

Abstract
A sample of 95 sib pairs affected with insulin-dependent diabetes and typed with their normal parents for 28 markers on chromosome 6 has been analyzed by several methods. When appropriate parameters are efficiently estimated, a parametric model is equivalent to the β model, which is superior to nonparametric alternatives both in single point tests (as found previously) and in multipoint tests. Theory is given for meta-analysis combined with allelic association, and problems that may be associated with errors of map location and/or marker typing are identified. Reducing by multipoint analysis the number of association tests in a dense map can give a 3-fold reduction in the critical lod, and therefore in the cost of positional cloning.
Success in mapping major loci has encouraged many researchers to search for genes in complex inheritance, using linkage and allelic association. Because we have a dense linkage map of highly polymorphic, codominant markers, it has become useful to consider three phenotypic classes of alleles. Major genes can be characterized by segregation analysis. They are usually rare and have megaphenic effects (measured as displacement between homozygotes) that are large relative to the standard deviation of liability. A major gene is sufficient to cause affection against almost any genetic background, and therefore interaction is negligible except for modifiers of expression. Major loci can be mapped rather easily. At the opposite extreme are polygenes, which are common and have microphenic effects much too small to be characterized, although they may perhaps be detected through allelic association at candidate loci. In the middle are oligogenes, also called leading factors (1), the object of study in complex inheritance. They are common and have mesophenic effects too small to be reliably characterized by segregation analysis, but in large samples they can be detected by nonparametric methods and elucidated by combined segregation and linkage analysis, which includes allelic association as coupling frequencies (2, 3). Small numbers of oligogenes interact to produce affection: this interaction is certainly not additive on penetrance, but may well be nearly additive on a probit or logistic scale. One locus may have all three allelic classes, and so small effects may be detected through allelic association at loci recognized as candidates through larger effects.
Mapping methods are termed parametric if gene frequency and penetrance must be estimated, and nonparametric otherwise. Within each class there are many models. We prefer the comds model for parametric analysis and the β model for nonparametric analysis. comds assumes one or two leading factors with effects additive on a probit or logistic scale (4). The unit of analysis is a nuclear family with pointers (affected relatives through whom the children were ascertained). Other ascertainment schemes are incorporated through probands. Phenotypes are polychotomized to minimize distributional assumptions. The program can estimate genetic parameters, including gene frequencies, penetrances, recombination, and linkage disequilibrium as coupling frequencies. Alternatives to comds ignore the second oligogene or replace it by polygenes or regressors, and they lack some of the other features of comds, which however has not been extended to multiple markers.
The β model has been shown to be the most powerful nonparametric method (5). Its single parameter (the logarithm of relative recurrence risk) is additive over loci if their effects are independent. Multipoint extension has been built on the mapmaker/sibs platform (6) to provide tests of significance and simultaneous estimates of effect and location on a marker map. We could not have written the beta program without this platform.
Here we compare various analyses of sib pairs affected with insulin-dependent diabetes mellitus (IDDM). This makes a good benchmark because the same data set has been analyzed previously (6–8).
Materials and Methods
The data consist of 95 pairs of affected sib pairs and their normal parents typed for 28 markers on chromosome 6. At each locus the alleles had been grouped into four classes corresponding to a mating ab × cd, with frequencies specified in the data file. Allelic association cannot be studied under this convention because allele a, for example, is seldom the same in different families (9). These loci were intended to be uniformly spaced, but this was not achieved because of low density and imprecise location in the map at the time the markers were chosen.
To apply comds the variables required by that program were created and population parameters specified as for segregation analysis. Estimates are biassed by omission of normal sibs, but linkage can still be tested on the two degrees of freedom (df) provided by three classes of identity by descent (Table 1). The β model uses 1 df, and the Δ model in the “possible triangle” uses 2 df (5). We defined three liability classes with population frequencies 0.0031, 0.0050, and 0.0070 for sons, daughters, and parents, respectively (10). We assumed single selection through multiplex probands and used likelihood of children conditional on parents. These assumptions affect estimates of parameters but not likelihood ratios. For comparison with the β model we estimated gene frequency and displacement under each hypothesis, assuming one locus and no dominance on the liability (probit) scale, with recombination 0.5 under H0 and 0 under H1. For comparison with the Δ model we estimated dominance simultaneously. The lod Z was calculated as (y0 − y1)/(2 ln 10), where y0, y1 are the values of −2 ln (likelihood) under the null and alternative hypotheses, respectively. In this material with both parents normal and all children affected the null hypothesis of no linkage is insensitive to changes in gene frequency and dominance, which are better determined in segregating families.
Table 1.
Alternative models of allelic interaction
| Model | Description | Probability of 0, 1, 2 alleles ibd in affected sib pair
|
|||||
|---|---|---|---|---|---|---|---|
| ζ0 | ζ1 | ζ2 | |||||
| β | No allelic interaction on logistic scale |
|
|
|
|||
| γ | No allelic interaction on penetrance scale |
|
|
|
|||
| Δ | Allelic interaction on logistic scale (Δ ≠ β) |
|
|
|
|||
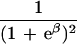
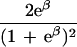
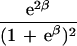
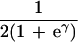
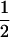
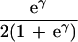
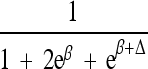
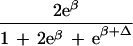
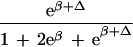
To apply beta we first estimated the effect β for zero recombination by placing a candidate at each marker in succession. We used the secant method to find β̂ with standard error 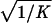 , where K = −∂ ln L/∂β2. For the Δ model we used the mapmaker/sibs program without modification, because maximization within the “permissible triangle” is equivalent to the Δ model (5). Given a location Si for the ith marker, the location S for a susceptibility locus can be estimated by maximum likelihood. We estimated β and S simultaneously over all markers, using different initial values for S to identify local maxima, and compared this with the multipoint option of the native program (6). We also applied the nonparametric option in the genehunter program (11). Marker locations (Table 2) were taken from the current sex-average genetic map in the location database ldb (12). When close markers are ordered differently in genetic and physical maps, the priority was physical > radiation hybrid > linkage. Location does not enter into single marker analyses.
, where K = −∂ ln L/∂β2. For the Δ model we used the mapmaker/sibs program without modification, because maximization within the “permissible triangle” is equivalent to the Δ model (5). Given a location Si for the ith marker, the location S for a susceptibility locus can be estimated by maximum likelihood. We estimated β and S simultaneously over all markers, using different initial values for S to identify local maxima, and compared this with the multipoint option of the native program (6). We also applied the nonparametric option in the genehunter program (11). Marker locations (Table 2) were taken from the current sex-average genetic map in the location database ldb (12). When close markers are ordered differently in genetic and physical maps, the priority was physical > radiation hybrid > linkage. Location does not enter into single marker analyses.
Table 2.
Single marker analyses (95 families, 28 markers)
| Marker | Location S, cM |
nopar
|
comds, ZΔ* | mls, ZΔ |
beta
|
M/S, Zγ | genehunter, ZGH | ||
|---|---|---|---|---|---|---|---|---|---|
| u | Zu | β | Z† | ||||||
| D6S470 | 26.13 | 0.27 | 0.02 | 0.05 | — | 0.05 | 0.02 | 0.01 | 0.01 |
| D6S259 | 41.21 | 2.52 | 1.38 | 1.45 | 1.4 | 0.48 | 1.45 | 1.41 | 0.90 |
| D6S260 | 42.37 | 3.88 | 3.27 | 3.88 | 3.9 | 0.64 | 3.30 | 3.88 | 2.64 |
| D6S276 | 52.42 | 4.38 | 4.16 | 4.33 | 4.3 | 0.78 | 4.29 | 3.87 | 3.09 |
| D6S258 | 55.50 | 5.97 | 7.74 | 8.41 | 8.0 | 1.21 | 8.41 | 7.27 | 5.14 |
| D6S273 | 56.46 | 5.41 | 6.36 | 7.03 | 7.6 | 1.10 | 6.89 | 5.63 | 4.12 |
| TNFA | 57.19 | 5.53 | 6.64 | 7.25 | 7.3 | 1.02 | 6.81 | 5.48 | 4.69 |
| D6S291 | 59.38 | 4.20 | 3.82 | 4.29 | 3.7 | 0.87 | 4.05 | 3.37 | 2.22 |
| D6S426 | 68.15 | 4.62 | 4.64 | 5.10 | 5.1 | 0.95 | 4.99 | 4.93 | 2.86 |
| D6S294 | 85.37 | 2.72 | 1.61 | 1.63 | 1.7 | 0.46 | 1.60 | 1.51 | 1.17 |
| D6S286 | 94.51 | 1.04 | 0.23 | 0.25 | — | 0.18 | 0.23 | 0.25 | 0.16 |
| D6S300 | 105.04 | 0.09 | 0.00 | 0.00 | — | 0.02 | 0.00 | 0.00 | 0.00 |
| D6S267 | 120.77 | −0.46 | −0.05 | 0.00 | — | −0.09 | 0.00 | 0.00 | |
| D6S287 | 121.04 | 0.25 | 0.01 | 0.14 | — | 0.04 | 0.02 | 0.01 | 0.01 |
| D6S407 | 124.08 | 0.00 | 0.00 | 0.00 | — | 0.00 | 0.00 | 0.00 | 0.00 |
| D6S262 | 126.89 | 0.35 | 0.03 | 0.03 | — | 0.06 | 0.03 | 0.03 | 0.02 |
| D6S270 | 132.46 | −0.29 | −0.02 | 0.00 | — | −0.05 | 0.00 | 0.01 | |
| D6S310 | 138.31 | 0.73 | 0.12 | 0.16 | — | 0.15 | 0.15 | 0.15 | 0.10 |
| D6S314 | 138.42 | 0.79 | 0.14 | 0.16 | — | 0.15 | 0.14 | 0.13 | 0.08 |
| D6S308 | 138.78 | 0.90 | 0.18 | 0.28 | — | 0.25 | 0.32 | 0.34 | 0.11 |
| D6S311 | 141.14 | 1.77 | 0.68 | 0.77 | — | 0.31 | 0.73 | 0.75 | 0.57 |
| ESR | 145.49 | 2.87 | 1.79 | 1.82 | 1.8 | 0.47 | 1.82 | 1.77 | 1.48 |
| D6S441 | 147.74 | 2.60 | 1.46 | 1.50 | 1.5 | 0.42 | 1.42 | 1.43 | 1.14 |
| D6S290 | 148.09 | 1.86 | 0.75 | 0.92 | — | 0.41 | 0.89 | 0.88 | 0.48 |
| D6S415 | 152.14 | 0.81 | 0.14 | 0.55 | — | 0.14 | 0.17 | 0.14 | 0.13 |
| D6S305 | 157.37 | 1.88 | 0.77 | 0.65 | — | 0.28 | 0.64 | 0.65 | 0.51 |
| D6S264 | 165.62 | 1.90 | 0.78 | 1.02 | 1.2 | 0.31 | 0.59 | 0.50 | 0.35 |
| D6S281 | 181.78 | 0.76 | 0.13 | 0.57 | — | 0.18 | 0.18 | 0.15 | 0.11 |
| Sum (Σ) | 46.85 | 52.24 | 49.14 | 44.54 | 32.13 | ||||
This is identical to the “possible triangle” lod in mapmaker/sibs (M/S)
This is identical to the β model in comds, except for β < 0 (comds gives 0). Z denotes a lod and subscripts β, γ, and Δ refer to corresponding models (Table 1). For other symbols see text.
For the values of u ≥ 0 in the nopar and genehunter programs we took as the equivalent lod Z = u2/(2 ln 10), where u = ∑x/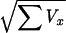 is an asymptotically normal deviate N (0,1) on the null hypothesis of no disease locus near the marker (13). The nopar program uses identity by descent as a variable, while genehunter uses a permutation score. In both programs the distribution is specified only on the null hypothesis of no linkage, and therefore is expected to have less power than a realistic model for the alternative hypothesis.
is an asymptotically normal deviate N (0,1) on the null hypothesis of no disease locus near the marker (13). The nopar program uses identity by descent as a variable, while genehunter uses a permutation score. In both programs the distribution is specified only on the null hypothesis of no linkage, and therefore is expected to have less power than a realistic model for the alternative hypothesis.
Single Marker Analyses
All analyses (Table 2) show a major peak in the HLA region (IDDM1) and minor peaks near ESR (IDDM5) and D6S264 (IDDM8). Differences among analyses reflect different assumptions. The score u in nopar is linear on the number of alleles identical by descent (ibd). It is an unbiased test (note negative values for D6S267 and D6S270, which give no evidence for linkage), and is therefore useful for meta-analysis of multiple samples. On the contrary, the lods in mapmaker/sibs and genehunter cannot be negative since they are constrained to the possible triangle in which the probability of 1 ibd is ζ1 ≤ 0.5 and the probability of 0 ibd is ζ0 ≤ ζ1/2. Within this triangle the lod is Z > 0, while other outcomes are assigned to a bound at which Z ≥ 0. comds gives the same result, since the displacement and gene frequency cannot be negative. Applied to a single sample these programs give a one-sided test, but they do not allow meta-analysis of multiple samples. The β model shares with nopar the property that an estimate of its parameter can be negative, and so it is applicable to meta-analysis as well as to single samples.
In the HLA region certain haplotypes interact so as to enhance the concordance of a pair with 2 ibd. This is well represented by the β model, but less well by the linearity assumption in nopar. Consequently the maximal lod at D6S258 is greater for the β model (8.41) than for nopar (7.74). This is not true for the minor peaks at ESR, where the lods of 1.79 and 1.82 are almost indistinguishable, and D6S264 where nopar gives a greater lod (0.78) than the β model (0.59). Where the Δ model gives a higher lod than the β model, the difference is too small to compensate for the extra degree of freedom, as has been found elsewhere (5). The MLS statistic is taken from Davies et al. (7) who reported 96 sib pair families, whereas the present sample has 95. MLS corresponds to the Δ model of mapmaker/sibs, with 2 df in the possible triangle. mapmaker/sibs also implements the γ model that fixes ζ1 at 0.5 on the hypothesis that genes act additively on penetrance. This is neither biologically plausible nor mathematically possible, since penetrance is limited to the 0,1 interval. Here and elsewhere the γ model is less powerful than the β model.
These results on 28 markers can be summarized by adding lods as at the bottom of Table 2. Of the tests with 1 df, the genehunter model gives the weakest evidence (∑ = 32.13), the γ model (∑ = 44.54) is next, the β model gives the strongest evidence (∑ = 49.14), and nopar is intermediate (∑ = 46.85). The Δ model appears to give the strongest evidence (∑ = 52.24), but the excess over the β model is associated with 28 superfluous df, and so the more parsimonious model wins. The same ordering holds for the most significant regional markers D6S258, ESR, and D6S264.
Although six different tests are presented in Table 2, only five are distinct. The two calculations for the Δ model differ only by 1 affected pair and the convergence criterion. mapmaker/sibs implements the Δ model and comds implements both the β and Δ models. The equivalence of combined segregation and linkage analysis to nonparametric analysis is remarkable, since restriction of the data to normal parents and a pair of affected sibs violates all ascertainment models on which the segregation analysis is based. Merely guessing parameters and selecting the highest likelihood in a finite set does not give equivalence. Even with true maximization of the likelihood by comds, equivalence must not hold for more complicated data structures, although we conjecture that the results would be nearly equivalent. This encourages use of multipoint nonparametric analysis to detect linkage, followed by combined segregation and linkage analysis with the nearest marker to determine gene frequency, dominance, and displacement, which are confounded in nonparametric analysis. This also removes ascertainment bias from these parameters, providing data on affection were collected according to an admissible ascertainment scheme that is incorporated in the analysis. There is no constraint on selective typing of markers, taking full advantage of extreme phenotypes within a sibship (14).
Multipoint Analyses
Likelihood for parametric models is a function of estimable quantities even under the null hypothesis H0. Therefore likelihoods and lods do not have the same maximum, and the maximum of the lod (MOD) has no known statistical properties. Under nonparametric models there are no nuisance parameters in H0, which enters the likelihood as a constant and so the likelihood under an alternative hypothesis H1 maximizes at the same point as the lod. We have seen that this is so for single marker analyses, where the programs that maximize lods (mapmaker/sibs and genehunter) give the same results as comds, which maximizes likelihood. We now use this property to obtain multipoint lods that are valid likelihood ratios,
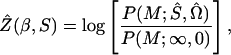 |
where M are the markers conditional on phenotypes, Ŝ is a location that maximizes the likelihood within a chromosome region, and Ω̂ is a vector of estimates jointly at Ŝ. A region is assumed to contain no more than one disease locus, but heterogeneity analysis tests this assumption. We are interested in the three regions corresponding to the possible loci IDDM1, IDDM5, and IDDM8. To maximize the likelihood for the β model (Ω = β) we use Newton–Raphson iteration with finite differences and backtracking (15). If K is the information matrix with elements KSS, KβS, Kββ and inverse K−1, the iteration is
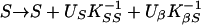 |
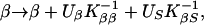 |
and at convergence the standard errors are 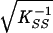 and
and 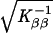 , respectively. At Ŝ the lod for β̂ is Uβ2/Kββ/(2 ln 10), where the term in square brackets in evaluated at β = 0. The other programs do not provide this logic, but the lod at Ŝ was approximated for mapmaker/sibs and genehunter by 4-point Lagrangian interpolation.
, respectively. At Ŝ the lod for β̂ is Uβ2/Kββ/(2 ln 10), where the term in square brackets in evaluated at β = 0. The other programs do not provide this logic, but the lod at Ŝ was approximated for mapmaker/sibs and genehunter by 4-point Lagrangian interpolation.
Table 3 summarizes these results. IDDM1 gives an overwhelming lod with a credible standard error (0.66 cM). IDDM5 and IDDM8 give suggestive lods that do not reach the critical value of 3, with standard errors that are implausibly small in the first case (0.13 cM) and large in the second (13.15 cM), corresponding to a very short and very broad local distribution, respectively. Errors in map location and/or typing are suggested, and may be obscuring signals. At this point existence of IDDM5 and IDDM8 cannot be asserted or denied with confidence. The evidence is not altered by using the map of Davies et al. (7). Efficiency of the maximal single point lod relative to the multipoint lod varies from 0.67 to 0.75. Other multipoint methods give similar results (Table 4). For IDDM1 the β lod exceeds the NPL lod of genehunter and the γ lod of mapmaker/sibs, while falling short of the Δ lod of the latter program by an amount too small to compensate for the extra degree of freedom. Despite difficulties in interpreting IDDM5 and IDDM8, the β model retains its superiority.
Table 3.
Multipoint analysis under the β model
| Locus | Interval | S | σS | β | σβ | KSS | KβS | Kββ | Ẑ(β) | Single locus efficiency |
|---|---|---|---|---|---|---|---|---|---|---|
| IDDM1 | pter-D6S267 | 55.89 | 0.66 | 1.16 | 0.18 | 2.33 | 0.00 | 32.76 | 10.77 | 0.75 |
| IDDM5 | D6S267-D6S415 | 145.49 | 0.13 | 0.51 | 0.15 | 59.21 | 0.28 | 42.41 | 2.46 | 0.73 |
| IDDM8 | D6S415-qter | 165.63 | 13.15 | 0.34 | 0.22 | 0.01 | −0.41 | 36.87 | 0.96 | 0.67 |
Table 4.
Multipoint lods Ẑ (β,S) under alternative models
| Locus | beta |
mapmaker/sibs
|
genehunter | |
|---|---|---|---|---|
| γ | Δ | |||
| IDDM1 | 11.28 | 8.88 | 11.61 | 9.73 |
| IDDM5 | 2.48 | 2.42 | 2.48 | 2.32 |
| IDDM8 | 0.96 | 0.90 | 1.03 | 0.76 |
Meta-Analysis
Ideally all data on a particular chromosome are kept together, however many samples are collected, and the same phenotypes and ascertainment scheme are used for each sample. The real world is messier even if all samples are assembled, because parameters may still vary among populations, and in the worst but most frequent case there are variations among samples in phenotype definition and mode of ascertainment. With multipoint analysis it is immaterial whether the same markers are used for different samples. Assuming that the samples are kept separate and only summary results are available in the form of Table 3, a meta-analysis is possible for the β model under large-sample theory. As discussed above, the possible triangle constraint makes biased alternatives invalid for meta-analysis, since in the limit for a large number of samples, the expected value of the lod is infinite even under H0.
To implement meta-analysis, let the estimates for the ith sample be subscripted. Although βi is a nuisance parameter sensitive to differences among populations, modes of ascertainment, and phenotype definitions, the parsimonious assumption that βi = β is likely to provide the most powerful test. For greater generality we consider the quadratic form
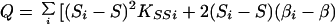 |
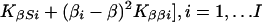 |
which is large-sample theory is a χ2 with 2I df if S and β are correctly specified a priori and Si = S, βi = β for all i. Since the corresponding likelihood is L = e−Q/2, maximum likelihood (ML) estimates of β and S are
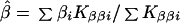 |
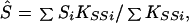 |
and the elements of the information matrix are ∑ Kββi, ∑ KSSi, and ∑ KβSi. Substituting the ML estimates, Q has 2 (I-1) df to test the above hypothesis, under which linkage is tested by
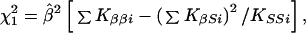 |
where the term in square brackets in evaluated at β = 0, with corresponding lod Ẑ (β) = χ2/(2 ln 10). However, if there is heterogeneity among samples, the error variance may be estimated by V = Q/df and χ2 replaced by χ2/V. Under a fixed effects model for βi,
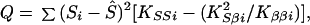 |
with I-2 df testing heterogeneity in Si whether or not there is heterogeneity in β. Under a random effects model Q = ∑ [(Si − Ŝ)2 KSSi + 2 (Si − Ŝ)(βi − β̂)KSβi + (βi − β̂)2 Kββi] with 2 (I-1) df testing heterogeneity in S and/or β. The main concern with this theory is that estimates of KSS are highly variable among regions. Does this mean that the sample is too small for large-sample theory to be reliable, or that likelihood is reflecting error in typing or map location? While the importance of these factors cannot be stressed too much, we need a theory that is robust to error. Possible approaches are being pursued.
If the appropriate lod exceeds the canonical level of 3, or whatever critical level may be chosen, a disease locus in the region is inferred. To protect against noise from a candidate in an adjacent region, it should be verified that the lod at both regional boundaries is substantially smaller than the maximal lod in the region. Subsequent observations will confirm or refute the regional locus. If it is real, its location can be refined by linkage, allelic association and ultimately sequencing.
Allelic Association
Lawrence et al. (16) derived the kinship between linked loci and concluded that efficient mapping by allelic association requires that distance between markers be much less than 1 Mb. Risch and Merikangas (17) showed that allelic association is more powerful than linkage in such dense maps. However, if extended to a genome screen, the critical lod for significance could be as much as 9, or 3 times the canonical lod for linkage. This approach can be applied to single base pair polymorphisms in a completely sequenced genome, typed by a nonfluorescent method. Under these conditions a great increase in throughput is possible, and even pooling of individuals with the same phenotype becomes feasible, although precision and haplotype information are lost.
If allelic association is to be an efficient adjunct to linkage, multilocus tests must be used in the same way—i.e., lods maximized with respect to an effect ∈ and location S, with information weights that give efficient combination with the information about S from linkage. A theory with these desirable properties was developed from Malecot (18) for isolation by distance (19), according to which
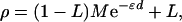 |
where ρ is a measure of linkage disequilibrium and
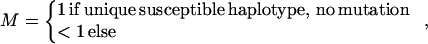 |
ɛ≥0 is dependent on duration of associated haplotypes, L= bias due to spurious association, d=δi(Si−SD), where D is the susceptibility locus, i is a marker at physical location Si, and
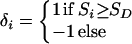 |
It appears that the most powerful definition of ρ is on the 2 × 2 table formed when marker alleles positively associated with disease susceptibility are pooled, and the residual alleles likewise (N.E.M., unpublished data). This approach reduces the number of tests to 1 per chromosome region, however many markers with any number of alleles are typed within that region, justifying the canonical lod of 3 for a dense map instead of 9 as contemplated by Risch and Merikangas (17). The cost of mapping is therefore reduced by a factor of 3, whether measured in dollars or time and effort. A trustworthy map at high resolution is the sine qua non for this method, illustrating a principle well known to geographers and classical geneticists, but not self-evident to molecular biologists, that exploration without a good map is possible but costly. Unfortunately, there is no international effort to create such a map.
Discussion
We have not applied the parametric model of genehunter, which lacks power because it makes no allowance for ascertainment and cannot estimate parameters. Such an extension would be useful to implement parametric multipoint analysis.
Although the conclusion from meta-analysis can be presented as a lod, we have invoked large-sample theory that is unnecessary for major loci. The reliability of this approach must be tested against the standard of pooled samples, which do not require quadratic forms but do use the probability transformation. Trials of meta-analysis for affection, polychotomies, and quantitative traits will be the next step, now that there is a general and empirically tested theory for mapping oligogenes.
The first generation of human geneticists included only a handful interested in mapping major genes. Perhaps as a consequence, it took 25 years after Bernstein introduced the problem to recognize lods as the method of choice (20). The next generation grew to several thousands interested in mapping oligogenes. Perhaps as a consequence, it took 60 years after Penrose introduced the problem to recognize lods as the method of choice. Methods that use means and regressions depend entirely on large-sample theory, are more dependent on distributional assumptions, and reflect gene action less credibly. They always have lower power and less reliability than lods, which are applicable to pairs of affected relatives, polychotomies, and quantitative traits.
We have seen that multilocus analysis increases power to detect linkage and efficiency to localize disease genes. It depends on an accurate and dense map of markers, integrating genetic with physical data. For IDDM1 the maximal multipoint lod is 11.28, while the greatest single-point lod is 8.41. Similar increases in power are expected for other disease genes, depending on location and heterozygosity.
Besides multipoint lods, efficient tests for oligogenic linkage require a credible model of gene expression. If χ1, χ2 are metrics in a pair of relatives, measured as deviations from the population mean, two similar values will give a large product χ1 χ2 if both are in either tail of the distribution. Conversely, an extremely discordant pair will give a small product, while a typical pair give a product near zero. Therefore a logit proportional to χ1 χ2 is an intuitive representation of gene action. On the contrary, the squared difference (χ1 − χ2)2 used in the test of Haseman and Elston (21) can take the same value for a similar pair drawn from any part of the distribution, and so cannot reflect the expectation that similarity is more informative in the tails of the distribution.
These results illustrate not only the superiority of multipoint tests, but also the advantages for meta-analysis of a theory that uses lods and makes effect β as well as location S estimable, efficient, and biologically meaningful parameters (5, 22). The Δ model with two parameters in the permissible triangle is less efficient, much clumsier for meta-analysis, and must be converted to an equivalent lod with 1 df. Now that we have methods based on the three principles of lods, multipoint analysis, and phenotype products, there is little interest in exploring the relative efficiency of inferior methods.
This paper has compared methods that differ in power and utility for meta-analysis. Qualitatively they all agree that IDDM1 is well established, while IDDM5 and IDDM8 fall in a grey zone where linkage has not been confirmed by a lod of 3 or greater nor excluded by a lod of −2 or less. A larger material has failed to increase the lods observed in this sample (8). However, when the sample size was increased more than 10-fold to about 1,070 sib pairs (23), IDDM5 became nearly significant (Ẑ = 2.92 for ESR) at β = 0.21 and IDDM8 became barely significant (Ẑ = 3.43 for D6S281) at β = 0.29. At least eight other regions previously identified by suggestive linkages have not been confirmed (24). Improvement in the map, increased sample size, denser markers, and especially evidence from allelic association will ultimately resolve these inconsistencies.
Acknowledgments
We are grateful to June Davies and John Todd for making IDDM data available to us.
ABBREVIATIONS
- IDDM
insulin-dependent diabetes mellitus
- ibd
identical by descent
References
- 1.Wright S. Evolution and the Genetics of Populations. Vol. 1. Chicago: Univ. of Chicago Press; 1968. pp. 411–417. [Google Scholar]
- 2.MacLean C J, Morton N E, Yee S. Comput Biomed Res. 1984;17:471–480. doi: 10.1016/0010-4809(84)90013-2. [DOI] [PubMed] [Google Scholar]
- 3.Shields D C, Ratanachaiyavong S, McGregor A M, Collins A, Morton N E. Am J Hum Genet. 1994;55:540–554. [PMC free article] [PubMed] [Google Scholar]
- 4.Morton N E, Shields D C, Collins A. Ann Hum Genet. 1991;55:301–314. doi: 10.1111/j.1469-1809.1991.tb00857.x. [DOI] [PubMed] [Google Scholar]
- 5.Collins A, MacLean C J, Morton N E. Proc Natl Acad Sci USA. 1996;93:9177–9181. doi: 10.1073/pnas.93.17.9177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krugylak L, Lander E S. Am J Hum Genet. 1995;57:439–454. [PMC free article] [PubMed] [Google Scholar]
- 7.Davies J L, Kawaguchi Y, Bennett S T, Copeman J B, Cordell H J, Pritchard L E, Reed P W, Gough S C L, Jenkins S C, Palmer S M, Balfour K M, Rowe B R, Farrall M, Barnett A H, Bain S C, Todd J A. Nature (London) 1994;371:130–135. doi: 10.1038/371130a0. [DOI] [PubMed] [Google Scholar]
- 8.Davies J L, Cucca F, Goy J V, Atta Z A A, Merriman M E, Wilson A, Barnett A H, Bain S C, Todd J A. Hum Mol Genet. 1996;5:1071–1074. doi: 10.1093/hmg/5.7.1071. [DOI] [PubMed] [Google Scholar]
- 9.Ott J. Ann Hum Genet. 1978;42:255–257. doi: 10.1111/j.1469-1809.1978.tb00657.x. [DOI] [PubMed] [Google Scholar]
- 10.Green A, Morton N E, Iselius L, Svejgaard A, Platz P, Ryder L P, Hauge M. Tissue Antigens. 1982;19:213–221. doi: 10.1111/j.1399-0039.1982.tb01442.x. [DOI] [PubMed] [Google Scholar]
- 11.Krugylak L, Daly M J, Reeve-Daly M P, Lander E S. Am J Hum Genet. 1996;58:1347–1363. [PMC free article] [PubMed] [Google Scholar]
- 12.Collins A, Frezal J, Teague J, Morton N E. Proc Natl Acad Sci USA. 1996;93:14771–14775. doi: 10.1073/pnas.93.25.14771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Collins A, Morton N E. Hum Hered. 1995;45:311–318. doi: 10.1159/000154299. [DOI] [PubMed] [Google Scholar]
- 14.Risch N, Zhang H. Science. 1995;268:1584–1589. doi: 10.1126/science.7777857. [DOI] [PubMed] [Google Scholar]
- 15.Press W H, Teukolsky S A, Vetterling W T, Flannery B P. Numerical Recipes in C: The Art of Scientific Computing. Cambridge: Cambridge Univ. Press; 1992. [Google Scholar]
- 16.Lawrence S, Beasley R, Doull I, Begishvili B, Lampe F, Holgate S T, Morton N E. Ann Hum Genet. 1994;58:359–368. doi: 10.1111/j.1469-1809.1994.tb00732.x. [DOI] [PubMed] [Google Scholar]
- 17.Risch N, Merikangas K. Science. 1996;273:1516–1517. doi: 10.1126/science.273.5281.1516. [DOI] [PubMed] [Google Scholar]
- 18.Malecot G. In: Les Deplacements Humains, Entretiens de Monaco en Sciences Humaines. Sutter J, editor. Paris: Hachette; 1962. pp. 205–212. [Google Scholar]
- 19.Morton N E, Klein D, Hussels I E, Dodinval P, Todorov A, Lew R, Yee S. Am J Hum Genet. 1973;25:347–361. [PMC free article] [PubMed] [Google Scholar]
- 20.Morton N E. Genetics. 1995;140:7–12. doi: 10.1093/genetics/140.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Haseman J K, Elston R C. Behav Genet. 1972;2:3–19. doi: 10.1007/BF01066731. [DOI] [PubMed] [Google Scholar]
- 22.Morton N E. Proc Natl Acad Sci USA. 1996;93:3471–3476. doi: 10.1073/pnas.93.8.3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Delepine M, Pociot F, Habita C, Hashimoto L, Froguel P, Rotter J, Cambon-Thomsen A, Deschamps I, Djoulah S, Weissenbach J, Nerup J, Lathrop M, Julier C. Am J Hum Genet. 1997;60:174–187. [PMC free article] [PubMed] [Google Scholar]
- 24.Luo D-F, Bui M M, Muir A, Maclaren N K, Thomson G, She J-X. Am J Hum Genet. 1995;57:911–919. [PMC free article] [PubMed] [Google Scholar]