

Abstract
The HIV-1 virion infectivity factor (Vif) is required during viral replication to inactivate the host cell anti-viral factor, APOBEC3G (A3G). Vif binds A3G and a Cullin5-ElonginBC E3 ubiquitin ligase complex which results in the proteasomal degradation of A3G. The Vif PPLP motif (amino acids 161–164) is essential for normal Vif function because mutations in this motif reduce the infectivity of virions produced in T-cells. In this report, we demonstrate that mutation of the Vif PPLP motif reduces Vif binding to A3G without affecting its interaction with ElonginC and Cullin5. We demonstrate that the failure of the Vif mutant to bind A3G resulted in A3G incorporation into assembling virions with loss of viral infectivity.
Keywords: HIV-1 Vif, APOBEC3G, Vif PPLP motif, Vif multimerization
Introduction
HIV-1 virion infectivity factor (Vif) functions during the viral life cycle to inactivate the host cytidine deaminase, APOBEC3G (A3G)(Sheehy et al., 2002). In the absence of Vif, A3G is efficiently incorporated into progeny virions. Following infection of target cells, A3G catalyzes the deamination of deoxycytidine residues in the viral cDNA minus strand during reverse transcription which leads to inhibition of viral replication (Lecossier et al., 2003;Mangeat et al., 2003;Zhang et al., 2003). Vif prevents A3G incorporation into assembling virions by inducing its degradation through the ubiquitin-proteasome pathway (Conticello et al., 2003;Marin et al., 2003;Mehle et al., 2004b;Sheehy et al., 2003;Stopak et al., 2003;Yu et al., 2003).
Deletion of the highly conserved Vif PPLP motif (amino acids 161–164) reduces the infectivity of progeny virions produced in non-permissive T-cells (Yang et al., 2001). Mutation of this motif also eliminates Vif-Vif interaction in vitro (Yang et al., 2003;Yang et al., 2001), and it has been suggested that Vif multimerization may be necessary for A3G binding (Miller et al., 2007). However, this region also has been identified as part of a novel “SOCS-box” motif implicated in Vif binding to ElonginC (Mehle et al., 2004a;Yu, et al., 2004) and to be involved in Vif interaction with the cellular Hck tyrosine kinase (Hassaine et al., 2001;Yang et al., 2003). Treatment of cells with PPLP motif peptides that disrupt Vif multimerization in vitro reduce viral replication (Yang et al., 2003), as well as increase virion incorporation of A3G and reduce the infectivity of those virions (Miller et al., 2007). However, the mechanism underlying this requirement for wild-type amino acids at Vif residues 161–164 was not investigated in these studies. In this report, we present data that adds to our understanding of the molecular mechanism of the requirement for an intact Vif PPLP motif for infectious virus production in non-permissive cells. We demonstrate that mutation of the Vif PPLP motif impaired the ability of Vif to bind A3G, and did not affect ElonginC and Cullin5 binding. As a consequence, the Vif mutant failed to induce A3G degradation and did not prevent A3G incorporation into virions.
Results
The Vif PPLP motif is necessary for A3G binding and degradation
To investigate whether an intact Vif PPLP motif (residues 161–164) is necessary for the functional interaction with A3G in vivo, we introduced substitution mutations in Vif which replaced PPL residues 161–163 with alanine residues [Vif(A 161 AA)]. We determined the effect of this mutation on the induction of A3G degradation by comparing the effect in 293T cells co-transfected with plasmids directing the expression of HA-tagged A3G and either wild type Vif or Vif(A 161 AA). Cell lysates were prepared 24 hours after transfection and A3G and Vif protein levels were analyzed by Western blotting. The results of this analysis showed that mutation of the PPLP motif impaired the ability of Vif to induce the degradation of A3G (Fig. 1A). To investigate the mechanism of this defect, the ability of Vif(A 161 AA ) to interact with A3G in vivo was determined by co-immunoprecipitation of A3G-Vif complexes with anti-HA antibody beads. The results of this analysis demonstrated that the binding of Vif(A 161 AA) to A3G-HA was reduced compared to wild type Vif binding (Fig. 1A). To confirm this result, 293T cells were co-transfected with plasmids encoding untagged A3G and either HA-tagged Vif wild type or PPL mutant and A3G-Vif complexes were immunoprecipitated using anti-HA antibody beads. The results of this analysis demonstrated A3G association with HA-tagged wild type Vif and reduced interaction with the HA-tagged Vif PPL mutant (Fig. 1B). In sum, these results demonstrated that an intact Vif PPLP motif is necessary for A3G binding and degradation.
Fig. 1.

The Vif PPLP motif is necessary for A3G binding and degradation. (A) HEK293T cells were co-transfected with plasmids expressing HA-tagged A3G (pA3G-HA) and either wild-type Vif (pHVif) or PPL mutant Vif [pHVif(A 161 AA)] as indicated. Cell lysates were prepared 24 hours after transfection and the levels of A3G and Vif determined by Western blotting (WB). A3G binding to wild type Vif or PPL mutant Vif was determined by co-immunoprecipitation of protein complexes from cell lysates using anti-HA antibody beads. (B) HEK293T cells were co-transfected with plasmids expressing A3G (pA3G) and HA-tagged Vif (pHVif-HA) or PPL mutant Vif [pHVif(A 161 AA)-HA] as indicated. Cell lysates were prepared 24 hours after transfection and A3G binding to wild type Vif or PPL mutant Vif was determined by co-immunoprecipitation of protein complexes using anti-HA antibody beads. Co-immunoprecipitated proteins were identified by Western blotting (WB) using the indicated antibodies as described in Materials and methods.
Vif(A 161 AA) is not defective in binding to ElonginC and Cullin5
It has been demonstrated that Vif interacts directly with ElonginC and Cullin5 in the assembly of an E3 ubiquitin ligase complex that is necessary for proteasomal degradation of A3G (Mehle et al., 2006;Xiao et al., 2006;Yu et al., 2004). Because of the requirement for E3 ubiquitin ligase in Vif induced A3G degradation, we sought to determine the effect of the PPL mutation on ElonginC and Cullin5 binding. 293T cells were co-transfected with plasmids encoding HA-tagged ElonginC or Myc-tagged Cullin5 and either wild type Vif or Vif(A 161 AA). Cell lysates were prepared 24 hours after transfection and protein complexes were immunoprecipitated either with anti-HA or anti-myc antibody beads. Co-immunoprecipitated proteins were identified by Western blotting (Fig. 2). The results of this analysis showed that Vif(A 161 AA) binding to ElonginC (Fig. 2A) and Cullin5 (Fig. 2B) was comparable to wild type Vif binding to these proteins. This result demonstrated that Vif recruitment and assembly of an E3 ubiquitin ligase complex does not require an intact Vif PPLP motif.
Fig. 2.

The Vif PPL mutant binds ElonginC and Cullin5. HEK293T cells were co-transfected with plasmids expressing HA-tagged ElonginC (pEloC-HA), Myc-tagged Cullin5 (pCul5-Myc), and either wild type Vif (pHVif) or PPL mutant Vif [pHVif(A 161 AA)] as indicated, and cell lysates were prepared 24 hours after transfection. Binding of wild-type Vif and PPL mutant Vif to ElonginC (A) and Cullin5 (B) was assessed by co-immunoprecipitation of protein complexes from cell lysates using either anti-HA antibody or anti-c-myc beads as indicated. Co-immunoprecipitated proteins were identified by Western blotting (WB) using the indicated antibodies as described in Materials and methods.
Mutation of the Vif PPLP motif affects the infectivity of virus produced in A3G expressing cells
We demonstrated that mutation of the Vif PPLP motif impaired Vif induction of A3G degradation in transient transfection assays (Fig. 1A). To confirm that the failure of the mutant Vif to induce A3G degradation resulted in the predicted reduction in viral infectivity, we measured the competency of the Vif(A 161 AA) mutant to rescue the infectivity of a vif -deleted virus. HEK293-APOBEC3G-HA cells, stably expressing HA tagged A3G, were co-transfected with pNL4-3(Δvif) provirus and either pcDNA3-HVif or pcDNA3-HVif(A 161 AA). Culture supernatants were collected 48 hours after transfection and viral infectivity was determined (Fig. 3A). The results showed that complementation of pNL4-3(Δvif) by wild type Vif resulted in a level of infectivity equivalent to virus produced from cells transfected with wild type pNL4-3 provirus. However, in comparison to wild-type Vif, the Vif(A 161 AA) mutant was unable to complement the vif-deleted provirus and infectivity was essentially equivalent to pNL4-3(Δvif) alone.
Fig. 3.
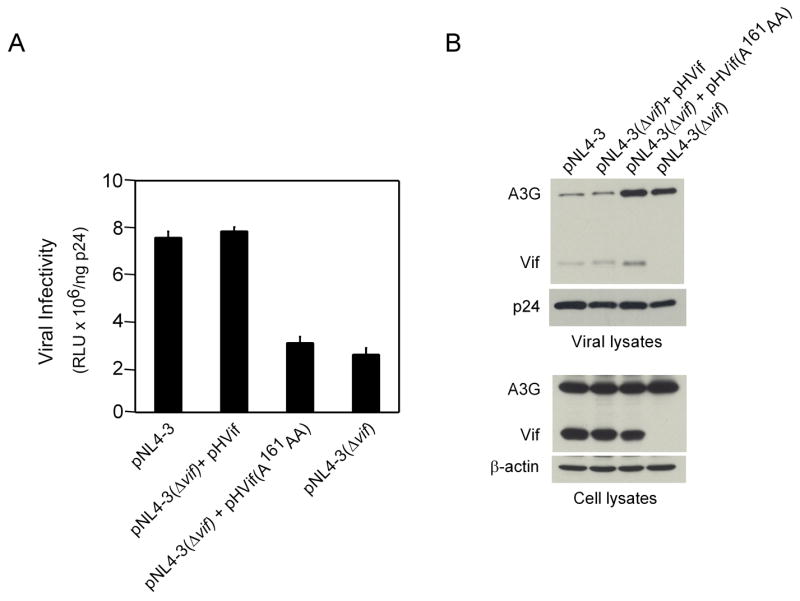
Mutation of the Vif PPLP motif affects the infectivity of virus produced in A3G expressing cells. HEK293-APOBEC3G-HA cells were co-transfected with proviral plasmid pNL4-3(Δvif) and plasmids expressing either wild-type Vif (pHVif) or PPL mutant Vif [pHVif(A 161 AA)]. Cells were also co-transfected with proviral plasmid pNL4-3 or pNL4-3(Δvif) plus empty vector, pcDNA3.1(−). (A) Viral infectivity of culture supernatants 48 hours post-transfection was determined using the TZM-bl indicator cell line as described in Materials and methods. Shown are Relative Light Units (RLU) plus standard deviation of triplicate determinations normalized to the viral p24 levels measured in culture supernatants. (B) Analysis of A3G and Vif levels in viral and cell lysates by Western blotting (WB). The antibodies used to detect A3G, Vif, p24 and β-actin are described in Materials and methods.
To correlate the observed infectivity data with the level of virion incorporation of A3G, virus was collected by centrifugation of culture supernatants through a 20% sucrose cushion and A3G content was analyzed by Western blotting. This analysis showed that Vif(A 161 AA) was defective in blocking the incorporation of A3G into virions (Fig 3B). The level of A3G incorporation was similar to that observed in the vif-deleted provirus alone. A low, but equivalent, level of A3G was also detected in virions produced by both wild-type Vif-complemented pNL4-3(Δvif) and Vif-positive pNL4-3 provirus. Finally, low levels of Vif were detected in virions produced by wild type Vif- and Vif(A 161 AA)-complemented pNL4-3(Δvif), as well as wild type Vif-positive pNL4-3 provirus. It has been demonstrated that incorporation of Vif into virions depends on Vif interaction with viral genomic RNA (Khan et al., 2001). Viral incorporation of Vif(A 161 AA) at levels comparable to wild-type Vif indicated that mutation of the Vif PPLP motif did not alter the RNA binding activity of Vif. Western blot analysis of cell lysates of the co-transfected cells demonstrated similar levels of expression of wild type Vif and Vif(A 161 AA), as well as A3G-HA. In these cells, A3G-HA is constitutively expressed at a high level, which may explain why the levels of this protein appear unaffected by wild-type Vif in this Western blot compared to the results presented in Fig 1A.
Discussion
In HIV-1 infected cells, Vif counteracts the activity of the host restriction factor A3G by inducing its proteasomal degradation. In this process, Vif functions as the substrate recognition subunit for an E3 ubiquitin ligase complex comprised of Cullin5, ElonginBC, and RbxI (Kobayashi et al., 2005;Mehle et al., 2004a;Yu et al., 2003;Yu et al., 2004). Vif binds A3G and assembles the E3 ubiquitin ligase complex through direct interaction with ElonginC and Cullin5. In addition, the production of infectious virions in non-permissive cells has been shown to require an intact PPLP motif which is implicated in mediating Vif multimerization in vitro. However, the molecular mechanism of the requirement for an intact PPLP motif in Vif has not yet been described. In this report, we provide a mechanistic basis for this requirement by demonstrating that mutation of the PPLP motif impaired the ability of Vif to bind A3G, but not to assemble an E3 ligase complex. As a consequence, the Vif mutant failed to induce A3G degradation and prevent A3G incorporation into virions resulting in loss of viral infectivity.
The majority of published reports have suggested that the binding site for A3G is located in the amino-terminal region of Vif and two recent studies presented evidence of the direct involvement of residues Y40RHHY in A3G binding (Mehle et al., 2007;Russell & Pathak, 2007). In the mutant we constructed, Vif PPL residues 161–163 were changed to alanine residues [Vif(A 161 AA)] which resulted in a reduction in A3G binding capacity. The loss of A3G binding by the Vif(A 161 AA) mutant probably was not caused by disruption of the native Vif structure as binding of ElonginC, Cullin5 and HIV-1 genomic RNA were all unaffected. The Vif binding site for HIV-1 genomic RNA maps between amino-terminal residues 1–64 (Zhang et al., 2000) and overlaps the Vif sequences recently demonstrated to bind A3G (Mehle et al., 2007;Russell & Pathak, 2007), further supporting the suggestion that the native structure of this region was unaffected by the carboxyl-terminal substitution mutations.
The results presented in this report suggest that Vif multimerization or post-translational modification involving the Vif PPLP motif is required for A3G binding, but not for binding to ElonginC and Cullin5. This indicates that the structural determinants of Vif recognition of A3G may be fundamentally different from those involved in Vif interaction with ElonginC and Cullin5. The potential importance of Vif multimerization, for which the PPLP motif has been demonstrated to be necessary in vitro and not yet in vivo, is supported by a recent study that probed Vif structure using chemical crosslinking, proteolysis and mass spectrometry of Vif monomers, dimers and trimers (Auclair et al., 2007). The results of that study demonstrated that Vif undergoes substantial changes in tertiary structure following protein multimerization in vitro. The changes in Vif tertiary structure as a consequence of protein multimerization, as well as the contribution of quaternary structure, may result in the formation and/or stabilization of an A3G binding site that is absent from the Vif monomeric structure. However, our data is also consistent with the Vif PPLP mutations abrogating a different structural or functional role of that motif, including the possibility that it contributes to post-translational modification of Vif needed for A3G binding.
Vif binding to ElonginC and Cullin5 was not affected by mutation of the PPLP motif in the current study. The binding sites for these proteins are well-defined and located in the carboxyl-terminal region of Vif. The highly conserved Vif S144LQYLAAL and downstream L163PxxxxL amino acid sequences share homology with the SOCS-box motifs of several cellular proteins that bind ElonginC (Kamura, Sato et al., 1998; Zhang, Farley et al., 1999). Mutational analysis has demonstrated the central role of the Vif S144LQYLAAL motif in binding ElonginC in the adaptor complex comprised of ElonginBC (Mehle et al., 2004a;Yu et al., 2004). This sequence is predicted to adopt an alpha helical structure, binding ElonginC through hydrophobic interactions. The downstream L163PxxxxL motif may not be necessary for Vif-ElonginC complex formation (Mehle et al., 2004a), although another report suggested that potential hydrophobic contacts with L163 and L169 may help stabilize ElonginC binding (Yu et al., 2004). In that report, substitution of L163 with serine within the PPLP motif impaired Vif binding to ElonginC but substitution of L163 with alanine had a lesser negative effect (Yu et al., 2004). This latter observation is consistent with the data presented in the current study demonstrating that the PPL161-163AAA mutation did not affect Vif binding to ElonginC. The binding site for Cullin5 involves a zinc stabilized HCCH motif encompassing Vif residues H108-H139 (Luo et al., 2005;Mehle et al., 2006;Xiao et al., 2006). This region is predicted to form a novel zinc finger domain containing a predicted alpha helix which contacts Cullin5 through hydrophobic interactions. The Vif binding sites for ElonginC and Cullin5 are contiguous and the secondary structure of the interacting surfaces appears similar. Our results showing that the Vif(A 161 AA) mutant retained binding to both ElonginC and Cullin5 suggests that the Vif monomer possesses the necessary secondary and tertiary structure to bind the E3 ubiquitin ligase complex. Validation of these conclusions awaits the determination of the three-dimensional crystal structures of Vif and the Vif-A3G and Vif-ElonginBC-Cullin5 complexes.
Materials and methods
Plasmids and cell lines
A full length infectious HIV-1 clone, pNL4-3 (Adachi et al., 1986), and isogenic vif-deleted clone, pNL4-3(Δvif), were obtained from Dr. Chris Aiken. The Vif expression plasmid pcDNA-HVif was obtained through the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH from Dr. Stephan Bour and Dr. Klaus Strebel. The vif mutant plasmid pcDNA-HVif(A 161 AA)], encoding Vif(PPL161-163AAA), was constructed from pcDNA-HVif by site directed mutagenesis using the Quick Change™ Site-directed Mutagenesis Kit (Stratagene) as described by the manufacturer. Plasmids pCul5-Myc (Yu et al., 2003) and pEloC-HA (Yu et al., 2004) were obtained from Dr. Bindong Liu. A plasmid encoding A3G with a single carboxyl-terminal HA tag was constructed by PCR from pCEM-15 (Sheehy et al., 2002) using appropriate primers and insertion in the NotI and HindIII sites of pcDNA3.1(-). HEK293-APOBEC3G-HA (Yu et al., 2003) cells were obtained through the NIH AIDS Research and Reference Reagent Program from Dr. Xiao-Fang Yu. TZM-bl indicator cells were obtained through the NIH AIDS Research and Reference Reagent Program from Dr. John C. Kappes, Dr. Xiaoyun Wu and Tranzyme, Inc. The TZM-bl cell line, a genetically engineered HeLa cell clone expressing CD4, CXCR4, CCR5 and Tat-responsive firefly luciferase and Escherichia coli β-galactosidase under the control of an HIV-1 long terminal repeat.
Antibodies
HIV-1HXB2 Vif antiserum and anti-ApoC17 antibody, which recognizes the 17 carboxyl-terminal residues of human APOBEC3G, were obtained through the NIH AIDS Research and Reference Reagent Program from Dr. Dana Gabuzda and Dr. Klaus Strebel, respectively. Rabbit anti-human c-myc and anti-hemagglutinin (HA tag) antibodies were from USBiological. Anti-HA (clone 3F10) affinity matrix was from Roche and anti-c-myc (clone 9E10) Sepharose- 4B beads were from the Vanderbilt University Antibody Core. Anti-β-actin monoclonal antibody (clone AC-74) was from Sigma. Anti-p24 monoclonal antibody 183-H12-5C was from Vanderbilt-Meharry Center for AIDS Research Virology Core.
Immunoprecipitation and Western blot analysis
HEK293T cells were plated at a density of 400,000 cells/well in a 6-well culture plate 24 hours prior to transfection. Cells were co-transfected with pcDNA-HVif, pcDNA-HVif(A 161 AA), pcDNA-A3G(HA), pCul5-Myc, and pEloC-HA plasmid DNA as indicated in individual figure legends using linear polyethylenimine (PEI; 25 kDa; Polysciences, Inc.) essentially as described before (Durocher et al., 2002). Cells were harvested 24 hours after transfection in ice-cold PBS and lysed in 250 μl of lysis/IP buffer [50 mM HEPES (pH 7.4), 150 mM NaCl, 1 mM Na2EDTA, 0.2% NP-40 (v/v), 0.1 mM PMSF and complete mini protease inhibitor cocktail without Na2EDTA (Roche)]. Cell lysates were incubated at 4°C for 30 minutes with constant mixing and then centrifuged at 10,000 x g for 5 minutes at 4°C. The supernatant fraction was removed and mixed with 10 μl (packed bead volume) of either mouse HA antibody conjugated agarose beads or mouse c-myc antibody conjugated Sepharose 4B beads. Samples were incubated at 4°C for 18 hours with constant mixing. Antibody beads were collected by centrifugation at 500 x g and washed successively with lysis/IP buffer and lysis/IP buffer with 500 mM NaCl. Captured protein was then eluted from the antibody beads by incubation in SDS-protein sample buffer [50 mM Tris-HCl (pH 6.8), 2 mM Na2EDTA, 2% SDS, 2% 2-mercaptoethanol, 10% glycerol, 0.05% bromophenol blue] at 100°C for 5 minutes and then separated by SDS-PAGE. Proteins were transferred to an Immobilon-P membrane (Millipore) and processed for Western blot analysis using specific antibodies, as indicated in figure legends, and chemiluminescent detection.
Vif complementation assay
HEK293-APOBEC3G-HA cells were plated at a density of 3 x 106 cells/100 mm culture dish 24 hours prior to transfection. Using the calcium phosphate precipitation method (Chen & Okayama, 1987), cells were co-transfected with 15μg of pNL4-3(Δvif) and either 5 μg of pcDNA-HVif or 5 μg of pcDNA-HVif(A 161 AA) to produce vif complemented virus. Cells were also co-transfected with 15 μg of pNL4-3 or pNL4-3(Δvif) and 5 μg of pcDNA3.1(−) as positive and negative controls, respectively. Culture supernatants were collected 48 hours after transfection and passed through a 0.45 μm filter to remove cellular debris. Viral particle content in culture supernatants was quantified by a capsid-specific (p24) ELISA as described (Wehrly & Chesebro, 1997). Virions were concentrated by centrifugation of culture supernatants through a 20% sucrose cushion at 125,000 x g for 45 minutes and normalized for their p24 content with viral lysis buffer [50 mM Tris (pH 8.0), 40 mM KCl, 50 mM NaCl, 5 mM Na2EDTA, 10 mM DTT and 0.1% (v/v) Triton X-100]. Transfected cells were lysed in 1 ml of lysis buffer [50 mM HEPES (pH 7.4), 150 mM NaCl, 1 mM Na2EDTA, 0.5% NP-40 (v/v), 0.1 mM PMSF and complete mini protease inhibitor cocktail without Na2EDTA (Roche)]. Cell lysates were incubated at 4°C for 30 minutes with constant mixing, centrifuged at 10,000 x g for 5 minutes (4°C) and the supernatant fraction removed for Western blot analysis as described above.
Viral infectivity assay
TZM-bl indicator cells were plated at a density of 15,000 cells/well in a 96 well culture plate 24 hours prior to infection and incubated at 37°C (5% CO2). The day of infection, the culture medium was removed and the cells inoculated in triplicate with 200 μl of 5-fold serial dilutions of viral supernatants in culture medium containing 20 μg/ml DEAE dextran and incubated at 37°C (5% CO2). After 60 hours incubation, 100μl of culture supernatant was removed from each well and replaced with 100 μl of Bright-Glo Luciferase Assay substrate (Promega). Following 2 minutes incubation at room temperature, 100μl of each cell lysate was transferred to a 96 well OptiPlate 96 (Perkin Elmer) and luminescence was measured in a TopCount scintillation counter (Packard/Perkin Elmer). Results are reported as Relative Light Units (RLU) recorded in the linear range of viral supernatant dilutions.
Acknowledgments
We thank Lorraine Sutton for help with p24 assays and DNA sequencing, Megan Johnson for plasmid pA3G-HA and Chisu Song for careful reading and critical evaluation of this manuscript. This work was supported by a Vanderbilt-Meharry Center for AIDS Research (CFAR) Developmental grant (J.P.D.) and National Institutes of Health grants R21 AI068490 (J.P.D.) and RO1 AI29193 (RTD). This work has been facilitated by the infrastructure and resources provided by the Vanderbilt-Meharry CFAR, funded by NIH program P30 AI54999.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol. 1986;59:284–291. doi: 10.1128/jvi.59.2.284-291.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auclair JR, Green KM, Shandilya S, Evans JE, Somasundaran M, Schiffer CA. Mass spectrometry analysis of HIV-1 Vif reveals an increase in ordered structure upon oligomerization in regions necessary for viral infectivity. Proteins. 2007;69:270–284. doi: 10.1002/prot.21471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Okayama H. High-efficiency transformation of mammalian cells by plasmid DNA. Mol Cell Biol. 1987;7:2745–2752. doi: 10.1128/mcb.7.8.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conticello SG, Harris RS, Neuberger MS. The Vif protein of HIV triggers degradation of the human antiretroviral DNA deaminase APOBEC3G. Curr Biol. 2003;13:2009–2013. doi: 10.1016/j.cub.2003.10.034. [DOI] [PubMed] [Google Scholar]
- Durocher Y, Perret S, Kamen A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002;30:e9. doi: 10.1093/nar/30.2.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassaine G, Courcoul M, Bessou G, Barthalay Y, Picard C, Olive D, Collette Y, Vigne R, Decroly E. The Tyrosine Kinase Hck Is an Inhibitor of HIV-1 Replication Counteracted by the Viral Vif Protein. J Biol Chem. 2001;276:16885–16893. doi: 10.1074/jbc.M009076200. [DOI] [PubMed] [Google Scholar]
- Kamura T, Sato S, Haque D, Liu L, Kaelin WGJ, Conaway RC, Conaway JW. The Elongin BC complex interacts with the conserved SOCS-box motif present in members of the SOCS, ras, WD-40 repeat, and ankyrin repeat families. Genes Dev. 1998;12:3872–3881. doi: 10.1101/gad.12.24.3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan MA, Aberham C, Kao S, Akari H, Gorelick R, Bour S, Strebel K. Human Immunodeficiency Virus Type 1 Vif Protein Is Packaged into the Nucleoprotein Complex through an Interaction with Viral Genomic RNA. The J Virol. 2001;75:7252–7265. doi: 10.1128/JVI.75.16.7252-7265.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi M, Takaori-Kondo A, Miyauchi Y, Iwai K, Uchiyama T. Ubiquitination of APOBEC3G by an HIV-1 Vif-Cullin5-Elongin B-Elongin C Complex Is Essential for Vif Function. J Biol Chem. 2005;280:18573–18578. doi: 10.1074/jbc.C500082200. [DOI] [PubMed] [Google Scholar]
- Lecossier D, Bouchonnet F, Clavel F, Hance AJ. Hypermutation of HIV-1 DNA in the absence of the Vif protein. Science. 2003;300:1112. doi: 10.1126/science.1083338. [DOI] [PubMed] [Google Scholar]
- Luo K, Xiao Z, Ehrlich E, Yu Y, Liu B, Zheng S, Yu XF. Primate lentiviral virion infectivity factors are substrate receptors that assemble with cullin 5-E3 ligase through a HCCH motif to suppress APOBEC3G. Proc Natl Acad Sci USA. 2005;102:11444–11449. doi: 10.1073/pnas.0502440102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mangeat B, Turelli P, Caron G, Friedli M, Perrin L, Trono D. Broad antiretroviral defence by human APOBEC3G through lethal editing of nascent reverse transcripts. Nature. 2003;424:99–103. doi: 10.1038/nature01709. [DOI] [PubMed] [Google Scholar]
- Marin M, Rose KM, Kozak SL, Kabat D. HIV-1 Vif protein binds the editing enzyme APOBEC3G and induces its degradation. Nat Med. 2003;9:1398–1403. doi: 10.1038/nm946. [DOI] [PubMed] [Google Scholar]
- Mehle A, Goncalves J, Santa-Marta M, McPike M, Gabuzda D. Phosphorylation of a novel SOCS-box regulates assembly of the HIV-1 Vif-Cul5 complex that promotes APOBEC3G degradation. Genes Dev. 2004a;18:2861–2866. doi: 10.1101/gad.1249904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehle A, Strack B, Ancuta P, Zhang C, McPike M, Gabuzda D. Vif overcomes the innate antiviral activity of APOBEC3G by promoting its degradation in the ubiquitin-proteasome pathway. J Biol Chem. 2004b;279:7792–7798. doi: 10.1074/jbc.M313093200. [DOI] [PubMed] [Google Scholar]
- Mehle A, Thomas ER, Rajendran KS, Gabuzda D. A zinc-binding region in Vif binds Cul5 and determines cullin selection. J Biol Chem. 2006;281:17259–17265. doi: 10.1074/jbc.M602413200. [DOI] [PubMed] [Google Scholar]
- Mehle A, Wilson H, Zhang C, Brazier AJ, McPike M, Pery E, Gabuzda D. Identification of an APOBEC3G Binding Site in Human Immunodeficiency Virus Type 1 Vif and Inhibitors of Vif-APOBEC3G Binding. J Virol. 2007;81:13235–13241. doi: 10.1128/JVI.00204-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH, Presnyak V, Smith HC. The dimerization domain of HIV-1 viral infectivity factor Vif is required to block APOBEC3G incorporation with virions. Retrovirol. 2007;4:81. doi: 10.1186/1742-4690-4-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell RA, Pathak VK. Identification of Two Distinct Human Immunodeficiency Virus Type 1 Vif Determinants Critical for Interactions with Human APOBEC3G and APOBEC3F. J Virol. 2007;81:8201–8210. doi: 10.1128/JVI.00395-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418:646–650. doi: 10.1038/nature00939. [DOI] [PubMed] [Google Scholar]
- Sheehy AM, Gaddis NC, Malim MH. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat Med. 2003;9:1404–1407. doi: 10.1038/nm945. [DOI] [PubMed] [Google Scholar]
- Stopak K, de Noronha C, Yonemoto W, Greene WC. HIV-1 Vif Blocks the Antiviral Activity of APOBEC3G by Impairing Both Its Translation and Intracellular Stability. Mol Cell. 2003;12:591–601. doi: 10.1016/s1097-2765(03)00353-8. [DOI] [PubMed] [Google Scholar]
- Wehrly K, Chesebro B. p24 antigen capture assay for quantification of human immunodeficiency virus using readily available inexpensive reagents. Methods. 1997;12:288–293. doi: 10.1006/meth.1997.0481. [DOI] [PubMed] [Google Scholar]
- Xiao Z, Ehrlich E, Yu Y, Luo K, Wang T, Tian C, Yu XF. Assembly of HIV-1 Vif-Cul5 E3 ubiquitin ligase through a novel zinc-binding domain-stabilized hydrophobic interface in Vif. Virology. 2006;349:290–299. doi: 10.1016/j.virol.2006.02.002. [DOI] [PubMed] [Google Scholar]
- Yang B, Gao L, Li L, Lu Z, Fan X, Patel CA, Pomerantz RJ, DuBois GC, Zhang H. Potent suppression of viral infectivity by the peptides that inhibit multimerization of human immunodeficiency virus type 1 (HIV-1) Vif proteins. J Biol Chem. 2003;278:6596–6602. doi: 10.1074/jbc.M210164200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Sun Y, Zhang H. The multimerization of human immunodeficiency virus type I Vif protein: a requirement for Vif function in the viral life cycle. J Biol Chem. 2001;276:4889–4893. doi: 10.1074/jbc.M004895200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Yu Y, Liu B, Luo K, Kong W, Mao P, Yu XF. Induction of APOBEC3G ubiquitination and degradation by an HIV-1 Vif-Cul5-SCF complex. Science. 2003;302:1056–1060. doi: 10.1126/science.1089591. [DOI] [PubMed] [Google Scholar]
- Yu Y, Xiao Z, Ehrlich ES, Yu X, Yu XF. Selective assembly of HIV-1 Vif-Cul5-ElonginB-ElonginC E3 ubiquitin ligase complex through a novel SOCS box and upstream cysteines. Genes Dev. 2004;18:2867–2872. doi: 10.1101/gad.1250204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Yang B, Pomerantz RJ, Zhang C, Arunachalam SC, Gao L. The cytidine deaminase CEM15 induces hypermutation in newly synthesized HIV-1 DNA. Nature. 2003;424:94–98. doi: 10.1038/nature01707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Pomerantz RJ, Dornadula G, Sun Y. Human Immunodeficiency Virus Type 1 Vif Protein Is an Integral Component of an mRNP Complex of Viral RNA and Could Be Involved in the Viral RNA Folding and Packaging Process. J Virol. 2000;74:8252–8261. doi: 10.1128/jvi.74.18.8252-8261.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JG, Farley A, Nicholson SE, Willson TA, Zugaro LM, Simpson RJ, Moritz RL, Cary D, Richardson R, Hausmann G, Kile BJ, Kent SB, Alexander WS, Metcalf D, Hilton DJ, Nicola NA, Baca M. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc Natl Acad Sci USA. 1999;96:2071–2076. doi: 10.1073/pnas.96.5.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]