

Abstract
Huntington’s disease (HD) is an autosomal dominant neurodegenerative disorder characterized by chorea, incoordination, and shortened life-span, and by huntingtin inclusions and neurodegeneration. We previously screened the 1040 FDA-approved compounds from the NINDS compound library and found that a compound, nipecotic-acid, significantly reduced mutant huntingtin aggregations and blocked cell toxicity in an inducible cell model of HD. Because nipecotic-acid does not cross the blood-brain barrier (BBB), we studied its analogue, tiagabine, which is able to cross the BBB, in both N171-82Q and R6/2 transgenic mouse models of HD. Tiagabine was administered intraperitoneally at 2 and 5 mg/kg daily in HD mice. We found that tiagabine extended survival, improved motor performance, and attenuated brain atrophy and neurodegeneration in N171-82Q HD mice. These beneficial effects were further confirmed in R6/2 HD mice. The levels of tiagabine at effective doses in mouse serum are comparable to the levels in human patients treated with tiagabine. These results suggest that tiagabine may have beneficial effects in the treatment of HD. Because tiagabine is an FDA-approved drug, it may be a promising candidate for future clinical trials for the treatment of HD.
Keywords: Huntington’s disease, Tiagabine, neuroprotection, Transgenic mouse model, Preclinical trials
Introduction
HD is an autosomal dominant neurodegenerative disease with expanding polyglutamine repeats in the protein huntingtin and abnormal folding of the huntingtin protein (Wanker 2000; Landles and Bates 2004; Ross and Poirier, 2004) that results in the formation of neuronal intranuclear inclusions in the striatum and cortex (DiFiglia et al., 1997). There is gross striatal atrophy with cell loss in the striatum, cortex, amygdala, and thalamus in patients with HD (Petersen et al., 1999). There is currently no therapy available to delay onset or prevent disease progression of HD patients.
In 2001, the National Institute of Neurological Disorders and Stroke (NINDS) compiled a drug library of 1040 compounds, selected in association with MicroSource Discovery Systems (Gaylordsville, CT), consisting mainly of FDA-approved drugs (Aiken et al., 2004; Wang et al., 2005) that were likely candidates for the treatment of neurodegenerative disorders. We applied this compound library to our inducible PC12 cell model of HD, and found nipecotic acid to be one of most effective compounds that protect cells against mutant htt toxicity (Wang et al., 2005). Nipecotic acid does not cross the blood brain barrier efficiently, however. We therefore tested its derivative tiagabine, which has an attached lipophilic group that enables the drug to cross the blood-brain barrier (Andersen et al., 2001; Krogsgaard-Larsen et al., 2000) in HD mouse models.
Tiagabine has been used as an anti-epilepsy drug (Luer and Rhoney 1998; Stefan and Feuerstein 2007). The exact mechanism of action of tiagabine in epilepsy is not fully understood, but it is believed to enhance activity of gamma-aminobutyric acid (GABA) in the central nervous system (Soudijn and van Wijngaarden 2000). GABAergic medium spiny neurons are the most vulnerable neurons and often degenerate in Huntington’s disease (Vonsattel et al., 1998; Albin et al., 1995, Reiner et al., 1988). Prevention of the neurodegenerative process could be achieved by enhancing GABAergic activity or by other yet unknown mechanism(s). We report here that tiagabine increased survival, improved motor performance, and attenuated brain atrophy in two mouse models of HD. The data obtained in the current studies may provide preclinical insight that could facilitate clinical trials of tiagabine in humans with HD.
Materials and methods
Mice and drug administration
N171-82Q transgenic HD mice were mated to hybrid mice (C3H/HEJ × C57BL/6J F1, Taconic, NY). All mice were housed under standard conditions with free access to food and water for 24 h, and 12-h light/dark cycle. We used male N171-82Q HD mice for all our studies since we found that there is significant variability in all phenotypes between male and female N171-82Q mice (Duan et al 2003). N171-82Q male mice usually develop motor performance deficit after 19 weeks of age; the average life-span in these mice is about 22 weeks. R6/2 mice with a CBA/C57Bl6 background were purchased from Jackson Laboratory (Bar harbor, ME). Mice were genotyped and CAG repeat size was determined by PCR. CAG repeat lengths of R6/2 mice are about 110–118. These mice develop motor phenotypes as early as 4 weeks of age. The average life-span for R6/2 mice is about 12–14 weeks. The animals were kept on a 12-h light/dark cycle, with food and water continuously available. There is no sex-dependent difference in R6/2 mice from our experience and other reports (Hersch and Ferrante 2004). therefore both male and female R6/2 mice were used in our studies. Experiments were carried out by procedures that minimized pain and discomfort. All animal experiments were performed according to procedures approved by the Institutional Animal Care and Use Committee.
Tiagabine hydrochloride (purity 99%) was purchased from R&S Pharmchem Co. Ltd (Hangzhou, China). Mice were treated daily with 2 or 5 mg/kg of tiagabine freshly prepared in saline by intraperitoneal injection.
HD cell model and drug treatments
Tet-off PC12 cells expressing tTA(Clontech), were stably transfected with htt-N63-148Q ( the first 63 N-terminal amino acid of huntingtin (htt) with 148 polyglutamine repeat). Htt gene expression was turned on when doxycycline (Dox; a tetracycline derivative) was removed from the culture medium (Wang et al., 2005). Cells were differentiated in the presence of NGF (50 ng/ml). Htt expression and cell differentiation were induced at the same time in order to determine htt toxicity in neuronal cells. Tiagabine was added to the medium once cells attached to the plate at about 4 hours after plating. Cells were trypsinized and washed with PBS, and 100 µl of cells at density of 1 × 105 cells/ml were plated in collagen I-coated plates (BD Biosciences) with differentiation medium.
Doxycycline (200 ng/ml) was added to the differentiation medium to suppress mutant htt expression in the control group. LDH release was used to measure cell toxicity at 4 days after induction of htt. Tiagabine was dissolved in sterile saline as a at 10 mM stock solution and added to medium at the indicated concentrations. Pan-caspase inhibitor z-VAD was purchased from R&D systems, Inc. and was used as a positive control compound since we had consistent protection of this compound in our culture system (Wang et al., 2005).
Behavioral test and Survival study
All mice were randomly divided into each group. Nontransgenic control mice are wild type littermates. Each group contained 15 mice at the beginning of the survival study and for motor behavioral analyses. We used the same set of animals for survival analyses and motor performance tests.
Survival was monitored twice daily, in the early morning and late afternoon, by two experienced investigators (NM and QP). The mice were euthanized when HD mice were unable to right themselves after being placed on their backs and initiate movement after being gently prodded for 30 sec.
Motor behavioral performance was assessed with a rotarod apparatus (Columbus Instrument, OH) in which the time that the mouse remained on the rod at accelerating speeds from 4 to 40 rpm was measured. Each mouse was trained for 5 min and the training session was followed by a 1-h rest period in the home cage. Mice were then placed back on the rotarod for three trials of maximal 5 min at accelerating speeds (4–40 rpm) separated by a 30-min rest period. Mice were tested for 3 consecutive days, by which time a steady baseline level of performance was attained.
Histology
Mice were perfused with 4% ice-cold paraformaldehyde following 1 × PBS perfusion transcardially, and brains were post-fixed in 4% paraformaldehyde overnight, and then transferred to 20% sucrose until the brains sank to the bottom of the tube. Coronal sections, 30 µm, were processed with a sliding microtome for histological staining. Silver staining for neurodegeneration was performed on free-floating sections by using the commercially available FD neurosilver Kit 1 (FD Neuro Technologies, Ellicott City, MD) according to the manufacturer's instructions. The degenerating neurons were identified by dark silver positive staining by using a computer via a CCD Digital camera (Hamamatus Photonics K.K., Hamamatus, Japan). Immunostaining for the mutant huntingtin protein was done by using EM48 (Chemicon, 1:200), a polyclonal antibody to mutant human huntingtin protein that labels mutant huntingtin aggregates (Li et al., 2001). The EM48-immunoreactive product was visualized with biotinylated goat anti-rabbit IgG (1:200 dilution) in ABC (avidin–biotin complex) / DAB (3,3 - diaminobenzidine-4HCl) kit (Vector Labs, Burlingame, CA) and DAB according to the manufacturer's guidelines.
Measurement of volume of striatum and size of lateral ventricles
The volume of the striatum and lateral ventricles by histological analysis was evaluated according to the principle of Cavalieri (Cyr et al., 2005) (volume = s1d1 + s2d2…sndn, where s = surface area and d = distance between two sections). We used different sets of mice for the volumetric measurement study. Each group contained four mice. The Cavalieri approach requires an initial random cut through the reference space of interest, with subsequent cuts at consistent intervals. Provided that the sections through the reference space are systematic-random, and that all sections through the reference space had an equal probability of being sampled, the Cavalieri method gives an unbiased total volume estimate from reference areas on sections. Using the Cavalieri principle, the mean total volume of an arbitrary-shaped reference space can be estimated without bias from the areas on systematic-uniform-random sections. The Cavalieri principle was applied to histological data because of the limited number of sections selected. We chose the first section when the striatum was first detected, and chose every four series sections for volumetric analysis. We considered approximately 11 anatomical levels of the striatum for the volume estimation of every brain by randomly selecting the first section. Image capturing was performed by using a light microscope coupled to a color digital camera (Retina 2000R, QImaging). The surface area was calculated in each of the 11 Nissl-stained sections by contour drawing the striatum and lateral ventricle on each brain hemisphere by using the Aperio Imagescope software (Aperio Technologies). Data were expressed as the average Cavalieri volume (mm3) ± SEM of four mice per group.
Quantification of Htt aggregates
The EM48-positive nuclei were evaluated with a40 × magnification in eight microscopic fields from each indicated brain region, including cortex, hippocampus, and striatum, and the mean number of aggregates per area was determined (DiFiglia et al., 2007).
Measurement of tiagabine by HPLC/MS/MS
Brain homogenates were mixed and centrifuged at 2500 × rpm for 10 min at 4 °C. A volume of 100 µl of the top organic layer was transferred to a disposable borosilicate glass culture tube (13×100 mm) and 100 µl of deionized water were added to this tube, mixed vigorously for 10 s, and centrifuged at 13,000 × rpm for 5 min. Volumes of 20 µl were injected onto the HPLC instrument for quantitative analysis. Serum samples were prepared as previously described (Zhao et al., 2005). Chromatographic analysis was performed using a Waters X-Terra™ MS (C18 3.5 µm, 50 × 2.1 mm i.d). Separation of the analyses from potential interfering material was achieved at ambient temperature by using the Waters X-Terra™ MS packed with a 3.5-µM ODS stationary phase, protected by a guard column packed with Waters Xterra TM (RP18, 3 µm). The mobile phase used for chromatographic separation was composed of 70% formic acid (0.1%) in CH3CN, 30% ammonium acetate (10 mM), and was isocratic at a flow rate of 0.2 ml/min. Method validation runs for serum and brain tissue calibration standard and quality controls were performed on 3 consecutive days and included a calibration curve processed in duplicate, and quality control samples, at four different concentrations, in triplicate. The accuracy and precision of the assay was assessed by the mean relative percentage deviation from the nominal concentrations and the within- and between-run, respectively.
Statistics
Data are expressed as the mean ± SEM. Data were analyzed by using Student's t test or Fisher's exact test. Survival data were assessed by log rank test.
Results
Tiagabine protects cells against mutant Htt induced toxicity
In order to determine whether tiagabine has the same degree of protection as nipecotic acid in HD cells, we used our inducible HD cell model (Wang et al., 2005). Cells inducibly expressed mutant htt and were differentiated by NGF. Tiagabine inhibited the LDH release induced by mutant htt in PC12 cells in a concentration-dependent manner, indicating that tiagabine protected cells from mutant htt-induced toxicity (Fig. 1).
Figure 1. Tiagabine (TGB) protects neurons against mutant huntingtin (htt)- induced toxicity in cultured cells.
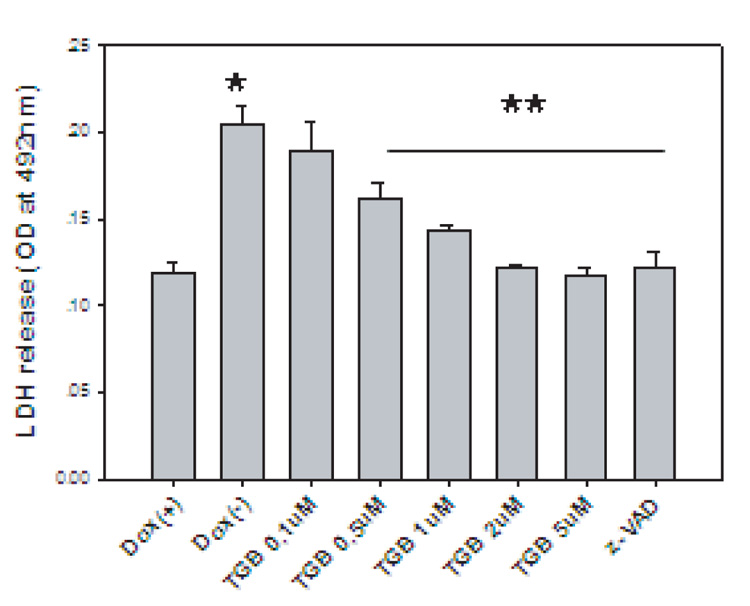
The N-terminal sequence of mutant htt with 148 polyglutamine repeats (N63-148Q-myc) was inducibly expressed in PC12 cells by withdrawal of doxycycline in culture medium in Tet-off system. Cells were differentiated by adding NGF (50 ng/ml) in culture medium. Cell toxicity was assessed by LDH release from dead cells. *p<0.05 compared to the values of Dox(+) group; **p<0.05 compared to the values of Dox(−) group. Three individual experiments with triplicates in each experiment. Standard Student’s t-test was used for statistical analysis.
Tiagabine extends survival, improves motor function but does not affect body weight in N171-82Q mice
To further determine whether tiagabine has neuroprotective effects in vivo, we used N171-82Q HD transgenic mice. HD-N171-82Q mice (line 81) express an N-terminal fragment of huntingtin with 82 glutamines (Schillings et al., 1999). The neuropathological features of N171-82Q mice more closely resemble human HD in that huntingtin aggregates are more prominent in cortex than in striatum, and neurodegeneration is more prominent in these mice than in the other HD mouse model (Yu et al., 2003). The rotarod deficit is usually detected after 10 weeks of age in N171-82Q mice. Beginning at 8 weeks old, tiagabine was administered to HD mice until the end of their life. The survival in tiagabine-treated HD mice was significantly extended. The average survival was 122.3 ± 5.0 (mean ±SE) in the HD-control group, 126.6 ± 2.7 in the HD-vehicle group, 134.0 ± 5.9 in the HD –tiagabine 2 mg/kg group, and 160.0 ± 5.8 in the HD-tiagabine 5 mg/kg group. (Fig. 2a). HD mice exhibited progressive body weight loss after 8 weeks of age. Tiagabine administration did not affect the body weight in wild type littermate control mice and did not affect body weight loss in HD mice (Fig. 2b).
Figure 2. Tiagabine extends survival and improves motor performance in N171-82Q HD mice.
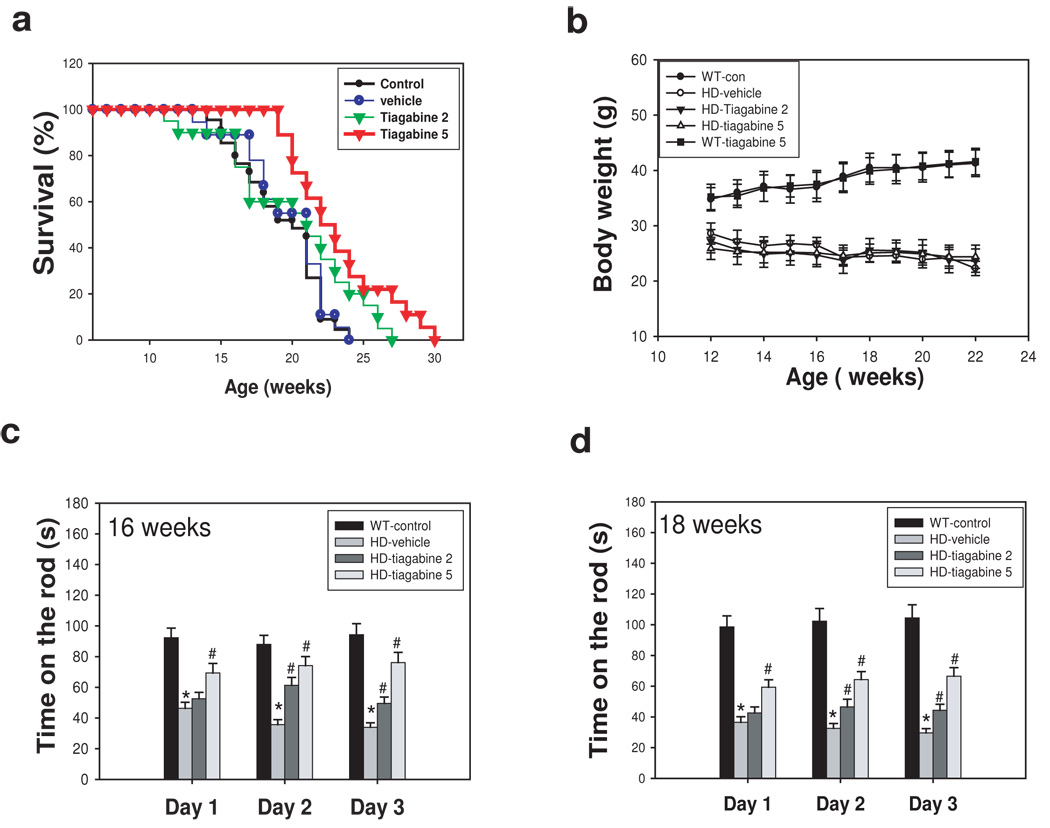
Tiagabine was administered to mice at doses of 2 and 5 mg/kg/d. Survival was monitored daily. (a) Tiagabine at 5 mg/kg (red line) significantly extended survival of N171-82Q mice. p=0.028 comparison between tiagabine 5mg/kg group and vehicle group by log-rank test. (b) tiagabine did not affect body weight in N171-82Q mice. Motor behavioral performance was evaluated by an accelerating rotarod apparatus in mice at 16 weeks of age (c) and 18 weeks of age (d). n=10–15. Values are the mean and SE. *p<0.05 compared to the value of WT-vehicle group; #p<0.05, compared to the value of HD-vehicle group by standard student t-tests.
We further evaluated the motor function of mice at indicated ages. There was significant impairment of motor function in N171-82Q HD mice at these ages, as indicated by significantly reduced running time on the rotarod (Fig 2c &d) compared to wild type littermate control mice. Tiagabine-treated mice showed superior dose dependent motor performance on the rotarod (Fig. 2c &d).
Tiagabine attenuates brain atrophy and neurodegeneration in N171-82Q HD mice
In order to determine whether the improved motor function and increased survival of HD mice treated with tiagabine resulted from slowed progression of the neurodegenerative process in the brain, we performed histological analyses of the brains of HD mice that had been treated with tiagabine or vehicle. As the disease progressed, brain atrophy occurred in the HD mice as indicated by an increase in the size of the lateral ventricles and decrease in the volume of striatum compared to those in nontransgenic mice (Fig. 3a–c). Tiagabine treatment attenuated brain atrophy, indicated by smaller ventricle size and larger striatal volumes in HD mice treated with tiagabine compared to those in HD mice treated with vehicle (Fig. 3a–c). Neurosilver staining was used to identify degenerative neurons. Silver positive neurons were found in HD mice after 18 weeks of age in both cortex and striatum (Fig. 3d). HD mice treated with tiagabine exhibited fewer silver positive neurons in both piriform cortex and striatal regions (Fig. 3d–f).
Figure 3. Tiagabine attenuates brain atrophy and neurodegeneration in N171-82Q HD mice.
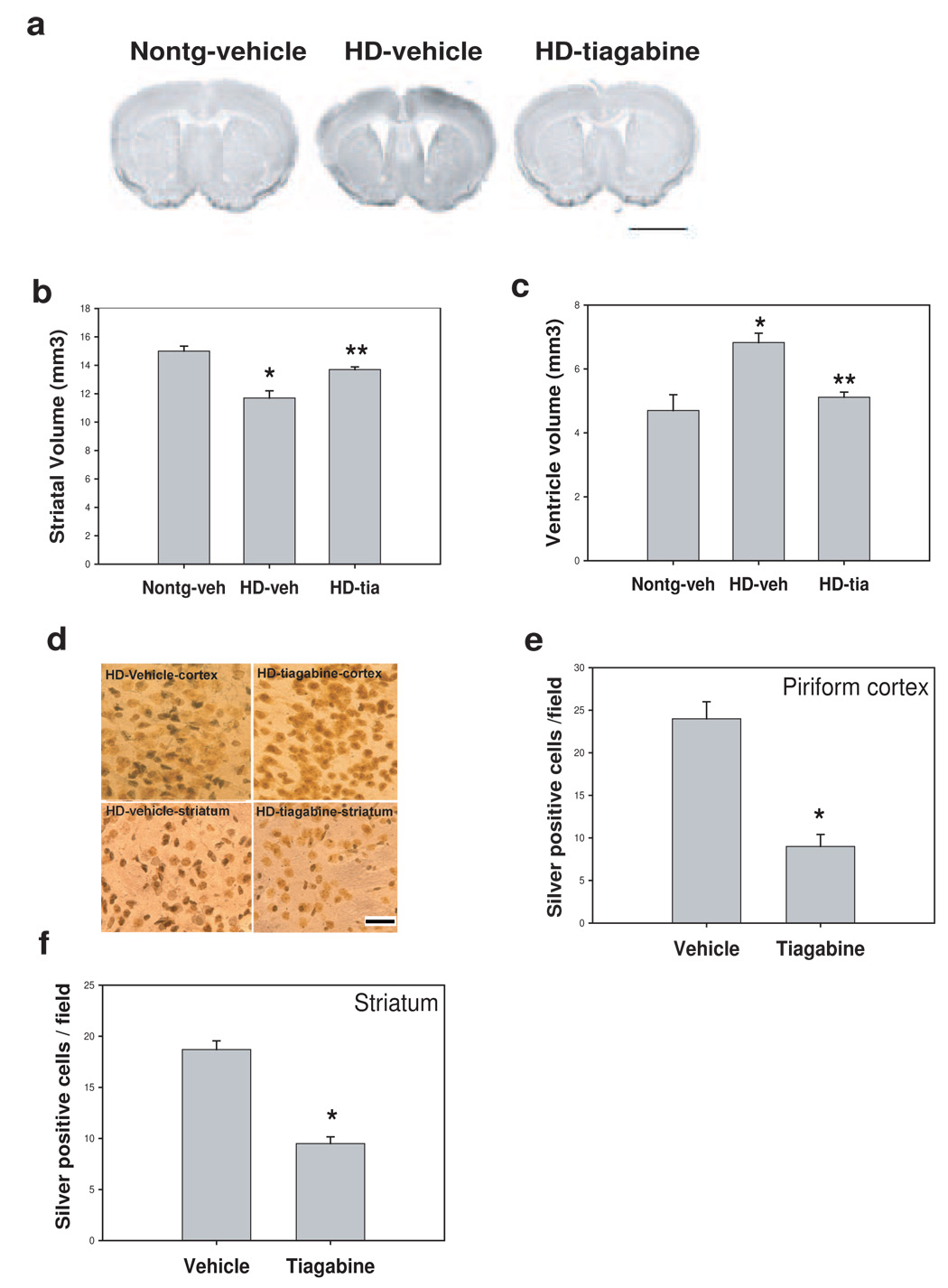
Tiagabine was administered to HD mice beginning at 8 weeks of age, mice were perfused at 18 weeks old, brain sections were processed for Nissl staining (a) and neurosilver staining for detecting degenerative neurons (b). (a) Representative photographs of brain sections of Nissl staining from indicated group. Scale bar=120 µm. note that N171-82Q mouse brain exhibited enlarged ventricles and shrunk striatum. Tiagabine treatment reversed the brain atrophy in HD mice. (b–c) Quantification data of brain atrophy. Tiagabine treatment significantly attenuated the brain atrophy in N171-82Q mice. n=4 mice. *p< 0.05, compared to the values of nontransgenic-vehicle group ( Nontg-veh); **p< 0.05, compared to the values of N171-82Q mice treated with vehicle (HD-veh) by standard student t-tests. (d) Representative silver staining results, the dark staining neurons represent cells undergoing neurodegeneration. Note more dark stained neurons were found in vehicle-treated brain compared to tiagabine-treated brain in both cortex and striatum regions. Scale bars = 20 µm. (e–f) Quantification of silver positive neurons in piriform cortex and striatum. There were fewer silver-positive neurons in both piriform cortex and striatum in tiagabine-treated samples. n=4 mice. *p< 0.05, compared to the values of vehicle group by standard student t-tests.
Tiagabine does affect huntingtin aggregates in N171-82Q HD mice
The extended polyglutamine repeat in huntingtin protein leads to abnormal folding and accumulation of aggregates in neurons (DiFiglia et al., 1997; Ross and Poirier, 2004). The EM48 antibody was used to determine the relative amount of aggregates in HD mouse brains. Interestingly, the most EM48-positive neurons were found in piriform cortex and hippocampus and there were fewer in striatum in N171-82Q HD mice. Treatment with tiagabine had no effect on htt aggregates in these brain regions (Fig. 4a–c).
Figure 4. Tiagabine did not affect huntingtin aggregates in N171-82Q mice.
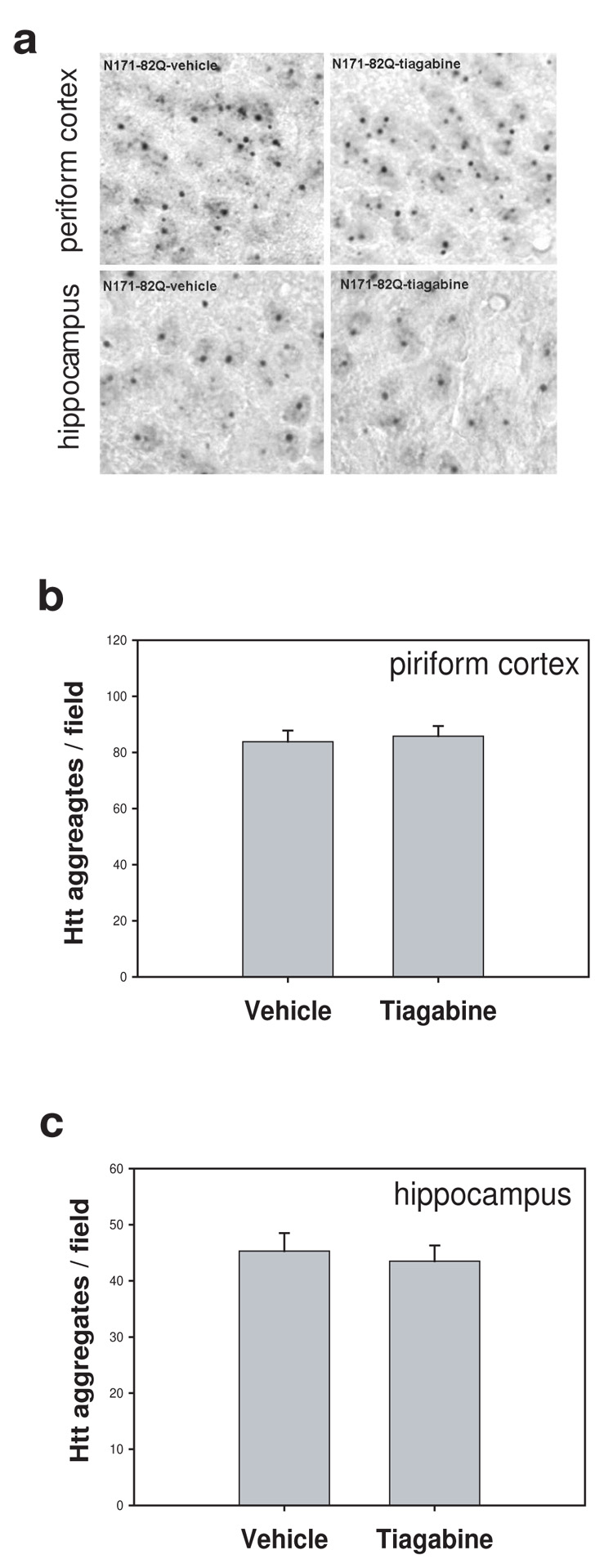
HD mice were treated with vehicle (saline) or tiagabine (5 mg/kg) from 8 weeks of age. Mice were perfused for EM48 immunostaining to detect the mutant huntingtin aggregates at 18 weeks of age. (a) Representative photographs of brain sections stained with EM48 antibody from indicated group. Scale bars=20 µm. (b–c) There was no difference in the density of cells with htt aggregates both in piriform cortex and hippocampus between N171-82Q mice treated with vehicle and tiagabine. n=4 mice.
Tiagabine decreases mortality and motor impairment, but does not prevent body weight loss in R6/2 mice
To further confirm the beneficial effects of tiagabine in HD, we examined R6/2 mice, which are widely used in preclinical trials (Hersch & Ferrante 2004). We found that R6/2 mice developed motor behavioral deficit as early as 4 weeks of age. Thus, tiagabine was administered to symptomatic R6/2 mice starting at 6 weeks of age. R6/2 mice treated with tiagabine at 5 mg/kg showed significantly decreased mortality compared to R6/2 mice treated with vehicle (Fig 5a). The average survival in HD mice treated with vehicle was 84.2 ± 3.5 (mean ±SE), but was 102.0 ± 2.9 in HD mice treated with tiagabine 5 mg/kg. We next evaluated the motor function in an accelerating rotarod test at 10 and 12 weeks of age since R6/2 mice showed severe deficit of motor function at these ages; however, R6/2 mice treated with tiagabine showed significantly improved motor performance at 10 weeks of age (Fig. 5c) and 12 weeks of age (Fig. 5d). R6/2 mice lost body weight with the progression of disease, and tiagabine administration did not prevent body weight loss in R6/2 mice (Fig. 5b).
Figure 5. Tiagabine increases survival and improves motor performance in R6/2 mice.
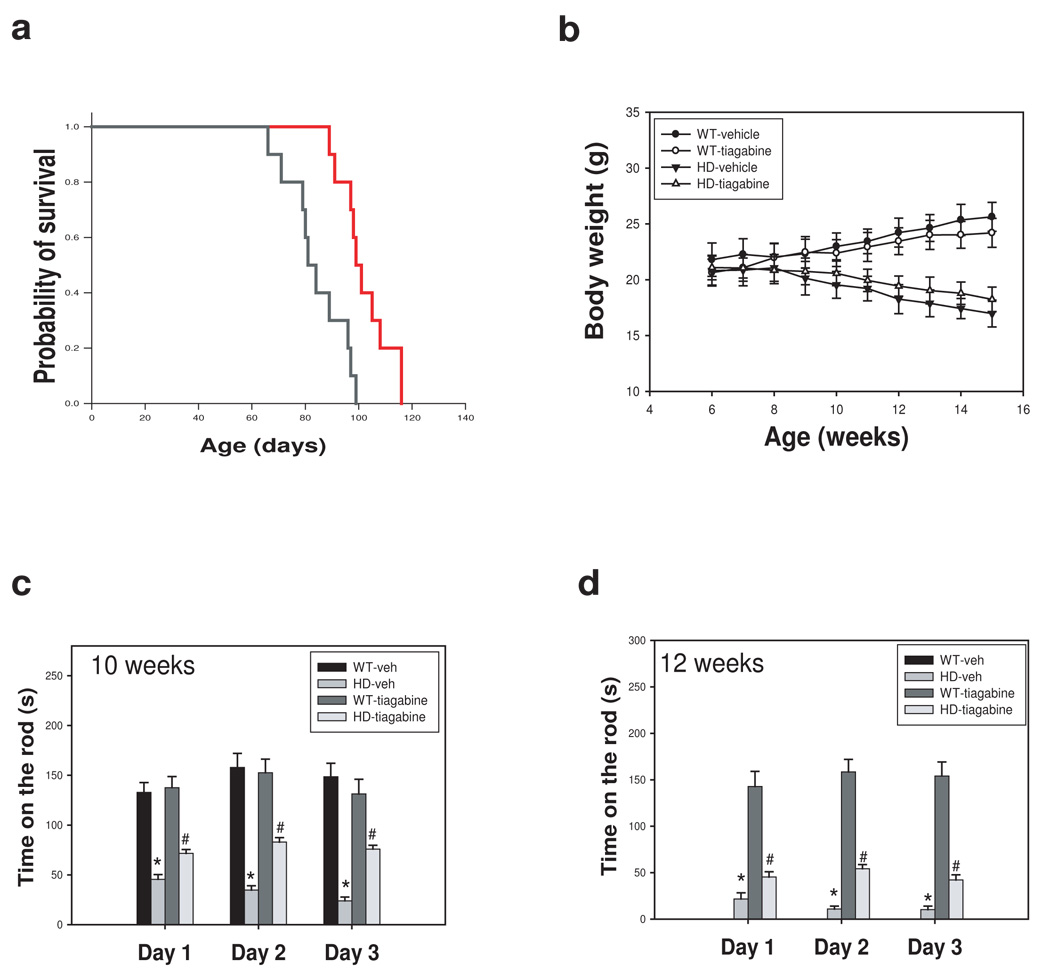
(a) 6-week-old R6/2 mice were injected with either vehicle (saline) or tiagabine (5 mg/kg) daily, and survival was monitored daily. Tiagabine at 5 mg/kg (red line) significantly increased survival. P<0.05 comparison between R6/2 –vehicle ( black line) and R6/2 tiagabine-treated group ( red line) by log rank test. (b) tiagabine did not affect body weight in R6/2 mice. (c) Motor behavioral performance was evaluated by an accelerating rotarod apparatus with mice at indicated ages, n=8–15. Values are the mean and SE. *p<0.05 compared to the value of WT-vehicle group; #p<0.05, compared to the value of HD-vehicle group by standard student t-tests.
Tiagabine attenuates brain atrophy but does not affect htt aggregation in R6/2 mice
There was severe brain atrophy indicated by enlarged ventricles, smaller striatum, thinner cortex, and reduced size of whole brain in R6/2 mice at 10 weeks old (Fig 6a). Tiagabine treatment attenuated brain atrophy (Fig. 6a–c). Widely distributed htt inclusions were detected by EM48 antibody in R6/2 mouse brains. Tiagabine treatment did not affect htt aggregates (Fig. 6d–f).
Figure 6. Tiagabine attenuates brain atrophy, but does not affect htt aggregates in R6/2 mice.
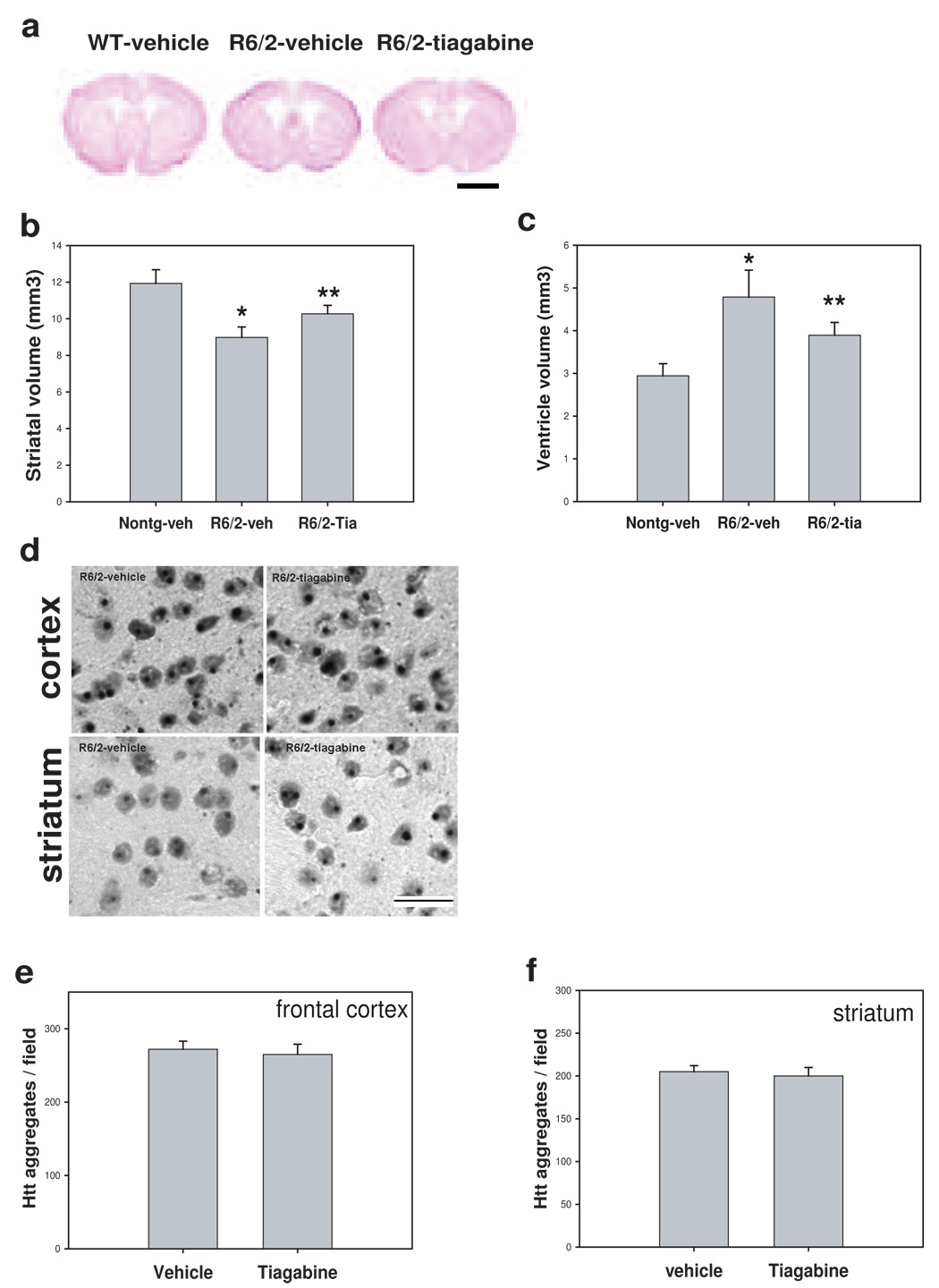
(a) Representative photographs of brain sections of Nissl staining from indicated group. Scale bar=120 µm. Note that R6/2 mouse brain showed enlarged ventricles and smaller striatum. Tiagabine treatment reversed the brain atrophy in R6/2 mice. (b–c) Quantification of brain atrophy. Tiagabine treatment significantly attenuated the brain atrophy in R6/2 mice. n=4 mice. *p< 0.05, compared to the values of nontransgenic-vehicle group (Nontg-veh); **p< 0.05, compared to the values of R6/2 mice treated with vehicle (R6/2-veh) by standard student t-tests. (d) Representative photographs of brain sections stained with EM48 antibody from indicated group. Scale bars=20 µm. (e–f) There was no difference in the density of cells with htt aggregates both in frontal cortex and striatum between R6/2 mice treated with vehicle and tiagabine. n= 4 mice.
Reinforcement of the GABA pathway does not appear to be involved in the mechanism of protection by tiagabine
In order to further determine whether protection of tiagabine in HD mice is due to its reinforcement of GABA pathway, we performed western blot analysis. There was no difference in the protein levels of GAD65/67 in HD mice treated with tiagabine compared to HD mice treated with vehicle (see suppl Fig), suggesting that the protection of tiagabine is not due to reinforcement of GABA signal.
Effective levels of tiagabine in mice are comparable to achievable levels in humans
In order to study whether effective doses in mice are comparable to doses achievable in humans, we used LC/MS/MS to measure the levels of tiagabine in both blood and brain tissues. Tiagabine was injected daily to mice for 1 week, and blood samples and brain tissues were collected for measuring drug concentrations. The blood concentrations were 17.2 ± 4.8 ng/ml (mean ± SD, n=10) in the 5 mg/kg group. The levels are comparable to the effective levels in patients who take 2 mg/day for 5 days (the plasma concentration of tiagabine is 20–50 ng/ml, Gustavson and Mengel, 1995). Drug concentrations in brain tissues were 177.0 ± 14.2 ng/g wet weight.
Discussion
There are currently no effective therapies either for preventing the onset or for slowing the progression of HD. Current therapies are symptomatic, and include the use of neuroleptics and psychotropic medications to decrease chorea, to address psychiatric symptoms including depression, obsessive compulsive symptoms, and psychosis. Our current studies demonstrated the beneficial effects of tiagabine in HD mice. We found that tiagabine prolonged survival, improved motor performance, and attenuated brain atrophy and neurodegeneration in both transgenic models of HD. Our initial studies were performed in N171-82Q mice that display a gradual onset of disease; we then confirmed the neuroprotective effect of tiagabine in R6/2 mice. It was important to test the effects of tiagabine in both N171-82Q and R6/2 models, because these two models have been used extensively in evaluating preclinical neuroprotective treatments of HD (Hersch & Ferrante, 2004), and we can easily compare the potency of tiagabine with that of reported compounds. Tiagabine treatment was started at the presymptomatic stage in N171-82Q mice and at the symptomatic stage of R6/2 mice; in both cases, tiagabine provided protection, suggesting that tiagabine may be given both to symptomatic patients and to gene-positive presymptomatic individuals.
In HD, the most vulnerable neurons are medium-sized spiny GABA projection neurons in striatum; consistent with the finding of loss of projection neurons was the early finding that GABA levels were markedly reduced in the caudate-putamen of HD patients (Perez-De La Cruz & Santamaria, 2006). The projection neurons are themselves divided into two classes. Both types contain GABA as a primary neurotransmitter, but of the two populations of striatal projection neurons, those of the indirect pathway (i.e., enkephalin/GABA-containing neurons) are affected first, thus providing an anatomical substrate for the increased movement that is the hallmark of HD (Reiner et al., 1988; Albin et al., 1992). In later stages of adult HD, both populations of striatal projection neurons are affected, with concomitant loss of markers of the direct pathway (i.e. , substance P/dynorphin/GABA-containing neurons), including dopamine D1 receptors and substance P (Reiner et al., 1988; Richfield et al., 1991). Tiagabine is one of the nipecotic acid analogues that successfully completed clinical trials and consequently was introduced to clinical practice. Although the exact mechanism of antiepileptic action of tiagabine is not fully understood, it is believed that this compound acts exclusively on GABAergic synapses by preventing GABA uptake by GAT-1, thus augmenting GABA-mediated inhibitory neurotransmission (Angehagen et al., 2003; Schachter 2001). Therefore, augmenting of GABAergic function may also be involved in neuroprotection by tiagabine in HD. Additionally, because tiagabine protected neurons against mutant huntingtin-induced toxicity in cultured cells, tiagabine may also provide direct neuroprotection in HD.
Tiagabine improved rotarod performance in both R6/2 and N171-82Q mice, but did not affect rotarod performance in nontransgenic control mice. These results suggest that tiagabine improved motor function by attenuating the pathological changes causing motor deficits induced by mutant huntingtin, not by improving the general activity and performance of mice.
Tiagabine reduced brain atrophy and neurodegeneration, but not huntingtin aggregates in HD mice. These data suggest that the neuroprotective effect of tiagabine does not appear to due to any effect on huntingtin aggregates. Interestingly, the brain regions that have most huntingtin aggregates are not those regions with large amounts of neurodegeneration. We found that, although there were large numbers of cells with aggregates in hippocampus of N171-82Q mice, neurodegeneration was not prominent in this region. There were very fewer cells with huntingtin aggregates in striatum, however, which is the region that has most neurodegeneration in HD. This is consistent with the finding that huntingtin protein that forms aggregates does not correlate positively with neuronal death and that it is the amount of diffuse intracellular huntingtin that leads to neuronal loss (Arrasate et al., 2004).
There is evidence of body weight loss in HD patients and mice, which may be due to the metabolic changes in HD. Tiagabine treatment could not prevent the body weight loss in both mouse models of HD, indicating that the neuroprotective effect of tiagabine is independent of a correction in metabolic change.
Our current studies suggest that tiagabine protects neurons in HD mice by mechanism(s) other than reinforcement of GABA pathway. Further study is needed to evaluate the neuroprotective mechanism of tiagabine in HD, and because tiagabine is a well-tolerated FDA-approved drug, it may be a good candidate for effective treatment of HD and other age-related neurodegenerative disorders.
Supplementary Material
Acknowledgements
We gratefully acknowledge the technical support of Laragen Inc. for genotyping service, and Dr. Pamela Talalay for her dedicated editorial assistance. This research was supported by the NS NINDS 055942 (to WD) and NS NINDS 16375 and NS38144 (to CAR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aiken CT, Tobin AJ, Schweitzer ES. A cell-based screen for drugs to treat Huntington's disease. Neurobiol Dis. 2004;16(3):546–555. doi: 10.1016/j.nbd.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Albin RL. Selective neurodegeneration in Huntington's disease. Ann Neurol. 1995;38(6):835–836. doi: 10.1002/ana.410380602. [DOI] [PubMed] [Google Scholar]
- Albin RL, Reiner A, Anderson KD, Dure LS, 4th, Handelin B, Balfour R, Whetsell WO, Jr, Penney JB, Young AB. Preferential loss of striato-external pallidal projection neurons in presymptomatic Huntington's disease. Ann Neurol. 1992;31(4):425–430. doi: 10.1002/ana.410310412. [DOI] [PubMed] [Google Scholar]
- Andersen KE, Sorensen JL, Lau J, Lundt BF, Petersen H, Huusfeldt PO, Suzdak PD, Swedberg MD. Synthesis of novel gamma-aminobutyric acid (GABA) uptake inhibitors. 5.(1) Preparation and structure-activity studies of tricyclic analogues of known GABA uptake inhibitors. J Med Chem. 2001;44(13):2152–2163. doi: 10.1021/jm990513k. [DOI] [PubMed] [Google Scholar]
- Angehagen M, Ben-Menachem E, Rönnbäck L, Hansson E. Novel mechanisms of action of three antiepileptic drugs, vigabatrin, tiagabine, and topiramate. Neurochem Res. 2003;28(2):333–340. doi: 10.1023/a:1022393604014. [DOI] [PubMed] [Google Scholar]
- Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature. 2004;431(7010):805–810. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- Cyr M, Caron MG, Johnson GA, Laakso A. Magnetic resonance imaging at microscopic resolution reveals subtle morphological changes in a mouse model of dopaminergic hyperfunction. NeuroImage. 2005;26:83–90. doi: 10.1016/j.neuroimage.2005.01.039. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intranuclear inclusions and dystrophic neurites in brain. Science. 1997;277(5334):1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, Aronin N. Aggregation of huntingtin in neuronal intracellular inclusions and dystrophic neuritis in brain. Science. 1997;277:1990–1993. doi: 10.1126/science.277.5334.1990. [DOI] [PubMed] [Google Scholar]
- DiFiglia M, Sena-Esteves M, Chase K, Sapp E, Pfister E, Sass M, Yoder J, Reeves P, Pandey RK, Rajeev KG, Manoharan M, Sah DW, Zamore PD, Aronin N. Therapeutic silencing of mutant huntingtin with siRNA attenuates striatal and cortical neuropathology and behavioral deficits. Proc Natl Acad Sci U S A. 2007;104(43):17204–17209. doi: 10.1073/pnas.0708285104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan W, Guo Z, Jiang H, Ware M, Li XJ, Mattson MP. Dietary restriction normalizes glucose metabolism and BDNF levels, slows disease progression, and increases survival in huntingtin mutant mice. Proc Natl Acad Sci U S A. 2003;100:2911–2916. doi: 10.1073/pnas.0536856100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustavson LE, Mengel HB. Pharmacokinetics of tiagabine, a gamma-aminobutyric acid-uptake inhibitor, in healthy subjects after single and multiple doses. Epilepsia. 1995;36(6):605–611. doi: 10.1111/j.1528-1157.1995.tb02575.x. [DOI] [PubMed] [Google Scholar]
- Hersch SM, Ferrante RJ. Translating therapies for Huntington's disease from genetic animal models to clinical trials. NeuroRx. 2004;1(3):298–306. doi: 10.1602/neurorx.1.3.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogsgaard-Larsen P, Frolund B, Frydenvang K. GABA uptake inhibitors. Design, molecular pharmacology and therapeutic aspects. Curr Pharm Des. 2000;6(12):1193–1209. doi: 10.2174/1381612003399608. [DOI] [PubMed] [Google Scholar]
- Landles C, Bates GP. Huntingtin and the molecular pathogenesis of Huntington's disease. Fourth in molecular medicine review series. EMBO Rep. 2004;5(10):958–963. doi: 10.1038/sj.embor.7400250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Li SH, Yu ZX, Shelbourne P, Li XJ. Huntingtin aggregate-associated axonal degeneration is an early pathological event in Huntington's disease mice. J Neurosci. 2001;21(21):8473–8481. doi: 10.1523/JNEUROSCI.21-21-08473.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luer MS, Rhoney DH. Tiagabine: a novel antiepileptic drug. Ann Pharmacother. 1998;32(11):1173–1180. doi: 10.1345/aph.18053. [DOI] [PubMed] [Google Scholar]
- Perez-De La Cruz V., Santamaria A. Integrative hypothesis for Huntington's disease: A brief review on experimental evidence. Physiol Res. Dec. 2006 doi: 10.33549/physiolres.931049. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Petersen A, Mani K, Brundin P. Recent advances on the pathogenesis of Huntington's disease. Exp Neurol. 1999;157(1):1–18. doi: 10.1006/exnr.1998.7006. [DOI] [PubMed] [Google Scholar]
- Reiner A, Albin RL, Anderson KD, D'Amato CJ, Penney JB, Young AB. Differential loss of striatal projection neurons in Huntington disease. Proc Natl Acad Sci U S A. 1988;85(15):5733–5737. doi: 10.1073/pnas.85.15.5733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richfield EK, O'Brien CF, Eskin T, Shoulson I. Heterogeneous dopamine receptor changes in early and late Huntington's disease. Neurosci Lett. 1991;132(1):121–126. doi: 10.1016/0304-3940(91)90448-3. [DOI] [PubMed] [Google Scholar]
- Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10 Suppl:S10–S17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- Schachter SC. Pharmacology and clinical experience with tiagabine. Expert Opin Pharmacother. 2001;2(1):179–187. doi: 10.1517/14656566.2.1.179. [DOI] [PubMed] [Google Scholar]
- Schilling G, Becher MW, Sharp AH, Jinnah HA, Duan K, Kotzuk JA, Slunt HH, Ratovitski T, Cooper JK, Jenkins NA, Copeland NG, Price DL, Ross CA, Borchelt DR. Intranuclear inclusions and neuritic aggregates in transgenic mice expressing a mutant N-terminal fragment of huntingtin. Hum Mol Genet. 1999;(3):397–407. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- Soudijn W, van Wijngaarden I. The GABA transporter and its inhibitors. Curr Med Chem. 2000;7(10):1063–1079. doi: 10.2174/0929867003374363. [DOI] [PubMed] [Google Scholar]
- Stefan H, Feuerstein TJ. Novel anticonvulsant drugs. Pharmacol Ther. 2007;113(1):165–183. doi: 10.1016/j.pharmthera.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57(5):369–384. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- Wanker EE. Protein aggregation and pathogenesis of Huntington's disease: mechanisms and correlations. Biol Chem. 2000;381(9–10):937–942. doi: 10.1515/BC.2000.114. [DOI] [PubMed] [Google Scholar]
- Wang W, Duan W, Igarashi S, Morita H, Nakamura M, Ross CA. Compounds blocking mutant huntingtin toxicity identified using a Huntington's disease neuronal cell model. Neurobiol Dis. 2005;20(2):500–508. doi: 10.1016/j.nbd.2005.03.026. [DOI] [PubMed] [Google Scholar]
- Yu ZX, Li SH, Evans J, Pillarisetti A, Li H, Li XJ. Mutant huntingtin causes context-dependent neurodegeneration in mice with Huntington's disease. J Neurosci. 2003;23(6):2193–2202. doi: 10.1523/JNEUROSCI.23-06-02193.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Hartke C, Jimeno A, Li J, He P, Zabelina Y, Hidalgo M, Baker SD. Specific method for determination of gefitinib in human plasma, mouse plasma and tissues using high performance liquid chromatography coupled to tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;819:73–80. doi: 10.1016/j.jchromb.2005.01.027. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.