

Abstract
Rhinoviruses are a frequent cause of the common cold. A series of antirhinoviral compounds have been developed that bind into a hydrophobic pocket in the viral capsid, stabilizing the capsid and interfering with cell attachment. The structures of a variety of such compounds, complexed with rhinovirus serotypes 14, 16, 1A, and 3, previously have been examined. Three chemically similar compounds, closely related to a drug that is undergoing phase III clinical trials, were chosen to determine the structural impact of the heteroatoms in one of the three rings. The compounds were found to have binding modes that depend on their electronic distribution. In the compound with the lowest efficacy, the terminal ring is displaced by 1 Å and rotated by 180° relative to the structure of the other two. The greater polarity of the terminal ring in one of the three compounds leads to a small displacement of its position relative to the other compounds in the hydrophobic end of the antiviral compound binding pocket to a site where it makes fewer interactions. Its lower efficacy is likely to be the result of the reduced number of interactions. A region of conserved residues has been identified near the entrance to the binding pocket where there is a corresponding conservation of the mode of binding of these compounds to different serotypes. Thus, variations in residues lining the more hydrophobic end of the pocket are primarily responsible for the differences in drug efficacies.
Human rhinovirus 16 (HRV16), like other picornaviruses (1), consists of 60 copies of four different viral proteins arranged to form an icosahedral shell around a single strand of positive sense RNA. The three major coat proteins, VP1, VP2, and VP3 (with molecular weights of approximately 30,000 Da) have the antiparallel β-barrel topology observed in many viral capsid proteins. The three-dimensional structure of HRV16 has been determined previously at 3.5-Å resolution (2) and subsequently refined by using data with a minimum Bragg spacing of 2.1 Å (3). HRV16 belongs to the major group of rhinoviruses (4) for which the cellular receptor is intercellular adhesion molecule-1 (ICAM-1) (5, 6). Other major-group rhinoviruses whose structures have been determined are HRV14 (7) and HRV3 (8). HRV1A (9) and HRV2 (N. Verdaguer, D. Blaas, and I. Fita, personal communication) are the only members of the minor group of rhinoviruses whose structures are known.
HRVs, of which there are at least 102 serotypes (1), are the major cause of the common cold. Because it is not possible to generate vaccines against so many HRVs, there is considerable interest in developing antiviral compounds that inhibit the virus infection. Compounds such as disoxaril (WIN 51711¶) (10, 11), which interact with the capsids of picornaviruses, have been known for a number of years. Although disoxaril entered phase I clinical trials, it failed toxicity tests. The crystallographic determination of the structure of WIN 51711, complexed with serotype 14 of HRV (12), was an essential step in the development of better therapeutic agents. A large number of antiviral compounds have been investigated crystallographically in complex with major-group rhinoviruses (8, 12–19), minor-group rhinoviruses (9, 20), polioviruses (21, 22), and coxsackievirus B3 (23). Structural studies have gone hand in hand with the development of more efficacious antiviral agents (24) against rhinoviruses and enteroviruses. A second compound, WIN 54954, reached phase II clinical trials, but was found to have low efficacy in vivo. Although this compound exhibited some activity when administered prophylactically, some undesirable side effects were observed (25). The third-generation compound, pleconaril (WIN 63843; Fig. 1) (26), which is closely related to the compounds discussed in this paper, is undergoing phase III clinical trials.
Figure 1.

Antiviral compounds. Full numbering of the atoms is given for compound 1 (corresponding to the Protein Data Bank file and topology file submitted to the Protein Data Bank). The other compounds are numbered in a corresponding fashion. Structural references are given in brackets under the compound names 4, 5, 6, and 7. MIC values are given when available.
These capsid-binding antiviral agents block uncoating of the viral particles (27, 28) and, in the case of some major-group rhinoviruses, also inhibit cell attachment (29). The antiviral compounds have been shown to bind into a hydrophobic pocket within VP1. In the absence of a compound, the pocket is either empty or is occupied by a cellular “pocket factor” proposed to be either a lipid or fatty acid (2, 30). When the pocket is filled with either an antiviral compound or a pocket factor, it has a bigger volume caused by an expansion of the β-barrel, which produces an “open” conformation.
The various serotypes of rhinoviruses also have been separated into two groups based on their cross-reactivity to a panel of antiviral compounds (31). Group A, which is more sensitive to longer compounds, includes HRV14 and HRV3. Group B, which is more sensitive to shorter compounds, includes HRV1A, HRV2, and HRV16. Although HRV16 shares a receptor with HRV14 and HRV3, it has greater sequence identity with HRV1A (80% in VP1) than with HRV14 or HRV3 (50% in VP1). Both HRV16 and HRV1A also have been observed to have small hydrophobic pocket factors, whereas the hydrophobic pocket was found to be empty in the native structures of HRV14 and HRV3.
The more efficacious WIN compounds generally contain three aromatic rings A, B, and C (Fig. 1). Ring C, an isoxazole ring, is separated from a phenoxy group (ring B) by an aliphatic chain. Ring A is linked to the phenoxy group by a single bond. The hydrophobic binding pocket in VP1 can be entered through a pore on the floor of the “canyon,” a depression on the viral surface that surrounds the 5-fold axis. A large variety of compounds have been studied when complexed to HRV14 (15, 32), HRV1A (20), and HRV3 (8). In HRV14, rings A and B generally will bind closer to the pore (the “heel” end of the pocket; Fig. 1) when the aliphatic linker in the compound is seven carbon atoms or more, although orientation is reversed for smaller compounds (17).
The minimum inhibitory concentration (MIC) of the antiviral compounds is defined as that concentration of the compound that is required to reduce the number of plaques formed by 50%. These measurements (plaque assays) are repeated at least three times, and the experimental results are reproducible within 2-fold accuracy for defined experimental conditions.
We report here refined structures of three WIN compounds, each complexed with HRV16. The compounds are similar in chemical structure (Fig. 1) but differ in the heteroatoms of the ring A, as well as in their MIC for different serotypes. These compounds correspond in length and binding orientation to the shorter compounds described by Badger et al. (17). The results provide a structural basis for the different efficacies of the compounds when complexed to HRV16 and other rhinovirus serotypes.
Materials and Methods
Virus and Crystal Preparation.
The virus was crystallized by using the hanging drop method. Viral particles at a concentration of 8–10 mg/ml in 0.25 mM Hepes, pH 7.5 were mixed with an equal volume of reservoir solution, 0.5–1.5% PEG 8000. Crystals were typically 0.2 mm × 0.2 mm × 0.2 mm, with cell dimensions a = 362.6 Å, b = 347.1 Å, and c = 334.9 Å in space group P22121. The antiviral compounds were provided by the former Sterling-Winthrop company and soaked into the crystals as described by Smith et al. (12) using a concentration of about 100 μg/ml in 0.2% DMSO.
Data Collection and Processing.
Data were collected at beamline F1, Cornell High Energy Synchrotron Source, by using x-rays with a wavelength of 0.908 Å. Crystals were cooled to 4°C. Images were recorded by using Fuji imaging plates at a distance of 295 mm. The crystal oscillation range was 0.3°, with exposure times ranging from 8 to 20 sec. Images were scanned by using a 100-μm raster step size with a Fuji BAS 2000 scanner. The films were indexed, integrated, and scaled by using the hkl package (33) (Table 1).
Table 1.
Summary of data collection and model refinement
| HRV16 in complex with: | Compound 1 | Compound 2 | Compound 3 |
|---|---|---|---|
| Resolution (Å) | 2.8 | 2.6 | 2.7 |
| Rmerge (%) (outer shell) | 7.8 (12.3) | 10.6 (18.8) | 11.0 (15.4) |
| Completeness (%) (outer shell) | 58.7 (44.5) | 48.8 (36.0) | 36.9 (32.6) |
| Corr. coef. (outer shell) | 91.6 (71.1) | 86.9 (59.3) | 86.4 (58.6) |
| Rw (outer shell) | 20.6 (27.0) | 23.1 (30.5) | 23.3 (29.5) |
| Rfree (outer shell) | 21.2 (28.2) | 23.5 (30.6) | 23.3 (29.0) |
| Protein residues/au | 797 | 797 | 797 |
| Solvent waters | 345 | 272 | 305 |
| Other ligands | 25 | 25 | 25 |
| rms dev. bonds (Å) | 0.006 | 0.01 | 0.006 |
| rms dev. angles (°) | 1.41 | 1.53 | 1.41 |
| Main chain (side chain) B (Å2) | 15.2 (17.2) | 19.0 (20.2) | 19.5 (21.0) |
| Solvent molecules B (Å2) | 25.8 | 25.6 | 21.8 |
| Antiviral compound B (Å2) | 14.4 | 20.6 | 22.1 |
Structure Determination.
The model derived from the open-pocket conformation of HRV16 (3) was used to phase the virus-compound complex. The pocket factor was omitted from the structure factor calculations, which were performed by using x-plor (34). Initial electron density maps were calculated for each compound based on model phases. These then were refined by iterative cycles of electron density averaging using programs written for parallel-processing computers (35). Convergence typically was achieved after 10–12 cycles. The compounds could be seen clearly in maps calculated with coefficients FobsWINexp[iαavgdWIN], where αavgdWIN is the phase derived by Fourier inversion of the icosahedrally averaged electron density map. Unit weights were used for all reflections in the first cycle of averaging to avoid bias toward the starting model. A geometric combination of Sim and Rayment weights (36) was used after two or three cycles. Because the antiviral compound-virus complex data were relatively incomplete, calculated structure factors were included in the electron density map after three cycles of averaging, with weights equal to the average in the resolution shell.
Crystallographic Refinement.
Model building was performed by using the program o (37). Strict noncrystallographic symmetry constraints were imposed throughout the crystallographic refinement procedures using the program x-plor (34). A free R factor was monitored by using 5% of the data. However, this factor was of limited use because of the high level of redundancy in the data as a result of 30-fold noncrystallographic symmetry (3). The Engh-Huber parameters were used for the refinement (38), as described for the native HRV16 structure (3).
Bond lengths and angles and partial charge assignments for ring C, the aliphatic chain, and ring A of compound 3 were based on parameters calculated for WIN 52084(S) (12). This compound has the same methyl isoxazole group at ring C, a longer aliphatic chain, an unsubstituted phenoxy group, and a terminal ring A as in compound 3, but with atoms 4 and 5 reversed (39). The equivalent parameters for the substituted phenoxy group (ring B), and ring A for compounds 1 and 2, were taken from calculations performed on WIN 58934 (40), a small compound very similar to rings A and B of compound 1.
A number of tests were carried out to validate the parameters and to confirm that there is a difference between the positions of ring A in a complex containing compound 1 and in a complex containing compound 3. A check showed that the difference in the final positions of compounds 1 and 3 was not simply a result of different starting positions. A model for compound 1 was built in the position observed for compound 3. After 40 cycles of standard crystallographic refinement, the atoms in ring A had moved by up to 1.2 Å from this displaced starting position, to be within 0.2 Å from their position in the previously refined structure. The orientation of ring A with respect to the methyl constituent also was determined from the refinement. Although all the compounds maintained good geometry in the absence of crystallographic data, initial attempts to refine compound 3 with the ring in the same orientation as in compound 1 resulted in distorted bond lengths and angles around the methyl group. After ring A was rotated by 180° in compound 3, the bond lengths and angles around this group maintained their expected values.
The compounds were modeled into the density manually before refinement. Low-temperature simulated annealing (highest temperature: 1,000°C; step size: 25°) was followed by Powell least-squares energy minimization. The final step in the crystallographic refinement was the calculation of individual isotropic B factors, justified by a data/parameter ratio of at least 16:1. The highest peaks in the final difference map, with Fourier coefficients FobsWIN-Fcalcexp[iαcalc], lie at the interface between the protein and the RNA. These probably correspond to those parts of the internal protein VP4 that could not be easily modeled (residues 8–22 and 45–67) and possibly to some icosahedrally ordered RNA, as was the case for the refined native HRV16 structure (3).
Structure Comparison.
HRV14 and HRV1A were superimposed on the refined model of HRV16 by using the LSQ_EXP and LSQ_IMP procedures in o (37). Using the refined structure of HRV16 as a basis for comparison, the rms distance between corresponding Cα atoms was 0.9 Å for HRV14 and HRV3 and 0.7 Å for HRV1A.
Results
Structure of Antiviral Complexes.
The structures of the complexes between HRV16 and each of compounds 1, 2, and 3 (Fig. 1) have been determined and refined to 2.8 Å resolution. The compounds could be seen clearly in electron density maps calculated with Fourier coefficients FobsWINexp[iαopen], where αopen are the phase angles calculated by using the protein atoms only with the drug binding pocket in the open conformation. FobsWIN are the observed structure factor amplitudes associated with a given HRV16-WIN compound complex. Subsequent to cycles of electron density averaging (see Materials and Methods), the final maps (two are shown in Fig. 2) show that all three compounds have close to full substitution in the 60 possible binding sites on each virion, as judged by the electron density for the compound being at roughly the same height as that of the surrounding protein. There is no evidence that some of the pockets are unoccupied and, therefore, in a closed conformation, as was seen in the native structure (3).
Figure 2.

The antiviral compound binding site. (A) An electron density section for the HRV16 + compound 1 complex contoured at a 2σ level, with the atomic model for compound 1 shown in the middle. (B) An electron density section for the HRV16 + compound 3 complex contoured at a 2σ level, with the atomic model for compound 3 shown in the middle. (C) Schematic representation of the WIN binding pocket in VP1 calculated with the molscript program (43). The side chains of all residues that have at least one atom lying within 4 Å of the WIN compounds are shown in black lines. Compound 1 is shown in ball-and-stick representation. A single water molecule, shown as a red sphere, lies within 4 Å of the compound. Two residues in the binding pocket are contributed by VP3. The green represents a portion of VP3 from a symmetry-related protomer.
The compounds lie slightly further into the hydrophobic binding pocket than the natural pocket factor (Fig. 3). There is no electron density corresponding to the position of the aliphatic chain of the pocket factor. Spherical density observed in the pocket entrance, at a position corresponding to the site of the pocket factor head group (a putative carboxylate) in the native virus, was, therefore, interpreted as a bound water molecule. This water molecule lies within hydrogen bonding distance of N2, the isoxazole ring nitrogen, and the main-chain nitrogen of Leu-1100. [Residues are given a four-digit notation. The first digit represents the chain identifier (1–4 for the viral proteins VP1, VP2, VP3, and VP4; 5 for water molecules; 6 for bound ions; 7 for antiviral compound); while the last three digits represent the amino acid sequence number or water oxygen identifier.] Four other water molecules are also in the WIN binding pocket at positions where water molecules were observed in the native structure, which contains a pocket factor. The most well-ordered of these is hydrogen-bonded to the side-chain hydroxyl groups of Tyr-1190 and Ser-1120. A water molecule was observed in an equivalent position in HRV14, both in the native structure (41) and in the presence of antiviral compounds (16). A water molecule closer to the pore hydrogen bonds to the side chain of Asn-1212 and to the main-chain nitrogen of Glu-1191. In HRV14-antiviral compound complexes, the equivalent asparagine commonly makes a hydrogen bond to polar groups on the compound (16, 17). Two further water molecules are involved in hydrogen bonding networks in the heel of the pocket.
Figure 3.

Conformational difference in residues surrounding ring A of the antiviral compound because of the displacement of the pocket factor by the compound. (A) Schematic representation of the WIN binding pocket in VP1, calculated with the molscript program (43), showing the pocket factor as a ball-and-stick model and compound 1 in magenta bonds. A single water molecule, shown as a red sphere, lies within 4 Å of the compound. (B) Electron density for the HRV16-compound 3 complex, with pocket factor model (black bonds) and WIN compound 3 (carbon, brown; nitrogen, blue; oxygen, red). A single water molecule, shown as a red sphere, lies within 4 Å of the compound. (C) Electron density for the HRV16-compound 3 complex, with HRV16-pocket factor model and WIN compound 3 colored as in B. Density is shown only around residues close to ring A, for clarity.
In each of the three virus-WIN compound complexes reported here, VP1 has a conformation similar to the “open pocket” observed in the native virus containing pocket factor (3). Very little rearrangement of the WIN binding pocket takes place on substitution of compounds 1, 2, or 3 for the pocket factor. The refined coordinates suggest that there is a small (<0.5 Å) systematic displacement of all eight of the β-strands in the β-barrel of VP1 away from the antiviral compounds. This displacement is largest in strands E (1143–1146) and G (1178–1186) where it is accompanied by some side-chain shifts of up to 0.8 Å around the toe of the pocket. The side chain of Tyr-1144 shows the biggest change in position on introduction of the antiviral compounds (Fig. 3), which, unlike the pocket factor, penetrate into the toe of the binding pocket. No significant changes were detected in the main-chain or side-chain conformations of the other viral proteins VP2, VP3, and VP4.
The distribution of temperature factors for the compound-virus complexes is similar to those of the virus by itself (3), with high values (>50 Å2) for some flexible side chains on the exterior of the virus and also for some disordered residues on the interior of the virus particle where the protein interacts with RNA. At the N terminus of VP3 and in parts of VP1 that interact with the antiviral compounds, the residue-averaged temperature factors were lower in the HRV16-compound complexes than in the native virus (3), consistent with binding of the WIN compounds decreasing the flexibility of the protein capsid.
Compounds 1 and 2.
The difference between compounds 1 and 2 is the position of attachment of the phenyl ring and methyl group to the tetrazole ring (ring A) (Fig. 1). There is no significant difference between the refined atomic coordinates of either of the two compounds or of the viral protein around the binding pocket. The MIC for compound 2 is twice that for compound 1. When each of these is complexed with HRV16, no significant difference was observed, with the two compounds binding in a similar fashion.
Compound 3.
Whereas compounds 1 and 2 have four nitrogens each and a single carbon in ring A, compound 3 has three carbons, an oxygen, and a nitrogen, with the methyl substituent remaining the same. The MIC against HRV16 is substantially larger for compound 3 than for compound 1 (25-fold) and compound 2 (12-fold). Compound 3 therefore might be expected to bind less well (42). The refined models of compounds 1, 2, and 3 differ only very slightly with respect to the isoxazole ring, the three-carbon chain, and the substituted phenoxy group. However, there is a small rotation about an axis perpendicular to the phenoxy group passing through the phenolic oxygen atom, which results in a shift of the position of all the atoms in ring A. Ring A also is rotated by 180o about the bond C4B-C2A in comparison to its orientation in compounds 1 and 2, as seen by the position of the methyl group on the ring (Fig. 4).
Figure 4.

Difference in binding of compounds 1 and 2 compared with compound 3. (A) Electron density for compound 1, contoured at a 1.5σ level. Compound 1 is shown in purple bonds and compound 3 is shown as black bonds. (B) Ball-and-stick representation of compound 1 (black sticks) and compound 3 (green sticks) viewed from (Lower) looking from the compound toward the outside of the virion and (Upper) looking from the compound toward the center of the virion. The insert localizes the site with respect to the symmetry axes in the icosahedral asymmetric unit. The labels X and Y define opposite sides of the WIN binding pocket. Side chains that are within 4 Å of the compounds are shown in thin black bonds. Water molecules are shown as red spheres with their hydrogen bonding environment in ball-and-stick representation. Compound 4 is overlaid in green dotted lines. The figure was prepared by using molscript (43).
Discussion
Comparison of Antiviral Compound-HRV16 Complex Structures.
Differences in the positioning of the compounds are localized to the hydrophobic toe of the pocket (Table 2, Fig. 4). There are two buried water molecules in this region lying within 7 Å of ring A of the compounds. The three residues that make the closest interactions with the compounds are Leu-1217, Tyr-1144, and Phe-1179 (Fig. 4). Atom CZ, at the tip of Phe-1179, lies approximately equidistant from all the atoms in the ring A of compounds 1 and 2. In contrast, the Phe ring lies off center compared with ring A of compound 3. On the other face of ring A, Tyr-1144 stacks parallel to the ring and makes the same extent of hydrophobic contact with all the compounds. There are 15 atoms that are situated less than 3.5 Å from any atom in ring A of compounds 1 and 2. In contrast, compound 3 makes 13 such contacts. The charge distribution in ring A for similar compounds (ref. 39; Don Phelps and Carol Post, personal communication) indicates this ring is more polar in compound 3 than in compounds 1 and 2, consistent with it making fewer close contacts in the hydrophobic pocket. The displacement of ring A in compound 3 is toward the side of the pocket where the buried water molecules are observed, which provides a more polar environment than the other side of the pocket.
Table 2.
Residues interacting with WIN compounds
| Position | HRV14 and HRV3 | HRV16 | HRV1A | |
|---|---|---|---|---|
| Ala-1150 | Tyr-1142 | Tyr-1145 | ||
| Pro-1174 | Ala-1166 | Met-1169 | ||
| Toe | Val-1176 | Val-1168 | Ile-1171 | |
| Leu-3014 | Met-3014 | Met-3014 | ||
| Ala-3024 | Ala-3024 | Ala-3024 | ||
| Ile-1130 | Met-1124 | Leu-1127 | ||
| Met-1224 | Leu-1217 | Ile-1220 | ||
| Phe-1186 | Phe-1179 | Phe-1182 | ||
| Val-1188 | Leu-1181 | Ile-1184 | ||
| Tyr-1152 | Tyr-1144 | Tyr-1147 | ||
| Val-1243 | Ile-1236 | Ile-1239 | ||
| Val-1077 | Ile-1077 | Val-1077 | ||
| Middle | Trp-1102 | Trp-1096 | Trp-1099 | |
| Ile-1104 | Ile-1098 | Ile-1098 | ||
| Tyr-1128 | Ile-1122 | Ile-1125 | ||
| Val-1191 | Leu-1184 | Leu-1187 | ||
| Met-1221 | Met-1214 | Met-1217 | ||
| Leu-1106 | Leu-1100 | Leu-1103 | ||
| Heel | Tyr-1197 | Tyr-1190 | Tyr-1193 | |
| Asn-1219 | Asn-1212 | Asn-1215 | ||
| Phe-1124 | Phe-1118 | Phe-1121 |
The residue(s) in bold are the most bulky residue in this position. The residues in italics are conserved between the serotypes in this position.
The location of the three compounds was compared with that of WIN 56291 (compound 4), which closely resembles compound 3 (Figs. 1 and 4). Although the structure of this compound was not refined, both the coordinates of the compound and the difference electron density coincide with the position of compound 3. Both compound 3 and WIN 56291 have A rings that are more polar and have higher MICs (Fig. 1) than compounds 1 and 2.
Comparison to HRV14, HRV3, and HRV1A.
The structures of various HRV14-, HRV1A-, and HRV3-WIN compound complexes (8, 14, 15, 17, 20) were superimposed on the refined model of HRV16, by minimizing the differences between equivalent Cα coordinates in the structurally conserved β-strands of VP1. The relative location of compounds in different serotypes of rhinoviruses then could be compared.
The side chains that interact with antiviral compounds in HRV16 were identified by selecting those residues that had at least one atom lying within 4 Å of any of the three compounds being discussed here. Side chains also were included that interacted with compound 7 (Protein Data Bank ID code 2RS5; ref. 17), a compound that has a similar chemical structure but a longer aliphatic chain (Fig. 1, ref. 17). Of the 21 residues thus identified, 10 residues are completely conserved in HRV16, HRV1A, HRV14, and HRV3. All of the residues that interact only with ring C in the heel of the pocket are in this category. Only one completely conserved residue is found to interact with ring A in the toe of the pocket. Of the six residues that interact only with ring A, HRV16 has three residues that protrude further into the pocket than those in HRV14 and two that are similar in size. HRV1A has four residues that are more protrusive. The decrease in the volume of the pocket in the group B viruses (HRV16 and HRV1A) correlates with the observation that shorter compounds are more efficacious against these serotypes (31).
Although there is a significant difference between the MICs of compounds 1 and 3 in HRV16, the difference between the MICs of these two compounds in either HRV14 or HRV3 (Fig. 1) is within experimental error. WIN 58768 (compound 6) is a partial structure of compound 1 and is missing the aliphatic chain and isoxazole ring. Crystallographic studies on this compound complexed with HRV14 (15) show that it binds in the toe, close to the position preferred in HRV16 for the binding ring A (Fig. 5). Complexes between HRV14 and other compounds with aliphatic chains of the same length as compounds 1 and 2, and with similar structures for rings A and B [e.g., compounds 4 (20) and 5 (14) in Fig. 1], also bind with ring A in the toe of the pocket (Fig. 5). In the central portion of the WIN binding pocket, two residues are bulkier in HRV14 and HRV3 than in HRV16 and HRV1A. Tyr-1128 (Ile-1122 in HRV16) and Met-1224 (Leu-1217 in HRV16) both are on the opposite side of the pocket closer to X in Fig. 5. On the side of the pocket closer to Y (Fig. 5), residues Leu-1181 and Leu-1184 in HRV16 protrude further into the pocket than the equivalent valines (1188 and 1191) in HRV14 and HRV3. The binding site of the antiviral compounds in HRV14 and HRV3 therefore is displaced compared with that in HRV16 (Fig. 5). At the heel end of the pocket, the isoxazole ring (ring C) of compounds 1 and 2 might be expected to bind to HRV14 in a similar position to that observed in HRV16, because the environment in that portion of the pocket is conserved (Table 2). Indeed, that is the case when the binding of ring C in compound 1 to HRV16 is compared with the binding of ring C in compound 5 to HRV14 (Fig. 5).
Figure 5.
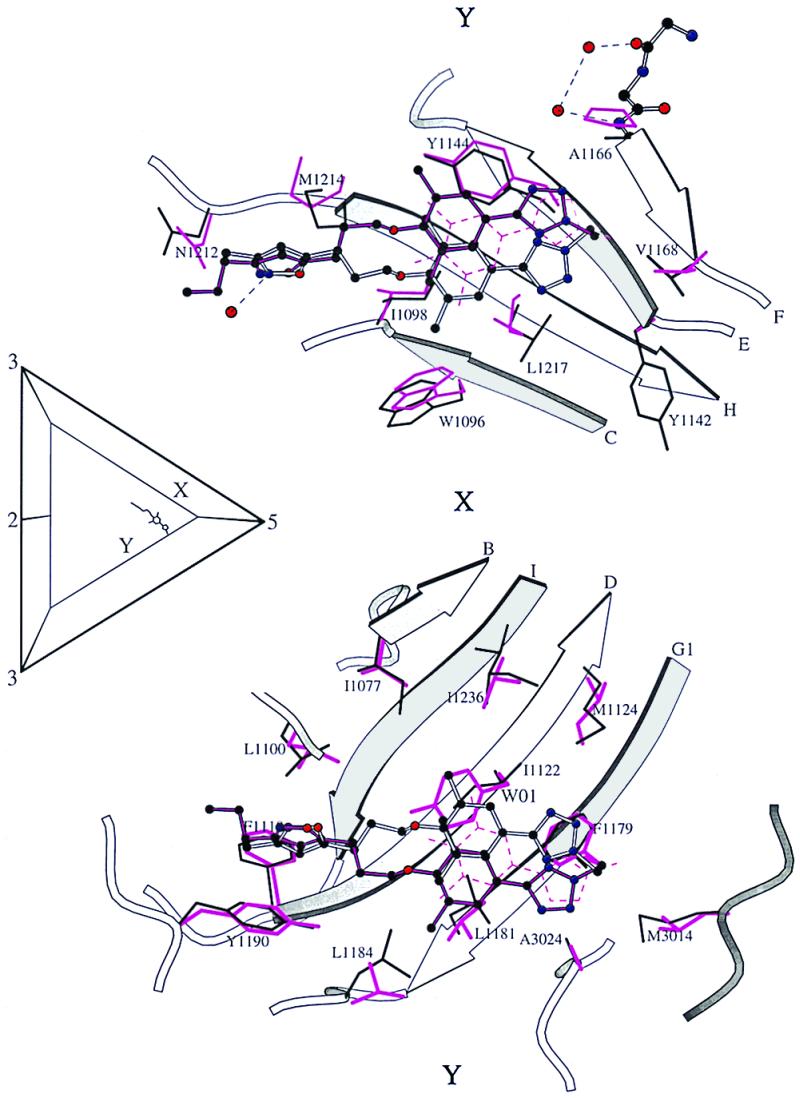
The same as Fig. 4B, but showing the comparison of compound binding sites in HRV16 and HRV14. Ball-and-stick representation of compound 1 (white sticks) bound to HRV16 (side chains within 4 Å shown in thin black bonds). The binding pocket of HRV14 with two compounds bound is superimposed. Compound 5 is shown in black ball-and-stick representation, while compound 6 is shown as dashed magenta lines (side chains within 4 Å of the compounds shown in magenta bonds). The comparison is based on a least-squares fit between Cα atoms in the β-barrel of VP1.
The less bulky side chains found in the toe of the pocket in HRV14 and HRV3 will have the effect of reducing the number of hydrophobic interactions around rings A and B of compounds 1 and 2 in these viruses compared with HRV16. The low efficacy of compounds 1 and 2 in HRV14 compared with their efficacy in HRV16 therefore can be explained in terms of presumably a lower binding energy. In HRV14, the positioning of the small compound 6 in the pocket (Fig. 5) was known to be determined by interactions with ring B and the conserved regions in the binding pocket around ring C. The table of MICs (Fig. 1) shows that HRV14 and HRV3 are less able to discriminate between compounds 1 or 2 as compared with compound 3 than is HRV16. This is possibly because ring A only partially fills the hydrophobic cavity in these serotypes and, hence, is not able to make the close interactions that favor the binding of ring A in compounds 1 and 2 compared with the more polar ring A in compound 3.
Conclusions
The structures of three HRV16-antiviral compound complexes have been determined to a resolution of 2.8 Å. The chemically similar compounds, closely related to a drug that is undergoing phase III clinical trials, were chosen to determine the structural impact of differences in one of the three rings. The compounds have binding modes that depend on their electronic distribution. In the compound with the lowest efficacy (compound 3), the terminal ring is displaced by 1 Å and rotated by 180° relative to the structure of the other two. The more polar nature of the terminal ring in that compound leads to a displaced position in the binding pocket. Its lower efficacy is likely to be caused by lower binding energy. Variations in the more hydrophobic end (the toe end) of the pocket are responsible for the differences in drug efficacies. A region of conserved residues is identified at the other, more hydrophilic, end (the heel end) of the drug binding pocket, resulting in a greater consistency of the antiviral compound binding mode at this region of the pocket. These results emphasize the importance of the toe end of the pocket in the design of antipicornavirus capsid-binding compounds.
Acknowledgments
We thank Iwona Minor for assistance with the processing of crystallographic data, Sharon Wilder and Cheryl Towell for help in preparation of this paper, and Dan Pevear for helpful discussions. The work was funded by grants to M.G.R. from the National Institutes of Health and the former Sterling-Winthrop Pharmaceutical Research Division. A Lucille P. Markey Foundation award and a Purdue reinvestment grant provided additional funding for the structural biology facility at Purdue University.
Abbreviations
- HRV
human rhinovirus
- MIC
minimum inhibitory concentration
Footnotes
Data deposition: The atomic coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID codes 1qju, 1qjx, and 1qjy for compounds 1, 2, and 3, respectively).
The projects initiated at Sterling Winthrop, involving compounds designated by the prefix WIN and certain serial numbers, have been taken over by ViroPharma, Inc.
References
- 1.Rueckert R R. In: Virology. Fields B N, Knipe D M, Howley P M, editors. New York: Lippincott–Raven; 1996. pp. 609–654. [Google Scholar]
- 2.Oliveira M A, Zhao R, Lee W M, Kremer M J, Minor I, Rueckert R R, Diana G D, Pevear D C, Dutko F J, McKinlay M A, Rossmann M G. Structure (London) 1993;1:51–68. doi: 10.1016/0969-2126(93)90008-5. [DOI] [PubMed] [Google Scholar]
- 3.Hadfield A T, Lee W, Zhao R, Oliveira M A, Minor I, Rueckert R R, Rossmann M G. Structure (London) 1997;5:427–441. doi: 10.1016/s0969-2126(97)00199-8. [DOI] [PubMed] [Google Scholar]
- 4.Abraham G, Colonno R J. J Virol. 1984;51:340–345. doi: 10.1128/jvi.51.2.340-345.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greve J M, Davis G, Meyer A M, Forte C P, Yost S C, Marlor C W, Kamarck M E, McClelland A. Cell. 1989;56:839–847. doi: 10.1016/0092-8674(89)90688-0. [DOI] [PubMed] [Google Scholar]
- 6.Staunton D E, Merluzzi V J, Rothlein R, Barton R, Marlin S D, Springer T A. Cell. 1989;56:849–853. doi: 10.1016/0092-8674(89)90689-2. [DOI] [PubMed] [Google Scholar]
- 7.Rossmann M G, Arnold E, Erickson J W, Frankenberger E A, Griffith J P, Hecht H J, Johnson J E, Kamer G, Luo M, Mosser A G, et al. Nature (London) 1985;317:145–153. doi: 10.1038/317145a0. [DOI] [PubMed] [Google Scholar]
- 8.Zhao R, Pevear D C, Kremer M J, Giranda V L, Kofron J, Kuhn R J, Rossmann M G. Structure (London) 1996;4:1205–1220. doi: 10.1016/s0969-2126(96)00128-1. [DOI] [PubMed] [Google Scholar]
- 9.Kim S, Smith T J, Chapman M S, Rossmann M G, Pevear D C, Dutko F J, Felock P J, Diana G D, McKinlay M A. J Mol Biol. 1989;210:91–111. doi: 10.1016/0022-2836(89)90293-3. [DOI] [PubMed] [Google Scholar]
- 10.Fox M P, Otto M J, McKinlay M A. Antimicrob Agents Chemother. 1986;30:110–116. doi: 10.1128/aac.30.1.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Otto M J, Fox M P, Fancher M J, Kuhrt M F, Diana G D, McKinlay M A. Antimicrob Agents Chemother. 1985;27:883–886. doi: 10.1128/aac.27.6.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Smith T J, Kremer M J, Luo M, Vriend G, Arnold E, Kamer G, Rossmann M G, McKinlay M A, Diana G D, Otto M J. Science. 1986;233:1286–1293. doi: 10.1126/science.3018924. [DOI] [PubMed] [Google Scholar]
- 13.Zhang A, Nanni R G, Oren D A, Rozhon E J, Arnold E. Semin Virol. 1992;3:453–471. [Google Scholar]
- 14.Giranda V L, Susso G R, Felock P J, Bailey T R, Draper T, Aldous D J, Suiles J, Dutko F J, Diana G D, Pevear D C. Acta Crystallogr D. 1995;51:496–503. doi: 10.1107/S0907444994011674. [DOI] [PubMed] [Google Scholar]
- 15.Bibler-Muckelbauer J K, Kremer M J, Rossmann M G, Diana G D, Dutko F J, Pevear D C, McKinlay M A. Virology. 1994;202:360–369. doi: 10.1006/viro.1994.1352. [DOI] [PubMed] [Google Scholar]
- 16.Chapman M S, Minor I, Rossmann M G, Diana G D, Andries K. J Mol Biol. 1991;217:455–463. doi: 10.1016/0022-2836(91)90749-v. [DOI] [PubMed] [Google Scholar]
- 17.Badger J, Minor I, Kremer M J, Oliveira M A, Smith T J, Griffith J P, Guerin D M A, Krishnaswamy S, Luo M, Rossmann M G, et al. Proc Natl Acad Sci USA. 1988;85:3304–3308. doi: 10.1073/pnas.85.10.3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Badger J, Minor I, Oliveira M A, Smith T J, Rossmann M G. Proteins. 1989;6:1–19. doi: 10.1002/prot.340060102. [DOI] [PubMed] [Google Scholar]
- 19.Badger J, Krishnaswamy S, Kremer M J, Oliveira M A, Rossmann M G, Heinz B A, Rueckert R R, Dutko F J, McKinlay M A. J Mol Biol. 1989;207:163–174. doi: 10.1016/0022-2836(89)90447-6. [DOI] [PubMed] [Google Scholar]
- 20.Kim K H, Willingmann P, Gong Z X, Kremer M J, Chapman M S, Minor I, Oliveira M A, Rossmann M G, Andries K, Diana G D, et al. J Mol Biol. 1993;230:206–226. doi: 10.1006/jmbi.1993.1137. [DOI] [PubMed] [Google Scholar]
- 21.Grant R A, Hiremath C N, Filman D J, Syed R, Andries K, Hogle J M. Curr Biol. 1994;4:784–797. doi: 10.1016/s0960-9822(00)00176-7. [DOI] [PubMed] [Google Scholar]
- 22.Hiremath C N, Grant R A, Filman D J, Hogle J M. Acta Crystrallogr D. 1995;51:473–489. doi: 10.1107/S090744499401084X. [DOI] [PubMed] [Google Scholar]
- 23.Muckelbauer J K, Kremer M, Minor I, Tong L, Zlotnick A, Johnson J E, Rossmann M G. Acta Crystallogr D. 1995;51:871–887. doi: 10.1107/S0907444995002253. [DOI] [PubMed] [Google Scholar]
- 24.Diana G D, Pevear D C. Antiviral Chem Chemother. 1997;8:401–408. [Google Scholar]
- 25.Turner R B, Dutko F J, Goldstein N H, Lockwood G, Hayden F G. Antimicrob Agents Chemother. 1993;37:297–300. doi: 10.1128/aac.37.2.297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fromtling R A, Castaner J. Drugs Future. 1997;22:40–44. [Google Scholar]
- 27.Diana G D, McKinlay M A, Otto M J, Akullian V, Oglesby C. J Med Chem. 1985;28:1906–1910. doi: 10.1021/jm00150a025. [DOI] [PubMed] [Google Scholar]
- 28.McSharry J J, Caliguiri L A, Eggers H J. Virology. 1979;97:307–315. doi: 10.1016/0042-6822(79)90342-8. [DOI] [PubMed] [Google Scholar]
- 29.Pevear D C, Fancher M J, Felock P J, Rossmann M G, Miller M S, Diana G, Treasurywala A M, McKinlay M A, Dutko F J. J Virol. 1989;63:2002–2007. doi: 10.1128/jvi.63.5.2002-2007.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Filman D J, Syed R, Chow M, Macadam A J, Minor P D, Hogle J M. EMBO J. 1989;8:1567–1579. doi: 10.1002/j.1460-2075.1989.tb03541.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andries K, Dewindt B, Snoeks J, Wouters L, Moereels H, Lewi P J, Janssen P A J. J Virol. 1990;64:1117–1123. doi: 10.1128/jvi.64.3.1117-1123.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Giranda V L. Structure (London) 1994;2:695–698. doi: 10.1016/s0969-2126(00)00069-1. [DOI] [PubMed] [Google Scholar]
- 33.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 34.Brünger A T. x-plor: A System for X-ray Crystallography and NMR. New Haven: Yale Univ. Press; 1992. , Version 3.1. [Google Scholar]
- 35.Cornea-Hasegan M A, Zhang Z, Lynch R E, Marinescu D C, Hadfield A, Muckelbauer J K, Munshi S, Tong L, Rossmann M G. Acta Crystallogr D. 1995;51:749–759. doi: 10.1107/S0907444995001399. [DOI] [PubMed] [Google Scholar]
- 36.Arnold E, Rossmann M G. Acta Crystallogr A. 1988;44:270–282. doi: 10.1107/s0108767387011875. [DOI] [PubMed] [Google Scholar]
- 37.Jones T A, Zou J Y, Cowan S W, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 38.Engh R A, Huber R. Acta Crystallogr A. 1991;47:392–400. [Google Scholar]
- 39.Lybrand T P, McCammon J A. J Comput Aided Mol Des. 1988;2:259–266. doi: 10.1007/BF01532989. [DOI] [PubMed] [Google Scholar]
- 40.Phelps D L, Rossky P J, Post C B. J Mol Sci. 1998;276:331–337. doi: 10.1006/jmbi.1997.1542. [DOI] [PubMed] [Google Scholar]
- 41.Arnold E, Rossmann M G. J Mol Biol. 1990;211:763–801. doi: 10.1016/0022-2836(90)90076-x. [DOI] [PubMed] [Google Scholar]
- 42.Fox M P, McKinlay M A, Diana G D, Dutko F J. Antimicrob Agents Chemother. 1991;35:1040–1047. doi: 10.1128/aac.35.6.1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kraulis P. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]