

Abstract
The immunosuppressant rapamycin inhibits Tor1p and Tor2p (target of rapamycin proteins), ultimately resulting in cellular responses characteristic of nutrient deprivation through a mechanism involving translational arrest. We measured the immediate transcriptional response of yeast grown in rich media and treated with rapamycin to investigate the direct effects of Tor proteins on nutrient-sensitive signaling pathways. The results suggest that Tor proteins directly modulate the glucose activation and nitrogen discrimination pathways and the pathways that respond to the diauxic shift (including glycolysis and the citric acid cycle). Tor proteins do not directly modulate the general amino acid control, nitrogen starvation, or sporulation (in diploid cells) pathways. Poor nitrogen quality activates the nitrogen discrimination pathway, which is controlled by the complex of the transcriptional repressor Ure2p and activator Gln3p. Inhibiting Tor proteins with rapamycin increases the electrophoretic mobility of Ure2p. The work presented here illustrates the coordinated use of genome-based and biochemical approaches to delineate a cellular pathway modulated by the protein target of a small molecule.
Cells have the remarkable ability to adapt to the dynamic pool of nutrients in their immediate environment. Mammalian cells respond continually to changes in available blood glucose and amino acids. Single cell organisms such as the budding yeast Saccharomyces cerevisiae are able to adapt to more extreme changes in nutrients such as a lack of fermentable carbon or of metabolically accessible nitrogen. The sensing and integration of such environmental cues require complex regulatory pathways that remain poorly understood.
Yeast cells treated with the Streptomyces hygroscopicus-derived small molecule rapamycin resemble ones deprived of nutrients in many ways. Rapamycin induces G1 cell cycle arrest, translation arrest, glycogen accumulation, sporulation, and autophagy and represses rRNA transcription (1–5). In addition, rapamycin was shown recently to reduce the level of mRNA transcripts corresponding to several yeast ribosomal proteins, independently of protein translation (6).
Rapamycin binds to Fpr1p (FKBP12 in mammalian cells), and this complex binds to and inactivates Tor1p and Tor2p (FRAP/RAFT1 in mammalian cells) with high specificity (7, 8). The primary function of Tor and FRAP proteins is currently thought to be the regulation of protein translation. These proteins are members of a family of high molecular weight phosphatidyl inositol kinase-related kinases that include ATM, ATR, and DNA-PK, mediators of DNA damage pathways (9, 10). Because Tor1p/2p and FRAP inhibition elicits cellular responses indicative of the physiologic state of starvation, these proteins are thought to be mediators of nutrient-sensing pathways (2).
Starvation, however, is a broad term that encompasses several diverse regulatory pathways. Prominent pathways in yeast include sporulation, general amino acid control, which regulates amino acid biosynthesis (11), glucose activation, which regulates hexose transport (12), the diauxic shift, which regulates the usage of fermentable carbon, nitrogen discrimination (also named nitrogen regulation, nitrogen catabolite repression), which responds to the quality of available nitrogen (13), and nitrogen starvation, which responds to a lack of available nitrogen. Often, activation of distinct nutrient-sensitive pathways results in gross physiologic similarities (nitrogen starvation or lack of fermentable carbon both induce autophagy, for example). Therefore, the physiologic response to rapamycin is insufficient to discriminate which nutrient-sensitive pathways are modulated by Tor proteins. To determine more accurately the cellular pathways affected by rapamycin, we used DNA microarrays to examine the genome-wide transcription program in yeast over a period of 2 h after treatment with rapamycin.
Materials and Methods
Construction of Microarrays.
A set of 6,220 verified ORFs from Saccharomyces cerevisiae was obtained from Research Genetics (Huntsville, AL). Each double-stranded ORF contains common 19-bp sequences at the 5′ and 3′ ends. Each ORF was amplified to levels required for arraying by PCR using primers corresponding to the common ends. Reactions (100 μl) containing 0.5 ng of ORF DNA, 50 pmol of each primer, 5 units of Taq polymerase, 0.25 units of Pfu polymerase, 200 μM each deoxynucleotide, 1.5 mM MgCl2, 1 mM DTT in Taq reaction buffer were subjected to 35 PCR cycles (1 min 92°C, 1 min 55°C, 4 min 72°C). Samples (3 μl) from each amplification reaction were analyzed by agarose gel electrophoresis to verify that the expected product was formed. Reactions that failed to produce single bands of the correct molecular weight were excluded from further analysis (≈5%). Amplified ORFs were precipitated with isopropanol in 96-well V-bottom polypropylene microtiter plates, pelleted by centrifugation, dried, and resuspended in 25 μl of Micro Spotting Solution (Telechem International, Sunnyvale, CA). A portion (12 μl) of each ORF was aliquoted to a second set of 96-well plates from which microarrays were printed. ORFs (6,220) as well as several internal quality controls were spotted onto poly-l-lysine-coated glass microscope slides (Labscientific, Livingston, NJ) with a microarraying robot constructed from plans published by the laboratory of Patrick Brown (http://cmgm.stanford.edu/pbrown/). One hundred and thirty-seven copies of the microarray were produced in 75 h. Each cDNA spot was ≈125 μm in diameter and was spaced 220 μm from its neighbors in the 1.8- × 1.8-cm arrays. Microarrays were stored in the dark at room temperature until they were processed immediately before hybridization according to published protocols (14).
Growth of Saccharomyces cerevisiae Cultures.
For expression profiling experiments with rapamycin, a single colony of S. cerevisiae strain BY4741 [MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) or BY4743 (diploid; BY4741/BY4742 (MATa his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0)] was used to inoculate 50 ml of YPD medium (2% glucose/2% peptone/1% yeast extract). The cells were grown at 30°C on a shaker at 225 rpm to a density of 1 × 108 cells/ml, were diluted 1:200 in YPD prewarmed to 30°C, and then were allowed to continue growing to a density of 1 × 107cells/ml (A600 = 1.0). Rapamycin was added to a final concentration of 100 nM, and 40-ml aliquots of cells were harvested at the indicated times. The harvested cells were pelleted by centrifugation for 10 min at 3,000 × g and 30°C, flash-frozen in liquid nitrogen, and stored at −80°C until RNA was extracted.
Preparation and Hybridization of cDNA Probes to Microarrays.
Total RNA was extracted with acidic phenol. Poly-A RNA was further isolated from total RNA by using oligo(dT) resin (Oligotex, Qiagen, Chatsworth, CA). Fluorescent cDNA probes were made by oligo(dT)-primed reverse transcription using Cy3-dUTP or Cy5-dUTP and poly-A RNA template essentially as described (14).
Data Acquisition and Analysis.
Fluorescent cDNA that bound to the microarrays was detected with an ArrayWoRx microarray scanner (Applied Precision, Issaquah, WA). The scanner uses a full spectrum illumination source in combination with user-selected excitation/emission filter sets and a cooled-CCD camera. Each microarray was scanned as a series of image panels at a resolution of 5 μm per pixel (81 Cy3 panels, 81 Cy5 panels). The image panels were stitched together in software to create high-resolution, two channel images. The arrayworx software package automatically locates individual spots in the microarray, quantitates the Cy3 and Cy5 fluorescence intensity at each spot, and determines background signal intensities (Applied Precision). Normalization between the Cy3 and Cy5 channels was achieved by calculating the ratio of total Cy3 signal from all spots in the microarray to total Cy5 signal from all spots. Data from spots that were marred by dust particles or hybridization artifacts were excluded from further analysis. Cy5/Cy3 intensity ratios were exported to genespring, a software package designed to display and analyze microarray data (Silicon Genetics, San Carlos, CA).
Anti-Ure2p Immunoblots.
Jk9–3da cells (15 ml of cells) (MATa trp1 his4 leu2 ura3 rme1 HMLa, kindly provided by Michael Hall, University of Basel) in early log phase (A600 = 0.3) growing in YPD at 30°C were treated with either no drug, 100 nM rapamycin, 100 nM FK506, or poor-quality nitrogen for 15 min. After 15 min of incubation at 30°C, cells were pelleted and resuspended in ice-cold lysis buffer A (0.5% Triton X-100/20 mM NaPO4, pH 7.2/2 mM EDTA/2 mM EGTA/25 mM NaF/100 μM Na3VO4/25 mM β-glycerophosphate/200 mM PMSF/1.5 mM β-mercaptoethanol). Cells were lysed by vortexing with 0.3 g glass beads (prerinsed with lysis buffer A) eight times for 20 seconds (chilling on ice between vortexing). Cellular debris was spun away, and equal volumes of lysate were resolved by SDS/PAGE through a 12.5% gel. Proteins were transferred to PVDF membrane (Immobilon-P, Millipore) and were blotted with antibodies against Ure2p (kindly provided by Reed Wickner, National Institutes of Health, Bethesda, MD) by using enhanced chemiluminescence (Amersham Pharmacia). CY5754 cells (MATa SSD1-v ura3–1 leu2–3, -112 his3–11, -15 trp1–1 ade2–1 can1–100 tap42∷TRP1 TAP42 on Leu2/Cen, kindly provided by Charles DiComo, Columbia University, New York) were processed identically except they were treated for 5 min with 20 nM rapamycin and were resuspended in lysis buffer B (20 mM Tris, pH 7.4/100 mM KCl/2 mM MgCl2/0.1 mM EDTA/1.5 mM β-mercaptoethanol), and lysates were resolved on a 7.5% gel. Cells in which the wild-type TOR1 gene was replaced with a rapamycin-resistant TOR1 allele (tor1-S1972T) and the parental strain from which they were derived were also treated similarly, except that they were treated for 30 min with 50 nM rapamycin.
Results and Discussion
DNA microarrays that simultaneously monitor genome-wide transcriptional responses provide a unique approach to understanding complex processes. Microarrays containing 5,910 (≈92%) individual ORFs from S. cerevisiae were manufactured in our laboratory following published methods (14–16). We exposed either haploid or diploid yeast growing in rich media to rapamycin and harvested cells 0, 15, 30, 60, and 120 min later.
During the 2-h time period examined, rapamycin increased the expression of 154 genes (61 of unknown function) more than four-fold and 78 genes more than five-fold relative to the t = 0 time point. (The complete data sets from all experiments are published as supplemental data on the PNAS web site, www.pnas.org.) One hundred and forty-seven genes (sixty-three of unknown function) were repressed at least four-fold at some time during the 2-h period of treatment. Seventy-six genes were repressed more than five-fold.
We applied a clustering algorithm from the genespring software package to 297 genes that were activated or repressed more than four-fold during the 2-h period of observation (Fig. 1). We found four general expression patterns (Fig. 1 A–D) for these highly regulated genes. Approximately 20% of the genes were repressed strongly in initial timepoints but were repressed less strongly after 2 h (Fig. 1A). An additional 25% of the genes were repressed rapidly and remained repressed for 2 h (Fig. 1B). Approximately 25% of the regulated genes were activated rapidly and remained activated through 2 h (Fig. 1C), and 20% of the genes were activated highly initially but became less activated or returned to untreated levels during the 2-h period (Fig. 1D). The identities of the genes in each of the four expression patterns are available in the online supplemental data.
Figure 1.

The 297 genes whose mRNA transcript levels changed more than four-fold after rapamycin exposure were clustered according to their expression profile over time by the genespring software package. The expression profile of each regulated gene is displayed as a row. Fold changes in expression are indicated by color value according to the color bar at the bottom of the figure (red, increased expression; yellow, no change in expression; green, decrease in expression). Fluorescence signal strength from the microarray is indicated by color brightness (saturated color, high fluorescence; black, low fluorescence). Gray bars indicate missing data points. Genes regulated similarly are labeled, and their general expression patterns over time are displayed (A–D).
The profile confirms the published finding (6) that the transcripts of ribosomal proteins are strongly and rapidly repressed by rapamycin (70 ribosomal genes repressed three-fold, 27 repressed four-fold) but also revealed many previously unknown effects. Unexpectedly, the general amino acid control pathway was not activated. This is surprising because the availability of amino acids to mammalian cells is sensed by FRAP, the mammalian homologue of the Tor proteins (17). The sporulation pathway was not activated in diploid cells despite the previous observation that rapamycin promotes sporulation in cells grown to saturation in rich media (3, 18). It is likely that sporulation is a late effect, only indirectly induced by treatment with rapamycin. In addition, the nitrogen starvation pathway was not activated. In contrast, the profile clearly implicates Tor proteins in other nutrient-sensitive pathways.
Yeast cells are atypical because they preferentially ferment glucose to ethanol rather than respire glucose to carbon dioxide. Fermentation, through glycolysis, provides most of the energy for yeast growing exponentially in glucose-containing media. As the media becomes depleted of fermentable carbon, cells begin respiration and metabolize accumulated ethanol through the citric acid cycle. This process is known as the diauxic shift. When the published profile of yeast undergoing the diauxic shift was compared with the rapamycin profile, significant similarities were observed (Fig. 2). In both cases, genes involved in glycolysis are increasingly repressed with time. Most glycolytic genes are controlled by the transcription factor Gcr1p (19, 20), and, interestingly, rapamycin (but not the diauxic shift) decreased GCR1 transcription 4.3-fold in 15 min. Regulation of GCR1 transcription has not previously been demonstrated. Nearly all of the citric acid cycle genes were induced in both profiles (Fig. 2). This induction suggests that Tor proteins may be upstream of the Hap2,3,4,5p complex known to regulate the transcription of citric acid cycle genes (21–23). However, rapamycin induced citric acid cycle gene expression slightly less strongly and more transiently than the diauxic shift, indicating that other complexities exist. Finally, rapamycin and the diauxic shift induce comparable expression of some genes in the glyoxylate cycle and in glycogen and trehalose synthesis (data not shown). Rapamycin did not induce genes containing the carbon source-responsive element, but we note that these genes are only activated late in the diauxic shift profile. We conclude that these genes are not under the direct control of the Tor proteins. Taken together, these data indicate that the Tor proteins at least partially regulate the switch from fermentation to respiration.
Figure 2.

Rapamycin modulates genes involved in glycolysis and the citric acid cycle. (A) Values are reported as fold changes. Diauxic shift data is taken from DeRisi et al. (16). The rapamycin data values represent the maximal fold change over the 2-h time course. (B) Expression of several glycolysis genes after treatment with rapamycin. (C) Expression of several citric acid cycle genes after treatment with rapamycin.
Rapamycin also controls glucose-sensitive signaling pathways not regulated during the diauxic shift. Glucose activates hexose transporter transcription via a membrane-to-nucleus signaling pathway. When glucose is present in the extracellular medium, the glucose receptors Rgt2p and Snf3p activate Grr1p that in turn inhibits Rgt1p, a transcriptional repressor for hexose transporter genes (12). The hexose transport gene HXT1, expressed only at high glucose concentrations, is repressed 3.6-fold in 60 min by rapamycin. Surprisingly, rapamycin activated RGT1 and GRR1 transcription 4.5-fold and 3.3-fold, respectively, in 15 min. These transcriptional effects (HXT1, RGT1, GRR1) are not seen in the diauxic shift, implying that the Tor proteins control multiple pathways involved in carbon metabolism.
The most striking rapamycin-induced transcriptional effects, however, are not in carbon metabolism. Rapamycin rapidly and strongly activated the nitrogen discrimination pathway that is normally repressed in the presence of high quality nitrogen sources (glutamine, asparagine, ammonia). When yeast cells lack such sources, they activate a genetic program enabling them to use poorer alternatives such as proline or allantoate. A variety of permeases and metabolic enzymes are up-regulated to import nitrogen metabolites and convert them into a useable form for biosynthesis. Vacuolar proteases are also induced as the cells presumably begin salvaging nutrients from their own macromolecules through autophagy (24). A complex regulatory network that is highly sensitive to the quality of nitrogen controls this concerted response.
Rapamycin quickly and comprehensively activates this nitrogen discrimination pathway in yeast cells despite the presence of high quality nitrogen sources in rich media. The membrane permeases GAP1 (general amino acid permease) and MEP2 (an ammonia sensor) are induced 27-fold and 19-fold, respectively, within 15 min of treatment. Several other permeases (AGP1, CAN1, DIP5, DUR3, MEP1, PTR2, ZRT1) are also induced. Genes in the allantoin utilization pathway (DAL1, DAL4, DAL5, DAL7, DAL80), the proline utilization pathway (PUT1, PUT2, PUT4), and the glutamine biosynthetic pathway (GDH2, GLN1) are also up-regulated, as they are when yeast cells are grown in the absence of a high quality nitrogen source. Consistent with published results that inhibiting the Tor proteins causes autophagy (4), rapamycin also induced expression of vacuolar proteases (PEP4, CPS1, LAP3, LAP4, PRB1) and APG1, a serine/threonine kinase required for autophagy.
The nitrogen discrimination pathway genes activated by rapamycin are under the direct control of the Ure2p/Gln3p transcription complex. In the presence of high quality nitrogen sources, Ure2p binds the GATA-binding factor Gln3p and thereby inhibits transcription. In a ure2Δ strain, the nitrogen discrimination genes become derepressed, a phenotype similar to that induced by treatment with rapamycin (25) (Fig. 3).
Figure 3.

Rapamycin modulates expression of genes in the nitrogen discrimination pathway. (A) A selection of genes regulated by nitrogen source quality and treatment with rapamycin. ND, nitrogen discrimination pathway (repressed on asparagine, derepressed on proline); R, repressed on proline (R* if remaining expression has been shown to be ND); D, derepressed on asparagine; –, not determined. (B) Expression of several nitrogen source-sensitive genes after treatment with rapamycin.
One nitrogen discrimination gene, PUT1, is expressed independently of Gln3p, implicating Ure2p over Gln3p as a possible downstream target of the Tor proteins (30). Because URE2 transcript levels are not affected by rapamycin treatment, we hypothesized that inhibiting Tor proteins might induce posttranslational modification of Ure2p. We found that the electrophoretic mobility of Ure2p increases within 15 min of treatment with rapamycin (Fig. 4A). To examine more precisely the kinetics of the shift and to verify that this was not a strain-specific effect, we tested a 5-min treatment with rapamycin in a strain of a different background. The Ure2p shift was again observed (Fig. 4B). The nature of the shift, and the fact that rapamycin is known to cause dephosphorylation of another downstream target, Npr1p (31), within 5 min, suggests that Ure2p is being dephosphorylated. The phosphatase subunit Tap42p is known to modulate Npr1p dephosphorylation and, in mammalian cells, inactivation of FRAP activates the cellular phosphatase PP2A, which subsequently dephosphorylates p70S6K (32). We confirmed that the rapamycin-induced shift in Ure2p mobility was completely dependent on Tor function by performing the experiment in cells in which TOR1 had been replaced with TOR1 (Ser1972→Thr), a mutant allele encoding a fully functional but rapamycin-resistant form of the protein. We found that Ure2p did not shift upon rapamycin treatment in these cells, indicating that Tor proteins dominantly control Ure2p function.
Figure 4.

Rapamycin increases the mobility of the transcriptional regulator Ure2p by inhibiting Tor proteins. Anti-Ure2p immunoblots of whole cell lysates were performed as described in Materials and Methods. (A) Jk9–3da cells were treated with either 100 nM rapamycin or 100 nM FK506 for 15 min. (B) CY5754 cells were treated with 20 nM rapamycin for 5 min. (C) Cells expressing a mutant form of TOR1 (Ser1972→Thr) that is unable to bind rapamycin and the wild-type parental strain were exposed to 50 nM rapamycin for 30 min.
When yeast are completely starved of nitrogen, they activate a pathway that is distinct from the quality-sensitive nitrogen discrimination pathway. Thus, nitrogen starvation does not merely result from extreme activation of the nitrogen discrimination pathway. The transcription profile revealed that rapamycin does not cause yeast growing in rich media to appear nitrogen-starved. Two genes that are normally induced by nitrogen starvation, YVH1 and PTP2 (33), are repressed or unchanged, respectively, after treatment with rapamycin. Also, of 52 genes of unknown function that were shown to be sensitive to nitrogen starvation (34), only 19% (10 genes) were appropriately modulated in the response to rapamycin. Thus, inactivation of the Tor proteins activates the nitrogen discrimination pathway, but not the nitrogen starvation pathway.
Before this work, the primary focus of research on the Tor and FRAP proteins had been on their roles in signaling pathways leading to the regulation of translation rather than transcription. In addition, the signaling pathways that they impinge on had been defined primarily in terms of an overall physiological response. We have shown that Tor inhibition by the small molecule rapamycin leads to modulation of at least three distinct pathways: the nitrogen discrimination pathway (likely via an alteration in the phosphorylation of a transcription factor complex), the glucose activation pathway, and pathways involved in the diauxic shift (Fig. 5). Our data also show that three other nutrient-sensitive pathways are not modulated by rapamycin in rich media: general amino acid control, sporulation, and the nitrogen starvation pathways. The strongest effects of rapamycin on transcription occurred between 15 and 30 min of treatment, with significant effects seen by 15 min. The kinetics of these results strongly suggest a translation-independent mechanism of rapamycin action. It remains to be determined whether the Tor proteins sense intracellular amino acids, charged or uncharged tRNAs, glucose, or some other indicator of nutrient availability before signaling to both translational and transcriptional complexes.
Figure 5.
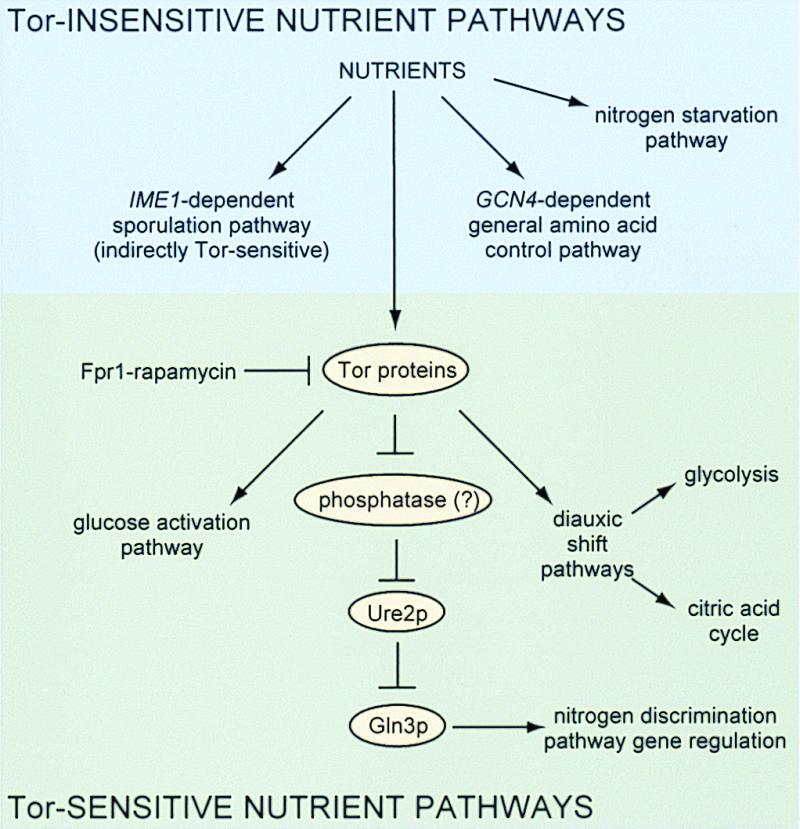
A summary of the nutrient-sensitive signaling pathways and their relation to signaling by Tor proteins.
The analysis of nutrient-sensitive signaling pathways described in this study suggests a general means for dissecting biological pathways. Small molecules of high specificity (35) can lead to modulation of pathways both rapidly and conditionally. Profiling provides a means to perform a broad search for the resulting transcriptional phenotype. Analyses of profiling data can suggest biochemical experiments to probe specific components of small molecule-sensitive pathways.
Supplementary Material
Acknowledgments
The authors thank the members of Patrick Brown's laboratory for their assistance with DNA microarraying technology, Brian McNeil and Mike Tyers for their generous assistance with the set of ORFs, Charles DiComo and Michael Hall for yeast strains, and Reed Wickner for Ure2p antibodies. J.S.H. is a Leukemia Society of America Fellow. F.G.K. is supported by the National Institutes of Health Medical Scientist Training Program. J.K.T. is supported by a National Science Foundation Predoctoral Fellowship and the Harvard-Markey Biomedical Scientist program. A.F.S is supported by the Harvard College Research Program. S.L.S. is an investigator at the Howard Hughes Medical Institute. This research was funded by a grant from the National Institute of General Medical Sciences (GM-52067).
References
- 1.Heitman J, Movva N R, Hall M N. Science. 1991;253:905–909. doi: 10.1126/science.1715094. [DOI] [PubMed] [Google Scholar]
- 2.Barbet N C, Schneider U, Helliwell S B, Stansfield I, Tuite M F, Hall M N. Mol Biol Cell. 1996;7:25–42. doi: 10.1091/mbc.7.1.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zheng X F, Schreiber S L. Proc Natl Acad Sci USA. 1997;94:3070–3075. doi: 10.1073/pnas.94.7.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Noda T, Ohsumi Y. J Biol Chem. 1998;273:3963–3966. doi: 10.1074/jbc.273.7.3963. [DOI] [PubMed] [Google Scholar]
- 5.Zaragoza D, Ghavidel A, Heitman J, Schultz M C. Mol Cell Biol. 1998;18:4463–4470. doi: 10.1128/mcb.18.8.4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Powers T, Walter P. Mol Biol Cell. 1999;10:987–1000. doi: 10.1091/mbc.10.4.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cafferkey R, Young P R, McLaughlin M M, Bergsma D J, Koltin Y, Sathe G M, Faucette L, Eng W K, Johnson R K, Livi G P. Mol Cell Biol. 1993;13:6012–6023. doi: 10.1128/mcb.13.10.6012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kunz J, Henriquez R, Schneider U, Deuter-Reinhard M, Movva N R, Hall M N. Cell. 1993;73:585–596. doi: 10.1016/0092-8674(93)90144-f. [DOI] [PubMed] [Google Scholar]
- 9.Keith C T, Schreiber S L. Science. 1995;270:50–51. doi: 10.1126/science.270.5233.50. [DOI] [PubMed] [Google Scholar]
- 10.Kuruvilla F G, Schreiber S L. Chem Biol. 1999;6:R129–R136. doi: 10.1016/S1074-5521(99)80070-2. [DOI] [PubMed] [Google Scholar]
- 11.Hinnebusch A G. In: The Molecular and Cellular Biology of the Yeast Saccharomyces. Jones E W, Pringle J R, Broach J R, editors. Vol. 2. Plainview, NY: Cold Spring Harbor Lab. Press; 1992. pp. 319–414. [Google Scholar]
- 12.Carlson M. Curr Opin Genet Dev. 1998;8:560–564. doi: 10.1016/s0959-437x(98)80011-7. [DOI] [PubMed] [Google Scholar]
- 13.Magasanik B. In: The Molecular and Cellular Biology of the Yeast Saccharomyces. Jones E W, Pringle J R, Broach J R, editors. Vol. 2. Plainview, NY: Cold Spring Harbor Lab. Press; 1992. pp. 283–317. [Google Scholar]
- 14.Eisen M B, Brown P O. Methods Enzymol. 1999;303:179–205. doi: 10.1016/s0076-6879(99)03014-1. [DOI] [PubMed] [Google Scholar]
- 15.Schena M, Shalon D, Davis R W, Brown P O. Science. 1995;270:467–470. doi: 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- 16.DeRisi J L, Iyer V R, Brown P O. Science. 1997;278:680–686. doi: 10.1126/science.278.5338.680. [DOI] [PubMed] [Google Scholar]
- 17.Hara K, Yonezawa K, Weng Q P, Kozlowski M T, Belham C, Avruch J. J Biol Chem. 1998;273:14484–14494. doi: 10.1074/jbc.273.23.14484. [DOI] [PubMed] [Google Scholar]
- 18.Chu S, DeRisi J, Eisen M, Mulholland J, Botstein D, Brown P O, Herskowitz I. Science. 1998;282:699–705. doi: 10.1126/science.282.5389.699. [DOI] [PubMed] [Google Scholar]
- 19.Clifton D, Weinstock S B, Fraenkel D G. Genetics. 1978;88:1–11. doi: 10.1093/genetics/88.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chambers A, Packham E A, Graham I R. Curr Genet. 1995;29:1–9. doi: 10.1007/BF00313187. [DOI] [PubMed] [Google Scholar]
- 21.Forsburg S L, Guarente L. Genes Dev. 1989;3:1166–1178. doi: 10.1101/gad.3.8.1166. [DOI] [PubMed] [Google Scholar]
- 22.Olesen J T, Guarente L. Genes Dev. 1990;4:1714–1729. doi: 10.1101/gad.4.10.1714. [DOI] [PubMed] [Google Scholar]
- 23.Rosenkrantz M, Kell C S, Pennell E A, Devenish L J. Mol Microbiol. 1994;13:119–131. doi: 10.1111/j.1365-2958.1994.tb00407.x. [DOI] [PubMed] [Google Scholar]
- 24.Coffman J A, Cooper T G. J Bacteriol. 1997;179:5609–5613. doi: 10.1128/jb.179.17.5609-5613.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coschigano P W, Magasanik B. Mol Cell Biol. 1991;11:822–832. doi: 10.1128/mcb.11.2.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coffman J A, el Berry H M, Cooper T G. J Bacteriol. 1994;176:7476–7483. doi: 10.1128/jb.176.24.7476-7483.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Daugherty J R, Rai R, el Berry H M, Cooper T G. J Bacteriol. 1993;175:64–73. doi: 10.1128/jb.175.1.64-73.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Coffman J A, Rai R, Loprete D M, Cunningham T, Svetlov V, Cooper T G. J Bacteriol. 1997;179:3416–3429. doi: 10.1128/jb.179.11.3416-3429.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coffman J A, Rai R, Cooper T G. J Bacteriol. 1995;177:6910–6918. doi: 10.1128/jb.177.23.6910-6918.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu S, Falvey D A, Brandriss M C. Mol Cell Biol. 1995;15:2321–2330. doi: 10.1128/mcb.15.4.2321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmidt A, Beck T, Koller A, Kunz J, Hall M N. EMBO J. 1998;17:6924–6931. doi: 10.1093/emboj/17.23.6924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Peterson R T, Desai B N, Hardwick J S, Schreiber S L. Proc Natl Acad Sci USA. 1999;96:4438–4442. doi: 10.1073/pnas.96.8.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Park H D, Beeser A E, Clancy M J, Cooper T G. Yeast. 1996;12:1135–1151. doi: 10.1002/(sici)1097-0061(19960915)12:11<1135::aid-yea11>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 34.Planta R J, Brown A J, Cadahia J L, Cerdan M E, de Jonge M, Gent M E, Hayes A, Kolen C P, Lombardia L J, Sefton M, et al. Yeast. 1999;15:329–350. doi: 10.1002/(SICI)1097-0061(19990315)15:4<329::AID-YEA360>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 35.Marton M J, DeRisi J L, Bennett H A, Iyer V R, Meyer M R, Roberts C J, Stoughton R, Burchard J, Slade D, Dai H, et al. Nat Med. 1998;4:1293–1301. doi: 10.1038/3282. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.