

Abstract
We have used molecular modeling techniques to design a dissociable covalently bonded base pair that can replace a Watson-Crick base pair in a nucleic acid with minimal distortion of the structure of the double helix. We introduced this base pair into a potential precursor of a nucleic acid double helix by chemical synthesis and have demonstrated efficient nonenzymatic template-directed ligation of the free hydroxyl groups of the base pair with appropriate short oligonucleotides. The nonenzymatic ligation reactions, which are characteristic of base paired nucleic acid structures, are abolished when the covalent base pair is reduced and becomes noncoplanar. This suggests that the covalent base pair linking the two strands in the duplex is compatible with a minimally distorted nucleic acid double-helical structure.
Keywords: nucleic acid double helix, covalent base pair, nonenzymatic template-directed ligation
The importance of the nucleic acids in biology and biotechnology has been the motivation for many studies of oligonucleotide analogues. The sugar-phosphate backbones of RNA and DNA have been replaced by a variety of alternative backbones (1–4), a number of alternative or additional base pairs have been described (5, 6), and the possibility of using shape analogues of the bases has been explored (7, 8). Cross-linking of nucleic acid strands has also been discussed in many publications (9–14), but, as far as we are aware, reversible cross-linking leading to a “covalent base pair” without distortion of the structure of the double helix has not been described.
An ideal covalent base pair would mimic closely the geometry of a standard base pair. In particular, it should be approximately planar, and the glycosidic angles and the distance between the glycosidic carbon atoms should be as close as possible to those in a standard nucleic acid double helix. In the context of molecular replication and prebiotic chemistry, it is also important that the covalent base pair can be formed and dissociated reversibly. Devadas and Leonard (15) have synthesized glycosidic derivatives of fused heterocyclic systems that resemble pairs of nucleosides in a DNA double helix—for example, structure A. These molecules are coplanar and have geometry fairly close to that of a standard base pair, but they cannot form and dissociate reversibly. Another interesting base pair (B) has been described by Cowart and Benkovic (16) and by Webb and Matteucci (17). This adduct, although not coplanar, can adopt a coplanar structure in the context of a nucleic acid double helix, but it cannot dissociate reversibly. ![]()
Here we describe the design and synthesis of a covalent base pair that can replace a normal Watson-Crick base pair in nucleic acid with a minimum of structural perturbation. We have incorporated it into a DNA double helix by nonenzymatic aqueous solution chemistry and have shown that the helix can be formed and dissociated reversibly.
Materials and Methods
De Novo Design of Covalently Bonded Base Pair.
We decided to study molecules in which two aromatic systems are joined by a C⩵N—N linker: 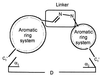
Systems of this kind are conjugated and, therefore, approximately coplanar and can be synthesized fairly easily from appropriate hydrazines and aldehydes. The hydrazone bond can be dissociated by treatment with excess hydrazine.
We investigated isolated five- and six-membered rings and fused bicyclic ring systems as possible aromatic components of the covalent base pair. For each possible structure, D, α1, and α2 were calculated by using standard geometry parameters. Structures with D in the range 10.8 ± 3.0 Å, and α1 and α2 in the range 50° ± 30° were selected for further consideration. In each of the selected structures, the simple aromatic rings were systematically replaced by possible pyrimidine and related heterocyclic systems. These structures were then optimized with molecular mechanics, CFF91 force field (18), using Discover module in insightii software (Biosym Technologies, San Diego). In a second screen, the structures with D in the range 10.8 ± 1.5 Å and α1 and α2 in the range 50° ± 15° were selected for a final refinement by using modified neglect of differential overlap semiempirical quantum chemistry (19). The bestfit structure I was used to suggest a closely related structure II for experimental investigation. We chose II because it would be expected to have a geometry similar to that of I and can be synthesized more easily. ![]()
Although II has several degrees of freedom because of its flexibility, the conformation shown here is always found to be the most stable by systematic search and optimization at PM3 semiempirical quantum chemistry level (20). The geometry of the proposed covalent base pair, calculated by using PM3 semiempirical quantum chemistry, closely resembles that of a standard base pair (Fig. 1). The estimated values of D, and α1 and α2 are 10.5 Å and 48.5°/57.1°, very close to those for the DNA double helix (10.8 Å and 52°). Dynamic simulation and optimization suggests that a double helix containing this covalent base pair should be stable.
Figure 1.

(A) The structure of the proposed covalently bonded base pair. (B) The structure of a standard base pair. (C) Superposition of the covalently bonded base pair (darker) with a Watson-Crick base pair (lighter).
Overview of the Synthesis of a Nucleic Acid Incorporating a Covalent Base Pair.
The structure of a single-stranded nucleic acid modified to include a covalently bonded base pair is illustrated in Fig. 2. The base pair in adduct Template⩵F is formed by condensing a nucleoside analogue F containing an aldehyde group (V) with a N4-aminocytidine residue in the target single-stranded nucleic acid. Outlines of the synthesis of the dialdehyde and details of the conversion of a cytidine residue in the target strand to a N4-aminocytidine residue are given below.
Figure 2.

Scheme for the synthesis of an oligonucleotide incorporating a covalent base pair (the hydrazone bond is indicated by the arrow). i, SnCl4/HMDS/TCS/CH3CN. ii, NaHCO3. iii, POCl3/DMF. iv, H2O. v, MeONa/MeOH. vi, NH2NH2/NaHSO3/phosphate buffer (pH 5). vii, F/phosphate buffer (pH 5). HMDS, hexamethyldisilazane; TCS, trimethylchlorosilane; Template, oligonucleotide template; Template′, oligonucleotide incorporating a N4-aminocytidine residue; Template⩵F, template covalently linked to the nucleoside analogue F by means of a hydrazone bond. Either T3CT3 or Tem (Table 1) can be used as the Template.
Synthesis of Compound V.
The synthesis of compound V is illustrated in Fig. 2. 4-Methyl-1-(2,3,5-tri-O-benzoyl-β-d-ribofuranosyl)-2-pyrimidinone (III) is synthesized efficiently by the reaction of silylated 4-methyl-2-pyrimidinone with 1-O-acetyl-2,3,5-tri-O-benzoyl-β-d-ribofuranose. The methyl group of the pyrimidinone is then converted to an α-diformylmethyl group by the Vilsmeier reaction (21). The product is deprotected to give the nucleoside analogue (compound V, designated as F) as a light yellow powder in 45% overall yield. The product was characterized by NMR, mass spectroscopy, and UV spectroscopy.
Incorporating a Covalent Base Pair into a Single-Strand DNA.
All standard oligodeoxynucleotides were synthesized by using an Applied Biosystems DNA Synthesizer, were deprotected in concentrated ammonia at 55°C, and were concentrated by using an OP 120 OligoPrep instrument (Savant). They were then purified on a 20% denaturing polyacrylamide gel (PAGE in 1× TBE buffer) and were eluted with TE buffer (0.01 M Tris⋅HCl/0.001 M EDTA, pH 8). A NENSORB 20 column (DuPont) was used to desalt the oligomers. 5′-32P-labeled oligomers were prepared by using T4 polynucleotide kinase (New England Biolabs) and [γ-32P]ATP (Amersham Pharmacia) (22). Enzymatic synthesis of 5′-32P-labeled oligonucleotide 3′,5′-diphosphates was carried out as described (23). Oligomers used in this work and various derivatives of them are listed in Table 1. Oligomers L3 and L5 are complementary to the 3′ and 5′ segments, respectively, of template Tem.
Table 1.
The oligodeoxynucleotides and their derivatives used in these experiments
| Oligodeoxynucleotides and their derivatives | Symbol |
|---|---|
| 5′TTTCTTT3′ | T3CT3 |
| 5′TTT-C-TTT3′ | |
| | | T3C′T3 |
| NH2 | |
| 5′TTT-C-TTT3′ | |
| ∥ | T3(C⩵F)T3 |
| F | |
| 5′TGTGTGTTGTG-C-GTTGGTGGTTTGGT3′ | Tem |
| 5′TGTGTGTTGTG-C-GTTGGTGGTTTGGT3′ | |
| ∥ | Tem⩵F |
| F | |
| 5′TGTGTGTTGTG-C-GTTGGTGGTTTGGT3′ | |
| | | Tem⩵F |
| F | |
| 5′CACAACACACA3′ | L5 |
| 5′GCACAACACACA3′ | GL5 |
| 5′CAAACCACCAAC3′ | L3 |
| 5′pCAAACCACCAACpGCACAACACACA3′ | pL3pGL5 |
| 5′TGTGTGTTGTG-C-GTTGGTGGTTTGGT3′ | |
| ∥ | Tem⩵L5pF |
| 3′ACACACAACACpF5′ | |
| 5′TGTGTGTTGTG-C-GTTGGTGGTTTGGT3′ | |
| ∥ | Tem⩵FpL3p |
| 3′FpCAACCACCAAACp5′ | |
| 5′TGTGTGTTGTG-C-GTTGGTGGTTTGGT3′ | |
| ∥ | Tem⩵L5pFpL3p |
| 3′ACACACAACACpFpCAACCACCAAACp5′ | |
| 5′pCAAACCACCAACpF3′ | FpL3p |
| 5′FpCACAACACACA3′ | L5pF |
In Fig. 3, we illustrate our general approach to the incorporation of a covalent base pair into a presynthesized oligodeoxynucleotide. The incorporation protocols were developed by using a short oligodeoxynucleotide T3CT3 as substrate. The conversion of a cytidine residue to N4-aminocytidine was achieved by using a slight modification of a published procedure (24). A reaction solution containing the oligonucleotide (1 nM to 1 mM), 4 M hydrazine, 0.3 M sodium bisulfite, and 0.1 M phosphate buffer at pH 5.0 (the pH was adjusted with concentrated HCl) in a 50-μl volume, was incubated at 50°C for 3 hours. The reaction was stopped by dilution with 500 μl TET buffer (0.1 M Tris⋅HCl/0.001 M EDTA/0.01 M triethylamine, pH 8) and was desalted on a NENSORB column. The N4-aminocytidine residue is unstable and decomposes slowly, even at −20°C. The freshly formed oligomer containing a N4-aminocytidine residue must be condensed immediately with the aldehyde.
Figure 3.

General approach to the synthesis of nucleic acid double helices incorporating a covalently bonded base pair by nonenzymatic template-directed ligation. The oligonucleotide segments are those listed in Table 1.
Oligomer T3C′T3 (0.5 nM to 5 mM), prepared as described above, was incubated with a 0.01 M solution of the nucleoside analogue F in 0.05 M phosphate buffer (pH 5.0) at room temperature for 36 hours (reaction volume 20 μl). The reaction was stopped by mixing with gel loading buffer (a mixture of 900 μl of deionized formamide, 25 μl of 2% xylene cyanol, 25 μl of 2% bromophenol blue, and 100 μl of 10× TBE buffer) and 10 M urea in the ratio of 2:1:2. The hydrazone derivative was purified by gel electrophoresis. The identity of the adduct, T3(C⩵F)T3, was confirmed by matrix-assisted laser desorption ionization (MALDI) mass spectrometry: C81H104N18O52P6 (calculated 2,346.45, found 2,346). The unmodified oligomer T3CT3 did not react with analogue F under identical conditions.
The protocols developed by using T3CT3 as described above were then used to synthesize a longer adduct, Tem⩵F, and the 32P-labeled adduct, p*Tem⩵F. The hydrazone bond in adduct p*Tem⩵F could be reduced to a C—N single bond by treatment with sodium borohydride. The adduct p*Tem⩵F (10,000 cpm in 10 μl of reaction volume) was treated with 0.1 M NaBH4 and 0.001 M EDTA in 0.025 M phosphate buffer (pH 6.8) at room temperature for 45 minutes. The reduction reaction was terminated by 20-fold dilution with TET buffer at 0°C. The product p*Tem—F was desalted immediately on a NENSORB column and was used in the ligation reactions without further purification.
Ligation Reactions.
The labeled oligomer p*L3p (1 × 103 to 8 × 105 cpm in 10 μl of reaction volume) was treated with 0.11 M 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDC) in 0.1 M imidazole·HCl solution (pH 6.0) at room temperature for 1 hour to give the phosphorimidazolide of p*L3p, Im(p*L3p). An aliquot of the solution containing Im(p*L3p) (4 μl), without further purification, was then added to the ligation reaction mixture (total reaction volume 40 μl), which contained the analogue Tem⩵F(Imp*L5:Tem⩵F ≈ 1:1.5), 0.1 M KCl, 0.05 M MgCl2, and 0.1 M Tris⋅HCl (pH 7.4) (see Fig. 3, a). The solution was first heated to 70°C, was allowed to cool to 25°C in 45 minutes, and then was left at room temperature for 4 to 7 days. The reaction was terminated by adding 0.5 M EDTA (10 μl). The reaction products were desalted and analyzed by gel electrophoresis. Experiments in which EDC or MgCl2 was omitted were used as controls. A standard ligation experiment with unmodified oligonucleotides GL5 and Im(p*L3p) aligned on the unmodified template Tem was carried out under identical conditions to permit comparison between a standard ligation reaction and ligation to a covalent base pair. A ligation reaction between Tem⩵F and p*L5 (Fig. 3, b) was carried out in the same way. In addition, we purified the product Tem⩵FpL3p* of the first ligation reaction by gel electrophoresis and used it in a second ligation reaction with p*L5 (Fig. 3, c).
The ligation products, Tem⩵L5p*F and Tem⩵FpL3p* (10,000 cpm), were dissociated by treating them with 0.1 M hydrazine at room temperature for 10 hours (5-μl reaction volume). A parallel reaction omitting hydrazine was used as a control.
The orientations of the products of the ligation reactions were determined by dephosphorylation with alkaline phosphatase (Boehringer Mannheim) (22). Ligation products, Tem⩵L5p*F and Tem⩵FpL3p*, were first dissociated with excess hydrazine, and the product oligomers, L5p*F and FpL3p*, which contain the nucleotide analogue F, were purified by gel electrophoresis. L5p*F and FpL3p* (1,000 cpm) were then treated with alkaline phosphatase (0.1 unit) in 50 mM Tris⋅HCl, and 0.1 mM EDTA in 10 μl (pH 8.5). The reaction products were analyzed by PAGE.
Results and Discussion
We used computer modeling techniques to design de novo a dissociable covalent base pair II that can be formed by reaction between two nucleoside analogues, the dialdehyde F and N4-aminocytidine. We synthesized F in 45% overall yield from 1-O-acetyl-2,3,5-tri-O-benzoyl-β-d-ribofuranose and 4-methyl-2-pyrimidinone. A N4-aminocytidine residue in an oligonucleotide was obtained from a cytidine residue by treating the oligomer with hydrazine in the presence of bisulfite as a catalyst. The nucleoside F reacts readily with the N4-aminocytidine residues in a single-stranded nucleic acid to form a hydrazone derivative (covalent base pair). Treatment of 5′-32P-labeled oligomer T3CT3 with hydrazine under conditions that convert cytidine to N4-aminocytidine does not change its mobility (Fig. 4, lane 2). However, subsequent treatment of the N4-aminocytidine-containing oligomer with the nucleoside analogue F results in the formation of the adduct p*T3(C⩵F)T3, which has a reduced electrophoretic mobility (Fig. 4, lane 3). The yield is ≈85%.
Figure 4.

Incorporation of a covalent base pair into a DNA strand. Oligomer p*T3CT3 (lane 1) was treated with hydrazine (lane 2) and then was condensed with nucleoside analogue F (lane 3).
Nonenzymatic template-directed ligation reactions can be used to join two oligonucleotides aligned on a complementary template. If the structure of the covalent base pair resembles the structure of a standard base pair closely enough, the F residue that is covalently linked to the template should be able to replace the complex of the template with one of the complementary oligomers in this reaction. If the presumed covalent base pair differs greatly in structure from a standard base pair, it would not be expected to ligate efficiently. The oligomer Tem is a 26-mer with a single C residue at position 12. Nonenzymatic template-directed ligation between the nucleoside analogue F covalently linked to the unique C residue of the template and complementary oligodeoxynucleotide sequences was used to test whether the covalent base pair disturbs the double helical structure of DNA. Fig. 3 depicts the general design of the experiments.
We preactivated the p*L3p by treating it with EDC and imidazole⋅HCl at pH 6. The yield of the 5′-32P-labeled phosphorimidazolide of p*L3p, Im(p*L3p) varied from 70% to 90% (data not shown). The formation of the ligation product Tem⩵Fp*L3p of Tem⩵F with Im(p*L3p) after 7 days at room temperature is shown in Fig. 5, lane 1 (≈44% yield). In a control reaction in which the activation step was omitted, no ligation product is observed (Fig. 5, lane 2). When Mg2+ is omitted from the reaction mixture, the yield of ligation product is much reduced (Fig. 5, lane 3). We carried out a parallel standard ligation reaction using ≈0.7 equivalent of p*L3p and a 10-fold excess of GL5 to guarantee that the 5′-end of the template Tem was fully occupied. The yield of the standard ligation product was only 7% (Fig. 5, lane 4). Ligation in the covalent template is clearly much more efficient (≈6×) than standard ligation between two oligonucleotides. Presumably, this is partly attributable to incomplete hybridization in the standard ligation reaction, but the efficiency of the reaction with the covalent base pair is nonetheless surprising. The corresponding ligation reaction involving the 5′-phosphate-terminated oligomer (p*L5) and Tem⩵F gave a similar yield (48%) of the ligation product Tem⩵L5p*F.
Figure 5.

Autoradiograms of the products of ligation reaction using the 26-mer template Tem. A compares ligation to the covalently bonded base pair to standard nonenzymatic template-directed ligation. Lanes: 1, ligation of Tem⩵F with p*L3p; 2, control omitting EDC; 3, control omitting Mg2+; 4, standard ligation of GL5 with p*L3p. B illustrates the preparation of the duplex containing an internal covalently bonded base pair (Tem⩵L5p*FpL3p*, lane 5). Lanes: 6, ligation of Tem⩵F with p*L5; 7, ligation of Tem⩵F with p*L3p and the ligation product Tem⩵FpL3p* was used to ligate to p*L5 shown in lane 5; 8, control experiment, in which Im(p*L3p) and Imp*L5 were incubated together in absence of template. C illustrates the effect of the reduction of the hydrazone bond on the yield of ligation products. Lanes: 9, ligation of L5 with p*Tem⩵F; 10, ligation of L5 with p*Tem—F, the reduction product of p*Tem⩵F.
Next, we synthesized a nucleic acid duplex containing an internal covalent base pair. We prepared Tem⩵FpL3p* as described above and purified it by gel electrophoresis. This product was then incubated with activated p*L5 under the standard ligation condition. Gel electrophoresis (Fig. 5, lane 5) revealed the formation of the higher molecular weight product, Tem⩵L5p*FpL3p*. A control ligation reaction of p*L3p and p*L5 in the absence of template gave no detectable ligation product (Fig. 5, lane 8). Tem⩵L5p*FpL3p* can also be formed by ligation of Tem⩵L5p*F with p*L3p, but with lower efficiency, presumably because an isolated 5′-hydroxyl group is a less good nucleophile than a cis-glycol (data not shown).
To establish that efficient ligation depends on the geometry of the covalent base pair and not only on its proximity to the activated phosphate group of its neighbor, we next attempted a ligation reaction to a covalently linked oligomer with altered structure. Reduction with NaBH4 converts Tem⩵F to Tem—F in which the C⩵N double bond (hydrazone bond) has been reduced to a C—N single bond. After reduction, the base pair is no longer coplanar. In Fig. 5, lane 9, we show the product p*Tem⩵L5p*F from a ligation reaction between 5′-32P-labeled Tem⩵F and activated 5′-32P-labeled L5 and, in Fig. 5, lane 10, the product from the corresponding reaction with the reduced form p*Tem—F. Clearly, reduction of the C⩵N double bond results in a complex that ligates with much decreased efficiency.
The hydrazinolysis of Tem⩵L5p*F is illustrated in Fig. 6. Tem⩵L5p*F can be dissociated by treatment with excess hydrazine. As anticipated, the product Fp*L5 of hydrazinolysis (Fig. 6, lane 2) has an electrophoretic mobility less than that of the labeled starting material p*L5 (Fig. 6, lane 3).
Figure 6.

Dissociation of covalent base pair with excess hydrazine. Lanes: 1, duplex Tem⩵L5p*F; 2, dissociation products from Tem⩵L5p*F after treatment with excess hydrazine; 3, p*L5.
Ligation of Tem⩵F with p*L5 should produce an internally labeled phosphodiester group. In Fig. 7, lane 2, we show that the dissociated product L5p*F is not dephosphorylated by alkaline phosphatase, an enzyme that cleaves phosphomonoesters but not phosphodiesters, confirming that L5p*F does indeed contain an internally labeled phosphate group. The corresponding ligation product, FpL3p*, includes a labeled 5′-terminal phosphomonoester group, which is, as anticipated, removed by alkaline phosphatase (Fig. 7, lane 4). These experiments confirm that the complementary pairing of the short oligomers on the template is essential for efficient ligation reaction.
Figure 7.

Dephosphorylation with alkaline phosphatase (AP) of products obtained by dissociating duplexes incorporating the covalent base pair with hydrazine. Lanes: 1, L5p*F; 2, attempted dephosphorylation of L5p*F with alkaline phosphatase; 3, FpL3p*; 4, dephosphorylation of FpL3p* with alkaline phosphatase.
The experiments described above establish that efficient ligation occurs between the covalently attached F residue and an adjacent activated oligonucleotide. We have shown that an oligodeoxynucleotide incorporating a single covalent base pair resembles an oligodeoxynucleotide-primer complex. Non-enzymatic ligation to an appropriate complementary oligonucleotide is more efficient than the corresponding ligation reaction between pairs of oligonucleotides. In an important series of controls, we find that reduction of the C⩵N double bond in the covalent base pair, which abolishes its planarity, greatly reduces the efficiency of the ligation reactions. We believe that these results establish that covalent cross-linking occurs to give a double helix, the structure of which is little different from the standard nucleic acid duplex structure, although, without structural data, we cannot quantitate the extent of distortion.
Minimally distorted, cross-linked nucleic acid duplexes should prove useful in exploring the mechanisms of enzymes that act on double-helical nucleic acids because they would help to establish the extent to which strand separation is an essential feature of the reaction mechanisms. The inclusion of one or more covalent base pairs should also permit that stabilization of normally inaccessible secondary and tertiary structure. The experimental results presented have established that it is possible to form covalent base pairs in a fairly simple way. This would justify the considerable effort that would be needed to develop protected nucleoside analogues for the routine solid-phase synthesis of cross-linkable oligomers. The present covalent base pair would not be useful in antisense applications because both the antisense oligomer and its target would need to be modified. However, the possibility of developing base-analogues that cross link reversibly to a normal base to generate a covalent base pair needs to be explored.
The covalent base pair that we have synthesized is not of direct relevance to prebiotic chemistry because there are no plausible prebiotic syntheses of the component hydrazine-containing and aldehyde-containing nucleotides. However, they do show in a more general way that a genetic system base on covalent rather than hydrogen bonding is possible (25). If life originated at high temperatures, this would be an attractive option.
Acknowledgments
We thank Aubrey R. Hill, Jr. for technical assistance, Bernice Walker for manuscript preparation, Senyon Choe for access to a workstation (Silicon Graphic, Inc.), and Gerald F. Joyce and Xiaochang Dai for assistance with mass spectroscopy. We also thank Dr. Alex Rich, Guoqiang Shi, Bin Zhou, Jian Li, Guangcheng Jiang, Shaokai Jiang, Barbara C. F. Chu, and Zhiyuan Zhang for helpful discussions. This work is supported by the National Aeronautics and Space Administration Grant NAGW-1660 and National Aeronautics and Space Administration Specialized Center of Research and Training/Exobiology Grant NAGW-2881.
Abbreviation
- EDC
1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride
References
- 1.Eschenmoser A, Leowenthal E. Chem Soc Rev. 1992;21:1–16. [Google Scholar]
- 2.Joyce G F, Orgel L E. In: The RNA World. Gesteland R F, Atkins J F, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1999. pp. 49–77. [Google Scholar]
- 3.Bolli M, Litten J C, Schutz R, Leumann C. J Chem Biol. 1996;3:197–206. doi: 10.1016/s1074-5521(96)90263-x. [DOI] [PubMed] [Google Scholar]
- 4.Koshkin A A, Nielsen P, Meldgaard M, Rajwanshi V K, Singh S K, Wengel J. J Am Chem Soc. 1998;120:13252–13253. [Google Scholar]
- 5.Lutz M J, Held H A, Hottiger M, Hubscher U, Benner S A. Nucleic Acids Res. 1996;24:1308–1313. doi: 10.1093/nar/24.7.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Strobel S A, Cech T R, Usman N, Beigelman L. Biochemistry. 1994;33:13824–13835. doi: 10.1021/bi00250a037. [DOI] [PubMed] [Google Scholar]
- 7.Morales J C, Kool E T. Nat Struct Biol. 1998;5:950–954. doi: 10.1038/2925. [DOI] [PubMed] [Google Scholar]
- 8.Guckian K M, Krugh T T, Kool E T. Nat Struct Biol. 1998;5:954–959. doi: 10.1038/2930. [DOI] [PubMed] [Google Scholar]
- 9.Knorre D G, Vlassov V V. Prog Nucleic Acid Res Mol Biol. 1985;32:291–320. doi: 10.1016/s0079-6603(08)60352-9. [DOI] [PubMed] [Google Scholar]
- 10.Webb T R, Matteucci M D. Nucleic Acids Res. 1986;14:7661–7664. doi: 10.1093/nar/14.19.7661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong Q, Barsky D, Colvin M E, Melius C F, Ludeman S M, Moravek J F, Colvin O M, Bigner D D, Modrich P, Friedman H S. Proc Natl Acad Sci USA. 1995;92:12170–12174. doi: 10.1073/pnas.92.26.12170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang H, Zhu L, Reid B R, Drobny G P, Hopkins P B. Science. 1995;270:1842–1845. doi: 10.1126/science.270.5243.1842. [DOI] [PubMed] [Google Scholar]
- 13.Coleman R S, Pires R M. Nucleic Acids Res. 1997;25:4771–4777. doi: 10.1093/nar/25.23.4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fagan P A, Spielmann P, Sigurdsson S T, Rink S M, Hopkins P B, Wemmer D E. Nucleic Acids Res. 1996;24:1566–1573. doi: 10.1093/nar/24.8.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Devadas B, Leonard N J. J Am Chem Soc. 1990;112:3125–3135. [Google Scholar]
- 16.Cowart M, Benkovic S J. Biochemistry. 1991;30:788–796. doi: 10.1021/bi00217a032. [DOI] [PubMed] [Google Scholar]
- 17.Webb T R, Matteucci M D. J Am Chem Soc. 1986;108:2764–2765. [Google Scholar]
- 18.Maple J, Dinur U, Hagler A T. Proc Nat Acad Sci USA. 1988;85:5350–5354. doi: 10.1073/pnas.85.15.5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dewar M J S, Thiel W. J Am Chem Soc. 1977;99:4899–4907. [Google Scholar]
- 20.Stewart J P. J Comp Chem. 1989;10:209–220. [Google Scholar]
- 21.Marson C M, Giles P R. Synthesis Using Vilsmeier Reagents. Boca Raton, FL: CRC; 1994. [Google Scholar]
- 22.Maniatis T, Fritsch E F, Sambrook J. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1982. [Google Scholar]
- 23.Chu B C F, Orgel L E. Methods Mol Biol. 1994;26:145–165. doi: 10.1007/978-1-59259-513-6_5. [DOI] [PubMed] [Google Scholar]
- 24.Negishi K, Nitta N, Yamashita Y, Takahashi M, Harada C, Oohara K, Nishizawa M, Wataya Y, Hayatsu H. Nucleic Acid Res. 1983;12:29–30. [PubMed] [Google Scholar]
- 25.Weber A L. Origins Life Evol Biosphere. 1989;19:179–186. doi: 10.1007/BF01808151. [DOI] [PubMed] [Google Scholar]