

Abstract
Inadequate trophoblast invasion and spiral artery remodeling leading to poor placental perfusion and hypoxia are believed to underlie preeclampsia (PE) and intrauterine growth restriction (IUGR). Recent studies implicate increased circulating endoglin as a contributor to the pathogenesis of PE. The objective of this study was to determine whether placental and circulating endoglin concentrations are altered in pregnancies complicated by intrauterine growth restricted (IUGR) infants and to address the role of hypoxia on the regulation of placental endoglin. We analyzed 10 placentas each from normal pregnant (NP), PE, and IUGR subjects. Endoglin levels were 2.5-fold higher in preeclamptic placentas compared to NP (15.4 ± 2.6 versus 5.7 ± 1.0, p < 0.01). In contrast, endoglin levels were similar in NP and IUGR placentas (5.7 ± 1.0 vs 5.9 ± 1.1, p = NS). Placentas from pregnancies with both PE and IUGR exhibited endoglin levels comparable to the PE group and significantly different from normotensive pregnancies with and without IUGR pregnancies (mean 14.9 ± 4.0, n = 9, p = 0.013). Soluble endoglin concentrations in maternal plasma were comparable in NP and IUGR, but higher in women with PE (n = 10 per group, p < 0.05). Despite a 2-fold increase in hypoxia inducible factor, HIF-1α, we did not observe endoglin upregulation in NP, PE, or IUGR placental villous explants exposed to hypoxia (2% oxygen). In contrast to PE, placental or circulating endoglin is not increased in normotensive women delivering small, asymmetrically grown (IUGR) infants at term. The placentas of women with IUGR appear to be fundamentally different from PE women with respect to endoglin, despite the proposed common pathology of deficient trophoblast invasion/spiral artery remodeling and poor placental perfusion.
Keywords: Soluble endoglin, Preeclampsia, Intrauterine growth restriction, Hypoxia
1. Introduction
Intrauterine growth restriction (IUGR), a pregnancy complication in which fetal growth fails to reach its theoretical potential, is associated with significant fetal and neonatal mortality and morbidity [1–6]. IUGR is often associated with abnormal placental development and/or function with suboptimal delivery of oxygen and nutrients to the fetus. Preeclampsia, characterized by new onset hypertension and proteinuria after 20 weeks of gestation, complicates 3–5 percent of pregnancies. Preeclampsia is associated with IUGR in approximately one-third of cases [7] and also results in substantial maternal as well as fetal/neonatal morbidity and mortality [8–10]. There is increasing evidence that perturbations in placental development may lead to compromised pregnancy outcomes in both IUGR and preeclampsia [11]. Poor placental perfusion and hypoxia as a result of inadequate trophoblast invasion and uterine spiral artery remodeling has been proposed as a common factor [12,13].
An imbalance of placentally-derived angiogenic growth factors has recently been implicated in the pathogenesis of preeclampsia and possibly IUGR [14–18]. Excess soluble vascular endothelial growth factor (VEGF) receptor-1 (also known as soluble fms-like tyrosine kinase receptor-1 or sFlt-1) in the maternal circulation, binds to VEGF and placental growth factor (PlGF), thereby preventing the action of these angiogenic growth factors on vascular tissues [14,19,20]. Endoglin, a co-receptor for transforming growth factor β1 and β3, is expressed by vascular endothelial cells and placental trophoblasts. Its soluble form has anti-angiogenic properties, possibly via impairment of TGFβ1 signaling in the vasculature [21–23]. In the rat model, soluble endoglin potentiates sFlt-1 to produce a preeclampsia-like syndrome including development of hemolysis, elevated liver enzymes, and low platelets (HELLP syndrome) and reduced fetal growth [16]. Furthermore, sFlt-1 and soluble endoglin concentrations are higher and PlGF lower, weeks prior to the clinical syndrome of preeclampsia [17].
Although most studies demonstrate consistently higher concentrations of endoglin in the placenta and circulation of preeclamptic women [15,17,24], the data are less clear with IUGR. Levine et al. as well as Romero et al. observed higher circulating soluble endoglin in women with small for gestational age babies less than the 10%ile [17,25]. Using abnormal uterine artery Doppler and birth weight less than the 5%ile to define IUGR, Stepan and colleagues also demonstrated higher soluble endoglin and sFlt-1 [26]. Yinon et al. observed increased endoglin mRNA and protein in the placenta of severely growth restricted and preterm babies and confirmed these findings in ten discordant twin pairs demonstrating increased placental endoglin mRNA and protein in the growth restricted twin compared to its normally grown counterpart [27]. However, there is also emerging evidence that placentas from preeclamptic and IUGR pregnancies may be different with respect to angiogenic factors and hypoxia; whether this is a function of the severity and/or gestational age at onset of growth restriction is unclear. Shibata and colleagues have shown that placental and maternal circulating sFlt-1 concentrations were not higher in normotensive pregnancies with small for gestational age, asymmetrically grown neonates compared to uncomplicated pregnancies [28]. In addition, our recent work using placental biopsies demonstrated that hypoxia inducible factors, which are highly sensitive to low oxygen tension in the placenta, and sFlt-1 are higher in preeclampsia, but not in normotensive pregnancies complicated by late onset IUGR [29]. Thus, further study of endoglin in IUGR is warranted.
Placental hypoxia, as a result of poor perfusion, has been proposed as a mechanism for PE and IUGR. Endoglin gene transcription is upregulated by hypoxia in a tissue specific manner [30,31]. One study demonstrated hypoxia-induced up-regulation of endoglin in villous explants from normal and preeclamptic placentas [32]; however, the association of hypoxia, endoglin expression, and development of preeclampsia and IUGR remains incompletely understood.
The objective of this study was to determine the concentrations of placental endoglin and maternal circulating endoglin in pregnancies complicated by late onset intrauterine fetal growth restriction. Women with preeclampsia with or without IUGR were used as positive control groups. Additionally, we addressed the role of hypoxia on endoglin concentrations using placental explants in vitro.
2. Methods
2.1. Study population and definitions
This was a retrospective cohort study of women with uncomplicated or normal pregnancies (NP), pregnancies complicated by intrauterine growth restriction (IUGR), preeclampsia (PE) with appropriately grown infants, and preeclampsia with growth restricted infants (PE/IUGR). The study was approved by the Institutional Review Board and informed consent was obtained from all subjects. Gestational age was determined by best obstetric estimate (first trimester ultrasound when available). Exclusion criteria included multiple gestation, prior preeclampsia, illicit drug use and preexisting medical conditions such as diabetes, chronic hypertension, and renal disease. Preeclampsia was diagnosed by the presence of gestational hypertension (an absolute blood pressure ≥140 mmHg systolic and/or 90 mmHg diastolic) and proteinuria (greater than 300 mg per 24-h urine collection, >2+ on a voided or >1+ on a catheterized random urine sample, or a protein/creatinine ratio of >0.3) beginning after the 20th week of pregnancy with resolution of blood pressure and proteinuria postpartum (Working Group Report on High Blood Pressure in Pregnancy) [33], as well as hyperuricemia (≥1 standard deviation above reference values for the gestational age the sample was obtained (e.g. term, >5.5 mg/dL)). We include hyperuricemia in our classification as it identifies a more homogeneous group of gestational hypertensive women with a greater frequency of adverse fetal outcomes [34]. Fetal intrauterine growth restriction (IUGR) was defined as normotensive women with infant birth weight less than the tenth percentile as well as an asymmetric growth profile and/or abnormal umbilical artery Doppler waveforms; thus, small babies with failure to achieve their growth potential. Percentiles for growth parameters were derived from nomograms based on race, gender, and gestational age from a reference population of over 10,000 neonates delivered at Magee-Womens Hospital. The umbilical artery Doppler was considered abnormal, if the systolic/diastolic ratio was greater than the 95%ile for gestational age or if the diastolic flow was absent or reversed. Asymmetric growth profile was defined by birth weight < length ≤ head circumference percentiles, when the weight percentile was at least two percentile categories below length and/or head circumference. Percentile categories were defined by cut points of 3, 5, 10, 25, 50, 75, 90, 95, 97% [25]. Classification of preeclampsia and fetal growth restriction was determined retrospectively based on medical chart review by a jury of research and clinical investigators (Table 1).
Table 1.
Characteristics of the entire study population
| Normal pregnancy (n = 24) | Preeclampsia† (n = 24) | IUGR‡ (n = 24) | Preeclampsia + IUGR (n = 9) | |
|---|---|---|---|---|
| Maternal age (years) | 25.8 ± 1.3 | 28.9 ± 1.3 | 25.0 ± 1.2 | 26.1 ± 1.8 |
| Gestational weeks at delivery | 39.2 ± 0.3 | 37.3 ± 0.7 | 38.5 ± 0.5 | 35.6 ± 0.9* |
| Maternal race (n) | ||||
| White | 19 | 21 | 14 | 7 |
| African-American | 5 | 3 | 8 | 2 |
| Others | 0 | 0 | 2 | 0 |
| Nulliparous (n) | 23 | 23 | 20 | 9 |
| Mode of delivery (n) | ||||
| C-section | 11 | 9 | 5 | 5 |
| Vaginal | 13 | 15 | 19 | 4 |
| Birth weight (g) | 3564.5 ± 95.6** | 2783.5 ± 163.1 | 2318.8 ± 87.3 | 1829.6 ± 154.2 |
| Birth weight <5th percentile | 0% (0/24) | 0% (0/24) | 58% (17/24) | 78% (7/9) |
| Birth weight <3rd percentile | 0% (0/24) | 0% (0/24) | 54% (13/24) | 44% (4/9) |
| Systolic BP (mmHg) | 119.4 ± 2.1 | 155.4 ± 2.7* | 114.0 ± 1.9 | 162.1 ± 3.8* |
| Diastolic BP (mmHg) | 72.9 ± 1.5 | 95.2 ± 1.5* | 71.7 ± 2.3 | 97.9 ± 4.2* |
| Proteinuriaa (n) | 0 | 24 | 0 | 9 |
| Uric acid | ND | 5.9 ± 0.04 | ND | 6.9 ± 0.2 |
| Smoking | 2 | 1 | 4 | 0 |
Mean ± SEM. ND = not determined.
Preeclampsia definition is based on the Working Group Report (2000) and hyperuricemia of 1SD above normal for gestational age,
IUGR was defined as normotensive women with infant birth weight <10%ile for gestational age at delivery, gender, and race and asymmetric growth profile.
p < 0.005 between groups and compared to normal pregnancy,
p < 0.005 between groups and compared to PE, IUGR and PE/IUGR.
1–3 + dip, protein/creatinine ratio > = 0.3, 24 h protein >300 mg.
2.2. Sample collection
Maternal venous serum samples were obtained prior to delivery from a cohort of subjects with normal and preeclamptic pregnancies, as well as from normotensive women with pregnancies complicated by fetal growth restriction (n = 10 from each group). Samples were stored at −70 °C until assayed. Placentas were obtained from a second cohort of women with pregnancies that were uncomplicated (n = 14), normotensive women with pregnancies complicated by IUGR (n = 14), preeclampsia with appropriately grown infants (n = 14) and preeclampsia with IUGR (n = 9). There was no overlap between the first and second cohorts with a total of 81 subjects. The placentas were sampled immediately after extraction from the uterus. To ensure systematic and unbiased sampling of the entire placenta, we fitted a circular plastic grid containing 16 holes over the maternal face of the placenta and obtained 16 biopsy samples [35,36]. Each 0.5 g sample contained the decidua basalis and villous placenta but not chorionic plate. After several rapid rinses in saline, the tissues were blotted dry and flash frozen in liquid nitrogen and stored at −70 °C until assayed. Equal mass from three such biopsy sites for each placenta were pooled for the analysis of placental endoglin.
2.3. Villous explant culture
Villous explants were prepared as described previously [37] with some modifications. Several cotyledons from third trimester placentas were excised at random and rinsed extensively in sterile saline to remove blood. Decidua and large vessels were removed from the villous placenta by blunt dissection. The villous tissue was then finely dissected into 5–10 mg pieces in a sterile saline bath. The pieces were washed two or three more times before culture.
Fifty mg of villous tissue was placed into each well of a 24 well plate (Becton Dickinson, Franklin Lakes, NJ) containing 1.0 ml of Medium 199 (Mediatech, Cellgro, Herndon, VA) supplemented with 10% Fetal Bovine Serum (FBS, Summit Technology, Ft. Collins, CO) and antibiotics. Explants were incubated at 37 °C for a 12 h preincubation period on an orbital shaker (60 rpm, Stovall Life Science Inc., Greensboro, NC) under standard tissue culture conditions of 5% CO2-balance room air (non-hypoxic condition, pO2 ~147 mmHg or 21% O2) in a cell culture incubator (Forma Scientific, Marietta, OH). After a medium change, the plates were again placed on the orbital shaker at 37 °C under either non-hypoxic or hypoxic conditions (see below) for 4 h.
Reduced O2 (‘‘hypoxia’’, pO2 ~15 mmHg or 2% O2+5% CO2-balance nitrogen) exposures were carried out in a Hypoxic chamber/Glove box (Coy Laboratory Products, Grass Lake, MI) with a probe for continuous oxygen monitoring and an orbital shaker. At the end of the incubation period, the explants were removed, excess medium blotted with cotton gauze and the explants flash frozen in liquid nitrogen and stored at −70 °C.
2.4. Western analysis
Total protein from placental tissues or villous explants were extracted using our published procedures with slight modifications [37]. Tissues were homogenized by sonication (Ultrasonic Processor, Tekmar, Cincinnati, OH, microprobe setting 70 for 30 s) in 4 volumes of 1× Laemmli buffer (50 mM Tris–HCl, pH 6.8, 2% SDS, 10% Glycerol) containing 5 mM DTT, 0.5 mM phenyl methyl sulfonyl fluoride (PMSF), 1 mM sodium vanadate and one μl per ml of protease inhibitors cocktail (1000×, Protease Inhibitor Cocktail Set III, Calbiochem, San Diego, CA). The crude homogenate was centrifuged at 13,400 × g at 4 °C for 15 min. Protein estimation was carried out using the Biorad assay. All preparations were boiled for 5 min and briefly centrifuged. The samples were loaded in 25 μl volume containing 50 μg of total protein and 5% β-mercaptoethanol and 0.005% pyronin (Sigma Chemical Co., St. Louis, MO) and separated on a SDS-containing 7.5% poly-acrylamide gel (20 × 20 cm, Owl Separations Systems Inc., Portsmouth, NH) at 100 V for 4 h. The proteins were next transferred to polyvinylidine fluoride membranes (Immobilon, Millipore, Bedford, MA) using a semidry transfer system (The Panther™ Semidry Electroblotter, Owl Separations Systems Inc., Portsmouth, NH) at a constant current of 1.0 mA per cm2. Detection of proteins was carried out after blocking the membranes with a 5% solution of non-fat dry milk for 1 h. Endoglin antibody (Santa Cruz Biotechnology, Santa Cruz, CA. Cat # sc-20632) was diluted 1:200 and used at a final concentration of one μg/ml. This endoglin antibody recognizes both the membrane-bound 90 kD form of endoglin and the 65 kD soluble endoglin. A hypoxia inducible factor (HIF)-1α monoclonal antibody (Transduction Laboratories, Lexington, KY, cat # H72320) was diluted to 1.25 μg/ml in Tris buffered saline (TBS) containing 0.05% Tween-20. Membranes were incubated with the primary antibody for 1 h at room temperature, then washed three times for 10 min each and incubated with alkaline phosphatase conjugated secondary antibody (1:2500 dilution, Promega, Madison, WI) for 30 min. The membranes were again washed three times in TBS-T for 10 min each. They were further incubated in buffer without Tween-20 for 10 min and then equilibrated in alkaline phosphatase buffer (100 mM Tris, pH 9.5, 150 mM NaCl) for 5 min. Chemiluminescent detection was carried out using the CDP-Star substrate (Boehringer Manheim, Indianapolis, IN) diluted 1:200 in the alkaline phosphatase buffer for 5 min.
For the analysis of β-actin, 10 μg of total protein was electrophoresed on separate gels and subjected to Western analysis and probed with a monoclonal anti-β-actin antibody (Clone AC-15, Sigma, St. Louis, MO; 2.2 μg/ml). In addition, Coomassie blue staining of gels and membranes indicated uniform loading and transfer (data not shown).
The bands on the developed Western blot films were scanned using a Hewlett Packard laser scanner (Scanjet 5370C, Hewlett Packard, Palo Alta, CA). Densitometry was carried out using automated digitizing software UN-SCA-NIT™ Gel Version 4.3 (Silk Scientific Inc., Orem, UT).
2.5. Quantitation of serum endoglin
The concentration of total soluble endoglin in human serum samples and conditioned media was measured in duplicate using a specific ELISA kit from R&D Systems according to the manufacturer’s instructions (Cat # DNDG00, Minneapolis, MN). Serum samples were diluted 1–5. The minimum detectable dose was 0.007 ng/ml with an inter-assay coefficient of variation of 6.7%.
2.6. Statistical analysis
Group summaries are presented as mean ± standard error unless otherwise indicated. Fisher’s exact test was used to compare discrete variables between groups. Group comparisons for continuous, normally distributed variables were made using one way analysis of variance. Kruskal–Wallis testing was used for non-parametric distributions. When appropriate, posthoc LSD testing was used to compare specific group differences. A p-value of less than 0.05 was considered to be significant.
3. Results
3.1. Clinical characteristics
Clinical characteristics of the 81 subjects are presented in Table 1. As expected based on the definitions, women with preeclampsia collectively had proteinuria, elevated uric acid, and higher systolic and diastolic blood pressures ( p < 0.005 compared to normal pregnancy). Birth weight was significantly higher in the group of women with uncomplicated pregnancies ( p < 0.005 compared to each of the other groups). There was no difference in maternal age, race, parity (all but six were nulliparous) and mode of delivery. Overall, the gestational age at delivery for preeclamptic women with associated IUGR (35.6 ± 0.9 weeks of gestation) was slightly lower than the other groups. Two subjects in the PE-IUGR group had features of HELLP syndrome.
All the IUGR placentas used in this study were classified based on asymmetric growth profile as an indicator of the failure to achieve full growth potential. Notably, most of these babies were delivered at or close to full term gestation. Only one subject in the PE-IUGR group had the additional features of both oligohydramnios and abnormal umbilical artery Doppler waveforms on ultrasound.
3.2. Placental endoglin levels
We used Western blot analysis for the detection of endoglin protein in placental tissues. A total of 39 placental samples from NP, PE, and IUGR (n = 10 in each group), as well as 9 women with both PE and IUGR were analyzed. Endoglin protein was detectable in all placental samples and quantified after normalizing to β-actin (Fig. 1, Table 2). There was significantly higher endoglin protein expression in preeclamptic placentas (endoglin to β-actin ratio of 14.4 ± 2.7, p = 0.01 using post-hoc testing) compared to normal pregnancy (5.7 ± 0.9) and IUGR (5.9 ± 1.1). Notably, there was no difference in the 90 kD form of placental endoglin in normotensive women with IUGR compared to uncomplicated pregnancies. The densitometric quantitation of placental endoglin in women with both PE and IUGR was 14.9 ± 4.0; this was comparable to the preeclamptic group and significantly different from the other two groups, however there was marked variability with a wide range (2.0–37.8). This suggests that these placentas may more closely resemble the placentas from preeclamptic women than those with isolated asymmetric IUGR with respect to placental endoglin protein.
Fig. 1.
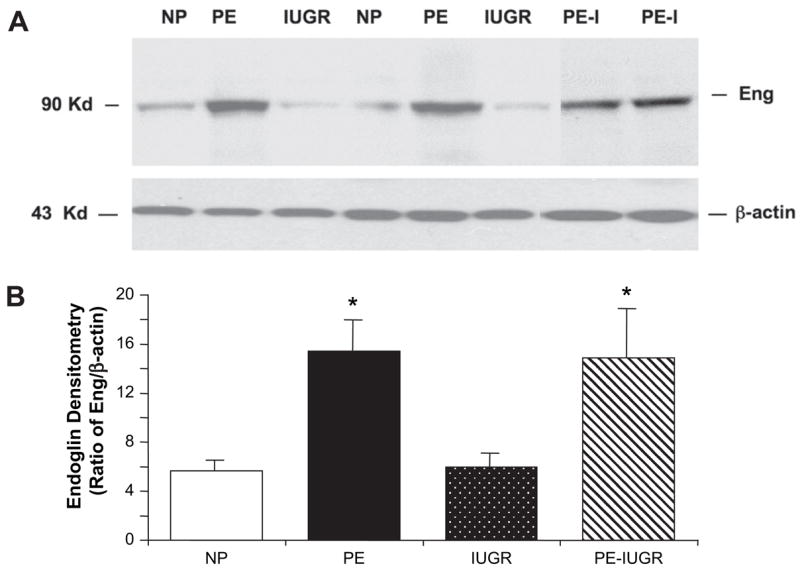
In contrast to preeclampsia, placental endoglin is not increased in normotensive pregnancies with late onset, asymmetric fetal growth restriction compared to uncomplicated pregnancies. (A) Representative Western blot of placental endoglin (90 kD) in women with normal pregnancy (NP), preeclampsia (PE), intrauterine growth restriction (IUGR) and PE with IUGR (PE-I); (B) Densitometric ratio of endoglin/β-actin for n = 10 placentas each for NP, PE, IUGR and n = 9 for PE with IUGR. Significant difference between groups using Kruskal–Wallis analysis with *p < 0.05 compared to NP by post-hoc LSD test.
Table 2.
Clinical characteristics of pregnancies with fetal growth restriction presented in Figure 1.
| Patient # | Birth weight percentile | Length percentile | Head circumference percentile | Gestational age at delivery | Oligohydramnios | Abnormal umbilical artery Doppler | Placental weight (g) | Placental wt %tile |
|---|---|---|---|---|---|---|---|---|
| 1 | 0.4 | <3 | 5–10 | 38 | – | – | 245 | <10 |
| 2 | 9.5 | 25–50 | 5 | 40 | – | – | 400 | 10–25 |
| 3 | 9.8 | 75–90 | 25 | 39 | – | NA | NA | NA |
| 4 | 2.0 | 10–25 | 10 | 37 | – | – | 390 | 10–25 |
| 5 | 2.4 | 3–5 | 5–10 | 39 | – | – | 190 | <10 |
| 6 | 1.5 | 5–10 | 5 | 37 | – | – | 325 | <10 |
| 7 | 0.6 | 3 | 25–50 | 38 | – | NA | 440 | 50–75 |
| 8 | 8.3 | 5–10 | 10–25 | 39 | – | – | NA | NA |
| 9 | 0.0 | <3 | <3 | 37 | – | – | 160 | <10 |
| 10 | 9.1 | 50 | 50–75 | 37 | – | – | 240 | <10 |
Although the endoglin antibody that we used recognizes both the membrane-bound 90 kD form of endoglin and the 65 kD soluble endoglin, we chose to focus on the 90 kDa form. The 90 kD form of endoglin is membrane-bound and therefore, unlikely to be affected by the placental sample collection and processing. However, the soluble, circulating 65 kD form is likely to be affected by placental handling, such as the extent of blood removal and may be falsely affected by processing techniques of the total placental extracts.
3.3. Circulating endoglin concentrations
Soluble endoglin concentrations were determined using serum from a second cohort: women with uncomplicated pregnancies, with preeclampsia and appropriately grown infants, and normotensive women with IUGR (n = 10) in each group (Fig. 2). We demonstrated significant differences between groups, specifically, higher soluble endoglin concentrations in women with preeclampsia (39.1 ± 1.0 ng/ml) compared to normal pregnancies (17.5 ± 1.0 ng/ml, p = 0.001) and IUGR (19.9 ± 0.5 ng/ml, p = 0.002). There was no difference in the serum endoglin concentrations between women with growth restricted babies and women with uncomplicated pregnancies ( p = 0.91).
Fig. 2.
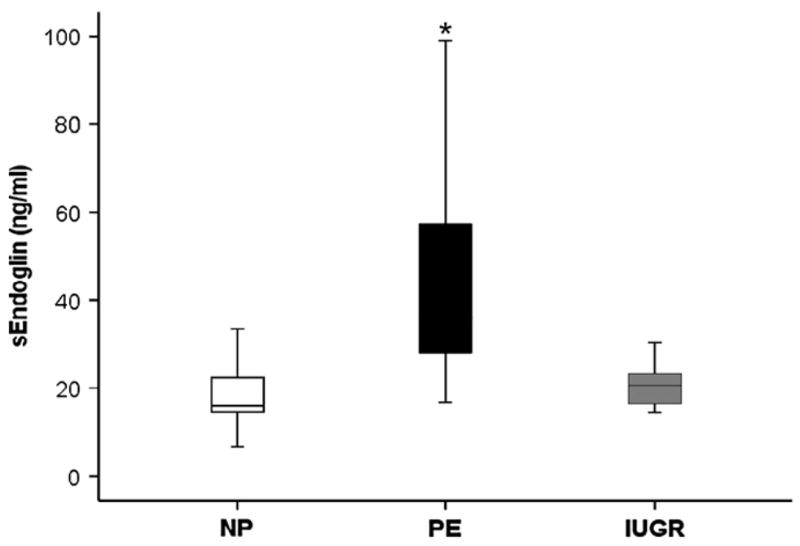
In contrast to preeclampsia, circulating soluble endoglin is not increased in normotensive pregnancies with late onset, asymmetric fetal growth restriction compared to uncomplicated pregnancies. N = 10 serum samples each from women with normal pregnancy (NP), preeclampsia (PE), and intra-uterine growth restriction (IUGR). Significant difference between groups using Kruskal–Wallis analysis with *p < 0.01 compared to NP by post-hoc LSD test.
3.4. Endoglin quantitation in villous explant cultures
Explants from normal, preeclamptic and IUGR placentas (n = 4) showed a robust induction of HIF proteins under hypoxic conditions (2% oxygen) compared to normoxia (21% oxygen, p = 0.02, in all three groups) (Figs. 3A top panel, B) which serves as a reliable positive control. None of the three groups of villous explants demonstrated significant up-regulation of endoglin protein in response to hypoxia compared to normoxic exposure of four hours (Figs. 3A,C). As previously demonstrated, endoglin protein expression was significantly higher in preeclamptic villous explants under normoxic conditions compared to normal pregnant or IUGR explants, 1.2 ± 0.1 vs 0.7 ± 0.1 and 0.7 ± 0.1, respectively. Additionally we collected conditioned media from explant cultures of normal and preeclamptic placentas after 24 h of incubation at either 21 or 2% oxygen and analyzed the soluble endoglin levels by ELISA. Despite the soluble endoglin increase in conditioned medium from preeclamptic placentas, both groups did not show increase in soluble endoglin under hypoxic incubations (results not shown), further supporting the lack of hypoxic upregulation of endoglin in term villous explant cultures in vitro.
Fig. 3.

Hypoxia inducible factor-1α and membrane-bound endoglin protein expression in term villous explants exposed to normoxia and hypoxia. Villous explants prepared from placentas of normal, preeclamptic and normotensive pregnancies with growth restricted babies (n = 4 each) were incubated under standard culture conditions at 21% oxygen and 2% oxygen for 4 h. (A) Representative Western blot for HIF-1α and membrane-bound endoglin (90 kD) protein detection and (B & C) associated densitometric analysis. *p < 0.05 compared to normoxia within each group, †p < 0.05 compared to villous explants from normal pregnancies exposed to normoxia, both using Kruskall–Wallis testing.
4. Discussion
Given the many shared and disparate characteristics of preeclampsia and IUGR [13], we sought to determine whether pregnancies complicated by late onset IUGR were comparable to preeclamptic pregnancies with respect to placental and circulating endoglin. We also questioned whether hypoxia may be a common mechanism regulating placental endoglin in these complicated pregnancies. Major findings of our study included: (1) placental and circulating endoglin concentrations were comparable in normotensive women with or without fetal growth restriction; and (2) hypoxia does not appear to increase endoglin concentrations in term placental villous explants from normal pregnancies or pregnancies complicated by preeclampsia or IUGR.
Although the published information regarding endoglin and IUGR is limited, our first major finding is in contrast to reports of elevated endoglin concentrations with IUGR. Other investigators have reported higher circulating soluble endoglin in women with small for gestational age babies with birth weight less than the 10%ile [17,25]. Similar findings were reported in a cohort of women with abnormal uterine artery Doppler and birth weight less that the 5%ile [26]. Increased placental endoglin protein and mRNA were also noted in severe IUGR and growth restricted twins with absent end diastolic flow on umbilical artery Doppler evaluation compared to their normally grown counterparts [27]. Although our findings are different from the studies mentioned above, these data are consistent with our earlier observation that placental hypoxia inducible factors and sFlt-1 are comparable in normotensive women with or without late onset IUGR, but are elevated with preeclampsia [29]. Additionally, Shibata and colleagues demonstrated that placental and maternal concentrations of sFlt-1, an anti-angiogenic factor, were not higher in normotensive pregnancies with asymmetric, small for gestational age neonates [28]. In sum, these findings suggest that placentas from normotensive women with late IUGR are different from preeclamptic placentas with respect to the anti-angiogenic factors, sFlt-1 and endoglin, as well as response to low oxygen tension as represented by the HIF proteins. Fundamental differences in placentas from pregnancies complicated by preeclampsia and IUGR have also been observed with respect to histopathology [12] as well as biochemical markers such as syncytialization and secretion of hormones such as human chorionic gonadotropin and human placental lactogen [38]. These important molecular differences may indicate differing pathophysiologic mechanisms for the development of preeclampsia and IUGR.
One explanation for the seemingly contradictory results of our findings compared to that of other investigators may be the heterogeneity of clinical and research definitions used for intrauterine growth restriction [39,40]. IUGR is generally agreed to be the failure of the fetus to achieve its growth potential due to associated placental or other pathology [39]. In addition to birth weight percentile adjusted for gestational age, race and gender, clinical characteristics such as abnormal umbilical artery Doppler waveforms in the fetus, abnormal maternal uterine artery Doppler waveforms, asymmetrical growth profile, and/or oligohydramnios support the diagnosis of intrauterine growth restriction [39]. These associated findings may also shed light on potential etiologies of the growth restriction, such as pre-placental, utero-placental, or post-placental [12]. Mayhew and colleagues specifically differentiate between IUGR with persistent end diastolic flow in the umbilical artery versus IUGR with absent end diastolic flow given the differences in placental pathologic findings [12]. Others have defined various placental phenotypes of IUGR [41]. Our cohort consisted of a late onset IUGR defined by an asymmetrical growth profile and, as such, may not be directly comparable to other studies of early onset growth restriction [26] or twins [27]. It is important to note that in this work, we specifically chose to focus on the subgroup of late onset IUGR as it is more frequent than early onset IUGR, representing 90% of IUGR cases and contributes significantly to fetal/neonatal morbidity and mortality [42].
Interestingly, we found that placental endoglin in the subgroup of preeclamptic pregnancies complicated by IUGR was comparable to that observed with preeclampsia with appropriately grown infants, but was significantly different from normotensive women with IUGR or uncomplicated pregnancies. However, the range of endoglin concentrations was quite variable. We speculate that this may represent a more severe form of placental pathology in combination with maternal disease and therefore, may have higher levels of placental endoglin. Conclusions regarding this group are limited by the small sample size, variability in gestational age at delivery, severity of IUGR and maternal disease.
Inadequate trophoblast invasion and remodeling of the uterine spiral arteries leading to poor placental perfusion and the resulting placental hypoxia has been proposed as a common factor in the development of both preeclampsia and IUGR [12,13]. Placental hypoxia, as indicated by the increased hypoxia inducible factor (HIF) protein levels, is likely a pathophysiologic mechanism in preeclampsia [43,44]. However, hypoxia as a common mechanism for both preeclampsia and IUGR has been brought into question, as hypoxia regulated genes are overexpressed in preeclampsia but not in late onset IUGR placentas [45] and HIF expression is comparable in normal and late onset IUGR placentas [29]. Endoglin expression is regulated by oxygen in certain vascular systems [30,31] and therefore, important to consider in the placenta. Yinon and colleagues demonstrated that exposure of first trimester villous explants to 3% oxygen resulted in elevated expression of endoglin compared to 20% oxygen [27]. Gu and colleagues studied cultured trophoblast cells from normal and preeclamptic term pregnancies and observed endoglin upregulation in response to hypoxia [32]. However, hypoxic exposure for four hours did not upregulate endoglin in the cultured explants of third trimester villous explants from placentas obtained from normal pregnant women, women with IUGR and preeclamptic subjects in our study. These differences may be explained by the use of different systems and/or differing gestational ages of the villous tissue—e.g., villous explant cultures in our investigation and cultured trophoblast cells by Gu et al., third trimester villous explants compared to first and early second trimester in Yinon et al.’s investigations [27].
Currently, the precise pathobiology underlying the increased endoglin expression in preeclamptic placentas is incompletely understood. During early placental development and particularly in the first trimester, placental endoglin is quite high; these tend to be lower in late first and second trimester placentas [27]. Based on these observations, one can speculate that the increased expression of endoglin in preeclamptic placentas may be due to failure of down regulating endoglin gene expression in the second trimester. Another possible explanation is that the increase in placental endoglin with preeclampsia may be a compensatory mechanism and pro-TGF effect [16]. In the absence of a unique mRNA for the soluble endoglin, it is possible that in both events the soluble endoglin may be an inadvertent N-terminal cleavage product by the membrane type 1 metalloproteinase [16,23]. This increase in soluble endoglin may, in turn, create an anti-angiogenic milieu in the circulation and lead to the maternal manifestations of preeclampsia [16]. It is possible that with fetal growth restriction, this compensatory effect may not occur leading to placental and circulating endoglin levels comparable to uncomplicated pregnancies. Unfortunately, much of this has been speculative and has not been proven.
While our findings are novel and exciting, we acknowledge some inherent limitations of the data. First, our cohort is limited to late onset IUGR and therefore, may represent a different subset from the early onset group. This important factor must be taken into account when comparing studies of pregnancies complicated by preeclampsia and/or IUGR. Second, because of our strict definitions of IUGR in this cohort, some of these conclusions are limited by the sample size; particularly, in the case of preeclamptic pregnancies complicated by IUGR. Third, villous explant studies are limited by the in vitro nature of the approach and may not adequately represent what occurs in an in vivo system. Lastly, use of first trimester villous explants is preferable to third trimester placental samples; however, most first trimester samples are obtained from pregnancy terminations in which pregnancy outcome is unknown.
Collectively, our data suggest that the pathogenic mechanisms underlying late onset IUGR may be very different from preeclampsia and more specifically, that hypoxia may not be a mechanism in late onset IUGR at least in term placental villous explants. Normotensive pregnancies complicated by late onset IUGR appear to be more comparable to uncomplicated pregnancies than to preeclamptic pregnancies with or without IUGR babies, with respect to placental and circulating endoglin. Further study is necessary to determine the mechanisms and precise pattern of circulating and placental endoglin expression in women with both preeclampsia complicated by IUGR.
Supplementary Material
Acknowledgments
This project was supported by national Institutes of Health grant numbers P01-HD30367, RO3-HD055219 and K12-HD043441-04.
References
- 1.Gardosi J, Kady SM, McGeown P, Francis A, Tonks A. Classification of stillbirth by relevant condition at death (ReCoDe): population based cohort study [see comment] BMJ. 2005;331:1113–7. doi: 10.1136/bmj.38629.587639.7C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Froen JF, Gardosi JO, Thurmann A, Francis A, Stray-Pedersen B. Restricted fetal growth in sudden intrauterine unexplained death. Acta Obstetricia et Gynecologica Scandinavica. 2004;83:801–7. doi: 10.1111/j.0001-6349.2004.00602.x. [DOI] [PubMed] [Google Scholar]
- 3.Garite TJ, Clark R, Thorp JA. Intrauterine growth restriction increases morbidity and mortality among premature neonates. American Journal of Obstetrics & Gynecology. 2004;191:481–7. doi: 10.1016/j.ajog.2004.01.036. [DOI] [PubMed] [Google Scholar]
- 4.Jarvis S, Glinianaia SV, Torrioli MG, Platt MJ, Miceli M, Jouk PS, et al. Cerebral palsy and intrauterine growth in single births: European collaborative study [see comment] Lancet. 2003;362:1106–11. doi: 10.1016/S0140-6736(03)14466-2. [DOI] [PubMed] [Google Scholar]
- 5.Gilbert WM, Danielsen B. Pregnancy outcomes associated with intrauterine growth restriction. American Journal of Obstetrics & Gynecology. 2003;188:1596–9. doi: 10.1067/mob.2003.384. discussion 1599–1601. [DOI] [PubMed] [Google Scholar]
- 6.Williams RL, Creasy RK, Cunningham GC, Hawes WE, Norris FD, Tashiro M. Fetal growth and perinatal viability in California. Obstetrics & Gynecology. 1982;59:624–32. [PubMed] [Google Scholar]
- 7.Eskenazi B, Fenster L, Sidney S, Elkin EP. Fetal growth retardation in infants of multiparous and nulliparous women with preeclampsia. American Journal of Obstetrics & Gynecology. 1993;169:1112–8. doi: 10.1016/0002-9378(93)90265-k. [DOI] [PubMed] [Google Scholar]
- 8.Sibai B, Dekker G, Kupferminc M. Preeclampsia. Lancet. 2005;365:785–99. doi: 10.1016/S0140-6736(05)17987-2. [DOI] [PubMed] [Google Scholar]
- 9.Roberts JM, Gammill HS. Preeclampsia: recent insights. Hypertension. 2005;46:1243–9. doi: 10.1161/01.HYP.0000188408.49896.c5. [DOI] [PubMed] [Google Scholar]
- 10.Redman CW, Sargent IL. Latest advances in understanding preeclampsia. Science. 2005;308:1592–4. doi: 10.1126/science.1111726. [DOI] [PubMed] [Google Scholar]
- 11.Benirschke K, Kaufmann P. Pathology of the human placenta. 4. New York: Springer Verlag; 2000. pp. 452–456.pp. 542–562. [Google Scholar]
- 12.Mayhew TM, Charnock-Jones DS, Kaufmann P. Aspects of human feto-placental vasculogenesis and angiogenesis. III Changes in complicated pregnancies [see comment] Placenta. 2004;25:127–39. doi: 10.1016/j.placenta.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 13.Ness RB, Sibai BM. Shared and disparate components of the pathophysiologies of fetal growth restriction and preeclampsia. American Journal of Obstetrics & Gynecology. 2006;195:40–9. doi: 10.1016/j.ajog.2005.07.049. [DOI] [PubMed] [Google Scholar]
- 14.Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S, et al. Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia [see comment] Journal of Clinical Investigation. 2003;111:649–58. doi: 10.1172/JCI17189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levine RJ, Maynard SE, Qian C, Lim KH, England LJ, Yu KF, et al. Circulating angiogenic factors and the risk of preeclampsia [see comment] New England Journal of Medicine. 2004;350:672–83. doi: 10.1056/NEJMoa031884. [DOI] [PubMed] [Google Scholar]
- 16.Venkatesha S, Toporsian M, Lam C, Hanai J, Mammoto T, Kim YM, et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nature Medicine. 2006;12:642–9. doi: 10.1038/nm1429. [DOI] [PubMed] [Google Scholar]
- 17.Levine RJ, Lam C, Qian C, Yu KF, Maynard SE, Sachs BP, et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia [see comment] [erratum appears in N Engl J Med. 2006 Oct 26; 355(17):1840] New England Journal of Medicine. 2006;355:992–1005. doi: 10.1056/NEJMoa055352. [DOI] [PubMed] [Google Scholar]
- 18.Ahmed A, Dunk C, Ahmad S, Khaliq A. Regulation of placental vascular endothelial growth factor (VEGF) and placenta growth factor (PIGF) and soluble Flt-1 by oxygen—a review. Placenta. 2000;21(Suppl A):S16–24. doi: 10.1053/plac.1999.0524. [DOI] [PubMed] [Google Scholar]
- 19.Kendall RL, Wang G, Thomas KA. Identification of a natural soluble form of the vascular endothelial growth factor receptor, FLT-1, and its heterodimerization with KDR. Biochemical & Biophysical Research Communications. 1996;226:324–8. doi: 10.1006/bbrc.1996.1355. [DOI] [PubMed] [Google Scholar]
- 20.Shibuya M. Structure and function of VEGF/VEGF-receptor system involved in angiogenesis. Cell Structure & Function. 2001;26:25–35. doi: 10.1247/csf.26.25. [DOI] [PubMed] [Google Scholar]
- 21.Toporsian M, Gros R, Kabir MG, Vera S, Govindaraju K, Eidelman DH, et al. A role for endoglin in coupling eNOS activity and regulating vascular tone revealed in hereditary hemorrhagic telangiectasia. Circulation Research. 2005;96:684–92. doi: 10.1161/01.RES.0000159936.38601.22. [DOI] [PubMed] [Google Scholar]
- 22.Bernabeu C, Conley BA, Vary CP. Novel biochemical pathways of endoglin in vascular cell physiology. Journal of Cellular Biochemistry. 2007;102:1375–88. doi: 10.1002/jcb.21594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ten Dijke P, Goumans MJ, Pardali E. Endoglin in angiogenesis and vascular diseases. Angiogenesis. 2008;11:79–89. doi: 10.1007/s10456-008-9101-9. [DOI] [PubMed] [Google Scholar]
- 24.Ahmad S, Ahmed A. Elevated placental soluble vascular endothelial growth factor receptor-1 inhibits angiogenesis in preeclampsia. Circulation Research. 2004;95:884–91. doi: 10.1161/01.RES.0000147365.86159.f5. [DOI] [PubMed] [Google Scholar]
- 25.Romero R, Nien JK, Espinoza J, Todem D, Fu W, Chung H, et al. A longitudinal study of angiogenic (placental growth factor) and anti-angiogenic (soluble endoglin and soluble vascular endothelial growth receptor-1) factors in normal pregnancy and patients destined to develop preeclampsia and deliver a small for gestational age neonate. Journal of Maternal-Fetal and Neonatal Medicine. 2008;21:9–23. doi: 10.1080/14767050701830480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stepan H, Kramer T, Faber R. Maternal plasma concentrations of soluble endoglin in pregnancies with intrauterine growth restriction. Journal of Clinical Endocrinology and Metabolism. 2007 doi: 10.1210/jc.2006-2774. epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 27.Yinon Y, Nevo O, Xu J, Many A, Rolfo A, Todros T, et al. Severe intrauterine growth restriction pregnancies have increased placental endoglin levels – hypoxic regulation via transforming growth factor β3. American Journal of Pathology. 2008;172:77–85. doi: 10.2353/ajpath.2008.070640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shibata E, Rajakumar A, Powers RW, Larkin RW, Gilmour C, Bodnar LM, et al. Soluble fms-like tyrosine kinase 1 is increased in preeclampsia but not in normotensive pregnancies with small-for-gestational-age neonates: relationship to circulating placental growth factor. Journal of Clinical Endocrinology & Metabolism. 2005;90:4895–903. doi: 10.1210/jc.2004-1955. [DOI] [PubMed] [Google Scholar]
- 29.Rajakumar A, Jeyabalan A, Markovic N, Ness R, Gilmour C, Conrad KP. Placental HIF-1{alpha}HIF-2{alpha}, membrane and soluble VEGF receptor-1 proteins are not increased in normotensive pregnancies complicated by late-onset intrauterine growth restriction. American Journal of Physiology – Regulatory Integrative & Comparative Physiology. 2007;293:R766–74. doi: 10.1152/ajpregu.00097.2007. [DOI] [PubMed] [Google Scholar]
- 30.Sanchez-Elsner T, Botella LM, Velasco B, Langa C, Bernabeu C. Endoglin expression is regulated by transcriptional cooperation between the hypoxia and transforming growth factor-beta pathways. Journal of Biological Chemistry. 2002;277:43799–808. doi: 10.1074/jbc.M207160200. [DOI] [PubMed] [Google Scholar]
- 31.Zhu Y, Sun Y, Xie L, Jin K, Sheibani N, Greenberg DA. Hypoxic induction of endoglin via mitogen-activated protein kinases in mouse brain microvascular endothelial cells [see comment] Stroke. 2003;34:2483–8. doi: 10.1161/01.STR.0000088644.60368.ED. [DOI] [PubMed] [Google Scholar]
- 32.Gu Y, Lewis DF, Wang Y. Placental productions and expressions of sEndoglin, sFlt-1, and PlGF in normal and preeclamptic pregnancies. Journal of Clinical Endocrinology & Metabolism. 2008;93(1):260–266. doi: 10.1210/jc.2007-1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gifford RW, August PA, Cunningham FG, Green LA, Lindheimer MD, McNellis D, et al. Report of the national high blood pressure working group on research on hypertension in pregnancy. American Journal of Obstetrics & Gynecology. 2000;183:S1–22. [PubMed] [Google Scholar]
- 34.Roberts JM, Bodnar LM, Lain KY, Hubel CA, Markovic N, Ness RB, et al. Uric acid is as important as proteinuria in identifying fetal risk in women with gestational hypertension [see comment] Hypertension. 2005;46:1263–9. doi: 10.1161/01.HYP.0000188703.27002.14. [DOI] [PubMed] [Google Scholar]
- 35.Rajakumar A, Conrad KP. Expression, ontogeny, and regulation of hypoxia-inducible transcription factors in the human placenta. Biology of Reproduction. 2000;63:559–69. doi: 10.1095/biolreprod63.2.559. [DOI] [PubMed] [Google Scholar]
- 36.Benyo DF, Smarason A, Redman CW, Sims C, Conrad KP. Expression of inflammatory cytokines in placentas from women with preeclampsia. Journal of Clinical Endocrinology & Metabolism. 2001;86:2505–12. doi: 10.1210/jcem.86.6.7585. [DOI] [PubMed] [Google Scholar]
- 37.Rajakumar A, Doty K, Daftary A, Harger G, Conrad KP. Impaired oxygen-dependent reduction of HIF-1alpha and -2alpha proteins in pre-eclamptic placentae [erratum appears in Placenta 2003 Jul;24(6):711] Placenta. 2003;24:199–208. doi: 10.1053/plac.2002.0893. [DOI] [PubMed] [Google Scholar]
- 38.Newhouse SM, Davidge ST, Winkler-Lowen B, Demianczuk N, Guilbert LJ. In vitro differentiation of villous trophoblasts from pregnancies complicated by intrauterine growth restriction with and without preeclampsia. Placenta. 2007;28:999–1003. doi: 10.1016/j.placenta.2007.04.008. [DOI] [PubMed] [Google Scholar]
- 39.Tan TY, Yeo GS. Intrauterine growth restriction. Current Opinion in Obstetrics & Gynecology. 2005;17:135–42. doi: 10.1097/01.gco.0000162181.61102.d7. [DOI] [PubMed] [Google Scholar]
- 40.Cetin I, Foidart JM, Miozzo M, Raun T, Jansson T, Tsatsaris V, et al. Fetal growth restriction: a workshop report. Placenta. 2004;25:753–7. doi: 10.1016/j.placenta.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 41.Sibley CP, Turner MA, Cetin I, Ayuk P, Boyd CA, D’Souza SW, et al. Placental phenotypes of intrauterine growth. Pediatric Research. 2005;58:827–32. doi: 10.1203/01.PDR.0000181381.82856.23. [DOI] [PubMed] [Google Scholar]
- 42.Boulet SL, Alexander GR, Salihu HM, Kirby RS, Carlo WA. Fetal growth risk curves: defining levels of fetal growth restriction by neonatal death risk. American Journal of Obstetrics and Gynecology. 2006;195:1571–7. doi: 10.1016/j.ajog.2006.03.069. [DOI] [PubMed] [Google Scholar]
- 43.Caniggia I, Winter JL. Adriana and Luisa Castellucci Award lecture 2001. Hypoxia inducible factor-1: oxygen regulation of trophoblast differentiation in normal and pre-eclamptic pregnancies—a review. Placenta. 2002;23(Suppl A):S47–57. doi: 10.1053/plac.2002.0815. [DOI] [PubMed] [Google Scholar]
- 44.Rajakumar A, Whitelock KA, Weissfeld LA, Daftary AR, Markovic N, Conrad KP. Selective overexpression of the hypoxia-inducible transcription factor, HIF-2alpha, in placentas from women with preeclampsia. Biology of Reproduction. 2001;64:499–506. doi: 10.1093/biolreprod/64.2.499. Erratum 64;1019-1020. [DOI] [PubMed] [Google Scholar]
- 45.Vaiman D, Mondon F, Garces-Duran A, Mignot T-M, Robert B, Rebourcet R, et al. Hypoxia-activated genes from early placenta are elevated in Preeclampsia, but not in Intra-Uterine Growth Retardation. BMC Genomics. 2005;6:111. doi: 10.1186/1471-2164-6-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.