

Synopsis
Proper immunologic balance between pro- and anti-inflammatory forces is necessary for recovery from critical illness. Persistence of a marked compensatory anti-inflammatory innate immune response following an insult such as sepsis or trauma is termed immunoparalysis. This state of acquired immunodeficiency can be quantified through the measurement of monocyte cell-surface HLA-DR expression or through analysis of the capacity of whole blood to produce TNFα upon ex vivo stimulation with endotoxin. Critically ill patients demonstrating prolonged, severe reductions in monocyte HLA-DR expression or ex vivo TNFα production are at high risk for the development of nosocomial infection and death. Reversal of immunoparalysis can be accomplished through the administration of immunostimulatory agents or tapering of exogenous immunosuppression. Intriguing evidence suggests that this may be associated with improved clinical outcomes. Immune monitoring protocols are needed to identify patients who may benefit from immunomodulatory trials in the pediatric intensive care unit.
Keywords: Immunoparalysis, monocyte, HLA-DR, TNFα, mortality, nosocomial infection
Introduction
Children can face a variety of inflammatory challenges ranging from the benign (otitis media) to the life-threatening (severe trauma, open heart surgery, septic shock). The immune system is of vital importance for the successful weathering of these challenges. A massive proinflammatory response without proper controls is pathologic and places the patient at risk for organ dysfunction and death. Conversely, an underactive immune system which is unable to detect pathogens, mount an inflammatory response, destroy microbial invaders, or repair damaged tissue places the patient at risk for death from secondary infection and persistent organ failure.
In the 1980s and 1990s, multiple therapies targeting proinflammatory mediators and aimed at reducing inflammation reached phase III clinical trials in adults with severe sepsis and septic shock [1–12]. Nearly all of these studies failed to demonstrate a survival benefit, suggesting that perhaps reducing inflammation is not the appropriate therapeutic goal in all cases. Subsequent studies have suggested that late mortality from surgery, sepsis, or trauma can be associated with an acquired immune deficiency state. If prolonged and severe, this state has been termed immunoparalysis. Characterized by markedly impaired innate immune function, immunoparalysis is now recognized as a predictor of morbidity and mortality for both children and adults [13–18]. Moreover, the phenomenon is often occult, is not heralded by any premorbid phenotype, and commonly occurs in patients previously thought to be immunocompetent. Although immunoparalysis cannot be detected by analysis of a patient’s complete blood count or white blood cell differential, methods of immune monitoring exist which can permit the diagnosis to be made in a same-day fashion. Recent investigations offer intriguing evidence that immunoparalysis can be reversed with benefit to the patient.
In order to provide a biologic framework, this discussion will begin with an overview of the immune system. Following this, the phenomenon of immunoparalysis will be reviewed in detail with attention paid to mechanisms of disease, clinical significance, and potential for therapeutic intervention. The overall goal of this review is to highlight the anti-inflammatory end of the spectrum of the immune response as an under-appreciated yet highly relevant contributor to outcomes in critically ill patients.
The Monocyte and the Inflammatory Response
The Innate Immune System
In general terms, the immune system can be divided into the innate and the adaptive arms (Table 1). The innate immune system is most easily thought of as the body’s first cellular line of defense. It includes members whose primary roles include phagocytosis and intracellular killing (polymorphonuclear cells), cytotoxic killing (natural killer cells), and antigen presentation (dendritic cells). Another innate immune cell, the monocyte, is thought to be a key determinant of the acute immune response. Its diverse roles (and those of its descendant, the tissue macrophage) include recognition and phagocytosis of pathogens, presentation of digested peptides on their cell surfaces in order to activate the adaptive immune response, and secretion of mediators which modulate the overall immune response (Figure 1). A characteristic feature of monocytes and other innate immune cells is their ability to consistently respond to pathogens regardless of prior exposure history. They do not require prior sensitization in order to mount a robust immune response. This is accomplished through the presence of constitutively expressed receptors present on the plasma membranes of innate immune effector cells. These receptors recognize broad classes of microbial constituents. For example, bacterial lipopolysaccharide (LPS) is recognized by monocytes through its interaction with the toll-like receptor (TLR)-4 on the monocyte’s cell surface. A monocyte which encounters LPS should vigorously produce proinflammatory cytokines regardless of whether or not it has been exposed to LPS in the past.
Table 1.
Elements of the innate and adaptive immune systems.
| Innate | Adaptive |
|---|---|
| Cellular Elements | Cellular Elements |
| Phagocytosis | Antibody production |
| Monocytes/Macrophages | B cells/Plasma cells |
| Polymorphonuclear leukocytes | Cytotoxic killing |
| Dendritic cells | CD8+ T cells |
| Antigen presentation | Cytokine/chemokine production |
| Monocytes/Macrophages | CD4+ T cells |
| Dendritic cells | TH1 cells (proinflammatory) |
| Cytotoxic killing | TH2 cells (anti-inflammatory) |
| Natural killer cells | Treg cells (anti-inflammatory) |
| Polymorphonuclear leukocytes | |
| Cytokine/chemokine production | |
| All of the above | |
|
| |
| Noncellular Elements | Noncellular Elements |
| Cytokines | Immunoglobulins |
| Chemokines | Cytokines |
| Complement | Chemokines |
Figure 1. Functions of the activated monocyte.
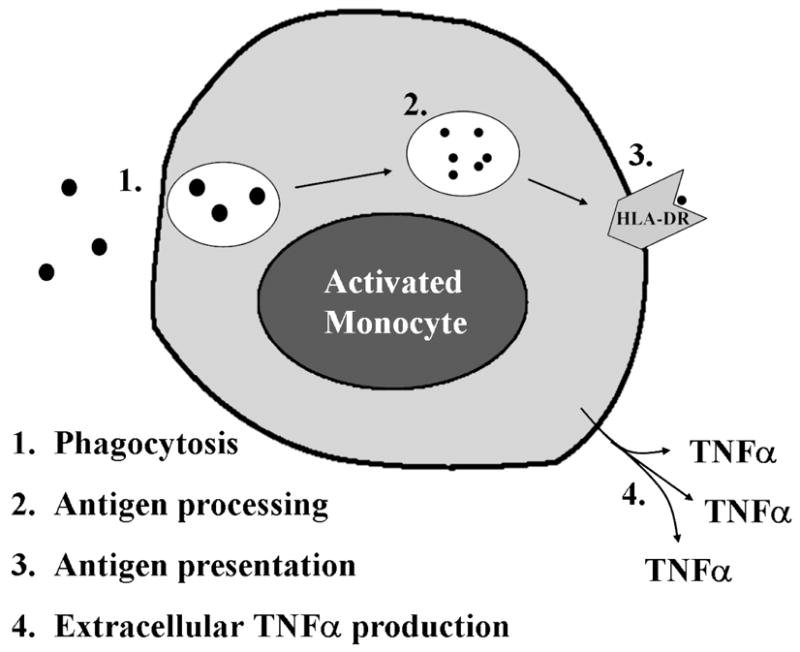
A normal monocyte, upon activation, is capable of ingesting pathogens; effecting intracellular killing and processing the foreign proteins into antigenic peptides; presenting these peptides on its cell surface in conjunction with HLA-DR molecules; and producing proinflammatory cytokines such as TNFα. An immunoparalyzed monocyte demonstrates reduced HLA-DR expression and impaired TNFα production capacity.
Once stimulated, monocytes engulf and destroy microbes which are then processed into specific antigenic peptides. These are presented on the external surfaces of innate immune cells in conjunction with class II Major Histocompatibility Complex (MHC) molecules such as Human Leukocyte Antigen (HLA)-DR. Antigens presented in this way, along with co-stimulatory input from the innate immune cells, activate the adaptive arm of the immune response. Additionally, innate immune cells secrete cytokines and chemokines which modulate the inflammatory response by recruiting and activating other immune effector cells and activating non-cellular aspects of the immune response such as the complement and coagulation cascades. Local production of proinflammatory cytokines such as tumor necrosis factor (TNF)-α results in activation of nearby immune cells and proinflammatory changes in vascular endothelium promoting cellular migration into the periphery. It is when this modulation becomes systemic that the clinical signs and symptoms of hyperinflammation (fever, hemodynamic instability, and capillary leak) become evident.
The Adaptive Immune System
While measures of immunoparalysis primarily reflect the innate immune response, it is important to realize that the innate and adaptive immune systems do not act in isolation. Adaptive immune elements respond to, and in turn affect, the pro- and anti-inflammatory balance of the patient as well.
T and B lymphocytes are adaptive immune cells. In contrast to innate immunity, the adaptive immune system produces responses that are highly antigen-specific. Adaptive immune responses typically require antigen presentation by innate immune cells. The initial lymphocyte response to a pathogen takes more time and is of smaller magnitude than the initial innate response. Repeat exposure to an antigen, however, provokes a more rapid and powerful adaptive immune response due to the presence of antigen-specific T and B cell clones that can be maintained for decades.
Upon stimulation, B cells mature into plasma cells which produce antibodies that opsonize invaders, thus marking them for clearance by lymphoid organs and phagocytes. Activated T cells regulate the immune response through the production of cytokines (CD4+) and via cytotoxic killing (CD8+). Naïve CD4+ T cells can differentiate into one of a number of T cell subtypes depending upon the cytokine milieu in which they are activated. While a detailed review of T cell biology is beyond the scope of this review, three major classes of CD4+ T cells merit general discussion here. These include the T-helper (TH)-1, TH2, and regulatory T cells (Treg). In the presence of proinflammatory cytokines, naïve T cells will differentiate into TH1 cells. These cells in turn produce proinflammatory cytokines such as interferon (IFN)-γ and serve to perpetuate the inflammatory response. TH2 cells, by contrast, arise when naïve T cells are activated in an anti-inflammatory environment. They themselves produce anti-inflammatory cytokines including interleukin (IL)-10 and transforming growth factor (TGF)-β with a resultant inhibitory effect on the inflammatory response. Treg cells represent a more recently describe subgroup of CD4+ T cells. Often (but not always) characterized by cell surface expression of CD25 and the transcription factor FOXP3, these cells exert an even more powerful anti-inflammatory effect through contact-mediated direct inhibition of other immune cells and production of high levels of TGFβ and interleukin (IL) -10 (reviewed in [19]).
Cytokines and Chemokines
To accomplish communication between cells of the innate and adaptive immune systems, a common language is needed. Cytokines and chemokines are the extracellular vocabulary of the immune system (Table 2). Certain cytokines (e.g. TNFα, IFNγ and IL-1β) have proinflammatory effects. Anti-inflammatory cytokines (e.g. IL-10 and TGFβ) deactivate effector cells and inhibit the proinflammatory response. Chemokines, such as IL-8, are chemoattractants which result in cellular migration into an inflamed area. When considering cytokines, it is important to appreciate that these molecules often have differing effects depending on the specific cell type being stimulated. Therefore, defining cytokines as either purely pro- or anti- inflammatory can be misleading. An example of this can be found in IL-6. This cytokine is induced by proinflammatory stimuli and plasma IL-6 levels are often used as a marker of systemic inflammation [20–23]. While IL-6 is known to stimulate the hepatic acute phase response, it also results in other, decidedly anti-inflammatory effects including the induction of an adrenal glucocorticoid response [24, 25]. It is in this setting of competing pro- and anti-inflammatory mediators that the concept of immunoparalysis becomes relevant.
Table 2.
Selected cytokines and the ir effects
| Cytokine | Primary Producer(s) | Action(s) | |
|---|---|---|---|
| Proinflammatory | IL-1β | Monocytes/macrophages | Fever, vasodilation, activation of T cells, monocytes/macrophages |
| TNFα | Monocytes/macrophages, T cells (TH1), NK cells | Fever, vasodilation, apoptosis, activation of T cells, monocytes/macrophages | |
| IL-18 | Macrophages | Activation of T cells and monocytes | |
| IL-12 | Macrophages, DCs | Activation of NK cells | |
| IFNγ | T cells (TH1), NK cells | Activation of monocytes/macrophages | |
| GM-CSF | T cells, macrophages | Increases production and promotes growth and activation of monocytes, macrophages, PMNs, DCs | |
| ? | IL6 | Monocytes, macrophages, Vascular endothelium | Promotes acute phase response (pro), activates adrenal axis (anti) |
| Anti-inflammatory | IL-10 | Monocytes/macrophages, T cells (TH2, Treg) | Inhibits monocyte/macrophage activation |
| TGFβ | Monocytes, T cells (TH2, Treg) | Inhibits monocyte/macrophage proliferation and activation | |
| IL-13 | T cells (TH2) | Inhibits monocyte/macrophage cytokine production | |
| IL-1ra | Hepatocytes, monocytes/macrophages, PMNs | Inhibits IL-1 action by blocking the IL-1 receptor |
DC: dendritic cell; PMN: polymorphonuclear cell; NK: natural killer
Immunoparalysis
SIRS, CARS and Immunoparalysis in Critical Illness
When a sufficiently severe proinflammatory insult occurs, the patient typically experiences a constellation of vital sign and laboratory changes (tachycardia, tachypnea, abnormal temperature, leukocytosis) that is termed the Systemic Inflammatory Response Syndrome (SIRS) [26]. It is notable that in the most severe cases of hyperinflammation (e.g. septic shock), the acute morbidity and mortality result not from overwhelming infection but from the systemic immune response to that infection. In short order (likely < 24 hours later) compensatory mechanisms come into play and shut down the immune system’s inflammatory response, often through the induction of anti-inflammatory cytokines. This phenomenon is referred to as the Compensatory Anti-inflammatory Response Syndrome (CARS). While a transient CARS state is a necessary countermeasure to regulate and protect against inflammatory injury it, like SIRS, must also be regulated. Immunoparalysis represents a persistent, severe form of a CARS state which has become pathologic (Figure 2). Given the importance of the immune system in responding to pathogens and remodeling injured tissue, it is intuitive that patients recovering from septic shock or major surgery who develop immunoparalysis would find themselves at increased risk for the development of nosocomial infection and death.
Figure 2. Immunologic homeostasis vs. immunoparalysis.
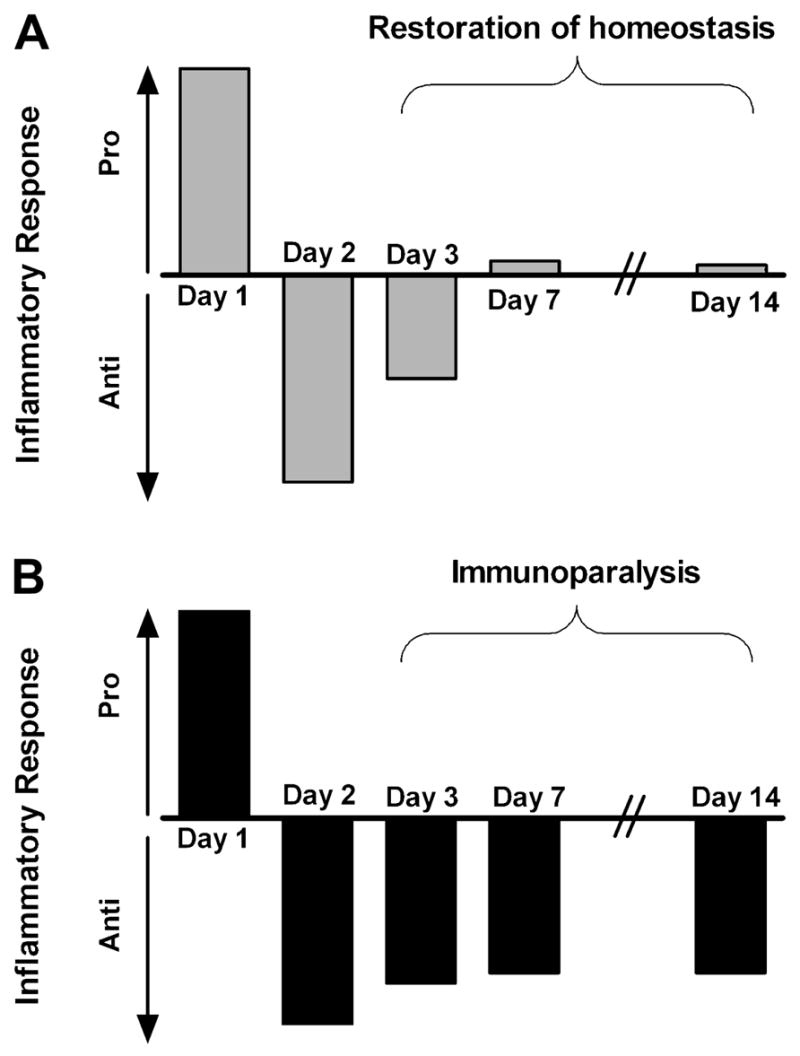
A transient compensatory anti-inflammatory response typically follows a proinflammatory insult, but the immunologic balance between pro- and anti-inflammatory forces should be restored within a few days (A). If the anti-inflammatory response is severe and prolonged it is termed immunoparalysis (B).
Diagnosis and Quantification of Immunoparalysis
The concept of innate immune dysfunction being associated with critical illness is hardly a new one. In the mid-1980’s Polk et al investigated the behavior of the immune system in adults following trauma, with an interest in identifying the immunologic basis for late mortality and nosocomial infections in their patients. Their systematic examination of the immune response demonstrated an association between reduction in monocyte antigen presenting capacity and the development of secondary infections [16]. Subsequent investigations have focused largely on two measures of innate immune competence in critical illness: monocyte class II MHC expression and whole blood ex vivo LPS-induced cytokine production capacity.
As noted above, monocytes activate the adaptive immune response through the presentation of antigens on cell-surface class II MHC molecules such as HLA-DR. HLA-DR expression on circulating monocytes can be quantified by flow cytometry. Flow cytometric analysis involves the staining of whole blood with fluorochrome-tagged antibodies. Comparison of anti-HLA-DR fluorescence in CD14+ cells (monocytes) with that of a non-specific control antibody enables the calculation of HLA-DR %-positivity. One can therefore interpret a monocyte HLA-DR expression level of < 30%, for example, to mean that < 30% of the subject’s circulating monocytes have anti-HLA-DR fluorescence which is greater than background fluorescence. Most of the literature on the subjects of HLA-DR expression and immunoparalysis in the ICU use this nomenclature. In fact, it is this threshold of < 30% HLA-DR+ monocytes which has become an accepted definition of immunoparalysis. In recent years, however, another option has become available that addresses the lack of standardization inherent in varying lots of antibodies and differences in cytometer calibration across centers. The Quantibrite system (Becton Dickinson, Franklin Lakes, NJ) is another flow cytometric tool for the quantification of monocyte HLA-DR expression. In this approach, a subject’s monocyte anti-HLA-DR fluorescence is compared with that of a set of standard beads which express a known quantity of fluorescent antibodies on their surfaces. Using this technology one can calculate the number of HLA-DR molecules per cell in a way which is reproducible across centers. While clinical investigators are beginning to use this method [27], it does not yet represent the standard approach in this field.
The other commonly used method for quantification of innate immune function in critical illness is through measurement of the capacity of whole blood to produce proinflammatory cytokines when stimulated ex vivo. While a normal monocyte will produce copious amounts of TNFα when exposed to LPS, an immunoparalyzed monocyte will not [28–33]. This approach suffers from a lack of standardization across investigators. There is no single protocol for defining immunoparalysis by ex vivo cytokine production capacity that has gained universal acceptance. The volume of blood used, the strain of bacteria from which the LPS originates, the method of LPS purification used, the incubation time, and the method of TNFα measurement are all variables which must be held constant to define thresholds for diagnosis of immunoparalysis in a given patient. In our laboratory, for example, a whole blood ex vivo LPS-induced TNFα response < 200 pg/ml after four hours of incubation at 37°C is the threshold for defining immunoparalysis [30]. This threshold value may be different in another laboratory if other reagents and experimental methods are used. It bears re-emphasizing that this type of measurement does not reflect circulating TNFα levels in the plasma, but rather reflects the ability of a patient’s blood to make TNFα when it is needed.
Mechanisms of immunoparalysis
The underlying causes of immunoparalysis are, to date, poorly understood. Experimental and clinical evidence suggests, not surprisingly, that regulation of inflammation is not normal in innate immune cells with an immunoparalyzed phenotype. The concept of “endotoxin tolerance” represents an experimental analogue to the clinical entity of immunoparalysis. Culturing monocytes or monocytic cells lines in the presence of IL-10 or TGFβ, for example, will induce a state of hyporesponsiveness to subsequent LPS challenge [34–36]. Perhaps the best inducer of experimental endotoxin tolerance is endotoxin itself. Pretreatment of cells or animals with a sublethal dose of LPS will result in a long-lasting refractory period during which the innate immune cells responds weakly to a second dose of LPS [37–39]. As many of our patients have no known LPS exposure associated with the development of immunoparalysis, it seems unlikely that this accounts for most episodes of innate immune dysfunction in the ICU. Possible intracellular mechanisms that have been associated with models of endotoxin tolerance include inhibition of the proinflammatory transcription factor NFκB through either alteration of its subunit composition [40, 41] or upregulation of its inhibitor IκBα [42]; upregulation of the NFκB pathway inhibitor IRAK-M [43–45]; or impairment of TLR4 signaling [46–48].
Mechanistic data from human subjects with immunoparalysis are rare, but Fumeaux et al demonstrated internalization of cell-surface HLA-DR molecules in normal human monocytes upon co-incubation with serum from septic patients [49]. This monocyte deactivation was partially reversed by the addition of anti-IL-10 neutralizing antibodies. Others have shown that septic adults with impaired innate immune function demonstrate upregulation of IRAK-M [44] and downregulation of the receptor for granulocyte-macrophage colony stimulating factor (GM-CSF) on monocytes [50]. Pachot et al examined the mRNA transcription characteristics of whole blood from 38 adult patients with septic shock who survived at least 48 hours in an effort to identify differences in gene expression between survivors and nonsurvivors [51]. The results of their analyses revealed 28 individual genes whose transcription levels predicted mortality. Survivors showed increased transcription of genes encoding chemokine receptors, cytokine receptors, and signal transducers, all involved in initiating and maintaining the innate immune response. Nonsurvivors did not demonstrate a similar upregulation of proinflammatory gene transcription. These results suggest that preservation of the ability to mount a proinflammatory response may be an important determinant of survival. Our laboratory recently studied monocyte mRNA from 28 children with multiple organ dysfunction syndrome (MODS) [52]. We found that expression of mRNA coding for IL-10, IRAK-M, and pyrin (a putative inhibitor of intracellular inflammatory pathways) was elevated in nonsurvivors compared with survivors, with nonsurvivors demonstrating lower whole blood ex vivo LPS-induced TNFα responses. These data therefore suggest that children dying from MODS are doing so with an anti-inflammatory innate immune phenotype.
Corticosteroids may play a role in the development or perpetuation of immunoparalysis. Le Tulzo et al in 2004 studied 48 septic patients and found an association between high levels of circulating cortisol and reductions in monocyte HLA-DR expression on Day 6 of illness [53]. The authors then demonstrated in vitro that dexamethasone caused a down-regulation of a key transcription factor for HLA-DR in normal monocytes. They suggest that glucocorticoid action may represent another mechanism for the development of innate immune dysfunction. Similarly, Volk et al demonstrated that the administration of methylprednisolone in the setting of cardiopulmonary bypass resulted in an exacerbation of innate immunosuppression over that which was seen with byass alone [54].
It is worth noting that acquired innate and adaptive immune dysfunction can coexist in critically ill patients. Monneret and others have recently demonstrated associations between immunoparalysis and the subsequent development of a Treg-dominant adaptive immune response [55, 56]. It has also been clearly shown in critically ill adults [57] and children [58] that lymphocyte apoptosis and lymphopenia are common in ICU nonsurvivors. The temporal and causal relationships between innate and adaptive immune dysfunction in critical illness remain incompletely understood.
It is likely that host genetic factors are important in determining who goes on to develop immunoparalysis following an inflammatory insult. To date, however, no single polymorphism (or group of polymorphisms) has been able to explain predisposition toward this phenotype. The importance of this issue was highlighted by the work of Westendorp et al who, in 1997, examined the ex vivo LPS-induced cytokine production capacity of 190 first degree relatives of patients with invasive meningococcal disease [59]. They found that family members of nonsurvivors produced less TNFα and more IL-10 upon ex vivo LPS stimulation than did the relatives of patients who survived, indicating that a predilection towards an anti-inflammatory phenotype may be heritable.
The Clinical Significance of Immunoparalysis
Trauma
Trauma surgeons were among the first clinicians to study the innate immune response in the setting of critical illness. In 1986, Polk et al [16] undertook a systematic evaluation of 20 adults with severe trauma in the hopes of elucidating the immunologic factors associated with the development of secondary infection and late mortality. The results of their analyses indicated that persistently impaired monocyte antigen presenting capacity was associated with the development of nosocomial sepsis. Interestingly, all of the traumatized patients had reduced monocyte HLA-DR expression compared to healthy controls when first evaluated upon ICU admission. It was those who exhibited prolonged HLA-DR down-regulation in whom secondary sepsis developed more often. These findings were confirmed by Livingston et al [18] in a separate series of adult trauma patients. It appears that the initial reduction in monocyte HLA-DR expression is indicative of the CARS state and is not itself pathologic. Rather, it is the failure of HLA-DR expression to recover to normal levels over time that places patients at risk for developing nosocomial sepsis.
The ability of HLA-DR expression profile to predict outcome in traumatized adults was strengthened by the work of Cheadle et al in 1989 [60] in a study of serial monocyte HLA-DR measurements in 60 trauma victims beginning the first 24 hours after injury. In their cohort, the depth and persistence of HLA-DR expression was predictive of the development of secondary infection. The study differed from previous work in that monocytes with low HLA-DR expression underwent ex vivo LPS-induced stimulation. Interestingly, patients who developed secondary sepsis but went on to survive had low initial monocyte HLA-DR expression which was reversible by LPS stimulation. In contrast, persistently low HLA-DR expression, even after ex vivo LPS stimulation, was characteristic of patients who died.
Ex vivo TNFα production has also been correlated with outcome in clinical trauma studies. Majetschak et al in 1999 [28] performed ex vivo LPS-induced stimulation of whole blood samples from 46 adult blunt trauma victims. They documented a profound reduction in patients’ TNFα production compared to healthy controls. The degree of reduction in the TNFα response was associated with the severity of injury and this reduction was detectable in samples obtained within 90 minutes after trauma. Patients requiring surgery to treat their injuries experienced a further impairment in their TNFα response.
Septic Disease
If innate immune dysfunction can follow the inflammatory insult of trauma then it stands to reason that the hyperinflammation associated with severe sepsis and septic shock should also be associated with the development of immunoparalysis. In the mid-1990s Volk et al studied monocyte HLA-DR expression in 247 adult surgical patients with septic disease [61]. In their cohort, an HLA-DR expression level < 30% for five days or more was associated with a 12% survival rate. This was in contrast to a survival rate of 88% in patients whose reduction in monocyte HLA-DR expression was transient or less severe (p < 0.01; χ2). Similarly, patients whose monocyte HLA-DR expression was < 30% were at increased risk for the development of nosocomial sepsis associated with postoperative peritonitis. We found similar results in children with MODS, in that those who demonstrated monocyte HLA-DR expression < 30% for more than three days had significantly increased risks for the development of nosocomial infection and death [62].
Monneret et al in 2006 followed 93 adult patients (83 with complete data sets) with a primary diagnosis of septic shock and evaluated HLA-DR expression [14]. Patients who survived demonstrated recovery of HLA-DR expression by Day 3 or 4, while those who eventually died failed to recover HLA-DR expression (Figure 3). Multivariate logistic regression analysis showed that a monocyte HLA-DR expression < 30% on day 3 or 4 was associated with mortality after adjusting for confounders including initial severity of illness and presence of co-morbidities (OR of 6.48 [95%CI: 1.62 – 25.93]).
Figure 3. Prolonged suppression of monocyte HLA-DR expression predicts outcome.
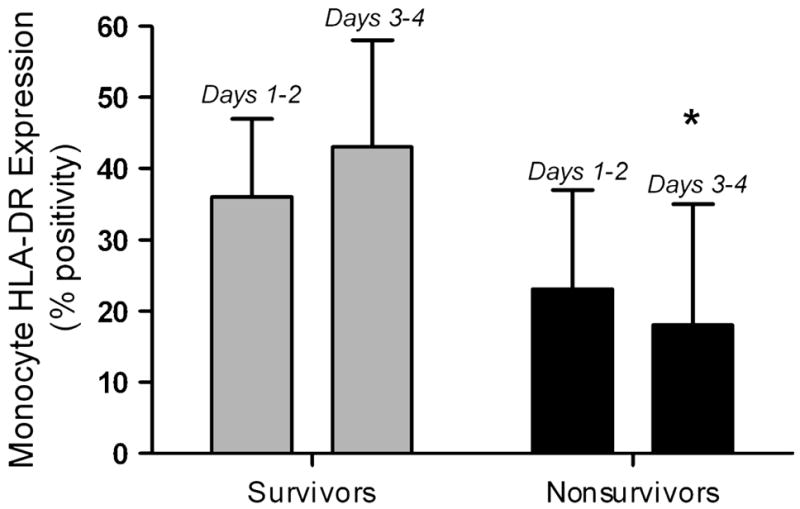
In 86 adult patients with septic shock, monocyte HLA-DR expression was suppressed on Days 1 – 2 in all patients, but to a greater degree in nonsurvivors. By Days 3 – 4, survivors had begun to recover HLA-DR expression whereas it remained low in nonsurvivors. *p < 0.001 vs. survivors, Mann – Whitney test. Data represent median (25th – 75th %). Data from Monneret G, Lepape A, Voirin N, et al. Persisting low monocyte human leukocyte antigen-DR expression predicts mortality in septic shock. Intensive Care Med. Aug 2006;32(8):1175–1183.
Not all studies of the association between monocyte function and outcome from critical illness have been positive. Perry et al in 2003 examined HLA-DR expression in 70 patients on Days 1 through 3 following the onset of septic shock [63]. In this study monocyte HLA-DR expression was not found to correlate with mortality. To look for such an association so early in a patient’s ICU course may, however, be problematic. A more longitudinal approach to immune monitoring is likely required to identify patients with prolonged innate immune dysfunction. It is these patients that appear to be at highest risk for the development of adverse outcomes.
As in the setting of trauma, impairment of the ex vivo LPS-induced TNFα response has also been shown to occur following septic disease and is also associated with adverse outcomes. In fact, Ploder et al recently described a series of 19 polytrauma patients with sepsis in whom the degree of depression of the ex vivo TNFα response was more predictive of death on the day of sepsis onset than was the level of monocyte HLA-DR expression [64]. Our recent cohort of 28 children with MODS (25 of whom had septic shock) also underwent measurement of their whole blood ex vivo LPS-induced TNFα responses [52]. These data showed that nonsurvivors demonstrated lower ex vivo LPS-induced TNFα production over the first two weeks of MODS compared to survivors (Figure 4).
Figure 4. The ex vivo LPS-induced TNFα response is lower over time in nonsurvivors of pediatric MODS.
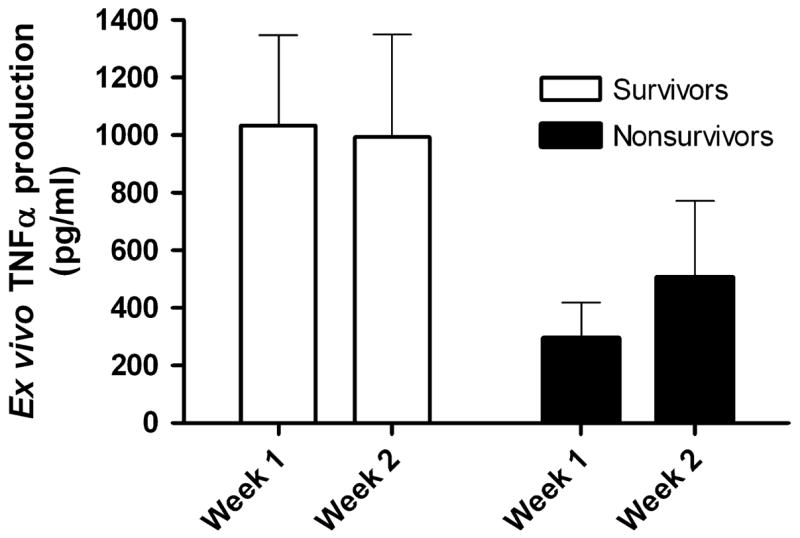
Thirty children with MODS underwent serial measurement of their whole blood ex vivo LPS-induced TNFα response in the first and second weeks of illness. Nonsurvivors demonstrated a reduced capacity to produce TNFα when stimulated ex vivo over time compared to survivors (p = 0.017; 2-way ANOVA on log transformed data). Data represent mean (standard error). Data from Hall MW, Gavrilin MA, Knatz, NL et al. Monocyte mRNA phenotype and adverse outcomes from pediatric multiple organ dysfunction syndrome. Pediatric Research. Nov 2007; 62(5):597–603.
Innate Immune Dysfunction in Other Settings
Other types of proinflammatory insults have been shown to be associated with innate immunodepression. Ho et al [15] showed that both suppressed monocyte HLA-DR expression and reduced ex vivo LPS-induced TNFα production on Day 10 of acute pancreatitis were predictive of mortality, as was the presence of an elevated serum IL-10 level. In fact, monocyte HLA-DR expression on Day 10 was better able to predict nosocomial infection and mortality than either the Ranson score or APACHE severity of illness score.
Allen et al 2002 [65] studied 82 infants and children undergoing cardiopulmonary bypass (CPB) for repair of congenital heart defects and documented reduced monocyte HLA-DR expression in all patients post-CPB compared with pre-CPB levels. Importantly, the authors correlated prolonged reduction of HLA-DR expression with the development of SIRS and sepsis after surgery. A multivariate analysis identified HLA-DR expression as an independent predictor of secondary sepsis after controlling for other variables including severity of illness, complexity of surgery, and length of CPB. These results were supported same group in a 2006 follow-up study [66] in which 36 children undergoing CPB were enrolled. Immune function was monitored serially by ex vivo LPS stimulation assays and IL-10 measurements. Higher levels of plasma IL-10 were associated with the greatest risk of developing more severe innate immune dysfunction. Patients with more profoundly reduced ex vivo TNFα responses as early as Day 2 following CPB had a greater likelihood of experiencing post-operative complications including nosocomial sepsis.
Reversibility of Immunoparalysis
As the natural history of innate immune dysfunction in critical illness becomes clearer, a question arises: Is immunoparalysis simply an epiphenomenon associated with critical illness or does it represent a reversible target for intervention in hopes of improving outcomes? The answer to this question is far from clear, but several lines of evidence suggest that the latter may be true.
Numerous investigators have described reversal of endotoxin tolerance in vitro with agents including anti-IL-10 neutralizing antibody [49], IL-12 [67], GM-CSF [37, 67, 68], and flt3-ligand [69]. Hershman et al reported in 1989 that monocytes collected from immunoparalyzed adult trauma victims could be made to upregulate their HLA-DR expression upon ex vivo culture with the TH1 cytokine IFNγ [70]. The effects of ex vivo culture with GM-CSF on monocyte function were reported by Flohe et al in 2003 [71]. In this study of 16 adult trauma patients, blood samples were obtained biweekly during the ICU stay. Individual samples were divided into two aliquots, one pretreated with GM-CSF and the other untreated. Monocyte HLA-DR expression and ex vivo LPS-induced TNFα production were then analyzed for each aliquot and compared. GM-CSF stimulation improved TNFα production in the treated aliquots and, to a lesser extent, resulted in upregulation of HLA-DR expression. In patients who went on to recover uneventfully, ex vivo GM-CSF treatment restored monocyte function to the level of normal volunteers. GM-CSF treatment was unable to improve monocyte function to normal levels in the most severely injured patients, who eventually developed sepsis and MODS. The blood from patients who eventually developed severe MODS with three or more failed organs did not respond to GM-CSF at all. More recently, Lendemans et al showed that ex vivo treatment with IFNγ or GM-CSF (but not granulocyte colony stimulating factor [GCSF]) resulted in improvement in both monocyte HLA-DR expression and whole blood LPS-induced cytokine production in blood samples from injured patients [72].
The administration of immune stimulating agents directly to patients with immunoparalysis has been done in a few small case series. In 1997 Kox et al demonstrated restoration of monocyte HLA-DR expression and ex vivo TNFα response in nine of 10 adult septic patients with immunoparalysis upon treatment with subcutaneous IFNγ, 100μg daily until monocyte HLA-DR expression remained > 50% for three days [73]. Eight of ten patients showed increased HLA-DR expression within one day of treatment. 28-day mortality in this cohort, with a median treatment duration of six days, was 40%. This mortality rate represented an improvement over the 58 – 88% mortality previously described by this group in adults with persistent immunoparalysis [74].
In 2002 Nakos et al conducted a small randomized placebo-controlled trial of inhaled interferon in 21 trauma patients who demonstrated reduced HLA-DR expression on alveolar macrophages obtained by bronchoalveolar lavage (BAL) [75]. Subjects randomized to the treatment group received 100 μg of inhaled recombinant human IFNγ three times daily. Although there was no difference in mortality between groups, the IFNγ-treated subjects showed increased HLA-DR expression on alveolar macrophages, decreased IL-10 levels in subsequent BAL fluid, and a reduced rate of ventilator-associated pneumonia compared to the placebo group.
Recombinant human (rh) GM-CSF has also been used to systemically treat adult septic patients with immunoparalysis. In 2003 Nierhaus et al reported a case series in which nine septic adults with immunoparalysis received 5 μg/kg rhGM-CSF by subcutaneous injection daily for three days [76]. All patients experienced increases in both monocyte HLA-DR expression and ex vivo LPS-induced TNFα production within 24 hours of initiation of rhGM-CSF treatment. Notably, there was no increase in circulating markers of inflammation (plasma IL-6, C-reactive protein) associated with rhGM-CSF therapy. Mortality in this series was 33%.
Some trials of immunostimulatory agents in critical illness have not employed an a priori measurement of the innate immune response. In 2001 Bilgin et al reported the results of a randomized placebo-control trial of rhGM-CSF therapy for neutropenic neonates with a diagnosis of sepsis [77]. Patients were assigned to experimental groups without measuring baseline innate immune function other than absolute neutrophil count. Treated infants received subcutaneous rhGM-CSF 5μg/kg/day for seven days. Neonates receiving GM-CSF demonstrated a significant improvement in mortality rate (10%) compared to those in the placebo group (30%; p < 0.05). In another GM-CSF treatment study which did not include measures of innate immune function as inclusion criteria, Rosenbloom et al randomized 40 adult ICU patients with infection-related SIRS to receive intravenous (IV) GM-CSF 120μg/m2 over 72 hours or placebo [78]. GM-CSF-treated patients demonstrated improved monocyte HLA-DR expression and shorter time to resolution of infection compared to the placebo group. There was no worsening of organ failure or other adverse drug effects in the GM-CSF group.
Not all studies of the use of colony stimulating factors in the ICU have been positive. For example, a 2003 Cochrane meta-analysis concluded that insufficient evidence existed to recommend either for or against the use of GM-CSF or G-CSF for prophylaxis or treatment of critically ill neonates [79]. It is worth noting that the studies reviewed for this analysis did not include measurement of parameters such as HLA-DR expression or ex vivo cytokine production in their protocols.
Transplant-related Immunosuppression
Patients who have undergone solid organ or hematopoetic stem cell transplantation and remain on exogenous immunosuppression find themselves at high risk for the development of secondary infection. While specific prophylaxis regimens have been recommended for this patient population [80], nosocomial infections are often difficult to prevent. At present, most transplant specialists follow drug levels and monitor end-organ function in order to titrate immunosuppressive therapy. This approach is most applicable to calcineurin inhibitors such as cyclosporine or tacrolimus. While these agents are classically thought of as T cell inhibitors, they also exert a polarizing effect, skewing both innate and adaptive immune systems toward a TH2-like phenotype. High plasma IL-10 levels have been associated with the development of nosocomial infection and death following bone marrow transplantation [81]. Some degree of TH2 polarization is likely necessary, however, as hepatic graft rejection has been associated with low plasma levels of IL-10 [82].
The innate immune monitoring methods discussed above have been applied to the transplant population with interesting results. In the early 1990s, Settmacher et al described an association between aggressive calcineurin inhibition and reduction in monocyte HLA-DR expression in the setting of induction therapy in adults following liver transplantation [83]. Among 91 patients, those whose monocyte HLA-DR expression dropped below 30% (n = 48) experienced increased rates of bacteremia, viremia, and fungemia compared to those whose HLA-DR levels remained > 30% (n = 43). Providing additional evidence for the reversibility of immunoparalysis, Reinke et al described a cohort of 45 adult kidney transplant recipients who developed nosocomial sepsis in the setting of immunoparalysis [84]. Patients who underwent rapid tapering of their calcineurin inhibition experienced a 90% survival rate (30/33) with 98% graft survival. In contrast, patients who did not undergo tapering had an 8% survival rate (1/12) with the sole survivor experiencing graft loss.
Pediatric data on this subject are limited, but Hoffman et al described a series of 13 pediatric lung transplant recipients who underwent monocyte HLA-DR monitoring weekly following transplantation [85]. Six of seven patients who developed post-transplant pneumonia demonstrated failure to recover monocyte HLA-DR expression within the first two weeks of monitoring. Those who developed pneumonia had lower monocyte HLA-DR expression over the four-week study period that those who remained infection-free.
Some investigators have employed immunostimulatory therapy with either IFNγ or GM-CSF for the treatment of post-transplant septic disease [86, 87], however prospective, standardized trials are lacking. Monocyte HLA-DR expression measurement and quantification of the ex vivo LPS-induced TNFα response both appear to have the potential to provide insight into the degree of functional immunosuppression following transplantation and are deserving of future study in this regard.
Other Determinants of the Immune Response in Critical Illness
While acquired monocyte dysfunction has been the subject of this review thus far, one must recognize that other factors can influence the immune response of patients in the PICU. As alluded to above, the adaptive immune response contributes importantly to the overall immunophenotpe of a given patient. A shift toward a TH2- or Treg-dominant adaptive immune state would likely result in anti-inflammatory changes in innate immune cells as well. Cell numbers are similarly important. The cancer literature and HIV literature have taught us that patients with low absolute neutrophil counts and/or lymphopenia are at high risk for the development of secondary infection and death [88–90]. Critical illness itself is frequently associated with unintended immunomodulation. Hyperglycemia [91] and hepatic failure [23] are frequently associated with a proinflammatory state while uremia [92, 93] and traumatic brain injury [94, 95] can be immunosuppressive. Also, many of the therapies which have become mainstays in the ICU have unintended immune effects including catecholamines (often proinflammatory) [96] and insulin, narcotics, and furosemide (anti-inflammatory) [97–99].
Conclusion
Historically the battle against hyper-inflammation the ICU has been of primary importance to the pediatric intensivist. The notion that many of our patients are suffering and dying from an occult anti-inflammatory phenotype requires a sea-change in clinicians’ philosophy. While some patients will continue to require anti-inflammatory treatments, others may need therapies that augment the immune response. It is likely that the maximum benefit from immunostimulation would be seen in children with underactive immune responses at the beginning of treatment. To give such agents to children with normal or high levels of proinflammatory responsiveness would at best be unhelpful and at worst, harmful. To that end, the use of immunomodulatory therapies for children in the ICU should be based upon the development and implementation of prospective, standardized immune monitoring regimens. This way specific children at high risk (e.g. those with immunoparalysis) can be identified as candidates for therapy, avoiding unnecessary or dangerous immunomodulation in low risk patients. To date, the vast majority of data outlining the natural history of innate immune function in critical illness come from the adult literature. There are only a handful of studies which hint at the developmental aspects of acquired immune dysfunction in neonates, infants, and small children [100–102]. The challenge before the pediatric intensive care community, therefore, is to design and implement multi-center prospective observational and interventional studies to address these important issues and work toward promoting immunologic homeostasis in the pediatric ICU.
Acknowledgments
This work was supported by Grant No. K08HL085525 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Prophylactic intravenous administration of standard immune globulin as compared with core-lipopolysaccharide immune globulin in patients at high risk of postsurgical infection. The Intravenous Immunoglobulin Collaborative Study Group. N Engl J Med. 1992 Jul 23;327(4):234–240. doi: 10.1056/NEJM199207233270404. [DOI] [PubMed] [Google Scholar]
- 2.Calandra T, Glauser MP, Schellekens J, Verhoef J. Treatment of gram-negative septic shock with human IgG antibody to Escherichia coli J5: a prospective, double-blind, randomized trial. J Infect Dis. 1988 Aug;158(2):312–319. doi: 10.1093/infdis/158.2.312. [DOI] [PubMed] [Google Scholar]
- 3.Bone RC, Balk RA, Fein AM, et al. A second large controlled clinical study of E5, a monoclonal antibody to endotoxin: results of a prospective, multicenter, randomized, controlled trial. The E5 Sepsis Study Group. Crit Care Med. 1995 Jun;23(6):994–1006. doi: 10.1097/00003246-199506000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Greenman RL, Schein RM, Martin MA, et al. A controlled clinical trial of E5 murine monoclonal IgM antibody to endotoxin in the treatment of gram-negative sepsis. The XOMA Sepsis Study Group. Jama. 1991 Aug 28;266(8):1097–1102. [PubMed] [Google Scholar]
- 5.McCloskey RV, Straube RC, Sanders C, Smith SM, Smith CR. Treatment of septic shock with human monoclonal antibody HA-1A. A randomized, double-blind, placebo-controlled trial. CHESS Trial Study Group. Ann Intern Med. 1994 Jul 1;121(1):1–5. doi: 10.7326/0003-4819-121-1-199407010-00001. [DOI] [PubMed] [Google Scholar]
- 6.Fisher CJ, Jr, Dhainaut JF, Opal SM, et al. Recombinant human interleukin 1 receptor antagonist in the treatment of patients with sepsis syndrome. Results from a randomized, double-blind, placebo-controlled trial. Phase III rhIL-1ra Sepsis Syndrome Study Group. Jama. 1994 Jun 15;271(23):1836–1843. [PubMed] [Google Scholar]
- 7.Opal SM, Fisher CJ, Jr, Dhainaut JF, et al. Confirmatory interleukin-1 receptor antagonist trial in severe sepsis: a phase III, randomized, double-blind, placebo-controlled, multicenter trial. The Interleukin-1 Receptor Antagonist Sepsis Investigator Group. Crit Care Med. 1997 Jul;25(7):1115–1124. doi: 10.1097/00003246-199707000-00010. [DOI] [PubMed] [Google Scholar]
- 8.Abraham E, Anzueto A, Gutierrez G, et al. Double-blind randomised controlled trial of monoclonal antibody to human tumour necrosis factor in treatment of septic shock. NORASEPT II Study Group. Lancet. 1998 Mar 28;351(9107):929–933. [PubMed] [Google Scholar]
- 9.Abraham E, Wunderink R, Silverman H, et al. Efficacy and safety of monoclonal antibody to human tumor necrosis factor alpha in patients with sepsis syndrome. A randomized, controlled, double-blind, multicenter clinical trial. TNF-alpha MAb Sepsis Study Group. Jama. 1995 Mar 22–29;273(12):934–941. [PubMed] [Google Scholar]
- 10.Cohen J, Carlet J. INTERSEPT: an international, multicenter, placebo-controlled trial of monoclonal antibody to human tumor necrosis factor-alpha in patients with sepsis. International Sepsis Trial Study Group. Crit Care Med. 1996 Sep;24(9):1431–1440. doi: 10.1097/00003246-199609000-00002. [DOI] [PubMed] [Google Scholar]
- 11.Panacek EA, Marshall JC, Albertson TE, et al. Efficacy and safety of the monoclonal anti-tumor necrosis factor antibody F(ab′)2 fragment afelimomab in patients with severe sepsis and elevated interleukin-6 levels. Crit Care Med. 2004 Nov;32(11):2173–2182. doi: 10.1097/01.ccm.0000145229.59014.6c. [DOI] [PubMed] [Google Scholar]
- 12.Fein AM, Bernard GR, Criner GJ, et al. Treatment of severe systemic inflammatory response syndrome and sepsis with a novel bradykinin antagonist, deltibant (CP-0127). Results of a randomized, double-blind, placebo-controlled trial. CP-0127 SIRS and Sepsis Study Group. Jama. 1997 Feb 12;277(6):482–487. [PubMed] [Google Scholar]
- 13.Docke WSU, Meinicke A, et al. Improvement in monocyte function: a new therapeutic approach in sepsis? In: Reinhart, editor. Update in intensive care and emergency medicine. Berlin: Springer-Verlag; 1994. pp. 473–488. [Google Scholar]
- 14.Monneret G, Lepape A, Voirin N, et al. Persisting low monocyte human leukocyte antigen-DR expression predicts mortality in septic shock. Intensive Care Med. 2006 Aug;32(8):1175–1183. doi: 10.1007/s00134-006-0204-8. [DOI] [PubMed] [Google Scholar]
- 15.Ho YP, Sheen IS, Chiu CT, Wu CS, Lin CY. A strong association between down-regulation of HLA-DR expression and the late mortality in patients with severe acute pancreatitis. Am J Gastroenterol. 2006 May;101(5):1117–1124. doi: 10.1111/j.1572-0241.2006.00495.x. [DOI] [PubMed] [Google Scholar]
- 16.Polk HC, Jr, Wellhausen SR, Regan M. A systematic study of host defense processes in badly injured patients. Ann Surg. 1986;204(3):282–297. [PMC free article] [PubMed] [Google Scholar]
- 17.Hershman MJ, Cheadle WG, Wellhausen SR, Davidson PF, Polk HC., Jr Monocyte HLA-DR antigen expression characterizes clinical outcome in the trauma patient. Br J Surg. 1990 Feb;77(2):204–207. doi: 10.1002/bjs.1800770225. [DOI] [PubMed] [Google Scholar]
- 18.Livingston DH, Appel SH, Wellhausen SR, Sonnenfeld G, Polk HC., Jr Depressed interferon gamma production and monocyte HLA-DR expression after severe injury. Arch Surg. 1988 Nov;123(11):1309–1312. doi: 10.1001/archsurg.1988.01400350023002. [DOI] [PubMed] [Google Scholar]
- 19.Chatila TA. Role of regulatory T cells in human diseases. J Allergy Clin Immunol. 2005 Nov;116(5):949–959. doi: 10.1016/j.jaci.2005.08.047. quiz 960. [DOI] [PubMed] [Google Scholar]
- 20.Calandra T, Gerain J, Heumann D, Baumgartner JD, Glauser MP. High circulating levels of interleukin-6 in patients with septic shock: evolution during sepsis, prognostic value, and interplay with other cytokines. The Swiss-Dutch J5 Immunoglobulin Study Group. Am J Med. 1991 Jul;91(1):23–29. doi: 10.1016/0002-9343(91)90069-a. [DOI] [PubMed] [Google Scholar]
- 21.Doughty LA, Kaplan SS, Carcillo JA. Inflammatory cytokine and nitric oxide responses in pediatric sepsis and organ failure. Crit Care Med. 1996 Jul;24(7):1137–1143. doi: 10.1097/00003246-199607000-00012. [DOI] [PubMed] [Google Scholar]
- 22.Ghani RA, Zainudin S, Ctkong N, et al. Serum IL-6 and IL-1-ra with sequential organ failure assessment scores in septic patients receiving high-volume haemofiltration and continuous venovenous haemofiltration. Nephrology (Carlton) Oct. 2006;11(5):386–393. doi: 10.1111/j.1440-1797.2006.00600.x. [DOI] [PubMed] [Google Scholar]
- 23.Pinsky MR, Vincent JL, Deviere J, Alegre M, Kahn RJ, Dupont E. Serum cytokine levels in human septic shock. Relation to multiple-system organ failure and mortality. Chest. 1993 Feb;103(2):565–575. doi: 10.1378/chest.103.2.565. [DOI] [PubMed] [Google Scholar]
- 24.Steensberg A, Fischer CP, Keller C, Moller K, Pedersen BK. IL-6 enhances plasma IL-1ra, IL-10, and cortisol in humans. Am J Physiol Endocrinol Metab. 2003 Aug;285(2):E433–437. doi: 10.1152/ajpendo.00074.2003. [DOI] [PubMed] [Google Scholar]
- 25.Diehl S, Rincon M. The two faces of IL-6 on Th1/Th2 differentiation. Mol Immunol. 2002 Dec;39(9):531–536. doi: 10.1016/s0161-5890(02)00210-9. [DOI] [PubMed] [Google Scholar]
- 26.Goldstein B, Giroir B, Randolph A. International pediatric sepsis consensus conference: definitions for sepsis and organ dysfunction in pediatrics. Pediatr Crit Care Med. 2005 Jan;6(1):2–8. doi: 10.1097/01.PCC.0000149131.72248.E6. [DOI] [PubMed] [Google Scholar]
- 27.Docke WD, Hoflich C, Davis KA, et al. Monitoring temporary immunodepression by flow cytometric measurement of monocytic HLA-DR expression: a multicenter standardized study. Clin Chem. 2005 Dec;51(12):2341–2347. doi: 10.1373/clinchem.2005.052639. [DOI] [PubMed] [Google Scholar]
- 28.Majetschak M, Flach R, Kreuzfelder E, et al. The extent of traumatic damage determines a graded depression of the endotoxin responsiveness of peripheral blood mononuclear cells from patients with blunt injuries. Crit Care Med. 1999 Feb;27(2):313–318. doi: 10.1097/00003246-199902000-00037. [DOI] [PubMed] [Google Scholar]
- 29.Majetschak M, Krehmeier U, Bardenheuer M, et al. Extracellular ubiquitin inhibits the TNF-alpha response to endotoxin in peripheral blood mononuclear cells and regulates endotoxin hyporesponsiveness in critical illness. Blood. 2003 Mar 1;101(5):1882–1890. doi: 10.1182/blood-2002-03-0918. [DOI] [PubMed] [Google Scholar]
- 30.Hall MW, Carcillo JA. Immune paralysis and the state of immunologic dissonance in pediatric multiple organ dysfunction syndrome. Crit Care Med. 2001;29(12):A20. [Google Scholar]
- 31.Docke WD, Randow F, Syrbe U, et al. Monocyte deactivation in septic patients: restoration by IFN-gamma treatment. Nat Med. 1997 Jun;3(6):678–681. doi: 10.1038/nm0697-678. [DOI] [PubMed] [Google Scholar]
- 32.Flach R, Majetschak M, Heukamp T, et al. Relation of ex vivo stimulated blood cytokine synthesis to post-traumatic sepsis. Cytokine. 1999 Feb;11(2):173–178. doi: 10.1006/cyto.1998.0412. [DOI] [PubMed] [Google Scholar]
- 33.Heagy W, Nieman K, Hansen C, Cohen M, Danielson D, West MA. Lower levels of whole blood LPS-stimulated cytokine release are associated with poorer clinical outcomes in surgical ICU patients. Surg Infect (Larchmt) Summer. 2003;4(2):171–180. doi: 10.1089/109629603766956960. [DOI] [PubMed] [Google Scholar]
- 34.Wolk K, Docke W, von Baehr V, Volk H, Sabat R. Comparison of monocyte functions after LPS- or IL-10-induced reorientation: importance in clinical immunoparalysis. Pathobiology. 1999;67(5–6):253–256. doi: 10.1159/000028104. [DOI] [PubMed] [Google Scholar]
- 35.Wolk K, Docke WD, von Baehr V, Volk HD, Sabat R. Impaired antigen presentation by human monocytes during endotoxin tolerance. Blood. 2000 Jul 1;96(1):218–223. [PubMed] [Google Scholar]
- 36.Randow F, Syrbe U, Meisel C, et al. Mechanism of endotoxin desensitization: involvement of interleukin 10 and transforming growth factor beta. J Exp Med. 1995 May 1;181(5):1887–1892. doi: 10.1084/jem.181.5.1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bundschuh DS, Barsig J, Hartung T, et al. Granulocyte-macrophage colony-stimulating factor and IFN-gamma restore the systemic TNF-alpha response to endotoxin in lipopolysaccharide-desensitized mice. J Immunol. 1997 Mar 15;158(6):2862–2871. [PubMed] [Google Scholar]
- 38.Flohe S, Dominguez Fernandez E, Ackermann M, Hirsch T, Borgermann J, Schade FU. Endotoxin tolerance in rats: expression of TNF-alpha, IL-6, IL-10, VCAM-1 AND HSP 70 in lung and liver during endotoxin shock. Cytokine. 1999 Oct;11(10):796–804. doi: 10.1006/cyto.1998.0490. [DOI] [PubMed] [Google Scholar]
- 39.Madonna GS, Peterson JE, Ribi EE, Vogel SN. Early-phase endotoxin tolerance: Induction by a detoxified lipid A derivative, monophosphoryl lipid A. Infect Immun. 1986;52:6–11. doi: 10.1128/iai.52.1.6-11.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bohuslav J, Kravchenko VV, Parry GC, et al. Regulation of an essential innate immune response by the p50 subunit of NF-kappaB. J Clin Invest. 1998 Nov 1;102(9):1645–1652. doi: 10.1172/JCI3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kastenbauer S, Ziegler-Heitbrock HW. NF-kappaB1 (p50) is upregulated in lipopolysaccharide tolerance and can block tumor necrosis factor gene expression. Infect Immun. 1999 Apr;67(4):1553–1559. doi: 10.1128/iai.67.4.1553-1559.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wahlstrom K, Bellingham J, Rodriguez JL, West MA. Inhibitory kappaBalpha control of nuclear factor-kappaB is dysregulated in endotoxin tolerant macrophages. Shock. 1999 Apr;11(4):242–247. doi: 10.1097/00024382-199904000-00003. [DOI] [PubMed] [Google Scholar]
- 43.Nakayama K, Okugawa S, Yanagimoto S, et al. Involvement of IRAK-M in peptidoglycan-induced tolerance in macrophages. J Biol Chem. 2004 Feb 20;279(8):6629–6634. doi: 10.1074/jbc.M308620200. [DOI] [PubMed] [Google Scholar]
- 44.Escoll P, del Fresno C, Garcia L, et al. Rapid up-regulation of IRAK-M expression following a second endotoxin challenge in human monocytes and in monocytes isolated from septic patients. Biochem Biophys Res Commun. 2003 Nov 14;311(2):465–472. doi: 10.1016/j.bbrc.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 45.del Fresno C, Otero K, Gomez-Garcia L, et al. Tumor cells deactivate human monocytes by up-regulating IL-1 receptor associated kinase-M expression via CD44 and TLR4. J Immunol. 2005 Mar 1;174(5):3032–3040. doi: 10.4049/jimmunol.174.5.3032. [DOI] [PubMed] [Google Scholar]
- 46.Dobrovolskaia MA, Vogel SN. Toll receptors, CD14, and macrophage activation and deactivation by LPS. Microbes Infect. 2002 Jul;4(9):903–914. doi: 10.1016/s1286-4579(02)01613-1. [DOI] [PubMed] [Google Scholar]
- 47.Alves-Rosa F, Vulcano M, Beigier-Bompadre M, Fernandez G, Palermo M, Isturiz MA. Interleukin-1beta induces in vivo tolerance to lipopolysaccharide in mice. Clin Exp Immunol. 2002 May;128(2):221–228. doi: 10.1046/j.1365-2249.2002.01828.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Medvedev AE, Lentschat A, Wahl LM, Golenbock DT, Vogel SN. Dysregulation of LPS-induced Toll-like receptor 4-MyD88 complex formation and IL-1 receptor-associated kinase 1 activation in endotoxin-tolerant cells. J Immunol. 2002 Nov 1;169(9):5209–5216. doi: 10.4049/jimmunol.169.9.5209. [DOI] [PubMed] [Google Scholar]
- 49.Fumeaux T, Pugin J. Role of interleukin-10 in the intracellular sequestration of human leukocyte antigen-DR in monocytes during septic shock. Am J Respir Crit Care Med. 2002 Dec 1;166(11):1475–1482. doi: 10.1164/rccm.200203-217OC. [DOI] [PubMed] [Google Scholar]
- 50.Pangault C, Le Tulzo Y, Tattevin P, Guilloux V, Bescher N, Drenou B. Down-modulation of granulocyte macrophage-colony stimulating factor receptor on monocytes during human septic shock. Crit Care Med. 2006 Apr;34(4):1193–1201. doi: 10.1097/01.CCM.0000207339.11477.62. [DOI] [PubMed] [Google Scholar]
- 51.Pachot A, Lepape A, Vey S, Bienvenu J, Mougin B, Monneret G. Systemic transcriptional analysis in survivor and non-survivor septic shock patients: a preliminary study. Immunol Lett. 2006 Jul 15;106(1):63–71. doi: 10.1016/j.imlet.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 52.Hall MW, Gavrilin MA, Knatz NL, Duncan MD, Fernandez SA, Wewers MD. Monocyte mRNA Phenotype and Adverse Outcomes From Pediatric Multiple Organ Dysfunction Syndrome. Pediatr Res. 2007 Aug 31; doi: 10.1203/PDR.0b013e3181559774. [DOI] [PubMed] [Google Scholar]
- 53.Le Tulzo Y, Pangault C, Amiot L, et al. Monocyte human leukocyte antigen-DR transcriptional downregulation by cortisol during septic shock. Am J Respir Crit Care Med. 2004 May 15;169(10):1144–1151. doi: 10.1164/rccm.200309-1329OC. [DOI] [PubMed] [Google Scholar]
- 54.Volk T, Schmutzler M, Engelhardt L, et al. Influence of aminosteroid and glucocorticoid treatment on inflammation and immune function during cardiopulmonary bypass. Crit Care Med. 2001 Nov;29(11):2137–2142. doi: 10.1097/00003246-200111000-00015. [DOI] [PubMed] [Google Scholar]
- 55.Monneret G, Debard AL, Venet F, et al. Marked elevation of human circulating CD4+CD25+ regulatory T cells in sepsis-induced immunoparalysis. Crit Care Med. 2003 Jul;31(7):2068–2071. doi: 10.1097/01.CCM.0000069345.78884.0F. [DOI] [PubMed] [Google Scholar]
- 56.Venet F, Pachot A, Debard AL, et al. Increased percentage of CD4+CD25+ regulatory T cells during septic shock is due to the decrease of CD4+CD25- lymphocytes. Crit Care Med. 2004 Nov;32(11):2329–2331. doi: 10.1097/01.ccm.0000145999.42971.4b. [DOI] [PubMed] [Google Scholar]
- 57.Hotchkiss RS, Tinsley KW, Swanson PE, et al. Sepsis-induced apoptosis causes progressive profound depletion of B and CD4+ T lymphocytes in humans. J Immunol. 2001 Jun 1;166(11):6952–6963. doi: 10.4049/jimmunol.166.11.6952. [DOI] [PubMed] [Google Scholar]
- 58.Felmet KA, Hall MW, Clark RS, Jaffe R, Carcillo JA. Prolonged lymphopenia, lymphoid depletion, and hypoprolactinemia in children with nosocomial sepsis and multiple organ failure. J Immunol. 2005 Mar 15;174(6):3765–3772. doi: 10.4049/jimmunol.174.6.3765. [DOI] [PubMed] [Google Scholar]
- 59.Westendorp RG, Langermans JA, Huizinga TW, et al. Genetic influence on cytokine production and fatal meningococcal disease. Lancet. 1997 Jan 18;349(9046):170–173. doi: 10.1016/s0140-6736(96)06413-6. [DOI] [PubMed] [Google Scholar]
- 60.Cheadle WG, Wilson M, Hershman MJ, Bergamini D, Richardson JD, Polk HC., Jr Comparison of trauma assessment scores and their use in prediction of infection and death. Ann Surg. 1989 May;209(5):541–545. 545–546. doi: 10.1097/00000658-198905000-00005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Volk HD, Reinke P, Krausch D, et al. Monocyte deactivation--rationale for a new therapeutic strategy in sepsis. Intensive Care Med. 1996 Oct;22(Suppl 4):S474–481. doi: 10.1007/BF01743727. [DOI] [PubMed] [Google Scholar]
- 62.Hall MW, Volk HD, Carcillo JA. Immune paralysis in pediatric multiple organ dysfunction syndrome. Pediatric Research. 2000;47(4):57A. [Google Scholar]
- 63.Perry SE, Mostafa SM, Wenstone R, Shenkin A, McLaughlin PJ. Is low monocyte HLA-DR expression helpful to predict outcome in severe sepsis? Intensive Care Med. 2003 Aug;29(8):1245–1252. doi: 10.1007/s00134-003-1686-2. [DOI] [PubMed] [Google Scholar]
- 64.Ploder M, Pelinka L, Schmuckenschlager C, et al. Lipopolysaccharide-induced tumor necrosis factor alpha production and not monocyte human leukocyte antigen-DR expression is correlated with survival in septic trauma patients. Shock. 2006 Feb;25(2):129–134. doi: 10.1097/01.shk.0000191379.62897.1d. [DOI] [PubMed] [Google Scholar]
- 65.Allen ML, Peters MJ, Goldman A, et al. Early postoperative monocyte deactivation predicts systemic inflammation and prolonged stay in pediatric cardiac intensive care. Crit Care Med. 2002 May;30(5):1140–1145. doi: 10.1097/00003246-200205000-00031. [DOI] [PubMed] [Google Scholar]
- 66.Allen ML, Hoschtitzky JA, Peters MJ, et al. Interleukin-10 and its role in clinical immunoparalysis following pediatric cardiac surgery. Crit Care Med. 2006 Oct;34(10):2658–2665. doi: 10.1097/01.CCM.0000240243.28129.36. [DOI] [PubMed] [Google Scholar]
- 67.Randow F, Docke WD, Bundschuh DS, Hartung T, Wendel A, Volk HD. In vitro prevention and reversal of lipopolysaccharide desensitization by IFN-gamma, IL-12, and granulocyte-macrophage colony-stimulating factor. J Immunol. 1997 Mar 15;158(6):2911–2918. [PubMed] [Google Scholar]
- 68.Borgermann J, Friedrich I, Scheubel R, et al. Granulocyte-macrophage colony-stimulating factor (GM-CSF) restores decreased monocyte HLA-DR expression after cardiopulmonary bypass. Thorac Cardiovasc Surg. 2007 Feb;55(1):24–31. doi: 10.1055/s-2006-924621. [DOI] [PubMed] [Google Scholar]
- 69.Wysocka M, Montaner LJ, Karp CL. Flt3 ligand treatment reverses endotoxin tolerance-related immunoparalysis. J Immunol. 2005 Jun 1;174(11):7398–7402. doi: 10.4049/jimmunol.174.11.7398. [DOI] [PubMed] [Google Scholar]
- 70.Hershman MJ, Appel SH, Wellhausen SR, Sonnenfeld G, Polk HC., Jr Interferon-gamma treatment increases HLA-DR expression on monocytes in severely injured patients. Clin Exp Immunol. 1989 Jul;77(1):67–70. [PMC free article] [PubMed] [Google Scholar]
- 71.Flohe S, Borgermann J, Dominguez FE, et al. Influence of granulocyte-macrophage colony-stimulating factor (GM-CSF) on whole blood endotoxin responsiveness following trauma, cardiopulmonary bypass, and severe sepsis. Shock. 1999 Jul;12(1):17–24. doi: 10.1097/00024382-199907000-00003. [DOI] [PubMed] [Google Scholar]
- 72.Lendemans S, Kreuzfelder E, Waydhas C, Schade FU, Flohe S. Differential immunostimulating effect of granulocyte-macrophage colony-stimulating factor (GM-CSF), granulocyte colony-stimulating factor (G-CSF) and interferon gamma (IFNgamma) after severe trauma. Inflamm Res. 2007 Jan;56(1):38–44. doi: 10.1007/s00011-007-6069-7. [DOI] [PubMed] [Google Scholar]
- 73.Kox WJ, Bone RC, Krausch D, et al. Interferon gamma-1b in the treatment of compensatory anti-inflammatory response syndrome. A new approach: proof of principle. Arch Intern Med. 1997 Feb 24;157(4):389–393. [PubMed] [Google Scholar]
- 74.Docke W, Syrbe U, Meinecke A, et al. Improvement in monocyte function: A new therapeutic approach? In: Reinhart K, Eyrich K, Sprung C, editors. Sepsis: Current perspectives in pathophysiology and therapy. New York, NY: Springer-Verlag; 1994. pp. 473–500. [Google Scholar]
- 75.Nakos G, Malamou-Mitsi VD, Lachana A, et al. Immunoparalysis in patients with severe trauma and the effect of inhaled interferon-gamma. Crit Care Med. 2002 Jul;30(7):1488–1494. doi: 10.1097/00003246-200207000-00015. [DOI] [PubMed] [Google Scholar]
- 76.Nierhaus A, Montag B, Timmler N, et al. Reversal of immunoparalysis by recombinant human granulocyte-macrophage colony-stimulating factor in patients with severe sepsis. Intensive Care Med. 2003 Apr;29(4):646–651. doi: 10.1007/s00134-003-1666-6. [DOI] [PubMed] [Google Scholar]
- 77.Bilgin K, Yaramis A, Haspolat K, Tas MA, Gunbey S, Derman O. A randomized trial of granulocyte-macrophage colony-stimulating factor in neonates with sepsis and neutropenia. Pediatrics. 2001 Jan;107(1):36–41. doi: 10.1542/peds.107.1.36. [DOI] [PubMed] [Google Scholar]
- 78.Rosenbloom AJ, Linden PK, Dorrance A, Penkosky N, Cohen-Melamed MH, Pinsky MR. Effect of granulocyte-monocyte colony-stimulating factor therapy on leukocyte function and clearance of serious infection in nonneutropenic patients. Chest. 2005 Jun;127(6):2139–2150. doi: 10.1378/chest.127.6.2139. [DOI] [PubMed] [Google Scholar]
- 79.Carr R, Modi N, Dore C. G-CSF and GM-CSF for treating or preventing neonatal infections. Cochrane Database Syst Rev. 2003;(3):CD003066. doi: 10.1002/14651858.CD003066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guidelines for preventing opportunistic infections among hematopoietic stem cell transplant recipients. MMWR Recomm Rep. 2000 Oct;49(RR10):1–125. CE121–127. [PubMed] [Google Scholar]
- 81.Hempel L, Korholz D, Nussbaum P, Bonig H, Burdach S, Zintl F. High interleukin-10 serum levels are associated with fatal outcome in patients after bone marrow transplantation. Bone Marrow Transplant. 1997 Sep;20(5):365–368. doi: 10.1038/sj.bmt.1700902. [DOI] [PubMed] [Google Scholar]
- 82.Gras J, Wieers G, Vaerman JL, et al. Early immunological monitoring after pediatric liver transplantation: cytokine immune deviation and graft acceptance in 40 recipients. Liver Transpl. 2007 Mar;13(3):426–433. doi: 10.1002/lt.21084. [DOI] [PubMed] [Google Scholar]
- 83.Settmacher U, Docke WD, Manger T, et al. Management of induction phase of immunosuppression in liver graft recipients: prevention of oversuppression by immune monitoring. Transplant Proc. 1993 Aug;25(4):2703–2704. [PubMed] [Google Scholar]
- 84.Reinke P, Volk HD. Diagnostic and predictive value of an immune monitoring program for complications after kidney transplantation. Urol Int. 1992;49(2):69–75. doi: 10.1159/000282398. [DOI] [PubMed] [Google Scholar]
- 85.Hoffman JA, Weinberg KI, Azen CG, et al. Human leukocyte antigen-DR expression on peripheral blood monocytes and the risk of pneumonia in pediatric lung transplant recipients. Transpl Infect Dis. 2004 Dec;6(4):147–155. doi: 10.1111/j.1399-3062.2004.00069.x. [DOI] [PubMed] [Google Scholar]
- 86.Denzel C, Riese J, Hohenberger W, et al. Monitoring of immunotherapy by measuring monocyte HLA-DR expression and stimulated TNFalpha production during sepsis after liver transplantation. Intensive Care Med. 1998 Dec;24(12):1343–1344. doi: 10.1007/s001340050775. [DOI] [PubMed] [Google Scholar]
- 87.Trindade E, Maton P, Reding R, et al. Use of granulocyte macrophage colony stimulating factor in children after orthotopic liver transplantation. J Hepatol. 1998 Jun;28(6):1054–1057. doi: 10.1016/s0168-8278(98)80356-5. [DOI] [PubMed] [Google Scholar]
- 88.Pizzo PA, Rubin M, Freifeld A, Walsh TJ. The child with cancer and infection. I. Empiric therapy for fever and neutropenia, and preventive strategies. J Pediatr. 1991 Nov;119(5):679–694. doi: 10.1016/s0022-3476(05)80281-1. [DOI] [PubMed] [Google Scholar]
- 89.Kaplan JE, Masur H, Holmes KK. Guidelines for preventing opportunistic infections among HIV-infected persons--2002. Recommendations of the U.S. Public Health Service and the Infectious Diseases Society of America. MMWR Recomm Rep. 2002 Jun 14;51(RR8):1–52. [PubMed] [Google Scholar]
- 90.Benson CA, Kaplan JE, Masur H, Pau A, Holmes KK. Treating opportunistic infections among HIV-exposed and infected children: recommendations from CDC, the National Institutes of Health, and the Infectious Diseases Society of America. MMWR Recomm Rep. 2004 Dec 17;53(RR15):1–112. [PubMed] [Google Scholar]
- 91.Marik PE, Raghavan M. Stress-hyperglycemia, insulin and immunomodulation in sepsis. Intensive Care Med. 2004 May;30(5):748–756. doi: 10.1007/s00134-004-2167-y. [DOI] [PubMed] [Google Scholar]
- 92.Jaber BL, Cendoroglo M, Balakrishnan VS, Perianayagam MC, King AJ, Pereira BJ. Apoptosis of leukocytes: basic concepts and implications in uremia. Kidney Int Suppl. 2001 Feb;78:S197–205. doi: 10.1046/j.1523-1755.2001.59780197.x. [DOI] [PubMed] [Google Scholar]
- 93.Massry S, Smogorzewski M. Dysfunction of polymorphonuclear leukocytes in uremia: role of parathyroid hormone. Kidney Int Suppl. 2001 Feb;78:S195–196. doi: 10.1046/j.1523-1755.2001.59780195.x. [DOI] [PubMed] [Google Scholar]
- 94.Woiciechowsky C, Schoning B, Lanksch WR, Volk HD, Docke WD. Mechanisms of brain-mediated systemic anti-inflammatory syndrome causing immunodepression. J Mol Med. 1999 Nov;77(11):769–780. doi: 10.1007/s001099900051. [DOI] [PubMed] [Google Scholar]
- 95.Woiciechowsky C, Volk HD. Increased intracranial pressure induces a rapid systemic interleukin-10 release through activation of the sympathetic nervous system. Acta Neurochir Suppl. 2005;95:373–376. doi: 10.1007/3-211-32318-x_76. [DOI] [PubMed] [Google Scholar]
- 96.Bergmann M, Sautner T. Immunomodulatory effects of vasoactive catecholamines. Wien Klin Wochenschr. 2002 Sep 30;114(17–18):752–761. [PubMed] [Google Scholar]
- 97.Dandona P, Aljada A, Mohanty P, et al. Insulin inhibits intranuclear nuclear factor kappaB and stimulates IkappaB in mononuclear cells in obese subjects: evidence for an anti-inflammatory effect? J Clin Endocrinol Metab. 2001 Jul;86(7):3257–3265. doi: 10.1210/jcem.86.7.7623. [DOI] [PubMed] [Google Scholar]
- 98.Singhal PC, Kapasi AA, Franki N, Reddy K. Morphine-induced macrophage apoptosis: the role of transforming growth factor-beta. Immunology. 2000 May;100(1):57–62. doi: 10.1046/j.1365-2567.2000.00007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yuengsrigul A, Chin TW, Nussbaum E. Immunosuppressive and cytotoxic effects of furosemide on human peripheral blood mononuclear cells. Ann Allergy Asthma Immunol. 1999 Dec;83(6 Pt 1):559–566. doi: 10.1016/S1081-1206(10)62870-0. [DOI] [PubMed] [Google Scholar]
- 100.Kanakoudi-Tsakalidou F, Debonera F, Drossou-Agakidou V, et al. Flow cytometric measurement of HLA-DR expression on circulating monocytes in healthy and sick neonates using monocyte negative selection. Clin Exp Immunol. 2001 Mar;123(3):402–407. doi: 10.1046/j.1365-2249.2001.01471.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Yerkovich ST, Wikstrom ME, Suriyaarachchi D, Prescott SL, Upham JW, Holt PG. Postnatal Development of Monocyte Cytokine Responses to Bacterial Lipopolysaccharide. Pediatr Res. 2007 Aug 31; doi: 10.1203/PDR.0b013e3181568105. [DOI] [PubMed] [Google Scholar]
- 102.El-Mohandes AA, Rivas RA, Kiang E, Wahl LM, Katona IM. Membrane antigen and ligand receptor expression on neonatal monocytes. Biol Neonate. 1995;68(5):308–317. doi: 10.1159/000244251. [DOI] [PubMed] [Google Scholar]