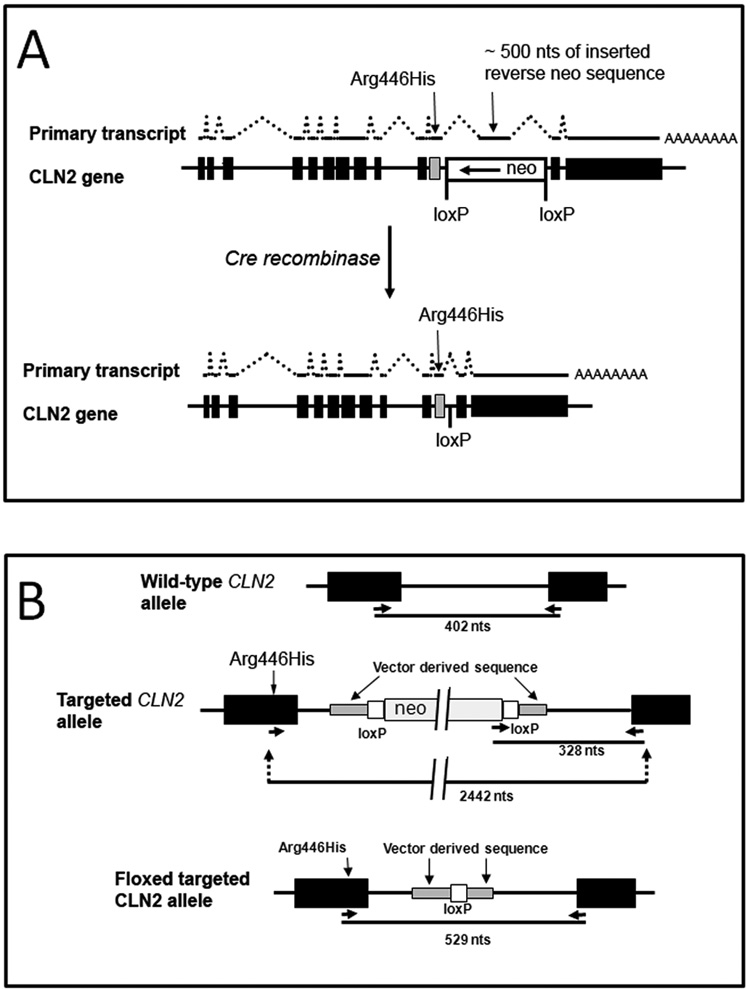
Panel A, The CLN2 targeted allele was originally generated by the synergistic combination of an Arg446His allele with the insertion of a neomycin selection cassette into intron 10 of the murine CLN2 gene which results in an unstable mRNA containing a ~ 500 nt reverse orientation neo insertion [9]. The targeted CLN2 alleles described in this study were generated by the cre-mediated excision of the neo marker allowing for transcription of a normally spliced and stable CLN2 mRNA that differs from the wild-type transcript only in the presence of the missense mutation. Panel B, Details of the targeted CLN2 allele and genotyping strategy.
Removal of the neo selection marker was achieved by crossing male CLN2 +/− mice with female Zp3-cre mice (The Jackson Laboratory, Bar Harbor, Maine) which is a transgenic line in a C57BL/6 background that expresses cre within the oocyte from the zona pellucida 3 gene promoter [13]. Progeny of this mating were screened for cre-mediated excision of neo by using primers (ATCTGATGGCTACTGGGTGG, CCCGGTAGAATTCCGATCAT and CCCCCAAACACTGGAGTAGA) that generate 402 nt and 328 nt products from the wild-type and neo-containing targeted alleles, respectively and 529 nt from the mutant allele after excision of neo (Fig. 1, Panel B). Founders that were heterozygous for the neo excision (+/f) were backcrossed against C57BL/6 mice and were selected for loss of the cre transgene using a primer set (GCGGTCTGGCAGTAAAAACTATC, GTGAAACAGCATTGCTGTCACTT) that generates a 100 nt product from this sequence. Complete annotated CLN2 sequences for all three alleles (wild-type, targeted and floxed-targeted) are provided as Online Supplementary Material. Data presented was obtained from −/− and f/− mice after 10 back crosses to C57BL/6 and +/−, f/+, +/+, and f/f mice after at least 5 backcrosses to C57BL/6.
Detection of CLN2 transcripts
Northern blotting for CLN2 transcripts were preformed on total RNA purified using an RNEasy kit (Qiagen, Valencia, CA) as described previously [9].
Subcellular fractionation
Brain and liver homogenates (H fraction) were prepared using a Potter homogenizer as described [14] in 0.25M sucrose. Nuclei and heavy mitochondrial components were pelleted by centrifugation for 3 minutes at 12,500 rpm in a 50 Ti rotor (Beckman Coulter, Fullerton, CA). The supernatant was then centrifuged for 6.42 minutes at 25,000 rpm in a 50 Ti rotor to pellet the light mitochondrial differential fraction (L fraction) which was washed and resuspended in 0.25 M sucrose.
Measurement of TPPI activity
TPPI was measured in the H and L fractions after dilution with cold 0.15M NaCl-0.1% Triton X-100 using 200 µM Arg-Ala-Phe-ACC (generously provided by Dr. John W. Taylor, Rutgers University) as substrate using the kinetic assay described previously [15]. For determination of KM, TPPI was measured using Arg-Ala-Phe-ACC and Ala-Ala-Phe-AMC (Sigma, St. Louis, MO). β-Galactosidase activity was measured using 4-methylumbelliferyl-β-D-galactopyranoside substrate [16]. For both enzymes, fluorescence was measured using a CytoFluor 4000 multiwell plate reader (PerSeptive Biosystems, Framingham, MA). Seven different dilutions of each sample, corresponding to ~0.15 g tissue equivalents/mL and six successive 2-fold dilutions, were used for enzyme activity measurements. TPPI activity values were calculated from 2–3 dilutions in the linear range using the lowest (most concentrated) dilutions for the f/f and −/− specimens and the highest dilutions for +/+ specimens. Activities were standardized to protein concentration measured with Advanced Protein Assay reagent (Cytoskeleton Inc., Denver, CO).
Behavioral testing
Mutant mice were analyzed for gait abnormalities and ataxia as reviewed [17]. Footprint patterns were visualized by painting the feet of mutant mice with non-toxic washable paint (forefeet, purple and hindfeet, orange) and placing the mice at the entrance of a dark tunnel (35cm long × 9cm wide × 6cm high) placed over white paper.
Pathology
For immunohistochemical detection of TPPI, mice were euthanized at 52–57 days of age and were transcardially infused with Bouin’s fixative. Brains were removed, bisected and drop-fixed at 4C for 16 hours. Samples were then washed three times with PBS and saturated with PBS containing 15% then 30% sucrose in consecutive 48hr incubations. Tissue samples were embedded in Shandon M-1 Embedding Matrix (Thermo Scientific) and 10 µm cryosections were prepared. Immunohistochemistry using a 1:100 dilution of a primary goat anti-rabbit polyclonal antibody (R72) was as described previously [18] with fluorescent staining using an Alexa Fluor 488 dye-labeled secondary antibody (Invitrogen) at a dilution of 1:400. For immunohistochemical detection of the subunit C of mitochondrial ATP synthase, tissues were fixed in a manner similar to the above but using 4.0% paraformaldehyde in 0.1 M phosphate buffer. Blocks of tissue were removed and sectioned at 35 microns on a Leica 9000S Vibratome. This was followed by staining overnight with a 1:250 dilution of a rabbit polyclonal antibody (generously provided by Dr. E. F. Neufeld) raised against a peptide corresponding to the amino terminus of the mature protein c subunit ([19]). After washing, sections were incubated in a biotinylated goat-anti-rabbit IgG (1:200) for one hour, followed by incubation in Vectastain ABC Elite complex (Vector Laboratories, Burlingame, CA). Antibody binding was visualized using a diaminaobenzidine substrate kit (Vector Laboratories). Sections were placed on gelatinized slides and dried overnight, followed by dehydration and cover-slipping, and examination on an Olympus research microscope. Autofluorescent storage was examined in 35 µm coronal vibratome sections that were cut from fixed brain tissue, washed with PBS and mounted on slides with Prolong Gold antifading reagent (Invitrogen: Molecular Probes). Sections were examined using the Zeiss Meta Duo V2 confocal system by simultaneous excitation with 458 and 488 nm lines of a multi-line Argon laser and collective emission capture using the Meta spectral scanner set for a band of 501–694 nm. This bandwidth was selected in part based on initial full spectrum scans to profile peaks of emission. Multiple sections from each of duplicate mice for each genotype and age were surveyed.
Survival analysis
Mice were handled gently as environmental disturbances can induce fatal seizures in the −/− animals [9]. Moribund animals (typically, severe ataxia and tremors restricting ability to feed) were euthanized under IACUC policy and were scored as dying from disease on the day of euthanasia. Kaplan-Meier analysis was conducted using Prism v 4.03 (GraphPad Software, San Diego).
Western blotting
Tissue homogenates were prepared in 250 mM sucrose and 10 or 20 ug protein equivalents fractionated by electrophoresis on 4–12 % Bis-Tris gradient gels (Invitrogen) with MES running buffer for SCMAS or 10% Bis-Tris gels with MOPS running buffer for TPPI. Proteins were transferred to nitrocellulose. SCMAS was detected using the rabbit polyclonal antibody described above which was diluted 1:3000-fold in PBS containing 0.2% Tween -20 and 5% BSA. TPPI was detected using an affinity-purified rabbit polyclonal antibody raised against recombinant TPPI which was diluted in PBS containing 10% fat free milk and 0.2% Tween-20. In both cases, the secondary antibody was a I125-labeled goat anti-rabbit polyclonal and signal was visualized using Typhoon scanner (GE Healthcare) and quantified using Imagequant 5.2 software.
RESULTS
Survival of CLN2-mutant mice is strain dependent
In our previous characterization of the CLN2-targeted mouse, we measured survival in either an isogenic 129Sv or a mixed C57BL/6, 129Sv strain background. We found that survival of the isogenic 129Sv mutant was longer (median, 164 days) than the mixed strain (median, 138 days). We interpreted these results to possibly indicate the presence of a strain-specific modifier(s) of the disease phenotype which could, for example, influence triggering of or susceptibility to fatal seizures. In this study, we have analyzed the effects of CLN2 mutations on phenotype by comparing congenic or, because of time constraints reflecting the life-span of the mutants, near-congenic (≥ 6 backcrosses) mice in a C57BL/6 genetic background. To determine the effect of this genetic background on LINCL phenotype, we compared the survival of −/− mutant mice that were in either a C57BL/6 or 129Sv background (Fig. 2). We find that survival of the CLN2 mutant in a 129Sv background (median 155 days) is extended (p=0.0008) compared to the mice in a congenic C57BL/6 background (median 132 days). Survival of the CLN2 mutant in a first generation cross between C57BL/6 and 129Sv was essentially the same as in the 129Sv background.
Figure 2. Strain-dependence of survival of the CLN2-mutant mice.
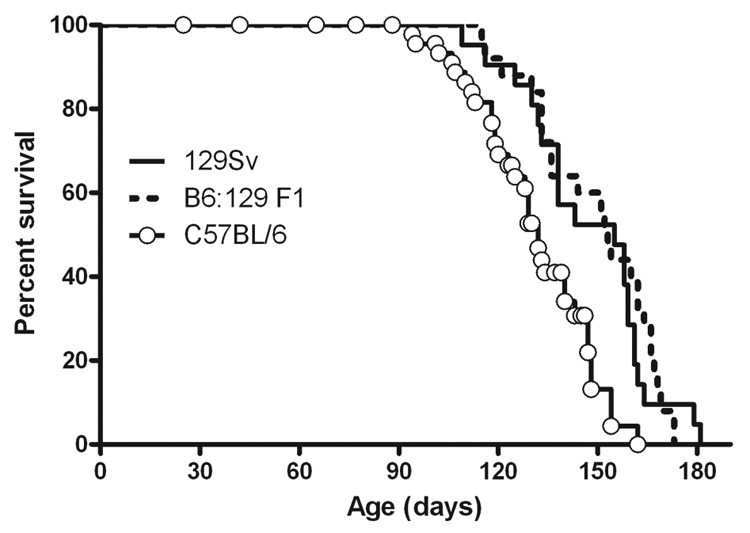
Mice were isogenic for either C57BL/6 or 129SvEv genetic background or were a first generation cross between these strains. 129Sv, n=21; C57BL/6, n=33, 129Sv × C57BL/6 F1, n=25.
Generation of mouse models expressing different levels of TPPI activity
The aim of this study was to generate mouse models of LINCL with hypomorphic mutations that express low levels of TPPI to determine thresholds of activity that provide therapeutic benefits. Our approach to achieving this has been to genetically modify our existing CLN2 mutant (technically designated neoinsArg446HisCLN2 allele (The Mouse Genome Initiative, http://www.informatics.jax.org) and designated here as −/− for brevity) to allow limited expression of TPPI. An explanation of this strategy requires a description of the molecular details by which our CLN2-targeted mouse model was generated and this is provided elsewhere [9]. In brief, CLN2 was disrupted by the synergistic effects of an Arg446His missense mutation in exon 11 and a splicing defect caused by the presence of cryptic splice sites within the neomycin selection marker inserted into intron 11 (Fig. 1, Panel A). Most transcripts from the targeted allele contained an antisense neo insertion within the CLN2 transcript but a small amount of mRNA (corresponding to ~4% of wild-type levels) was correctly spliced. Together, these mutations resulted in <1% (the lower limit of detection of our previous assay) of the normal levels of TPPI activity in homozygous targeted mice. In order to generate a TPPI hypomorph mutant with residual TPPI activity, we used cre-mediated recombination to remove the disruptive neo insertion to allow for normal splicing of a transcript containing the Arg446His missense mutation (Fig. 1, Panel B). Mice that are homozygous for the mutant allele in which the neo selection marker is removed are designated neodelArg446HisCLN2 and are referred to here as f/f. The compound heterozygote of these mutant CLN2 alleles is referred to as f/−.
We investigated the effect of the removal of neo on CLN2 transcription and splicing by northern blotting. Wild-type mouse RNA have two transcripts for CLN2 of ~3.5 and 4.5 kb (Fig. 3, Panel A) that result from transcription termination at two alternate polyadenylation sites as observed previously [9]. Levels of TPPI mRNA in the CLN2 −/− mutant were greatly reduced and transcripts were larger than observed in the wild-type mice, reflecting the insertion of ~ 500 nts corresponding to an antisense neo transcript. As predicted, size and abundance of the transcript from the mutant allele after excision of neo was indistinguishable with the wild-type mRNA (Fig. 3, Panel A). These results indicate that the CLN2 mRNA is stable when encoding the Arg446His missense mutation and that splicing of the primary transcription product is unaffected by the 175-nt insertion within intron 11 that results from targeting vector sequence and the single loxP site that remains after recombination (Fig. 1, Panel B).
Figure 3. Expression of TPPI in the CLN2-mutant mice.
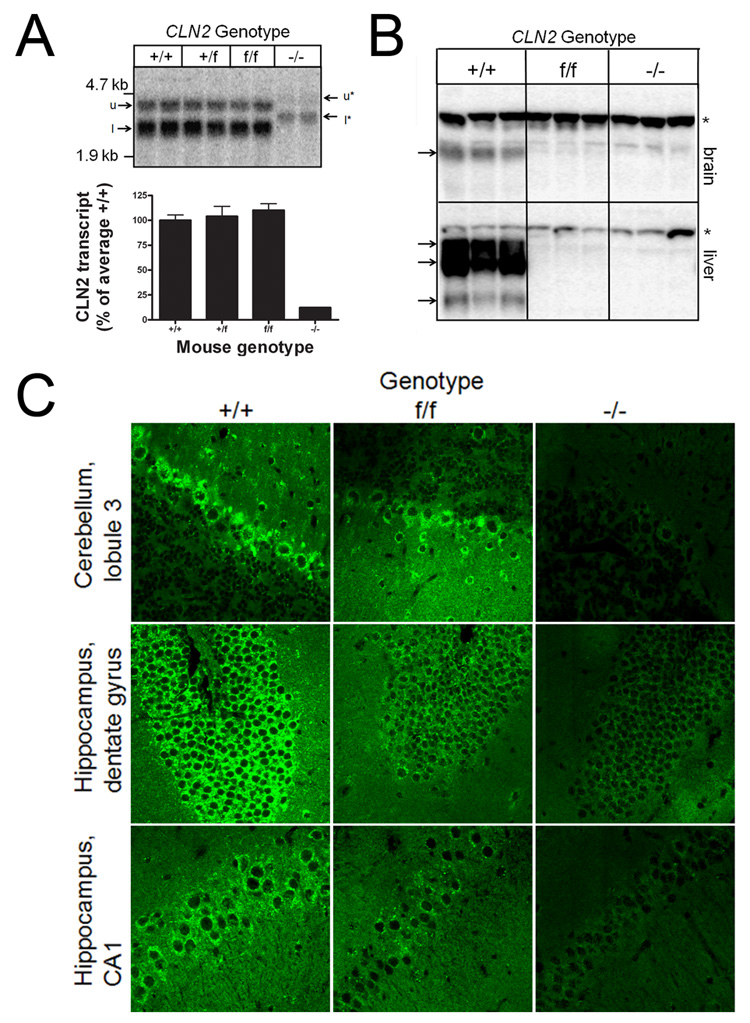
Panel A, CLN2 mRNA was detected by northern blotting in 10ug total RNA purified from mouse brain. Note the two sizes of transcript resulting from the use of alternate polyadenylation sites, designated u (upper) and l (lower) for transcripts from the wild-type and floxed mutant alleles, or u* and l* for the misspliced transcript from mutant allele containing the neo insertion (Fig. 1). Quantitation of correctly spliced CLN2 transcripts (u and l) as a percentage of the average levels in the wild-type control. Error bars represent the range of duplicate determinations. Panel B, Detection of TPPI by western blotting in L fractions prepared from brain or liver. Protein extracts (20µg equivalents) were fractionated by SDS-PAGE, transferred to nitrocellulose and TPPI visualized using a rabbit polyclonal antibody and radiolabeled secondary antibody. Bands corresponding to TPPI are indicated with arrows and non-specific bands found in all samples are indicated by asterices. Mice were 56–68 days old. Panel C, Immunohistochemical detection of TPP 1 in sections from cerebellum and hippocampus (CA1 region and dentate gyrus) using a rabbit polyclonal anti-TPPI antibody and visualized using a fluorescent-labeled secondary antibody. Mice were between 52 and 57 days of age and multiple sections were examined from independent duplicates for each genotype before selection of representative images. Images were acquired using a Zeiss LSM-410 confocal laser scanning microscope using a 63x oil objective. All mice were congenic for the C57BL/6 strain background.
TPPI is expressed in the CLN2-targeted hypomorphs
Given that a precise determination of TPPI levels in the CLN2 mutants is key to the interpretation of this study, TPPI expression in the CLN2-targeted mice was measured both by functional enzymatic assay and by immunological approaches. Enzyme activity measurements were conducted using a new substrate that is highly specific for TPPI on total homogenates and differential centrifugation light mitochondrial (L) fractions from both liver and brain (Table 1). The average TPPI specific activities in brain homogenates of f/f and −/− mutants are 5.9% and 0.3%, respectively, of wild type, with the values for the −/− samples approaching background levels of the assay. The L fractions are enriched in lysosomes and TPPI specific activities of f/f and −/− mutants are 4.7% and 0.2% of wild type. While the differential centrifugation step increases the specific activity of lysosomal enzymes and thus the signal to noise of the TPPI assay, the relative enrichments for the different samples differ: when considering the ratio of β-galactosidase specific activity in a given L fraction and homogenate (“enrichment factor”), the average enrichment factors are 2.28, 1.86 and 1.09 for the +/+, f/f and −/− brain samples, respectively. When correcting for this and using data from analysis of L fractions, we estimate that the relative TPPI activities in f/f and −/− mutant brain are 5.7% and 0.46%, respectively, of wild type.
Table 1. TPPI activity in the CLN2-mutant mice.
TPPI and β-galactosidase were measured in crude homogenates or lysosome-enriched subcellular fractions (L fraction) prepared from either liver or brain. Data are expressed as average arbitrary units of fluorescent product produced per hour per microgram of protein. Errors represent the standard deviation of measurements from three animals per genotype. All animals were 56–68 days of age and were in a congenic C57BL/6 strain background.
| GENOTYPE | |||||
|---|---|---|---|---|---|
| TISSUE | FRACTION | ENZYME | wild type | f/f | −/− |
| Liver | Homogenate |
β-gal |
352 ± 34 | 194 ± 17 | 205 ± 19 |
| TPPI | 1140 ± 108 | 12 ± 1 | 1 ± 0 | ||
| L Fraction |
β-gal |
2120 ± 529 | 1460 ± 273 | 898 ± 277 | |
| TPPI | 9340 ± 2370 | 102 ± 19 | 5 ± 4 | ||
| Brain | Homogenate |
β-gal |
209 ± 23 | 178 ± 4 | 202 ± 9 |
| TPPI | 490 ± 101 | 27 ± 1 | 1 ± 1 | ||
| L Fraction |
β-gal |
476 ± 70 | 331 ± 21 | 221 ± 29 | |
| TPPI | 1110 ± 239 | 52 ± 10 | 2 ± 1 | ||
The above analysis yields a consistent estimate of 6% for the relative activity of TPPI in f/f brain. The values for the −/− mutant are less certain given the residual activity is near the detection limit of our assay. We have previously used rtPCR to determine that the levels of the correctly spliced Arg447His mutant transcript in −/− brain are 4% of the corresponding normal transcript in wild type brain [9]. Given that the f/f and+/+ mice have indistinguishable levels of correctly spliced transcript (Fig. 3, Panel A) and that “normal” levels of the Arg447His transcript yield 6% of TPPI enzymatic activity, then a realistic estimate of the activity in −/− brain is ~0.2 % (6% × 4%) of wild type. In an independent experiment, levels of TPPI activity in crude homogenates from the f/− compound heterozygote were found to be 2.9% of wild-type levels (data not shown) which, as predicted, is intermediate between the f/f and −/− mutants.
Analysis of liver homogenates and differential centrifugation L fractions indicates that the TPPI activity of the f/f mutant is ~1% of wild type, while the value for the −/− mutant is below the limit of detection of our assay (Table 1). The reason for the lower relative levels of TPPI activity in f/f and −/− liver compared to brain is not clear but it could potentially reflect differences in the stability of the mutant protein in these different tissues. Interestingly, the specific activity of beta-galactosidase in the mutant liver samples are decreased ~50% compared to wild-type while the beta-galacosidase levels of the brain samples are essentially indistinguishable (Table 1).
Given that levels of the correctly spliced CLN2 transcript in the f/f mutant are similar to wild-type, there are two possible explanations for the reduced TPPI activity in this mutant. First, TPPI protein may be present at normal levels but the mutation could result in reduced activity of the enzyme. Second, the mutant protein may have normal activity but could be unstable and present at reduced levels. To distinguish between these possibilities, we compared the activity of TPPI in wild-type and f/f samples using two different synthetic substrates (Fig. 4). It is clear that the KM for both control and mutant TPPI is essentially identical when measured with either substrate, strongly suggesting that while transcription is normal, levels of TPPI protein are reduced in correspondence with activity.
Figure 4. Substrate saturation curves for normal and mutant TPPI.
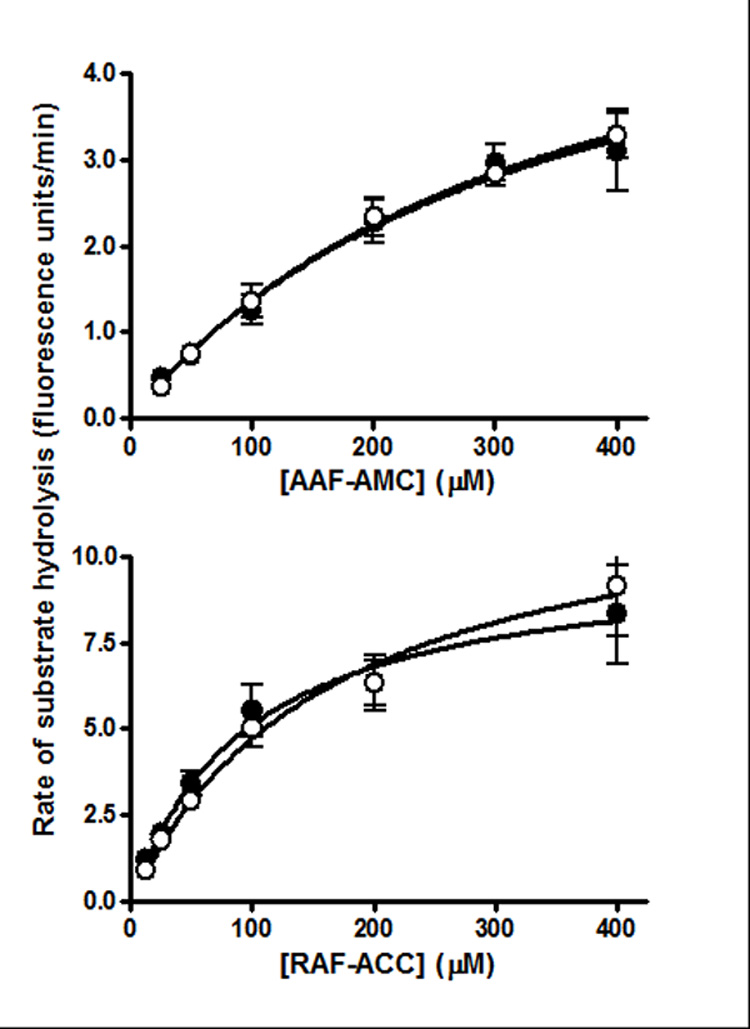
TPPI activity was measured using two synthetic substrates Arg-Ala-Phe-ACC (RAF-ACC) or Ala-Ala-Phe-AMC (AAF-AMC) in mouse liver L fractions. L fraction dilutions were adjusted such that the Vmax measured for both wild-type and mutant TPPI was identical. Open and filled circles represent wild-type and f/f mutant samples, respectively
Western blotting for TPPI in L fractions (Fig. 3, Panel B) as well as total homogenates (data not shown) revealed significant protein levels in the wild-type samples. TPPI levels were higher in liver than brain extracts which is consistent with the results of the enzymatic assay. TPPI levels in brain were below the limits of detection in the f/f and −/− samples. In liver, close inspection of the blot indicates a low but detectable level of TPPI protein in the f/f samples. Quantitation of the phosphorimager signal revealed protein levels corresponding to 1.0 ± 0.34% of the wild-type level, a value which is consistent with the enzyme assay data. TPPI expression was below limits of detection in the liver of the −/− samples.
We also investigated expression of TPPI in the CLN2-mutants by immunofluorescence microscopy (Fig. 3, Panel C). In the wild-type controls, staining for TPPI was widespread throughout the brain but was particularly strong in the cell bodies of pyramidal and granule neurons of the CA1 and dentate gyrus regions of hippocampus, respectively, and the Purkinje cells of the cerebellum. Staining was punctate and cytoplasmic which is consistent with lysosomal localization. TPPI was detectable in the same regions of the brain in the f/f hypomorph but was less intense than observed in the wild-type control. Essentially no staining for TPPI was detected in the −/− mutant. TPPI was also detected within other regions of the brain (cortex and thalamus) and in the liver of the wild-type and f/f mice but not in that of the −/− mutant (data not shown). Taken together, these data indicate that the TPPI transcript containing the Arg446His mutation is stable and present at normal levels but enzyme activity and levels are greatly reduced, most probably indicating the synthesis of an unstable but active protein product.
Expression of low levels of TPPI in CLN2-mutant mice attenuates disease
The CLN2 −/− mutant has a severe phenotype that results in a greatly shortened lifespan [9] and here, the median survival of the −/− C57BL/6 congenic mice was 135–138 days (Fig. 2 and Fig. 5), with the longest lived animal surviving to 162 days. The life-span of the f/f CLN2 hypomorph is greatly extended compared to the −/− mice, with a median and maximal survival of 603 and 734 days, respectively. While the lifespan of this mutant approaches that of a normal mouse, the f/f mutant does die somewhat earlier than the apparently unaffected controls (grouped +/+, f/+ and −/+ animals, median survival 822 days) (Fig. 5). The f/− compound heterozygotes have an intermediate phenotype, with a median and maximal survival of 269 and 438 days, respectively.
Figure 5. Survival analysis of CLN2 mutants.
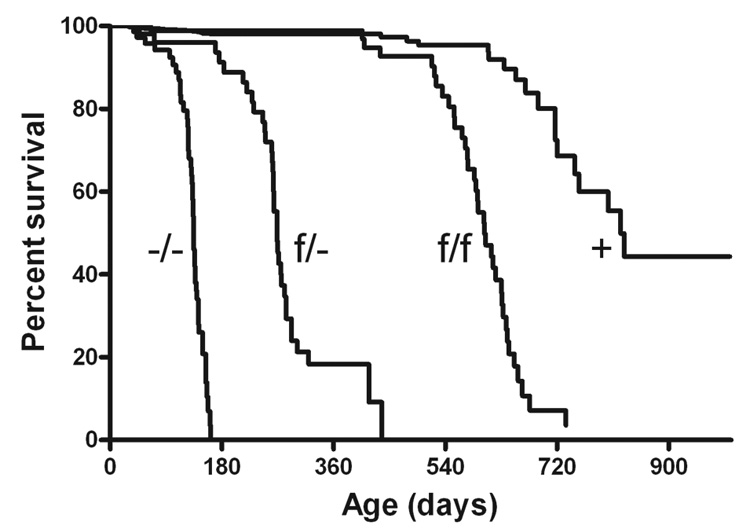
Mice were in either congenic or near congenic (≥ 6 backcrosses) C57BL/6. Data represent the following numbers of mice: −/−, n=47; f/−, n=35; f/f, n=36; control +, n=33 (+/−, n=15; +/+, n=5; +/f, n=13).
At around 7 weeks of age, a constant tremor is detected in the −/− mice which becomes more pronounced as they age and is eventually accompanied by ataxia and an abnormal gait. Both of the CLN2 hypomorphs described here show a similar disease progression but with a later onset and slower progression that is in proportion to the overall survival of the mice. As a general indicator of disease progression from a locomotor perspective, we measured the footprint patterns of the mutant mice at different time-points to detect gait abnormalities (Fig. 6).
Figure 6. Gait analysis of CLN2 mutant mice.
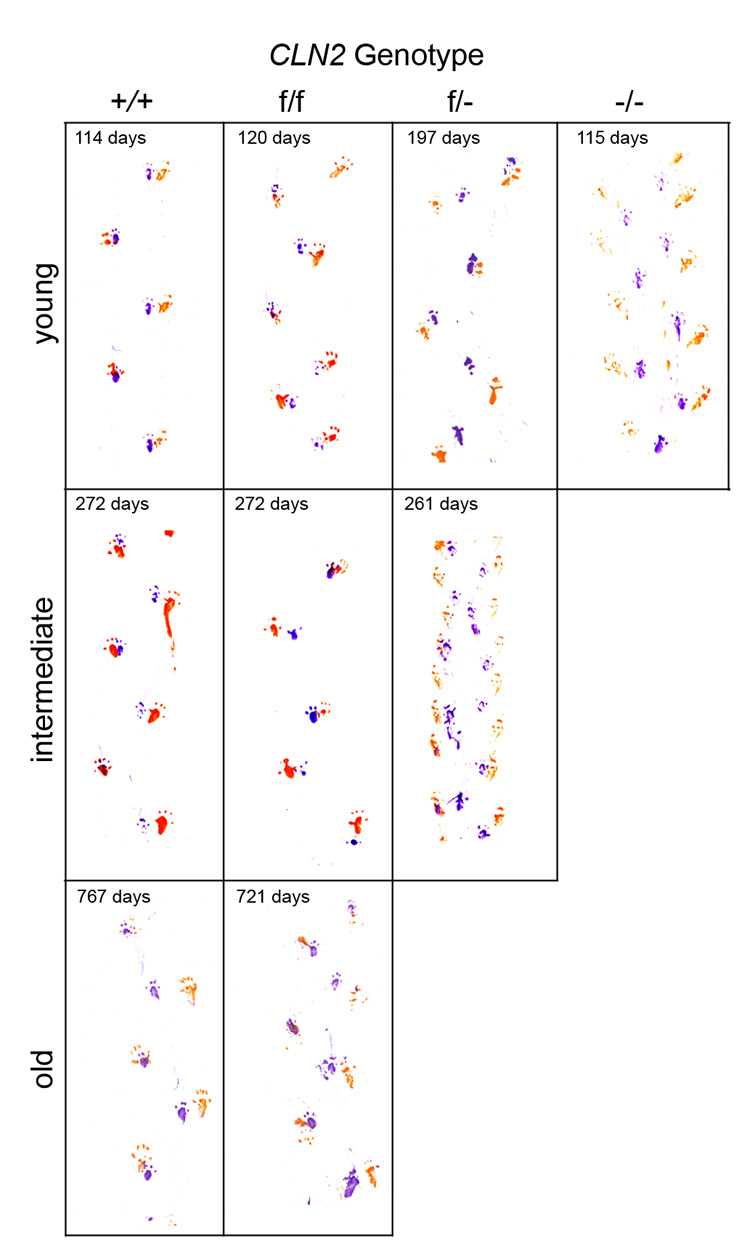
Gait analysis was conducted on mutant and control mice (n=2 for each genotype at each timepoint) and representative patterns shown. Mice were either congenic or near congenic (≥ 6 backcrosses) C57BL/6.
At ~4 months, gait of both the wild type controls and the f/f mutant was normal with regularly spaced strides and overlapping placement of footprints from the hind and forelimbs (Fig. 6). In contrast, the −/− mice have a shortened stride with a pronounced splaying of the rear limbs as described previously [9]. The gait pattern of the f/− compound heterozygote mice is similar to control at as late as ~7 months but by around 9 months, these mice develop an abnormal gait pattern that is very similar to that of the −/− mice at the earlier time-point. Like the −/− mice, the f/− mice grew increasingly ataxic and eventually developed a hunched appearance and noticeable side-to-side sway as they walked. As also reflected by their shortened lifespan compared to controls, the residual activity in the f/f mutant, while greatly ameliorating the phenotype, was not sufficient to prevent disease and at > 1 year of age, the onset of tremor was observed and eventually these mice did develop noticeable locomotor deficits. However, disease progression was slow and these mice appeared relatively healthy for most of their life-span when compared to the −/− mutants. Disease phenotype at the end of the lifespan of the f/f mutant was not as severe as observed in the end-stage −/− or f/− mutants. This is illustrated by the gait pattern of these mice at ~ 2 years of age, which is not dissimilar to that of wild-type controls of similar age.
Lysosomal storage is reduced by low levels of TPPI activity
Several of the genetically-distinct forms of neuronal ceroid lipofuscinosis, including the late-infantile form, accumulate a small proteolipid, subunit c of mitochondrial ATP synthase (SCMAS), as a major component of the storage material [20]. Accumulation of SCMAS in control and mutant mouse brain was measured by western blotting and immunodetection (Fig. 7, Panel A). At 60 days of age, significant accumulation of SCMAS was already detectable in the −/− mutant by western blotting even though the neurodegenerative phenotype was still mild. As the −/− mice continued to age (143 days), levels of SCMAS continued to increase. In contrast, there was no significant accumulation of SCMAS in the f/f hypomorph even when the mice were ~ 1 year of age. Immunohistochemical analysis revealed that levels of SCMAS were significantly elevated in the Purkinje cell layer of the cerebellar cortex in the −/− mouse at ~ 4 months of age (Fig. 7, Panel B). At the same age, levels of SCMAS were slightly elevated in the f/− mutant but staining in the f/f mutant appeared to be the same as observed in the wild-type controls. At ~ 22 months, staining for SCMAS was slightly increased in the f/f mutant compared to control. In cerebral cortex (Fig. 7, Panel C), accumulation of SCMAS followed the same pattern with storage being proportional to the severity of disease in the different models.
Figure 7. Accumulation of SCMAS in the brain of CLN2-mutant mice.
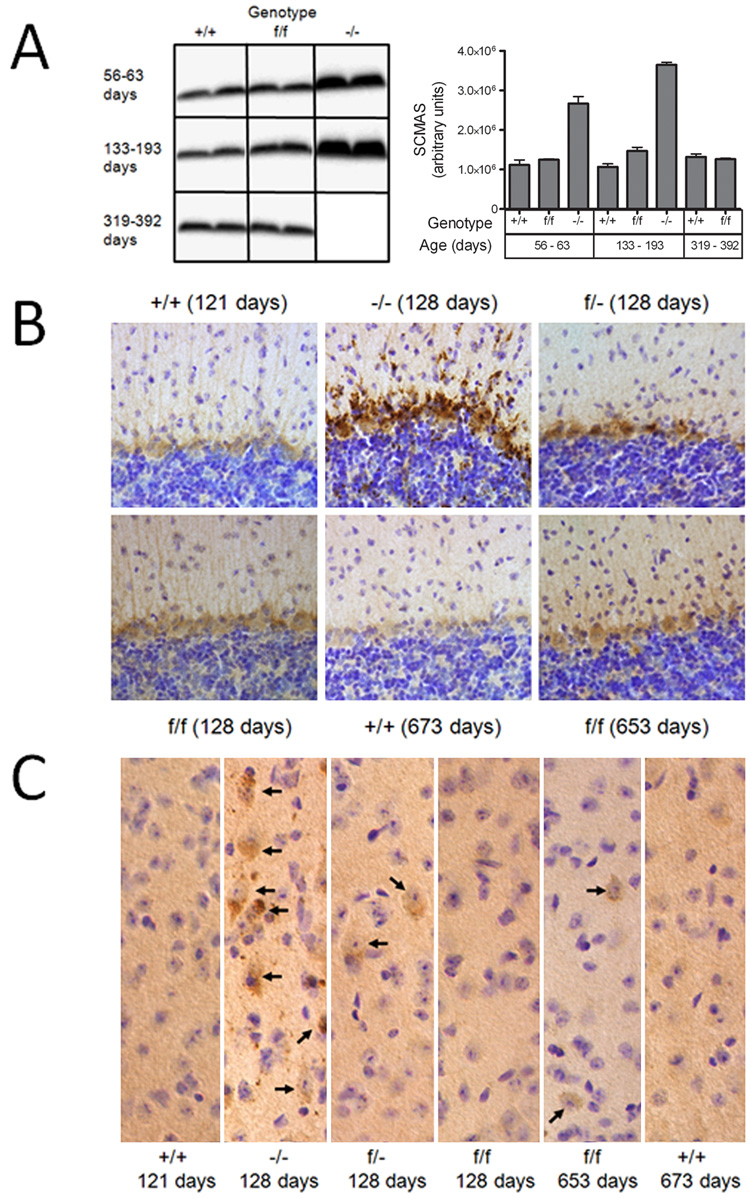
Panel A, Homogenates of mouse brain (10 ug protein equivalents) were fractionated by SDS-PAGE, transferred to nitrocellulose and SCMAS detected with a rabbit polyclonal antibody and visualized with an I125-labeled secondary antibody. Ages of congenic or near-congenic C57BL/6 mice were: +/+, 60, 63, 133 and 344 days; f/f, 59, 193, 319 and 392 days; −/−, 56 and 143 days. SCMAS levels in the mutant mice were determined from duplicate animals and error bars represent the range between these independent measurements. SCMAS was also detected by immunohistochemistry in the Purkinje cell layer of the cerebellum (Panel B) or cerebral cortex (Panel C). Arrows depicted individual neuronal cell bodies that stain for SCMAS. Age and genotype of animals are indicated for each panel.
One of the clinical hallmarks of LINCL and other NCLs is the presence of autofluorescent lysosomal storage material within the cells of affected individuals and this phenotype is recapitulated in the −/− mouse model [9]. At 128 days, there is a significant accumulation of punctuate cytoplasmic autofluorescent material in the cerebral cortex of the −/− mouse compared to the wild-type control (Fig. 8). In contrast, autofluorescence in the f/f and f/− mice was markedly less and similar to the wild-type mice at this age. There is an increase in autofluorescence in the f/f mouse at ~660 days compared to younger ages but it is essentially indistinguishable from that in the wild-type mice and presumably reflects the accumulation of normal aging lipofuscin rather than disease-related storage material.
Figure 8. Autofluorescent storage material within the cerebral cortex of CLN2 mutant mice.
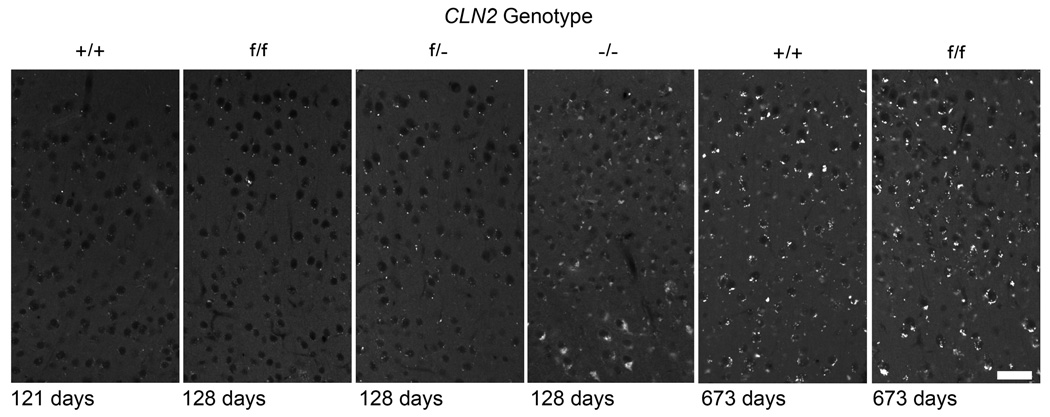
Age and genotype of animals are indicated for each panel. Note that the autofluorescent storage material in the wild-type mice is significantly increased in the older compared to younger mice, presumably reflecting the contribution of normal lipofuscin aging pigment. Images oriented with the pial surface just beyond the top of each panel. Mice were either congenic or near congenic (≥ 6 backcrosses) C57BL/6. Scale bar = 40µm.
DISCUSSION
Promising treatment approaches for lysosomal storage diseases (reviewed in [21] aim to either replace the defective protein with a functional recombinant version (e.g. gene, stem-cell and enzyme replacement therapies) or stimulate the production of a functional protein from an endogenous mutant gene (e.g. nonsense suppression and chemical chaperone therapy). For these, understanding the therapeutic benefits that are likely to be associated with any given level of a restored activity in preclinical models is critical in justifying whether such approaches should be extended to patients. However, it is generally accepted that the restoration of relatively low levels of the deficient activity will be useful and for most LSDs, <10% of normal activity is generally thought to have therapeutic benefits [6]. This is threshold is has arisen from measurements of residual activity that is present in patients with LSDs with attenuated, late-onset and/or slowly progressing disease and is supported by in vitro experiments demonstrating that the turnover of lysosomal enzyme substrates is normal in that cells contain as low as 15% of the normal activity of the respective enzymes [22].
Clinical correlates can certainly provide useful clues to the levels of lysosomal activities that are likely to be necessary for successful treatment but there are significant limitations to this approach. Although progression of LINCL tends to be fairly uniform, progression of other lysosomal storage diseases is typically very variable even when considering null alleles, reflecting environmental influences as well as possible epigenetic factors and modifying genes. The range of enzyme activities measured in different classes of severity of a given lysosomal storage disease are often overlapping and as a result, clinical genotype-phenotype correlations are frequently difficult or uninformative. In addition, clinical correlation can, by definition, only provide an indication of the residual levels of a lysosomal enzyme activity that are associated with the least severe variant of a given disorder, which is not equivalent to the minimum activity that is associated with an absence of disease.
A few studies have directly investigated the effect of residual lysosomal activities on respective disease phenotype. Several missense mutations were engineered into the β-glucuronidase gene in mouse models of mucopolysaccharidosis VII [23], resulting in low but measurable levels of β-glucuronidase activities (0.1–0.7% of normal levels, depending on tissue source). These levels were associated with a milder disease phenotype and decreased storage. Missense mutations of β-glucosidase have also been generated in mouse models of Gaucher disease [24] where null mutations are neonatally lethal. Most of the missense mutants were viable and these, regardless of whether they were homozygotes or compound heterozygotes with a null allele, retained ~ 25% normal activity within the brain. This was associated with greatly ameliorated disease. Another mouse model of Gaucher disease that retained 15–20% of normal β-glucosidase activity also presented a greatly attenuated phenotype [25]. While these studies do not identify minimum activities required to prevent the respective diseases, taken together, they do indicate that a small amount of residual lysosomal activity can attenuate disease and that ~25% of normal levels can result in a substantially corrected phenotype.
In this study, we have determined the effect of different residual levels of TPPI on the phenotype of gene-targeted mouse models of LINCL. Understanding the levels of enzymatic activity for useful therapeutic benefits is of particular relevance in LINCL because this is one of the first lysosomal storage diseases for which viral-mediated gene therapy trials has been undertaken with affected individuals (Clinicaltrials.gov NCT00151216, [26]). In addition, clinical trials are also underway for the use of human central nervous system stem cell transplantation in LINCL (identifier NCT00337636, [27]). We find that a hypomorphic mutant with 3% of wild-type activity in brain survived for twice as long as a mutant lacking detectable TPPI (median survival, ~ 9 months compared to 4.5 months). Albeit delayed, the disease phenotype of this hypomorph was severe and resembled that of the mouse that lacked detectable TPPI. However, we also find that 6% residual activity of TPPI in the brain of another hypomorph, while not sufficient to completely cure disease, elicit an improvement in phenotype that is dramatic when compared to the rapid progression of disease and death of the mouse lacking detectable TPPI. This hypomorph survives with a median life-span of 20 months and while this is somewhat shortened compared to wild-type (median survival ~ 25 months), the phenotype in terms of pathology and locomotor dysfunction was relatively mild, even at the later stages of disease. As the KM for TPPI in the wild-type and f/f specimens are indistinguishable (Fig. 4) and steady state TPPI protein levels are dramatically decreased in the f/f specimens (Fig. 3), it is highly likely that 6% of wild-type activity can be equated to 6% of the normal levels of protein.
If our results can be extrapolated from the mouse model to the clinic (and there is no reason to suspect otherwise given the accuracy with which the LINCL mouse recapitulates the human disease), then ≤6% of normal TPPI activity, even if it were possible to achieve this level in every cell of the CNS, will not be sufficient to cure or halt disease. It should be noted than an average CNS activity of 6% of wild-type may not be therapeutically equivalent to 6% in every cell. This is an important consideration for stem-cell or gene therapies given the chimeric nature of these approaches where individual corrected cells cross-protect other mutant cells. Thus, as an example, 18% of wild-type activity in one out of every three cells is likely not to be equivalent to 6% activity in every cell. Higher levels of TPPI (10% of normal ?) in each cell will likely be needed to completely prevent the onset of LINCL and whether this target can be achieved in patients using current recombinant approaches remains to be determined.
Finally, the observation that residual levels of TPPI can result in a late-onset, slowly progressing neurodegenerative phenotype in mice suggests that CLN2 defects may be worth considering in some human neurological disorders of unknown molecular etiology. One obvious candidate is the adult form of NCL, which can share many of the clinical features of LINCL including storage of SCMAS and curvilinear storage bodies [1].
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health Grants NS37918 (P.L.) and HD045561 (S.U.W.). We would like to thank Drs. Elizabeth Neufeld (UCLA) and John Taylor (Rutgers University) for generously providing the SCMAS antibody and substrate for TPPI assay, respectively.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Goebel HH, Mole SE, Lake BD. Neuronal Ceroid Lipofuscinoses (Batten Disease) Ios Pr Inc; 1999. [Google Scholar]
- 2.Sleat DE, Donnelly RJ, Lackland H, Liu CG, Sohar I, Pullarkat RK, Lobel P. Association of mutations in a lysosomal protein with classical late-infantile neuronal ceroid lipofuscinosis. Science. 1997;277:1802–1805. doi: 10.1126/science.277.5333.1802. [DOI] [PubMed] [Google Scholar]
- 3.Rawlings ND, Barrett AJ. Tripeptidyl-peptidase I is apparently the CLN2 protein absent in classical late-infantile neuronal ceroid lipofuscinosis. Biochim Biophys Acta. 1999;1429:496–500. doi: 10.1016/s0167-4838(98)00238-6. [DOI] [PubMed] [Google Scholar]
- 4.Sleat DE, Gin RM, Sohar I, Wisniewski K, Sklower-Brooks S, Pullarkat RK, Palmer DN, Lerner TJ, Boustany RM, Uldall P, Siakotos AN, Donnelly RJ, Lobel P. Mutational analysis of the defective protease in classic late-infantile neuronal ceroid lipofuscinosis, a neurodegenerative lysosomal storage disorder. Am J Hum Genet. 1999;64:1511–1523. doi: 10.1086/302427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maire I. Is genotype determination useful in predicting the clinical phenotype in lysosomal storage diseases? J Inherit Metab Dis. 2001;24 Suppl 2:57–61. doi: 10.1023/a:1012419823739. discussion 45–56. [DOI] [PubMed] [Google Scholar]
- 6.Sands MS, Davidson BL. Gene therapy for lysosomal storage diseases. Mol Ther. 2006;13:839–849. doi: 10.1016/j.ymthe.2006.01.006. [DOI] [PubMed] [Google Scholar]
- 7.Hackett NR, Redmond DE, Sondhi D, Giannaris EL, Vassallo E, Stratton J, Qiu J, Kaminsky SM, Lesser ML, Fisch GS, Rouselle SD, Crystal RG. Safety of direct administration of AAV2(CU)hCLN2, a candidate treatment for the central nervous system manifestations of late infantile neuronal ceroid lipofuscinosis, to the brain of rats and nonhuman primates. Hum Gene Ther. 2005;16:1484–1503. doi: 10.1089/hum.2005.16.1484. [DOI] [PubMed] [Google Scholar]
- 8.Haskell RE, Hughes SM, Chiorini JA, Alisky JM, Davidson BL. Viral-mediated delivery of the late-infantile neuronal ceroid lipofuscinosis gene, TPP-I to the mouse central nervous system. Gene Ther. 2003;10:34–42. doi: 10.1038/sj.gt.3301843. [DOI] [PubMed] [Google Scholar]
- 9.Sleat DE, Wiseman JA, El-Banna M, Kim KH, Mao Q, Price S, Macauley SL, Sidman RL, Shen MM, Zhao Q, Passini MA, Davidson BL, Stewart GR, Lobel P. A mouse model of classical late-infantile neuronal ceroid lipofuscinosis based on targeted disruption of the CLN2 gene results in a loss of tripeptidyl-peptidase I activity and progressive neurodegeneration. J Neurosci. 2004;24:9117–9126. doi: 10.1523/JNEUROSCI.2729-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Passini MA, Dodge JC, Bu J, Yang W, Zhao Q, Sondhi D, Hackett NR, Kaminsky SM, Mao Q, Shihabuddin LS, Cheng SH, Sleat DE, Stewart GR, Davidson BL, Lobel P, Crystal RG. Intracranial delivery of CLN2 reduces brain pathology in a mouse model of classical late infantile neuronal ceroid lipofuscinosis. J Neurosci. 2006;26:1334–1342. doi: 10.1523/JNEUROSCI.2676-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cabrera-Salazar MA, Roskelley EM, Bu J, Hodges BL, Yew N, Dodge JC, Shihabuddin LS, Sohar I, Sleat DE, Scheule RK, Davidson BL, Cheng SH, Lobel P, Passini MA. Timing of Therapeutic Intervention Determines Functional and Survival Outcomes in a Mouse Model of Late Infantile Batten Disease. Mol Ther. 2007 doi: 10.1038/sj.mt.6300249. [DOI] [PubMed] [Google Scholar]
- 12.Sondhi D, Hackett NR, Peterson DA, Stratton J, Baad M, Travis KM, Wilson JM, Crystal RG. Enhanced Survival of the LINCL Mouse Following CLN2 Gene Transfer Using the rh.10 Rhesus Macaque-derived Adeno-associated Virus Vector. Mol Ther. 2007;15:481–491. doi: 10.1038/sj.mt.6300049. [DOI] [PubMed] [Google Scholar]
- 13.de Vries WN, Binns LT, Fancher KS, Dean J, Moore R, Kemler R, Knowles BB. Expression of Cre recombinase in mouse oocytes: a means to study maternal effect genes. Genesis. 2000;26:110–112. [PubMed] [Google Scholar]
- 14.Della Valle MC, Sleat DE, Sohar I, Wen T, Pintar JE, Jadot M, Lobel P. Demonstration of lysosomal localization for the mammalian ependymin-related protein using classical approaches combined with a novel density shift method. J Biol Chem. 2006;281:35436–35445. doi: 10.1074/jbc.M606208200. [DOI] [PubMed] [Google Scholar]
- 15.Tian Y, Sohar I, Taylor JW, Lobel P. Determination of the substrate specificity of tripeptidyl-peptidase I using combinatorial peptide libraries and development of improved fluorogenic substrates. J Biol Chem. 2006;281:6559–6572. doi: 10.1074/jbc.M507336200. [DOI] [PubMed] [Google Scholar]
- 16.Sleat DE, Sohar I, Lackland H, Majercak J, Lobel P. Rat brain contains high levels of mannose-6-phosphorylated glycoproteins including lysosomal enzymes and palmitoyl-protein thioesterase, an enzyme implicated in infantile neuronal lipofuscinosis. J Biol Chem. 1996;271:19191–19198. doi: 10.1074/jbc.271.32.19191. [DOI] [PubMed] [Google Scholar]
- 17.Crawley JN. What's wrong with my mouse? : behavioral phenotyping of transgenic and knockout mice. New York: Wiley-Liss; 2000. [Google Scholar]
- 18.Lin L, Lobel P. Production and characterization of recombinant human CLN2 protein for enzyme-replacement therapy in late infantile neuronal ceroid lipofuscinosis. Biochem J. 2001;357:49–55. doi: 10.1042/0264-6021:3570049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ryazantsev S, Yu WH, Zhao HZ, Neufeld EF, Ohmi K. Lysosomal accumulation of SCMAS (subunit c of mitochondrial ATP synthase) in neurons of the mouse model of mucopolysaccharidosis III B. Mol Genet Metab. 2007;90:393–401. doi: 10.1016/j.ymgme.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palmer DN, Fearnley IM, Medd SM, Walker JE, Martinus RD, Bayliss SL, Hall NA, Lake BD, Wolfe LS, Jolly RD. Lysosomal storage of the DCCD reactive proteolipid subunit of mitochondrial ATP synthase in human and ovine ceroid lipofuscinoses. Adv Exp Med Biol. 1989;266:211–222. doi: 10.1007/978-1-4899-5339-1_15. discussion 223. [DOI] [PubMed] [Google Scholar]
- 21.Beck M. New therapeutic options for lysosomal storage disorders: enzyme replacement, small molecules and gene therapy. Hum Genet. 2007;121:1–22. doi: 10.1007/s00439-006-0280-4. [DOI] [PubMed] [Google Scholar]
- 22.Leinekugel P, Michel S, Conzelmann E, Sandhoff K. Quantitative correlation between the residual activity of beta-hexosaminidase A and arylsulfatase A and the severity of the resulting lysosomal storage disease. Hum Genet. 1992;88:513–523. doi: 10.1007/BF00219337. [DOI] [PubMed] [Google Scholar]
- 23.Tomatsu S, Orii KO, Vogler C, Grubb JH, Snella EM, Gutierrez MA, Dieter T, Sukegawa K, Orii T, Kondo N, Sly WS. Missense models [Gustm(E536A)Sly, Gustm(E536Q)Sly, and Gustm(L175F)Sly] of murine mucopolysaccharidosis type VII produced by targeted mutagenesis. Proc Natl Acad Sci U S A. 2002;99:14982–14987. doi: 10.1073/pnas.232570999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu YH, Quinn B, Witte D, Grabowski GA. Viable mouse models of acid beta-glucosidase deficiency: the defect in Gaucher disease. Am J Pathol. 2003;163:2093–2101. doi: 10.1016/s0002-9440(10)63566-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mizukami H, Mi Y, Wada R, Kono M, Yamashita T, Liu Y, Werth N, Sandhoff R, Sandhoff K, Proia RL. Systemic inflammation in glucocerebrosidase-deficient mice with minimal glucosylceramide storage. J Clin Invest. 2002;109:1215–1221. doi: 10.1172/JCI14530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arkin LM, Sondhi D, Worgall S, Suh LH, Hackett NR, Kaminsky SM, Hosain SA, Souweidane MM, Kaplitt MG, Dyke JP, Heier LA, Ballon DJ, Shungu DC, Wisniewski KE, Greenwald BM, Hollmann C, Crystal RG. Confronting the issues of therapeutic misconception, enrollment decisions, and personal motives in genetic medicine-based clinical research studies for fatal disorders. Hum Gene Ther. 2005;16:1028–1036. doi: 10.1089/hum.2005.16.1028. [DOI] [PubMed] [Google Scholar]
- 27.Taupin P. HuCNS-SC (StemCells) Curr Opin Mol Ther. 2006;8:156–163. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.