

Abstract
Recent findings intriguingly place DNA double-strand break repair proteins at chromosome ends in yeast, where they help maintain normal telomere length and structure. In the present study, an essential telomere function, the ability to cap and thereby protect chromosomes from end-to-end fusions, was assessed in repair-deficient mouse cell lines. By using fluorescence in situ hybridization with a probe to telomeric DNA, spontaneously occurring chromosome aberrations were examined for telomere signal at the points of fusion, a clear indication of impaired end-capping. Telomeric fusions were not observed in any of the repair-proficient controls and occurred only rarely in a p53 null mutant. In striking contrast, chromosomal end fusions that retained telomeric sequence were observed in nontransformed DNA-PKcs-deficient cells, where they were a major source of chromosomal instability. Metacentric chromosomes created by telomeric fusion became even more abundant in these cells after spontaneous immortalization. Restoration of repair proficiency through transfection with a functional cDNA copy of the human DNA-PKcs gene reduced the number of fusions compared with a negative transfection control. Virally transformed cells derived from Ku70 and Ku80 knockout mice also displayed end-to-end fusions. These studies demonstrate that DNA double-strand break repair genes play a dual role in maintaining chromosomal stability in mammalian cells, the known role in repairing incidental DNA damage, as well as a new protective role in telomeric end-capping.
Telomeres are unique structures at the physical ends of linear eukaryotic chromosomes that were first described over sixty years ago by Muller (1) in the fruit fly Drosophilia melanogaster based on their chromosome-end protection function. Shortly thereafter, McClintock's (2) cytogenetic studies in maize demonstrated that broken chromosomes were subject to end fusions. These studies demonstrated that a cell's ability to respond differently to natural chromosome ends than it does to ends created by spontaneous or induced breakage is critical to preserving a stable genetic inheritance (3–5). Modern molecular analysis has revealed that telomeric DNA consists of tandem arrays of short, repetitive G-rich (6, 7) sequence oriented 5′-to-3′ toward the end of the chromosome, terminating in a 3′ single-stranded G-rich overhang (8). Together with associated telomeric-binding proteins, a dynamic terminal structure is created that “caps” each end of linear chromosomal DNA molecules, providing protection from illegitimate recombination, exonucleolytic attack, and degradation.
In contrast to natural chromosome ends, DNA double-strand breaks (dsb) are highly recombinogenic and represent a major threat to the integrity of the cell's genome because of their potential for causing chromosome aberrations, mutagenesis, and carcinogenesis if misrepaired or left unrepaired. In mammalian cells, the majority of dsb are rejoined through repair pathways known as nonhomologous end-joining (NHEJ) (9). The Ku70, Ku80, and DNA-PKcs genes are part of the most studied NHEJ pathway. The Ku heterodimer, composed of 70-kDa and ≈80-kDa subunits, is the most abundant DNA end-binding protein in mammalian cells. It recognizes DNA ends whether blunt, overhanging, or hairpin in structure and binds with high affinity in a DNA sequence-independent manner. Mutant cells lacking Ku are deficient in the repair of dsb, as well as in recombination of the Ig V(D)J region (10). DNA-PK is a nuclear serine/threonine kinase made up of two components, a ≈465-kDa catalytic subunit (DNA-PKcs) and the Ku heterodimer (11). Yeast also possess NHEJ pathways, crucial components of which appear to be conserved between yeast and mammalian cells (12). For example, in Saccharomyces cerevisiae (baker's yeast), homologues of both human Ku70 (yKu70 or Hdf1) and Ku80 (yKu80 or Hdf2) have been identified (13, 14). Yeast has no close homologue of DNA-PKcs, but other kinases may play a similar role.
In addition to functioning in the vital process of DNA repair, NHEJ proteins are required for yeast telomere function, maintenance, and telomere-associated transcriptional silencing. The absence of the Ku heterodimer affects the perinuclear clustering of telomeres normally seen in wild-type yeast cells (15). Furthermore, strains defective in Ku70 or Ku80 lose the majority, but not all, of their terminal telomere repeats (14, 16). The roles these proteins play in telomere maintenance and function are currently unclear, but they may contribute to a telomere end-binding complex that recruits other proteins. The recent finding that yKu70 interacts with the yeast silencing protein Sir4p, which in turn interacts with Rap1p, a key regulator of telomere length, supports this idea (17, 18). Sir proteins (2p, 3p, and 4p) function in transcriptional silencing at telomeres through induction of a condensed, inaccessible heterochromatic state (19). Disruption of Ku function debilitates telomere-associated silencing, and, interestingly, mutation of Sir genes disrupts Ku-dependent NHEJ. Ku and Sir proteins present at yeast telomeres relocalize to sites of DNA damage, and this movement is controlled by DNA damage-checkpoint genes (20, 21). It also appears that Ku is critical for maintaining the Sir proteins, and thus transcriptional silencing, at telomeres.
Binding of Ku to mammalian telomeric DNA has been demonstrated also (22). Two human telomere-specific DNA-binding proteins, telomeric repeat-binding factor (TRF)-1 and TRF-2 have also been identified (23). TRF-2 has been shown to protect chromosome termini from end-to-end fusions and is the first telomere-associated protein implicated in the maintenance of the correct terminal DNA structure necessary for proper telomere function (24). An important clue as to what that structure might be was uncovered by the discovery that TRF-2 can remodel mammalian telomeric DNA into large duplex loops, termed t loops (25). A t loop is created when a telomere end loops back and the single-stranded G-rich tail invades an interior segment of duplex telomeric DNA. By sequestering natural chromosome ends, t loop formation may be an effective end-capping mechanism in mammalian cells.
Telomeres, as the physical ends of linear DNA duplexes, run the risk of being identified as dsb in need of repair and ligation. With this in mind, the recent evidence locating several proteins of the cell's repair arsenal at telomeres presents an interesting paradox. Why are DNA repair proteins, capable of binding and joining double-stranded ends, present at the telomere, and how are they involved in normal telomere function and maintenance?
Materials and Methods
Cell Culture.
Early-passage nontransformed lung-fibroblast cultures were derived from the following male mice: a repair-proficient mouse (C57BL/6TacfBR-[KO]p53N4 WT), two mice having severe combined immunodeficiency (Tac:Icr:Ha(ICR)-scid), a p53 knockout mouse (C57BL/6TacfBR-[KO]p53N4 HO), and two mice with a double p53−/−/scid mutation that were obtained by crossing p53 knockout mice with scid homozygous mice (26). These same cell lines were allowed to undergo spontaneous immortalization by simply maintaining cultures until they overcame senescence. Ku70- and Ku80-deficient mouse fibroblast cell lines were established from C57BL/6 knockout mice (27, 28). These cell lines and an isogenic control were transformed with the Abelson murine leukemia retrovirus. Some cells derived from the lung of a scid mouse were transformed with simian virus 40 (SV40), as previously described (29). These scid cells were transfected with the pPGDP-8 plasmid, which has an intact cDNA copy of the human DNA-PKcs gene, plus the pSV2neo plasmid (30). SV40-transformed scid cells were also transfected with the pSV2neo plasmid only. As expected, resistance to radiation was restored in cells transfected with the DNA-PKcs gene, but not in the negative-transfection control (30). All cells were incubated at 37°C, 5% CO2 and cultured in α-MEM supplemented with 20% FBS and antibiotics. A 1:10 split ratio was used at each passage.
Fluorescence in Situ Hybridization (FISH).
Near-confluent cultures were subcultured into fresh medium and incubated at 37°C for 24 hr. Colcemid (0.2 μg/ml) was added for 4 hr to accumulate mitotic cells. Cultures were trypsinized, and cells were suspended in 75 mM KCl for 15 min before fixing in 3:1 methanol/acetic acid. Fixed cells were dropped onto cold wet glass microscope slides. A probe to telomeric DNA was made by synthesizing an oligomer having the sequence (CCCTAA)7 and was labeled by terminal deoxynucleotidyltransferase tailing (Roche Molecular Biochemicals) with Cy3-dCTP, according to the manufacturer's instructions. A hybridization mixture containing 0.4 μg/ml probe DNA in 30% formamide, and 2× SSC (1× SSC is 0.15 M NaCl and 0.015 M sodium citrate) was applied to slides that had been denatured in 70% formamide and 2× SSC at 70°C for 2 min. After an overnight hybridization at 37°C, the slides were washed in 2× SSC at 42°C and mounted in a glycerol solution containing 1 mg/ml of the antifade compound p-phenylenediamine HCl and 0.1 μg/ml 4′,6-diamidino-2-phenylindole (DAPI) (31, 32). Cells were viewed with a Zeiss Axiophot Fluorescence Microscope. Images were obtained with a Photometrics (Tucson, AZ) CH250 charge-coupled device camera.
Suspected telomere involvement in a chromosome aberration was scored according to three progressively more stringent criteria. The first criterion is that commonly used for scoring telomeric associations, i.e., two chromosome termini are separated by no more than the width of a chromatid (i.e., close). Because no separation would be expected in a true end fusion, we felt that this criterion might be too lax and so devised two more restrictive standards. The second criterion allowed no visible separation between FISH telomere signals, but did score as telomere associations instances where telomeres were touching but still visible as distinct bodies. The third criterion scored as true end fusions only those events where the telomeres of adjoining chromosomes had fused into a single signal. In our experience, events scored as telomeric associations occurred in all cell types examined. However, scoring by the second and third criteria resulted in the expected zero background level of events in repair-proficient control samples. Few cases were observed where telomere signals were touching. Therefore, we chose to utilize the third, most stringent, criterion to reduce false positives to a minimum. We call aberrations meeting this standard “telomeric fusions” (TF) to distinguish them from events scored by other criteria. DAPI bright regions visible on most chromosomes correspond to the mouse major satellite (33) and are a useful indicator of centromere position.
Results
The effectiveness of mammalian telomeric end-capping was evaluated with FISH. By using a telomere probe, spontaneously arising chromosome aberrations were inspected for telomere signal at the point of fusion between two chromosomes, the presence of which gives an unambiguous indication that telomeric end-capping has failed. The appearance of such aberrations, it was reasoned, in a DNA repair-deficient cell line would be a clear demonstration that the mutated gene was required for the normal capping function of telomeres. Mouse cells are especially suitable for this type of analysis because their chromosomes have exceptionally long telomeres, thus yielding bright signals, and are without cytogenetically detectable interstitial blocks of TTAGGG repeats. The murine model has the additional advantage of genetic diversity, including several DNA repair-gene mutants.
Examination of early-passage nontransformed lung fibroblasts revealed chromosome aberrations having centrally located telomere signals, indicating a failure of telomeric capping, in each of the dsb repair-deficient cell lines analyzed. Examples of TF are shown in Fig. 1. Both chromosome- and (more rarely) chromatid-type TF were observed. In contrast, TF were not observed in repair-proficient controls. At least two independent sets of experiments were conducted with similar findings. Additionally, scid and p53−/−/scid cells were obtained from two different mice. Combined results are summarized in Table 1.
Figure 1.
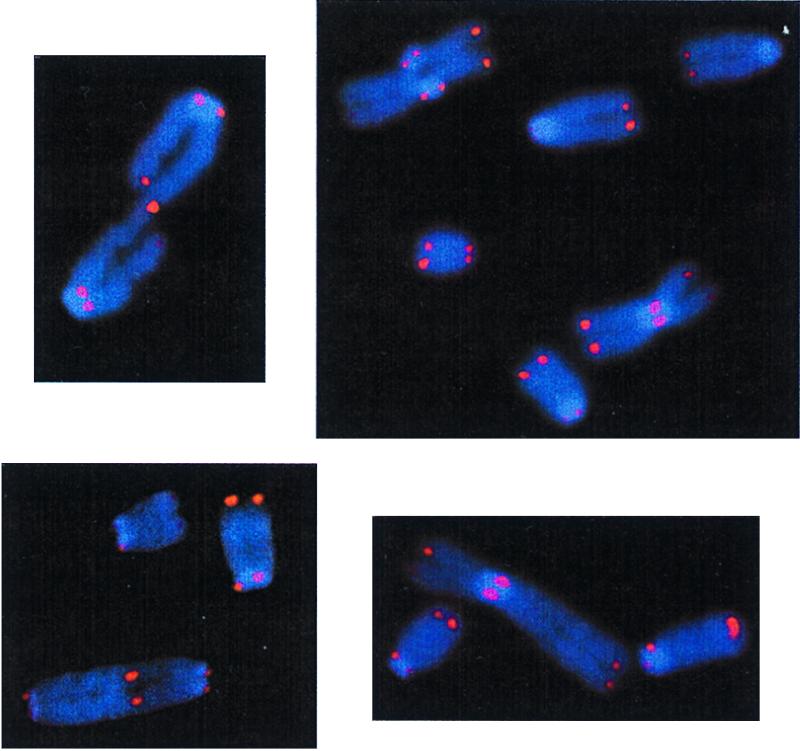
Chromosome aberrations illustrating telomeric fusion (TF). (Upper Left) An image of a chromatid dicentric observed in a scid cell. (Lower Left) A dicentric chromosome seen in a Ku70−/− cell. (Right) Two examples of metacentric chromosomes seen in scid cells. In the upper left corner of the top image is a tetra-radial, an ordinary chromatid-type aberration.
Table 1.
Spontaneous chromosome aberrations in nontransformed cells
| Cell type | No. cells scored | Gaps, breaks, and deletions* | Total exchange-type aberrations† | Telomeric fusions‡ | Telomeric fusions per cell |
|---|---|---|---|---|---|
| WT (p3,4)§ | 100 | 26 | 3 | 0 | 0.000 |
| scid (p5,6) | 91 | 47 | 22 | 15 | 0.165 |
| p53−/− (p3,5) | 100 | 57 | 10 | 1 | 0.010 |
| p53−/−/scid (p5,6) | 100 | 12 | 10 | 8 | 0.080 |
*Nonexchange-type aberrations are combined into a single category.
† All identifiable exchange-type aberrations, i.e., chromosome and chromatid dicentrics, metacentrics, and rings are included in this category. Many Ku−/− cells had small, difficult to classify, yet clearly aberrant, chromosomes. Their frequency, and the fact that some appear to be clonal in origin, indicates they are not lethal to the cells possessing them. They are not included in aberration counts.
‡ Number of exchange-type aberrations having telomere sequence at points of fusion.
§Passage numbers.
In the scid and p53−/−/scid cell lines, TF outnumbered ordinary exchange-type aberrations. Dicentric and metacentric TF occurred with about equal frequency. Oddly, chromosomal rings were never seen. This may indicate that interphase chromosome architecture is such as to keep the tips of the long and short arms of a chromosome well separated, thus reducing the probability of their fusing to form a ring. The metacentric chromosomes may have been either isochromosomes or Robertsonian translocations. In mouse, Robertsonian translocations are common, and the DNA sequence at the point of exchange has been studied previously (34, 35). The point of joining always appears to lie within mouse minor satellite DNA, the sequence thought to serve as the mouse centromere (36), whereas the telomeres on the short arms of the two fusing telocentric chromosomes are invariably lost. Thus, the TF Robertsonians (or isochromosomes) observed in DNA-PKcs mutant cells are distinctly different.
Hypothetically, TF might be expected to occur at a rate proportional to telomere length and were observed in our experiments simply because repair-deficient cells spawn numerous aberrations, a significant fraction of which might be TF because of the extraordinary length of mouse telomeres. That this is not the case can be seen from the following argument. The average mouse telomere length has been estimated to be 20–150 kbp by Southern blot analysis (37) and 10–60 kbp by quantitative FISH (38). Assuming an average mouse telomere length of 100 kbp and haploid G1 genome size of 3 × 109 bp, the fraction of the mouse genome that is composed of telomeric DNA is ≈1.4 × 10−3. If spontaneous TF occur in proportion to total telomere length, then about 1 in 700 aberrations would be expected to show telomere involvement. Contrary to expectation, TF were observed ≈500 times more frequently than predicted in both scid and p53−/−/scid cell lines, a difference that is statistically highly significant (χ2 test, P ≪ 0.001).
A single TF chromatid dicentric was seen in 100 p53−/− cells examined. Thus, p53 plays a far less significant role in telomeric capping. In comparison to the p53−/−/scid double mutant, scid cells had more than twice as many exchange-type aberrations per cell. However, the proportion of exchange-type aberrations that were created by fusion between telomeres was not significantly different (χ2 test, P > .05). Thus, loss of p53 gene function does not enhance the effect of the scid mutation on end-capping, implying that TF do not trigger p53-dependent apoptosis or a cell-cycle checkpoint.
Following spontaneous immortalization, these same cell lines were reexamined. Wild-type and p53−/−-transformed cells did not exhibit TF. However, the numbers of TF were found to have increased sharply in the transformed scid and p53−/−/scid cells (Table 2). This increase is attributable to a high frequency of a single type of fusion product, metacentrics created by fusion between telomeres (Fig. 2). The number of these aberrations increased still further in scid cells observed seven passages later. On the basis of gross chromosomal morphology, two metacentrics appeared to reoccur in nearly all of the scid cells examined. Fewer TF were seen in immortalized p53−/−/scid cells as compared with scid cells, even after adjusting for the lower average number of chromosomes per cell. This finding further indicates that loss of p53 does not enhance the end-capping deficiency of the scid mutation.
Table 2.
Chromosome aberrations in spontaneously immortalized cells
| Cell type | No. cells scored | Gaps, breaks, and deletions | Total exchange-type aberrations | Telomeric fusions | ITS* | TF per cell | Chromosomes per cell† |
|---|---|---|---|---|---|---|---|
| WT (p34) | 50 | 52 | 7 | 0 | 0 | 0.000 | 73.3 ± 3.7 |
| scid (p28) | 50 | 12 | 226 | 225 | 1 | 4.50 | 71.2 ± 5.2 |
| scid (p35) | 50 | 13 | 264 | 254 | 8 | 5.08 | 74.0 ± 3.1 |
| p53−/− (p26) | 50 | 17 | 28 | 0 | 0 | 0.000 | 65.7 ± 7.5 |
| p53−/−/scid (p23) | 50 | 26 | 69 | 58 | 3 | 1.16 | 57.5 ± 8.4 |
*ITS, interstitial telomeric sequence.
†Average number of chromosomes per cell ± SD.
Figure 2.

Representative metaphase spread of a spontaneously immortalized scid mouse fibroblast. Five Robertsonian translocations created by end-to-end fusion are visible in the photograph and are indicated by arrows.
A new type of TF appeared in immortalized DNA-PKcs-deficient cells. This new aberration was distinguishable by its having a block of telomeric sequence located within a chromosome arm but not adjacent to a centromere. The frequencies of this aberration, which was called interstitial telomeric sequence, are recorded separately in Table 2.
Spontaneously occurring aberrations were analyzed in a virally transformed scid cell line restored to repair proficiency by transfection with the DNA-PKcs gene (SC-A1) and a negative transfection control (SC-EM). SC-A1 cells had statistically fewer TF than the repair-deficient SC-EM cells (Student's t test, P < 0.001) (Table 3). Six passages later, TF in repair-proficient SC-A1 cells had decreased significantly (P < 0.001) by nearly 3-fold, whereas no significant change occurred in SC-EM cells (P > 0.05). These results indicate that restoration of DNA-PKcs function to scid cells improves the efficiency of end-capping. Those TF observed in SC-A1 cells were presumably preexisting in the population at the time of transfection.
Table 3.
Chromosome aberrations in virally transformed cells
| Cell type | No. cells scored | Gaps, breaks, and deletions | Total exchange-type aberrations | Telomeric fusions | ITS* | TF per cell | Chromosomes per cell |
|---|---|---|---|---|---|---|---|
| WT | 100 | 19 | 16 | 0 | 0 | 0.000 | 62.8 ± 7.6 |
| Ku 70−/− | 76 | 20 | 50 | 8 | 0 | 0.105 | 54.6 ± 5.1 |
| Ku 80−/− | 61 | 13 | 118 | 8 | 0 | 0.131 | 74.4 ± 9.1 |
| SC-A1 | 50 | 13 | 68 | 54 | 10 | 1.08 | 56.5 ± 6.6 |
| SC-EM | 50 | 21 | 95 | 89 | 3 | 1.78 | 57.5 ± 6.2 |
| SC-A1 (p + 6)† | 50 | 14 | 55 | 19 | 21 | 0.380 | 57.3 ± 6.5 |
| SC-EM (p + 6) | 50 | 41 | 81 | 70 | 5 | 1.40 | 57.0 ± 4.8 |
*ITS, interstitial telomeric sequence.
†Reexamination six passages later.
A deficiency in either Ku70 or Ku80 clearly results in an overall increase in chromosome aberrations (Table 3); these included TF. Thus, both Ku genes are required for effective end-capping. TF were not seen in the virally transformed repair-proficient control cells, indicating that virus transformation by itself does not compromise end-capping.
Discussion
Collectively, these experiments demonstrate a vital, if paradoxical, requirement for three members of the Ku NHEJ repair pathway in mammalian telomere biology. The need for both Ku70 and Ku80 suggests that they function in end-capping as a heterodimer just as they do in dsb repair. Yet the Ku heterodimer alone is insufficient to protect telomeres. The data are consistent also with a requirement for DNA-PKcs, which could serve either a structural or enzymatic role, e.g., posttranslational modification by DNA-PK of proteins affecting telomeric end-capping.
There was no noticeable reduction in signal brightness between those telomeres that entered into fusions as compared with those that did not. In fact, the brightest observed telomeric signals frequently occurred at the points of joining, as might be expected if no telomeric DNA was lost during fusion. This observation implies that joining is tip-to-tip, and argues against telomere-length erosion as the primary cause of impaired capping. This contrasts sharply with the end-to-end associations observed in mouse cells lacking the telomerase RNA component (mTR) (39). These cells have undetectable telomerase activity and exhibit a progressive decrease in telomere length. In mTR−/− mice, Robertsonian chromosomal rearrangements lack pericentromeric telomeric DNA, as observed by FISH. An inability to create or maintain a special end structure may be involved in both cases, i.e., mTR−/− cells eventually lose too much of the DNA sequence from which the end structure is formed, while dsb repair mutants lack one or more proteins that participate in its construction.
The end-capping function of human telomeres is known to require at least one other protein, the telomere-specific DNA-binding protein TRF-2 (24), apparently through its ability to form t loops at the ends of chromosomes (25). Ku70, Ku80, and DNA-PKcs may function together with TRF-2 in creating t loops. For example, DNA-PK may recruit and/or influence the activity of a 5′-to-3′ exonuclease at chromosome ends to remake 3′ overhangs after DNA synthesis. The mammalian MRE11/RAD50/NBS1 complex is a candidate exonuclease because its yeast counterpart participates in telomere function, and it has been shown to influence 5′-to-3′ exonucleolytic activity in vivo (40–42). TRF-2 might then complete the process by using 3′ overhangs to remodel linear chromosome ends into a looped configuration.
Ku70 and Ku80 knockout mice are small in size and have a short lifespan, whereas scid mice have a normal lifespan. This observation implies that, in addition to its role in NHEJ, the Ku heterodimer may have another function not requiring DNA-PKcs. Based on the recently discovered role of Ku in yeast telomere biology, it was proposed that Ku also is required in mammalian telomere biology (9). Our results confirm the proposed role of Ku, but further demonstrate a need for DNA-PKcs, thus they do not support a Ku-related deficiency in telomere function as the sole cause of the shortened lifespan observed in Ku knockout mice.
By impairing end-capping, DNA-PKcs deficiency greatly increases illegitimate recombination involving telomeres. Ineffective telomeric capping is an important source of spontaneous instability in a DNA-PKcs-deficient background, as can be seen from the large contribution that TF make to total exchange-type aberrations. Fewer TF were observed in the virally transformed Ku knockout cell lines compared with the transformed scid and p53−/−/scid cell lines. Though tempting, this finding does not necessarily indicate a lesser role for the Ku genes, but rather may reflect differences in the cell culture history of the respective cell lines, such as the number of population doublings that occurred before analysis, or genetic background.
The large increase in TF metacentric chromosomes that occurred in spontaneously immortalized scid cells was unexpected. These aberrations, which averaged about five per cell, are not lethal to the cells possessing them, despite their being dicentrics. However, their progressive loss in scid cells restored by transfection to repair proficiency suggests TF metacentrics are only quasi-stable. Whether or not a particular TF metacentric is maintained in a growing culture is likely to depend on the potential of that aberration to confer a growth advantage. Interestingly, telomeric associations have been observed previously in a transformed scid cell line (43). However, under the less restrictive scoring requirements employed, similar events were recorded in repair-proficient controls as well. Under our more stringent scoring criteria, actual fusions were restricted to dsb repair-deficient cells, thus clarifying the dependence of these events on genetic background.
The impairment of end-capping associated with dsb-repair deficiency is similar to that caused by expression of a mutant form of TRF-2, but is much less severe. Curiously, the severity of TRF-2 dysfunction precludes long-term cellular consequences, since the condition is most likely lethal. In contrast, the milder phenotype associated with dsb-repair deficiency allows cells to survive with a genomic instability that is transmitted to future generations. Thus, through their contribution to efficient telomeric end-capping, NHEJ repair genes help to preserve the fidelity of genetic inheritance. It is clear that a complete understanding of the mammalian DNA repair-deficient phenotype, and in particular how this phenotype relates to cancer proneness, will need to encompass the role of repair genes at the telomere.
Acknowledgments
We thank Dr. Stefan Burde for assistance with image analysis and Dr. Merl Hoekstra for the gift of several DNA repair-deficient mice. This work was supported by grants from the U.S. Army (DAMD 17-97-1-7165), Department of Energy (ERWF645), and National Institutes of Health (CA50519, CA56909, and CA78497).
Abbreviations
- dsb
double-strand break
- NHEJ
nonhomologous end-joining
- TF
telomeric fusion
- scid
severe combined immunodeficiency
- FISH
fluorescence in situ hybridization
- TRF
telomeric repeat-binding factor
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Muller H J. The Collecting Net. 1938;13:181–195. [Google Scholar]
- 2.McClintock B. Genetics. 1941;26:234–282. doi: 10.1093/genetics/26.2.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blackburn E H, Szostak J W. Annu Rev Biochem. 1984;53:163–194. doi: 10.1146/annurev.bi.53.070184.001115. [DOI] [PubMed] [Google Scholar]
- 4.Zakian V A. Annu Rev Genet. 1989;23:579–604. doi: 10.1146/annurev.ge.23.120189.003051. [DOI] [PubMed] [Google Scholar]
- 5.Preston R J. Radiat Res. 1997;147:529–534. [PubMed] [Google Scholar]
- 6.Biessmann H, Mason J M. Adv Genet. 1992;30:185–249. doi: 10.1016/s0065-2660(08)60321-1. [DOI] [PubMed] [Google Scholar]
- 7.Moyzis R K, Buckingham J M, Cram L S, Dani M, Deaven L L, Jones M D, Meyne J, Ratliff R L, Wu J R. Proc Natl Acad Sci USA. 1988;85:6622–6626. doi: 10.1073/pnas.85.18.6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wellinger R J, Sen D. Eur J Cancer. 1997;33:735–749. doi: 10.1016/S0959-8049(97)00067-1. [DOI] [PubMed] [Google Scholar]
- 9.Jeggo P A. Radiat Res. 1998;150:S80–S91. [PubMed] [Google Scholar]
- 10.Gu Y S, Jin S F, Gao Y J, Weaver D T, Alt F W. Proc Natl Acad Sci USA. 1997;94:8076–8081. doi: 10.1073/pnas.94.15.8076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gottlieb T M, Jackson S P. Cell. 1993;72:131–142. doi: 10.1016/0092-8674(93)90057-w. [DOI] [PubMed] [Google Scholar]
- 12.Critchlow S E, Jackson S P. Trends Biochem Sci. 1998;23:394–398. doi: 10.1016/s0968-0004(98)01284-5. [DOI] [PubMed] [Google Scholar]
- 13.Boulton S J, Jackson S P. EMBO J. 1996;15:5093–5103. [PMC free article] [PubMed] [Google Scholar]
- 14.Boulton S J, Jackson S P. Nucleic Acids Res. 1996;24:4639–4648. doi: 10.1093/nar/24.23.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Laroche T, Martin S G, Gotta M, Gorham H C, Pryde F E, Louis E J, Gasser S M. Curr Biol. 1998;8:653–656. doi: 10.1016/s0960-9822(98)70252-0. [DOI] [PubMed] [Google Scholar]
- 16.Porter S E, Greenwell P W, Ritchie K B, Petes T D. Nucleic Acids Res. 1996;24:582–585. doi: 10.1093/nar/24.4.582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsukamoto Y, Kato J, Ikeda H. Nature (London) 1997;388:900–903. doi: 10.1038/42288. [DOI] [PubMed] [Google Scholar]
- 18.Marcand S, Gilson E, Shore D. Science. 1997;275:986–990. doi: 10.1126/science.275.5302.986. [DOI] [PubMed] [Google Scholar]
- 19.Grunstein M. Curr Opin Cell Biol. 1997;9:383–387. doi: 10.1016/s0955-0674(97)80011-7. [DOI] [PubMed] [Google Scholar]
- 20.Martin S G, Laroche T, Suka N, Grunstein M, Gasser S M. Cell. 1999;97:621–633. doi: 10.1016/s0092-8674(00)80773-4. [DOI] [PubMed] [Google Scholar]
- 21.Mills K D, Sinclair D A, Guarente L. Cell. 1999;97:609–620. doi: 10.1016/s0092-8674(00)80772-2. [DOI] [PubMed] [Google Scholar]
- 22.Bianchi A, de Lange T. J Biol Chem. 1999;274:21223–21227. doi: 10.1074/jbc.274.30.21223. [DOI] [PubMed] [Google Scholar]
- 23.Broccoli D, Smogorzewska A, Chong L, de Lange T. Nat Genet. 1997;17:231–235. doi: 10.1038/ng1097-231. [DOI] [PubMed] [Google Scholar]
- 24.Vansteensel B, Smogorzewska A, Delange T. Cell. 1998;92:401–413. [Google Scholar]
- 25.Griffith J D, Comeau L, Rosenfield S, Stansel R M, Bianchi A, Moss H, de Lange T. Cell. 1999;97:503–554. doi: 10.1016/s0092-8674(00)80760-6. [DOI] [PubMed] [Google Scholar]
- 26.Xie G, Habbersett R, Jia Y, Peterson S R, Lehnert B E, Bradbury E M, D'Anna J A. Oncogene. 1998;16:721–736. doi: 10.1038/sj.onc.1201793. [DOI] [PubMed] [Google Scholar]
- 27.Ouyang H, Nussenzweig A, Kurimasa A, Soares V, Li X, Cordon-Cardo C, Li W, Cheong N, Nussenzweig M, Iliakis G, et al. J Exp Med. 1997;186:921–929. doi: 10.1084/jem.186.6.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nussenzweig A, Sokol K, Burgman P, Li L, Li G C. Proc Natl Acad Sci USA. 1997;94:13588–13593. doi: 10.1073/pnas.94.25.13588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kurimasa A, Nagata Y, Shimizu M, Emi M, Nakamura Y, Oshimura M. Hum Genet. 1994;93:21–26. doi: 10.1007/BF00218907. [DOI] [PubMed] [Google Scholar]
- 30.Kurimasa A, Kumano S, Boubnov N V, Story M D, Tung C S, Peterson S R, Chen D J. Mol Cell Biol. 1999;19:3877–3884. doi: 10.1128/mcb.19.5.3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meyne J, Moyzis R K. In: Methods in Molecular Biology. Choo K H A, editor. Totowa, NJ: Humana; 1994. pp. 63–74. [DOI] [PubMed] [Google Scholar]
- 32.Bailey S M, Meyne J, Cornforth M N, McConnell T S, Goodwin E H. Cytogenetics and Cell Genetics. 1996;75:248–253. doi: 10.1159/000134494. [DOI] [PubMed] [Google Scholar]
- 33.Goodwin E H, Meyne J, Bailey S M, Quigley D. Chromosoma. 1996;104:345–347. doi: 10.1007/BF00337223. [DOI] [PubMed] [Google Scholar]
- 34.Garagna S, Broccoli D, Redi C A, Searle J B, Cooke H J. Chromosoma. 1995;103:685–692. doi: 10.1007/BF00344229. [DOI] [PubMed] [Google Scholar]
- 35.Nanda I, Schneider-Rasp S, Winking H, Schmid M. Chromosome Res. 1995;3:399–409. doi: 10.1007/BF00713889. [DOI] [PubMed] [Google Scholar]
- 36.Wong A K C, Rattner J B. Nucleic Acids Res. 1988;16:11645–11661. doi: 10.1093/nar/16.24.11645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kipling D, Cooke H J. Nature (London) 1990;347:400–402. doi: 10.1038/347400a0. [DOI] [PubMed] [Google Scholar]
- 38.Zijlmans J M, Martens U M, Poon S S S, Raap A K, Tanke H J, Ward R K, Lansdorp P M. Proc Natl Acad Sci USA. 1997;94:7423–7428. doi: 10.1073/pnas.94.14.7423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Blasco M A, Lee H-W, Hande M P, Samper E, Lansdorp P M, DePinho R A, Greider C W. Cell. 1997;91:25–34. doi: 10.1016/s0092-8674(01)80006-4. [DOI] [PubMed] [Google Scholar]
- 40.Nugent C I, Bosco G, Ross L O, Evans S K, Salinger A P, Moore J K, Haber J E, Lundblad V. Curr Biol. 1998;8:657–660. doi: 10.1016/s0960-9822(98)70253-2. [DOI] [PubMed] [Google Scholar]
- 41.Trujillo K M, Yuan S S F, Lee E, Sung P. J Biol Chem. 1998;273:21447–21450. doi: 10.1074/jbc.273.34.21447. [DOI] [PubMed] [Google Scholar]
- 42.Haber J E. Cell. 1998;95:583–586. doi: 10.1016/s0092-8674(00)81626-8. [DOI] [PubMed] [Google Scholar]
- 43.Slijepcevic P, Hande M P, Bouffler S D, Lansdorp P, Bryant P E. Chromosoma. 1997;106:413–421. doi: 10.1007/s004120050263. [DOI] [PubMed] [Google Scholar]