

Abstract
The accelerated growth of proteomics data presents both opportunities and challenges. Large-scale proteomic profiling of biological samples such as cells, organelles or biological fluids has led to discovery of numerous key and novel proteins involved in many biological/disease processes including cancers, as well as to the identification of novel disease biomarkers and potential therapeutic targets. While proteomic data analysis has been greatly assisted by the many bioinformatics tools developed in recent years, a careful analysis of the major steps and flow of data in a typical highthroughput analysis reveals a few gaps that still need to be filled to fully realize the value of the data. To facilitate functional and pathway discovery for large-scale proteomic data, we have developed an integrated proteomic expression analysis system, iProXpress, which facilitates protein identification using a comprehensive sequence library and functional interpretation using integrated data. With its modular design, iProXpress complements and can be integrated with other software in a proteomic data analysis pipeline. This novel approach to complex biological questions involves the interrogation of multiple data sources, thereby facilitating hypothesis generation and knowledge discovery from the genomic-scale studies and fostering disease diagnosis and drug development.
Key Words: Proteomic profiling, high-throughput analysis, biomarkers, bioinformatic tools, iProXpress, sequence library, pathway discovery, stage specific proteins
INTRODUCTION
The human genome project has revolutionized the practice of biology and the future potential of medicine. The traditional one-gene-at-a-time approach, though effective in revealing detailed molecular functions of individual genes, does not provide a global view of gene function and temporal and spatial regulation for all genes at different physiological or pathological states or developmental stages. Researchers have begun to systematically tackle gene functions and complex regulatory processes by studying organisms on a global scale of genomes: transcriptomes (gene expression) [1], proteomes (protein expression) [2], metabolomes (metabolic networks) [3], and interactomes (protein-protein interactions) [4].
Proteomics aims to identify, characterize and quantify all proteins expressed in cells grown under a variety of conditions [5]. As most biological and disease processes are manifest at the protein level, proteomics has unique and significant advantages as an important complement to genomics and transcriptomics approaches. As a result, there is intense interest in applying proteomics to foster a better understanding of basic biology and disease processes, develop new biomarkers for diagnosis and early detection of disease, and accelerate drug development [6, 7]. Indeed, proteomics has been applied to the discovery of new diagnostic, prognostic and therapeutic targets for numerous diseases, including heart and cardiovascular disorders, cancer, infectious diseases, and diseases of the nervous system [8].
The accelerated growth of proteomics and other large-scale “omics” data presents both opportunities and challenges. The collective richness of the data allows researchers to ask complex biological questions and to gain new scientific insights, as illustrated in the integrated global profiling approach for studying the molecular basis of human cancer [9]. One major challenge, however, lies in the voluminous, complex, and dynamic data being maintained in heterogeneous and distributed sources. Advanced bioinformatics methods are needed for data integration and functional interpretation of large-scale proteomic data.
Due to its robustness, sensitivity and efficiency, tandem mass spectrometry (MS/MS) has become the method of choice for identification of proteins in high-throughput pro-teomics studies [10]. This approach subjects protein mixtures to proteolytic digestion prior to liquid chromatography separation and MS/MS analysis of the resulting peptides. Many bioinformatics tools have been developed for the management and analysis of proteomics data, such as the online tools listed at http://www.proteomecommons.org/tools.jsp. A number of database search programs (e.g., SEQUEST [11], Mascot [12] and X!Tandem [13] are used to assign probable peptide sequences to MS/MS spectra and to infer protein identities. Utility programs such as PeptideProphet [14] and ProteinProphet [15] are designed to improve the accuracy of peptide and protein identification using statistical models, while DBParser [16] employs a parsimony principal to consolidate redundant protein assignments. Several publicly available search algorithms were evaluated and benchmarked for sensitivity and specificity recently [17]. Furthermore, an entire pipeline has been developed for experiment annotation, database searching, peptide mining, and protein identification [18].
Once proteins are identified from the biological samples, they need to be analyzed for functional involvement in metabolic and signaling pathways and cellular functions and processes. Many programs have been developed for the biological interpretation of large lists of genes from genome-scale experiments, mostly for microarray gene expression data, with a few being extended to proteomics data. As the Gene Ontology (GO) [19] has become the common standard for genome annotation, most of these programs provide functional analysis in the context of GO (for examples, see http://www.geneontology.org/ GO.tools.microarray.shtml). A few examples include: (i) GoMiner [20], which presents gene lists in GO hierarchical views, (ii) MAPPFinder [21], integrating GO annotations with GenMAPP pathways, (iii) NetAffx [22], which renders a GO graph to display Affy-metrix probe sets, (iv) DAVID [23], which includes additional information on Pfam domains [24] and KEGG pathways [25], and (v) Babelomics [26], which includes InterPro motifs [27], KEGG pathways, and Swiss-Prot keywords [28].
CHALLENGES IN PROTEOMICS RESEARCH
A major issue in proteomics and tandem mass spectroscopy protein identification is that the general purpose protein sequence databases leave out many alternative splice iso-forms or include them only in the text comments. As a result, proteomic analysis may fail to identify bona fide protein products of alternative splice isoforms because the target sequence was not present in the database being searched. The absence of real protein sequences in the sequence library may further lead to incorrect peptide and protein identification due to the presence of degenerate peptides corresponding to more than one protein in the sequence database.
Another common problem when dealing with a large list of proteins annotated in different places is the lack of standardization. Different protein IDs and names may be used for the same proteins if a different underlying target database is used for MS/MS protein search. Even different versions of the same protein database may result in different IDs if the database identifier is not stable. The lack of common protein identifiers and naming standards presents a challenge for integrating annotations from multiple heterogeneous sources for biological interpretation of proteomic data. Consequently, expression data analysis is often carried out in an ad hoc manner, resulting in a fragmented and inefficient use of rich annotations available in numerous information resources.
SOLUTIONS.
Built upon the infrastructure developed by the investigative team at the Protein Information Resource [29], iProXpress facilitates protein identification using a comprehensive sequence library and functional interpretation using integrated data. iProXpress has been integrated with other programs in a proteomic data analysis pipeline that includes:
i) A comprehensive sequence library downloadable for use by proteomic search engines,
ii) Protein ID mapping web services,
iii) Protein annotation web services (returning a richly annotated “protein information matrix” from a protein list),
iv) Sequence analysis and protein curation services, and
v) A customized interactive web view for comparative profile analysis of multi-datasets/data types.
iProXpress System Overview
The iProXpress integrated protein expression analysis system is designed for function and pathway discovery from large-scale proteomic data, in a systems biology context, providing rich functional descriptions for individual proteins and detecting functional relationships among them. The system consists of three major software modules to support functionalities in protein mapping, functional annotation and expression profiling Fig. (fig. 1).
Fig. (1).
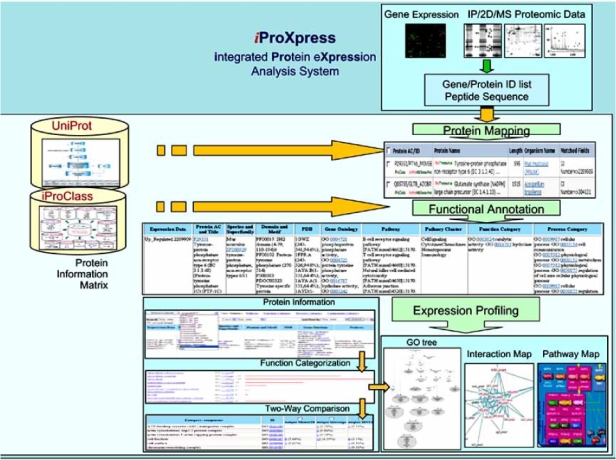
iProXpress system design.
A) Protein Mapping
The protein mapping module is designed to map user-submitted data to corresponding UniProt entries for data analysis in the protein-centric framework. The major functionalities are summarized in Table 1.
Table 1.
Functionalities of Protein Mapping Module
| Functionality | Current | Future |
|---|---|---|
| Input Data | Protein ID list, Peptide sequence | Protein or gene ID list, Peptide sequence |
| Data Type | Mass spectrometry (MS) | MS, 2D gel electrophoresis, immuno-precipitation (IP), gene expression |
| Sequence Library | UniProt (UniRef100 + unique UniParc) | UniProt (UniRef100 + unique UniParc), Genomic (SNP, EST, gene model), Protein identification DB (PRIDE, gpmDB) |
| Mapping Method | Protein ID, Peptide | Protein and gene ID, Peptide, Protein and gene name |
Input Data and Data Types
Currently, the accepted input data are protein IDs and, optionally, their associated peptide sequences, which may be generated from search programs such as SEQUEST or MASCOT. In the future, iProXpress will accept other user-supplied high-throughput data types.
Protein Sequence Library
iProXpress uses a comprehensive sequence library consisting of all known proteins and isoforms from the UniProt databases, namely UniRef100 and UniParc. UniRef100 is a non-redundant reference database maintained at PIR that provides complete non-redundant sequence coverage by combining identical sequences and sub-fragments into single entries. Currently containing about 3.1 million entries, Uni-Ref100 consists of all published protein sequences from several database sources, including UniProtKB (the primary source with 2.8 million entries), and unique sequences from RefSeq, GenPept, IPI, Ensembl and PDB. The sequence space is further expanded by representing the variant sequences (such as splice variants and isoforms annotated in UniProtKB) as separate UniRef100 entries. This data list is extended on a regular basis to guarantee full coverage of sequence space. UniRef100 is the most comprehensive database of its kind due to the curation activities of the UniProt consortium. UniParc, currently containing over 5.5 million sequences, provides a complete sequence archive, including conceptual translations from gene models of newly sequenced genomes. Though many UniParc entries may still undergo sequence revisions, UniParc sequences not present in UniRef100 nevertheless are useful as a supplement to the sequence library for complete coverage.
This UniProt sequence library, consisting of UniRef100 and unique UniParc sequences, has several advantages over NCBI’s nr database for use in the MS/MS database search engine because it will have (i) more sequences, (ii) stable identifiers for UniProtKB entries (NCBI’s GI number many change from version to version), (iii) more extensive functional annotation, and (iv) less redundancy. While the sequence library covers proteins in source organisms from the entire taxonomic range, options will be provided for users to select sub-datasets based on taxonomy (e.g., human proteins only, or human and mouse proteins only).
In the future, the sequence library will be further expanded to improve the sensitivity of protein identification. New data sources to be integrated will include: (i) the genomic evidence for protein sequence variants, such as coding SNPs, ESTs and gene models, and (ii) protein identification databases such as gpmDB [30] and PRIDE [31] for comparison and cross-reference of spectra and protein identifications.
Protein Mapping Methods
For proteomic data interpretation, a high level of annotation, minimal level of redundancy, and high degree of data integration is critical. iProXpress maps protein lists and their associated peptide sequences to UniProt entries to facilitate functional and pathway analysis. Proteins with various database identifiers are mapped using the PIR ID mapping service, which maps protein and gene IDs from about 30 data sources to UniProt. All major NCBI identifiers, such as GI number and Entrez Gene and RefSeq IDs are included. To cross-validate the ID mapping, the peptide sequence of each mapped protein is matched against the cross-referenced U-niProt sequence to confirm the correct assignment. For many-to-one mapping, where multiple IDs map to the same UniProt protein, as is often the case for GI numbers, this mapping removes redundancy effectively.
For proteins that cannot be mapped to UniProt entries through ID mapping, iProXpress searches their peptide sequences against the entire UniRef100 sequence set, or against a species-specific subset if appropriate. There are several mapping scenarios. In the case of one-to-one mapping, where the peptide matches exactly one UniProt protein, that distinct protein receives the assignment. In the case of one-to-many mapping, where the peptide sequence matches to more than one UniProt entry, sequence variations are identified by UniRef90 clusters, in which members share at least 90% sequence identity to the representative sequence. When all matched protein entries are in the same UniRef90 cluster, the peptides are mapped to the representative sequence from that cluster or to the member protein from the same species. Otherwise, if the proteins belong to different UniRef90 clusters; reliable protein assignments are made manually with retro-inspection of the original MS/MS protein identification results.
Proteins in the input list that are not mapped after ID and peptide mapping to UniRef100 are mapped to the unique UniParc sequence library. Since UniParc is a sequence archive with no functional annotation, the best-hit homologous protein in UniProtKB with at least 90% sequence identity is chosen as the sequence representative. All proteins mapped with sequence variations are flagged for manual validation.
To demonstrate the ability of iProXpress to process large-scale proteomic datasets, we analyzed the proteome of a human embryonic carcinoma stem cell line, NTera2, using our robust offline multidimensional protein identification strategy together with automated linear ion trap mass spectrometry. iProXpress functionally classified and physio-chemically characterized approximately 7000 proteins. This large scale proteomic profiling of the Ntera2 cell and its comparison with DNA microarray data contribute to a better understanding of gene regulation at a global level.
B. Functional Annotation
After the protein mapping, rich annotation is presented in a protein information matrix based on sequence analysis and integration of information from the iProClass database. iProClass also includes pre-computed sequence analysis results (e.g., BLAST related sequences) to support reliable annotation transfer from well-curated homologs to poorly characterized proteins, which is useful because an estimated 40-50% of proteins from complete genomes are “hypothetical”, and only a small fraction of proteins have experimentally validated annotations. We pre-compute and regularly update sequence features of functional significance for UniProt proteins, and make the sequence analysis tools available for online analysis of proteins/sequence variants not in UniProt. Pre-computed sequence features include homologous proteins in KEGG, BioCarta and other curated pathway databases to populate pathway annotation, InterProScan for family, domain and motif identification, and Phobius for trans-membrane helix and signal peptide prediction. Properties derived from homology-based inference are presented in the information matrix with evidence attribution.
C. Expression Profiling
Functional profiling analysis aims at discovering the functional significance of expressed proteins, the plausible functions and pathways, and the hidden relationships and interconnecting components of proteins, such as proteins sharing common functions, pathways, or cellular networks. As shown in Fig. (2), the extensive annotation in the protein information matrix (A) allows functional categorization and detailed analysis of expressed proteins in a given dataset, as well as cross-comparison of co-expressed or differentially-expressed proteins from multiple datasets. For functional categorization, proteins are grouped based on annotations such as GO terms and KEGG and BioCarta pathways, and then correlated with sequence similarity to identify relationships among individual proteins or protein groups. The functional categorization chart (B) displays the frequency (number of occurrences) of proteins in each functional category. Categorization and sorting of proteins based on functions, pathways, and/or other attributes in the information matrix generate various protein clusters, from which interesting unique or common proteins in different datasets can be identified in combination with manual examination. The cross-comparison matrix (C) shows the comparative distribution of functional categories in multiple datasets.
Fig. (2).
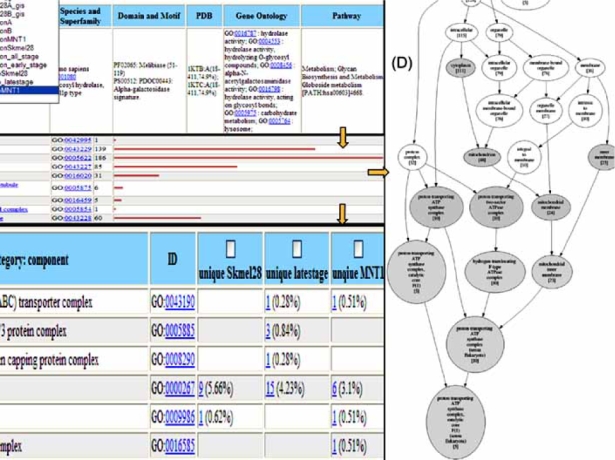
Functional profiling: (A) protein information matrix, (B) functional categorization chart, (C) cross-comparison matrix, (D) graphical GO hierarchy.
To correlate functional associations of expressed proteins in different samples, the relative enrichment of a given functional category in each sample will be calculated to identify all samples that contain a statistically significant proportion of proteins that are associated with the given category. Likewise, the system will point to groups of proteins that show a statistically significant correlation with certain pathways or functions, thus enabling characterization of biological pathways. Evidence on differential protein expression, protein interactions, pathway membership, and other attributes is combined to provide the evidence for pathway and network participation. This allows relative ranking of the proteins involved in the biological response to identify the critical nodes in the response pathway and hidden relationships.
ANALYSIS OF PROTEOMIC DATA USING IPROX- PRESS
The iProXpress system (http://pir.georgetown.edu/iprox press/) has also been applied to the expression profile analysis for hCG-induced changes in MA-10 mouse Leydig tumor cells [32], organelle proteome analysis of various melano-some stages from human melanoma cell lines [33]∗, and the comparative analyses of lysosome-related organelle (LRO) proteomes [34]∗. Here we use the melanosome proteome analyses to illustrate the integrative approach for function and pathway exploration and knowledge discovery using iProXpress.
Melanosomes are membrane-bound organelles special- ized in the production and distribution of melanin pigment and are conserved in structure from primitive organisms to mammals. Dysfunctions in pigmentation and melanosome biogenesis are associated with a wide variety of inherited genetic disorders and pigmentary diseases, including oculo-cutaneous albinism and Hermansky–Pudlak syndrome. Me-lanosome-specific proteins also provide important markers for malignant melanoma. In mammals, melanosomes mature from undifferentiated vesicles (stage I) to an elongated form with internal fibrils (stage II). In the presence of tyrosinase and other enzymes, melanin is synthesized and deposited on the internal fibrils (stage III) and can become uniformly dense (stage IV) in heavily-pigmented melanocytes. As melano-somes mature, they are gradually transported to the peripheries of the melanocytes in which they form and, in human skin, they are transferred to neighboring keratinocytes. A detailed understanding of how melanosomes mature and move within and between cells requires a comprehensive knowledge of the proteins comprising them. A combination of immunoblotting, immunofluorescence microscopy, and bioinformatics analysis was used to characterize the protein profiles of melanosomes at various biogenic stages.
The bioinformatics analysis of the melanosome proteo-mic data using the iProXpress system first involved mapping peptide sequences from the MS data and protein lists (NCBI gi numbers from the nr database) to UniProtKB entries. From a total of 2,298 gi numbers, 1,253 (55%) were mapped to UniProtKB following ID mapping, and 1,506 (66%) mapped based on peptide sequences. When the results from both mappings were combined, 1,936 (84%) gi numbers were mapped to 1438 UniProtKB sequences. The mapping reveals that the NCBI gi is an unstable database identifier, with many gi numbers changing from version to version or becoming obsolete. The result also indicates that the nr database is more redundant, with many gi numbers representing identical proteins that map to the same UniProtKB entries.
The determined melanosome proteomes contain ~1,500 proteins combined from all stages of melanosomes, with ~600 in any given stage. Protein information matrices were generated for corresponding UniProtKB entries of identified melanosome proteins, summarizing salient features retrieved from the underlying PIR data warehouse or inferred based on sequence homology (Table 2). Iterative categorization and sorting of proteins were carried out to generate various protein clusters, from which interesting unique or common proteins at different stages of melanosome biogenesis were identified through manual examination. The stage-related proteins provide direct evidence of protein sorting and trafficking to this organelle and provide information about melanosome biogenesis as lysosome-related organelles. Approximately 100 proteins shared by melanosomes from pigmented and non-pigmented melanocytes at all stages define the essential melanosome proteome. These common proteins are considered constituent or resident proteins throughout melanosome biogenesis. Using the functional information matrices, mela-nosome stage-specific proteins were proposed, some of which have been subsequently validated for their melanoso-mal localization, including PEDF (pigment-epithelium derived factor) and SLC24A5 (sodium/potassium/calcium exchanger 5, NCKX5).
Table 2.
Stage-Related Melanosome Proteins (Partial List)
| Stages | UniProtKB | Gene Name | Protein Name | Notes∗ |
| P36955 | PEDF | Pigment epithelium-derived factor precursor | ||
| P51148 | RAB5C | Ras-related protein Rab-5C (RAB5L) (L1880) | Validated | |
| P05556 | ITGB1 | Integrin beta-1 | ||
| Stage I | ||||
| Q9UMX9 | Matp | Membrane-associated transporter protein (SLC45A2) | Homolog | |
| O14880 | MGST3 | Microsomal glutathione S-transferase 3 | ||
| Proposed | ||||
| Q14254 | FLOT2 | Flotillin-2 (Epidermal surface antigen) | ||
| Q9UMX9 | Matp | Membrane-associated transporter protein (SLC45A2) | ||
| Homolog | ||||
| Q04656 | ATP7A | Copper-transporting ATPase 1 | ||
| Stage II | Q9P0L0 | VAPA | Vesicle-associated membrane protein-associated protein A | |
| P53992 | SEC24C | Protein transport protein Sec24C | Proposed | |
| O95782 | AP2A | Adapter-related protein complex 2 alpha- 1 subunit | ||
| O95670 | ATP6G2 | Vacuolar ATP synthase subunit G 2 | ||
| Validated | ||||
| Q71RS6 | SLC24A | Sodium/potassium/calcium exchanger 5 precursor | ||
| P57729 | Rab-38 | Ras-related protein Rab-38 | ||
| P51159 | RAB27A | Ras-related protein Rab-27A (Rab-27) | ||
| Homolog | ||||
| Q9Y4I1 | Myo5a | Myosin-5A (Myosin Va) | ||
| Stage IV | Q99698 | LYST | Lysosomal trafficking regulator | |
| Q16643 | DBN1 | Drebrin | ||
| P59998 | ARPC4 | Actin-related protein 2/3 complex subunit 4 | ||
| P18206 | VCL | Vinculin (Metavinculin) | Proposed | |
| P63000 | RAC1 | Ras-related C3 botulinum toxin substrate 1 (p21-Rac1) | ||
| P51148 | RAB5C | Ras-related protein Rab-5C (RAB5L) |
All proteins listed are identified in the melanosome proteomes. Validated –shown to be localized in melanosomes by immunostaining; Homolog – homologous to known mouse coat color genes; Proposed – proposed as protein of functional interest for validation.
Based on the functional profiling, a more detailed mela-nosomal biogenic pathway has been proposed that will facilitate understanding of the dynamic process of melanosome biogenesis, including the contribution of elements and complex membrane protein traffic input from several other organelles. Besides proteins previously known as melano-some-specific proteins (e.g., Pmel17, TYR, Tyrp1), this study provided a comprehensive list of proteins comprising this dynamic organelle. Table 2 selectively lists three groups of proteins that are functionally important at each stage of me-lanosome differentiation: 1) newly identified and validated in this study (e.g., PEDF and SLC24A5); 2) human homologs of mouse color genes identified in this study (e.g., Atp7a and MyoVa); 3) proposed stage-related proteins newly identified (e.g., Sec24 and vinculin); 4) proteins known as melanosome proteins from previous studies (e.g., Pmel17 and TYR). Many proteins detected in stage IV melanosomes are molecular motor- and cytoskeleton-related proteins (not listed), which may be necessary for directing fully pigmented melanosomes towards the cell periphery and for their eventual transfer to keratinocytes. Some proteins are found in all stages and are also common to other organelles, e.g., LAMP1 in lysosome. While it is obvious that multiple sources of cellular components contribute to the biogenesis of melanosomes, proteins more abundant in specific stages may define unique functions in that stage (e.g., the ion transporters VATPase and SLC24A5).
Therefore, it is possible to deduce a set of signature proteins for melanosomes that will consist of previously known melanosome-specific proteins, the proposed melanosome stage-specific proteins, and other constituent proteins commonly found in several other organelles. Thus, the proteomic analysis of melanosomes using iProXpress illustrates that bioinformatics characterization of melanosome proteomes facilitates a better understanding of the biogenesis and function of melanosomes.
SUMMARY
Advances in proteomic technology in combination with state-of-the-art bioinformatics tools have greatly facilitated proteomic data analysis and pathway discovery. iProXpress, a unique bioinformatics tool designed to facilitate functional annotation and pathway discovery from large scale proteo-mic data, will help researchers to answer complex biological questions by searching multiple data sources. The integration of iProXpress with high-throughput genomics and proteo-mics technology will expedite the prediction of pathway and functional relationships, thereby supporting hypothesis generation and knowledge discovery from genome wide studies.
ACKNOWLEDGEMENT
This research was supported in part by the Intramural Research Program of the NIH, National Institute on Aging and the NIH grants U01-HG02712 and HHSN2662004000- 61C to the Protein Information Resource (PIR) bioinforma-tics resources.
REFERENCES
- 1.Brentani RR, Carraro DM, Verjovski-Almeida S, Reis EM, Neves EJ, de Souza SJ, Carvalho AF, Brentani H, Reis LF. Gene expression arrays in cancer research: methods and applications. Crit Rev Oncol Hematol. 2005;54:95–105. doi: 10.1016/j.critrevonc.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 2.Anderson NL, Anderson NG. Gene expression arrays in cancer research: methods and applications. Crit Rev Oncol Hematol. 2005;54:95–105. doi: 10.1016/j.critrevonc.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 3.Bono H, Nikaido I, Kasukawa T, Hayashizaki Y, Okazaki Y. Comprehensive analysis of the mouse metabolome based on the transcriptome. Genome Res. 2003;13:1345–9. doi: 10.1101/gr.974603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Walhout AJ, Reboul J, Shtanko O, Bertin N, Vaglio P, Ge H, Lee H, Doucette-Stamm L, Gunsalus KC, Schetter AJ, Morton DG, Kemphues KJ, Reinke V, Kim SK, Piano F, Vidal M. Integrating interactome, phenome, and transcriptome mapping data for the C. elegans germline. Curr Biol. 2002;12:1952–1958. doi: 10.1016/s0960-9822(02)01279-4. [DOI] [PubMed] [Google Scholar]
- 5.Pandey A, Mann M. Proteomics to study genes and genomes. Nature. 2000;405:837–846. doi: 10.1038/35015709. [DOI] [PubMed] [Google Scholar]
- 6.Srivastava S, Srivastava RG. Proteomics in the Forefront of Cancer Biomarker Discovery. J Proteome Res. 2005;4:1098–1103. doi: 10.1021/pr050016u. [DOI] [PubMed] [Google Scholar]
- 7.Hanash S. Disease proteomics. Nature. 2003;422:226–232. doi: 10.1038/nature01514. [DOI] [PubMed] [Google Scholar]
- 8.Burkhard PR, Rodrigo N, May D, Sztajzel R, Sanchez JC, Hochstrasser DF, Schiffer E, Reverdin A, Lacroix JS. Assessing cerebrospinal fluid rhinorrhea: a two-dimensional electrophoresis. Electrophoresis. 2001;22:1826–1833. doi: 10.1002/1522-2683(200105)22:9<1826::AID-ELPS1826>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 9.Hanash S. Integrated global profiling of cancer. Nat Rev Cancer. 2004;4:638–644. doi: 10.1038/nrc1414. [DOI] [PubMed] [Google Scholar]
- 10.Aebersold R, Goodlett DR. Mass spectrometry in proteomics. Chem Rev. 2001;101:269–295. doi: 10.1021/cr990076h. [DOI] [PubMed] [Google Scholar]
- 11.Eng JK, McCormack AL, Yates JRI. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 12.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 13.Fenyö D, Beavis RC. A Method for Assessing the Statistical Significance of Mass Spectrometry-Based Protein Identifications Using General Scoring Schemes. Anal Chem. 2003;75:768–774. doi: 10.1021/ac0258709. [DOI] [PubMed] [Google Scholar]
- 14.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. An explanation of the Peptide Prophet algorithm developed. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 15.Nesvizhskii AI, Keller A, Kolker E, Abersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 16.Yang X, Dondeti V, Dezube R, Maynard DM, Geer LY, Epstein J, Chen X, Markey SP, Kowalak JA. DBParser: web-based software for shotgun proteomic data analyses. J Proteome Res. 2004;3:1002–1008. doi: 10.1021/pr049920x. [DOI] [PubMed] [Google Scholar]
- 17.Kapp EA, Schutz F, Connolly LM, Chakel JA, Meza JE, Miller CA, Fenyo D, Eng JK, Adkins JN, Omenn GS, Simpson RJ. An evaluation, comparison, and accurate benchmarking of several publicly available MS/MS search algorithms: sensitivity and specificity analysis. Proteomics. 2005;5:3475–3490. doi: 10.1002/pmic.200500126. [DOI] [PubMed] [Google Scholar]
- 18.Rauch A, Bellew M, Eng J, Fitzgibbon M, Holzman T, Hussey P, Igra M, Maclean B, Lin CW, Detter A, Fang R, Faca V, Gafken P, Zhang H, Whiteaker J, States D, Hanash S, Paulovich A, McIntosh MW. Computational Proteomics Analysis System (CPAS): An Extensible, Open-source Analytic System for Evaluating and Publishing Proteomic Data and High throughput Biological Experiments. J Proteome Res. 2006;5:112–121. doi: 10.1021/pr0503533. [DOI] [PubMed] [Google Scholar]
- 19.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology: The gene ontology consortium. Nat Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zeeberg BR, Feng W, Wang G, Wang MD, Fojo FT, Sunshine M, Narasimhan S, Kane DW, Reinhold WC, Lababidi S, Bussey KJ, Riss J, Barrett JC, Weinstein JN. GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biology. 2003;4:R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doniger SW, Salomonis N, Dahlquist KD, Vranizan K, Lawlo SC, Conklin BR. MAPPFinder: using Gene Ontology and GenMAPP to create a global gene-expression profile from microarray data. Genome Biology. 2003;4:R7. doi: 10.1186/gb-2003-4-1-r7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng J, Sun S, Tracy A, Hubbell E, Morris J, Valmeekam V, Kimbrough A, Cline MS, Liu G, Shigeta R, Kulp D, Siani-Rose MA. NetAffx gene ontology mining tool: a visual approach for microarray data analysis. Bioinformatics. 2004;20:1462–1463. doi: 10.1093/bioinformatics/bth087. [DOI] [PubMed] [Google Scholar]
- 23.Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:R60. [PubMed] [Google Scholar]
- 24.Bateman A, Coin L, Durbin R, Finn RD, Hollich V, Jones SG, Khanna A, Marshall M, Moxon S, Erik L, Sonnhammer L, Studholme DJ, Yeats C, Eddy SR. The Pfam protein families' database. Nucleic Acids Res. 2004;32:D138–D141. doi: 10.1093/nar/gkh121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanehisa M, Goto S, Kawashima S, Nakaya A. The KEGG databases at GenomeNet. Nucleic Acids Res. 2002;30:42–46. doi: 10.1093/nar/30.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Al-Shahrour F, Minguez P, Vaquerizas JM, Conde L, Dopaz J. BABELOMICS: a suite of web tools for functional annotation and analysis of groups of genes in high-throughput experiments. Nucleic Acids Res. 2005;33:W460–W464. doi: 10.1093/nar/gki456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mulder NJ, Apweiler R, Attwood TK, Bairoch A, Bateman A, Binns D, Bradley P, Bork P, Bucher P, Cerutti L, Copley R, Courcelle E, Das U, Durbin R, Fleischmann W, Gough J, Haft D, Harte N, Hulo N, Kahn D, Kanapin A, Krestyaninova M, Lonsdale D, Lopez R, Letunic I, Madera M, Maslen J, McDowall J, Mitchell A, Nikolskaya AN, Orchard S, Pagni M, Ponting CP, Quevillon E, Selengut J, Sigrist CJA, Silventoinen V, Studholme DJ, Vaughan R, Wu CH. InterPro, progress and status in 2005. Nucleic Acids Res. 2005;33:D201–D205. doi: 10.1093/nar/gki106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Boeckmann B, Bairoch A, Apweiler R, Blatter MC, Estreicher A, Gasteiger E, Martin MJ, Michoud K, O’Donovan C, Phan I, Pilbout S, Schneider M. The SWISS-PROT protein knowledgebase and its supplement TrEMBL. Nucleic Acids Res. 2003;31:365–370. doi: 10.1093/nar/gkg095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu CH, Yeh ASL, Huang H, Arminski L, Castro-Alvean J, Chen Y, Hu Z, Kourtesis P, Ledley RS, Suzek BE, Vinayaka CR, Zhang J, Barker CW. The Protein Information Resource. Nucleic Acids Res. 2003;31:345–347. doi: 10.1093/nar/gkg040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Craig R, Cortens JP, Beavis RC. Open source identification data.system for analyzing, validating, and storing protein. J Proteome Res. 2004;3:1234–1242. doi: 10.1021/pr049882h. [DOI] [PubMed] [Google Scholar]
- 31.Martens L, Vandekerckhove J, Gevaert K. DBToolkit: processing protein databases for peptide-centric proteomics. Bioinformatics. 2005;21:3584–3585. doi: 10.1093/bioinformatics/bti588. [DOI] [PubMed] [Google Scholar]
- 32.Li W, Amri H, Huang H, Wu C, Papadopoulos V. Gene and protein profiling of the response of MA-10 Leydig Tumor Cells to Human Chorionic Gonadotropin. J Andrology. 2004;25:900–913. doi: 10.1002/j.1939-4640.2004.tb03160.x. [DOI] [PubMed] [Google Scholar]
- 33.Chi A, Valencia JC, Hu ZZ, Watabe H, Yamaguchi H, Mangini N, Huang H, Canfield VA, Cheng K, Shabanowitz J, Hearing VJ, Wu C, Appella E, Hunt DF. A proteomics and bioinformatics approach to define the biogenesis and function of melanosomes. J Prot Res. 2006;5:3135–3144. doi: 10.1021/pr060363j. [DOI] [PubMed] [Google Scholar]
- 34.Hu ZZ, Valencia JC, Huang H, Chi A, Shabanowitz J, Hearing VJ, Appella E, Wu CH. Comparative Bioinformatics Analyses and Profiling of Lysosome-Related Organelle Proteomes. J Int Mass Spec. 2007;259:147–160. doi: 10.1016/j.ijms.2006.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]