

Abstract
Cytosolic phospholipase A2 (cPLA2)α responds to the rise in cytosolic Ca2+ ([Ca2+]i) attending cell stimulation by moving to intracellular membranes, releasing arachidonic acid (AA) from these membranes, and thereby initiating the synthesis of various lipid mediators. Under some conditions, however, cPLA2α translocation occurs without any corresponding changes in [Ca2+]i. The signal for such responses has not been identified. Using confocal microscopy to track fluorescent proteins fused to cPLA2α or cPLA2α’s C2 domain, we find that AA mimics Ca2+ ionophores in stimulating cPLA2α translocations to the perinuclear ER and to a novel site, the lipid body. Unlike the ionophores, AA acted independently of [Ca2+]i rises and did not translocate the proteins to the Golgi. AA’s action did not involve its metabolism to eicosanoids or acylation into cellular lipids. Receptor agonists also stimulated translocations targeting lipid bodies. We propose that AA is a signal for Ca2+-independent cPLA2α translocation and that lipid bodies are common targets of cPLA2α and contributors to stimulus-induced lipid mediator synthesis.
Keywords: cPLA2α, C2 Domain, Translocation, Arachidonic Acid, Lipid Bodies, Cellular Calcium, Cancer Cells
1. Introduction
Cytosolic phospholipase A2 (cPLA2α)1 is critical for the production of various active lipids [1]. The rise in cytosolic Ca2+ ([Ca2+]i) attending cell stimulation causes cPLA2α to move from the cytosol to the perinuclear ER and Golgi, deacylate resident phospholipids (PLs), and thereby generate arachidonic acid (AA) and lyso-PLs; these products are then metabolized to eicosanoids, platelet-activating factor, and other mediators [2–6]. cPLA2α thereby converts [Ca2+]i signals into the synthesis of lipids controlling various biological reactions. However, some stimuli cause cPLA2α to translocate and lipid mediators to form without altering [Ca2+]i [7–11]. Moreover, many cell stimulating conditions lead to [Ca2+]i. rises and cPLA2α translocations that rapidly reverse. Such fleeting changes do not lead to lipid mediator synthesis. Other stimulating conditions, contrastingly, cause cPLA2α translocations that persist long after [Ca2+]i normalizes [9,12,13] and evoke the synthesis of lipid mediators. The persistent translocation of cPLA2α appears due to an irreversible change in its structure that may or may not result from the stimulus-induced formation of phosphatidylinositol polyphosphates [7,14],[15] or from subtle differences in the duration or sub-cellular location of Ca2+ rises [3,9,12,13,13,16]. Using cPLA2α fluorescent fusion proteins to track the movements of cPLA2α, we show here that a product of cPLA2α’s action, AA, causes Ca2+-independent and sustained cPLA2α translocation responses. Thus, AA may serve as a signal for the Ca2+-independent and irreversible translocation of cPLA2α. In the course of these studies, we also found that AA, [Ca2+]i rises, and receptor agonists stimulate cPLA2α fluorescent proteins to translocate to a novel site, the lipid body. Lipid bodies, we suggest, are targets for translocating cPLA2α and provide substrates for lipid mediator synthesis under diverse cell stimulating conditions.
2. Materials and methods
2.1 Materials
Stock solutions of AA (NuChek Prep, Elysian, MN) and α-linolenic and 11Z, 14Z-eicosadienoic acids (Matreya LLC, Pleasant Gap, PA) (>99% purity) were stored in ethanol at −70° C under argon. Triacsin C (Biomol International, Plymouth Meeting, PA) was stored in DMSO at −70° C under argon. Sphingosine-1-phosphate (S1P) was taken up in saline-BSA just before use as instructed by Avanti Polar Lipids, Alabaster, AL; ionomycin (Io, Squibb & Sons, Inc., Cranbury, NJ), A23187 (Calbio-chem, San Diego, CA) and GTPγS (Sigma-Aldrich, St. Louis, MO) were dissolved in ethanol and water, respectively; nordihydroguairetic acid and indomethacin (Sigma-Aldrich) were dissolved in ethanol and incubated with cells for 30 min; pertussis toxin (List Biological Laboratories, Inc., Campbell, CA) was dissolved in water and incubated with cells for 18 hr before stimulation. At the final concentrations used, ethanol, DMSO, and saline-BSA (0.1%), nordihydroguairetic acid and indomethacin (10 μM), and pertussis toxin (1 μg/ml) did not alter the location of the fluorescent proteins examined here.
2.2 Cells and transfections
HEK-293, MDA-MB-231, and PC3 cells (ATCC, Manassus, VA) were cultured (37° C, 5% CO2) in DMEM containing penicillin, streptomycin, L-glutamine, and 10% FBS (Invitrogen, Carlsbad, CA). Cells were transfected stably with pEGFP-cPLA2 or transiently with pEYFP-cPLA2, ± pECFP-golgi subcellular localization vector (Clontech, Palo Alto, CA), using Fugene 6 Transfection Reagent as directed by the manufacturer (Roche Diagnostics, Indianapolis, IN). Primers for the C2 domain of cPLA2α (IDT, Greensboro, NC) were used to obtain cPLA2α’s nucleotide sequence (49–444) encoding amino acids 17–148. The sequence was inserted into the HindIII and SmaI restriction sites of pEYFP-C2 (Clontech); the resulting vector (pC2-EYFP) was verified to have the same sequence as pC2-EGFP between the appropriate restriction sites by the Wake Forest University Health Sciences DNA Sequencing Laboratory. Transfected cells were plated for 18 hr in single chamber cover glasses (Fisher Scientific, Pittsburgh, PA) and incubated in serum-free DMEM for 30–60 minutes before challenge. Where specified, the cells were treated with 10 μg/ml of BODIPY 493/503 for 10 min to stain lipid bodies as recommended by the supplier (Molecular Probes), washed twice, incubated in serum-free DMEM for 120 min, and challenged.
2.3 [Ca2+]i Measurements
Cells were loaded with 1 μM of X-rhod-1 Ca2+ indicator (Invitrogen) in 0.01% Pluronic F127 (Invitrogen) for 20 minutes, washed twice, and incubated in DMEM for 30 minutes before challenge. The intensity of X-rhod-1 fluorescence was monitored in a predetermined cytosolic region of cells each minute for 0–120 min after challenge. Results are reported as the fractional changes in fluorescent emissions or maximal fractional rises in emission relative to t=0 values. For some studies, cells were incubated in Ca2+-free medium and 10 μM of BAPTA-AM (Invitrogen) for 30 min.
2.4 Fluorescent microscopy
Fluorescence was monitored with a Zeiss LSM 510 laser scanning microscope (Carl Zeiss, International, Oberkochen, Germany) equipped with a 63X water corrected lens and external heating source (stage temperature, 37° C). ECFP, EGFP, and EYFP were detected by argon (respective emissions 458,488, and 514), X-rhod-1 by helium neon (emission 563 nm), and BODIPY by argon (emission 458 nm) lasers. We used filters to distinguish the fluorescent emission of EYFP from that of BODIPY and eliminate bleed-through: a longpass filter selecting excitation at ≥560 nm captured EYFP, a dichroic filter for excitation at 480–520 nm captured BODIPY emissions. Since the BODIPY emission closely overlapped that of EGFP, experiments on BODIPY tracked EYFP but not EGFP fusion proteins. DIC and fluorescent images were taken concurrently. Pinhole size was 1 Airy unit, zoom was at 2X. Cells were scanned at 1 min intervals from 0–120 min after challenge. For 3-dimensional images, scans of cells were taken in successive increasing slices at 0, 30, 60, 90, or 120 min after challenge. Photo-bleaching of fluorescent probes and movement or detachment of cells limited experiments to 2 hr. In studies using ≥10μM of a Ca2+ ionophore, cells often detached after 30 min of observation. These studies were ended after cell detachment.
2.5 Phase contrast microscopy
Cells were visualized at 37° C by phase contrast microscopy to identify refractive bodies and, within 1 min thereafter, fluorescence microscopy (Zeiss Axioplan 2 fluorescent microscope) to determine the fluorescence of these bodies.
3. Results
293 cells expressing EGFP-cPLA2 emitted fluorescence homogeneously throughout the cytoplasm. On exposure to Io, the cells shifted this fluorescence to the perinuclear area and an irregularly-shaped cytosolic site, as found by others, but also to sites not previously described as targets for this protein viz., small, spherical, cytoplasmic sites (Fig. 1, panels A–F). Responses began within 5, 10, 15, 20, and 40 min of exposure to 33, 10, 1, 0.1, and 0.01 μM of Io, respectively, and did not reverse during 2 hours of observation. Ca2+ ionophore A23187 had similar actions (not shown). AA also caused the translocation of EGFP-cPLA2. However, these responses began only after ~20, 40, 60, 90, and 100 min of exposure to 33, 10, 1, 0.1, and 0.01 μM of AA, respectively, and targeted the protein to the perinuclear and spherical but not irregular sites (Fig. 1, panels G–L). AA and Io exhibited the same differential targeting action in cells expressing EYFP-cPLA2 and a Golgi marker: Io caused EYFP-cPLA2 to translocate to the perinuclear, spherical, and irregular sites while AA caused it to move to the perinuclear and spherical but not irregular sites; the irregular but not other sites overlap our Golgi marker (Fig 2). Finally, the vehicle for AA and the ionophores, ethanol, did not alter EGFP-cPLA2’s location (Fig. 1, panels M–O) and neither Io [17] nor AA (Fig. 1, panels P–R) altered the distribution of EGFP not fused to cPLA2α. These results agree with reports [2–6] that [Ca2+]i rises like those caused by ionophores induce cPLA2α fluorescent proteins to translocate to the perinuclear ER and Golgi. They also show that AA stimulates irreversible translocation responses that target the perinuclear ER although not Golgi and that AA and the ionophores stimulate the proteins to move to sites 0.2–2 μm in diameter and more or less evenly distributed throughout the cytoplasm.
Fig. 1.
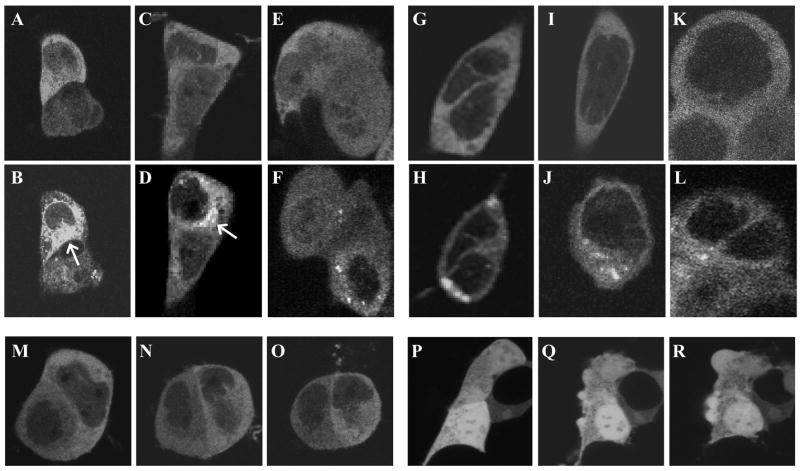
EGFP-cPLA2 translocation responses. Panels A–O: 293 Cells expressing EGFP-cPLA2 were scanned by confocal microscopy for EGFP just before (A, C, E, G, I, and K) or after challenge with 1 μM of Io for 4 min (B); 100 nM of Io for 15 min (D); 10 nM of Io for 120 min (F); 1 μM of AA for 45 min (H); 100 nM of AA for 90 min (I); 10 nM of AA for 120 min (L); or the solvent for AA and Io (ethanol) for 0 (M), 60 (N) or 120 (O) min. Panels P–R: Cells expressing EGFP not fused to cPLA2 were challenged with 33 μM of AA for 0 (P), 60 (Q), or 120 (R) min. Arrows point to the irregular site referred to in the text. Results are representative of ≥3 studies.
Fig. 2.

Translocations to the Golgi. Panels A–S: 293 Cells expressing EYFP-cPLA2 and ECFP-golgi marker were scanned for: EYFP (A) and ECFP (B) after an 8 min challenge with 1 μM of Io (panel C superimposes A and B); EYFP (D, F, H, J, L) and ECFP (E, G, I, K, M) after challenge with 1 μM of AA for 0 (D, E), 30 (F, G), 60 (H, I), 90 (J, K), or 120 (L, M) min; or EYFP (N, Q), ECFP (O, R), and DIC (P, S) images after challenge with 10 μM of Io for 30 min (N–P) or 33 μM of AA for 60 min (Q–S). Arrows indicate the Golgi marker; circles enclose the spherical sites. Results are typical of ≥3 studies.
DIC microscopy identified the spherical cytoplasmic sites to which EGFP-cPLA2α translocated as discrete spherical organelles distinct from the Golgi (Fig. 2, panels N–S). After challenge with AA or Io, EYFP-cPLA2 layered onto the surface of these organelles (Fig. 3, panels A–C). This surface-layering effect is most clearly seen in Supplementary Figures 2S, panels C, E, I, and M and 3S, panel S. The organelles were highly refractive on phase-contrast microscopy (not shown) and, in cells pre-stained with BODIPY, emitted the fluorescent signature of this lipid body marker (Fig. 3, panels D–O). 3-Dimensional analysis of such responses revealed that EYFP-cPLA2 and BODIPY fluorescent emissions exactly overlapped and that virtually all EYFP-tagged spherical organelles had incorporated BODIPY (not shown). AA and the ionophores thus stimulate PLA2α fluorescent proteins to translocate to the surface of lipid bodies.
Fig. 3.
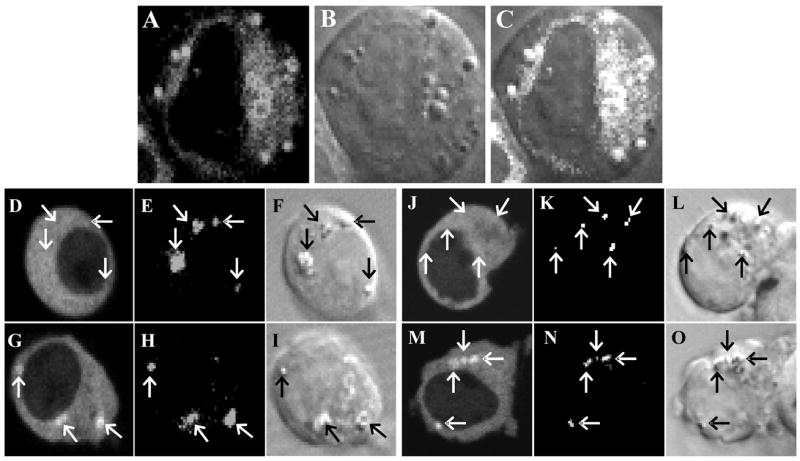
Translocations to lipid bodies. Panels A–C: 293 Cells expressing EYFP-cPLA2 were scanned for EYFP (A) and DIC (B) images after challenge with 10 μM of AA for 80 min (panel C superimposes A and B). Panels D–O: Cells expressing EYFP-cPLA2 and stained with BODIPY were scanned for EYFP (D, G, J, M), BODIPY (E, H, K, N), and DIC (F, I, L, O) images before (D–F, J–L) or after challenge with 1 μM of Io for 4 min (G–I) or 10 μM of AA for 60 min (M–O). Arrows point to BODIPY-stained bodies. Results are typical of ≥5 studies. Studies with 0.1 and 1μM of AA or Io gave results similar to these (not shown).
AA’s failure to target cPLA2α fluorescent proteins to the Golgi implies that it acts by a different mechanism than Ca2+ ionophores. Other studies supported this. Io and AA stimulated 293 cells to raise [Ca2+]i, as defined by the emissions of [Ca2+]i reporter X-rhod-1. All translocating concentrations (≥10 nM) of Io raised X-rhod emissions (Fig. 1S, panel A, interrupted line in Appendix A, Supplementary data). In contrast, at 10–30 μM, AA caused ≤1.2-fold rises and at <10 μM no rise in these emissions (Fig. 4, panels A and B, interrupted lines). Since AA’s translocating action occurs at ≥10 nM, these results tend to dissociate AA’s [Ca2+]i-raising from its translocating action. This was confirmed in cells loaded with Ca2+ chelator BAPTA and maintained in Ca2+-free medium. These cells held [Ca2+]i at pre-stimulatory levels after challenge with 33 μM of AA (Fig. 4, panels A and B, solid lines) or 10 μM of Io (Fig. 1S, panel A, solid line in Appendix A, Supplementary data) and, as expected, failed to translocate EGFP-cPLA2 in response to Io (Fig. 1S, panels B–J in Appendix A, Supplementary data). The [Ca2+]i- fixed cells nonetheless mounted full translocation responses to the perinuclear ER (Fig. 4, panels C and D) and lipid bodies (Fig. 4, panels E–J) in response to AA.
Fig. 4.

Effect of [Ca2+]i on translocation. Panels A–B: X-rhod-1-loaded 293 cells were [Ca2+]i-fixed (closed circles) or not (open circles), challenged, and assayed for X-rhod-1 fluorescent emissions. Data are the fractional rise in emissions relative to pre-challenge levels in cells stimulated with 33 μM of AA for 0–120 min (A) or the mean (±SEM, N≥3) of the highest fractional rise (B) in emissions in cells following challenge with 0.1–33 μM of AA. Panels C–D: Ca2+-fixed cells expressing EGFP-cPLA2 were scanned for EGFP after challenge with 10 μM of AA for 0 (C) or 60 min (D). Panels E–J: Ca2+-fixed cells expressing EYFP-cPLA2 and stained with BODIPY were scanned for DCI (E, H), BODIPY (F, I), and EYFP (G, J) images after challenge for 0 (E–G) or 55 min (H–J) with 10 μM of AA. Arrows indicate lipid bodies. Results for panels C–J are typical of ≥3 studies.
Studies on cPLA2α’s C2 domain fused to EYFP (C2-EYFP) indicated that AA did have at least one key mechanistic similarity with the ionophores: A23187 (Fig. 5, panels A–D) and Io (not shown) translocated C2-EYFP to the perinuclear ER, lipid bodies, and Golgi; AA translocated it to perinuclear ER and lipid bodies (Fig. 5, panels E–H); and AA (Fig. 5, panels I–L) but not A23187 (Fig. 1S, panels K-S in Appendix A, Supplementary data) or Io (not shown) was active in Ca2+-fixed cells. The C2 domain thus houses the triggering and membrane-recognizing motifs responsible for initiating and targeting the translocation of cPLA2α fluorescent proteins whether elicited by [Ca2+]i,, as found previously [2,14,15,18–22], or by AA.
Fig. 5.

C2-EYFP translocation responses. Panels A–H: 293 Cells expressing C2-EYFP and stained with BODIPY were scanned for EYFP (A, C, E, G, I, K) and BODIPY (B, D, F, H, J, L) after challenge with 10 μM of A23187 for 0 (A, B) or 5 (C, D) min or with 10 μM of AA for 0 (E, F) or 90 (G, H) min. Panels I–L: Ca2+-fixed cells were challenged with 10 μM of AA for 0 (I, J) or 95 (K, L) min. Arrows indicate lipid bodies. Results are typical of ≥3 studies; ethanol did not alter C2-EYFP’s location (not shown).
Cells pre-treated with nordihydroguairetic acid at 10 μM, a concentration inhibiting the cyclooxygenase, lipoxygenase, and cytochrome P450 enzymes which metabolize AA into eicosanoids, translocated EGFP-cPLA2 normally in response to AA (Fig. 6, panels A–C). Cells treated with 10 μM of indomethacin for 30 min likewise did not translocate cPLA2-EYFP when challenged with ethanol but mounted normal translocations responses to 1 and 10 μM of AA (not shown), In addition, 11Z, 14Z-eicosadienoic acid, α-linolenic acid, and triacsin C (a straight-chain undecene with cis double bonds at C2-3 and C5-6 and hydroxytriazine double-bonded to C1) also translocated EGFP-cPLA2 to the perinuclear ER and spherical sites but not Golgi (Fig. 6, panels D–I, and Fig 2S, panels A–F in Appendix A Supplementary data). These unsaturated compounds were active at ≥0.1–1 μM while the saturated analog of AA, eicosanoic acid, was inactive at 100 μM (Fig. 6, panels J–L). Neither 11Z, 14Z-eicosadienoic acid nor triacsin C are known to be metabolized to active products by mammalian AA oxygenases. Finally, cells treated with pertussis toxin demonstrated full translocation responses to AA (Fig. 6, panels M–O). Pertussis toxin would block these responses if AA acted after conversion to leukotriene B4, 5-hydroxyicosatetraenoate, or other metabolite acting through receptors linked to pertussis toxin-sensitive G proteins [23]. Taken together, these results render it unlikely that AA’s translocating activity requires it to be oxygenation by the cited enzymes.
Fig. 6.
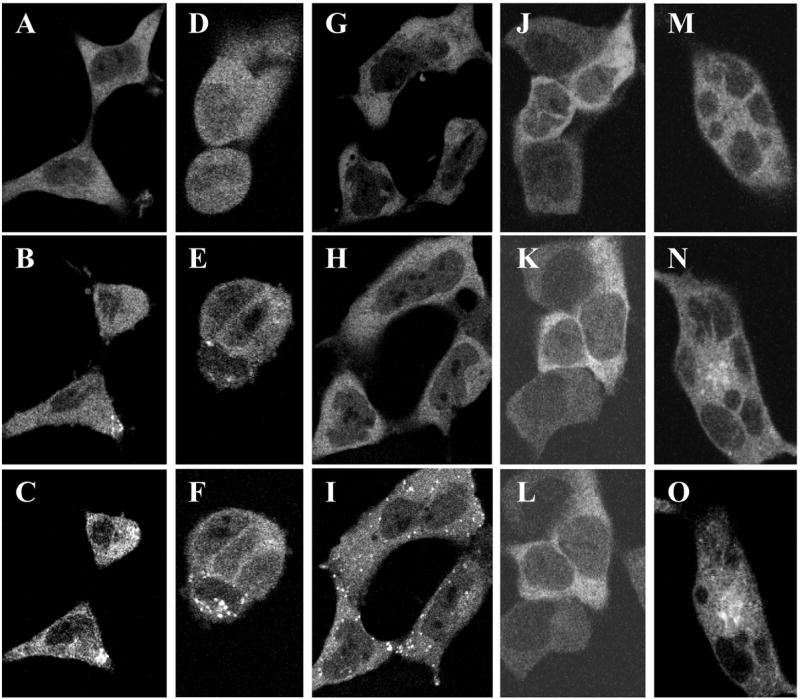
Translocating effects of selected agents. 293 Cells expressing EGFP-cPLA2 were scanned for EGFP. Panels A–C: Cells incubated with 10 μM of nordihydroguairetic acid for 30 min were challenged with 10 μM of AA for 0 (A), 30 (B), or 45 (C) min. Panels D–L: Cells were challenged with 10 μM of α-linolenate for 0 (D), 60 (E), or 90 (F) min; 33 μM of eicosadienoate for 0 (G), 90 (H), or 115 min (I); or 100 μM of eicosanoate for 0 (J), 90 (K), or 120 (L) min. Panels M–O: Cells were incubated with 1 μg of pertussis toxin for 18 hr and challenged with 1 μM of AA for 0 (M), 45 (N), or 90 (O) min. Nordihydroguairetic acid, pertussis toxin, and the vehicles for these drugs did not alter EGFP-cPLA2’s location (not shown). Results are representative of ≥3 studies.
Our final studies probed the generality of lipid body targeting by cPLA2α. Two agonists that act via different surface membrane receptors, S1P and GTPγS, stimulated 293 cells to translocate EYFP-cPLA2 to among other sites lipid bodies (Fig. 2S, panels G–N in Appendix A, Supplementary data). In addition, Io and AA stimulated MBA-MB-231 breast cancer cells to translocate EYFP-cPLA2 to lipid bodies although modest levels of EYFP-cPLA2 were on lipid bodies prior to stimulation in some of these cells (Fig 3S, panels A–P in Appendix A, Supplementary data). The latter type of localization was far more evident in resting PC3 prostate cancer cells, virtually all of which presented with EYFP-cPLA2 almost exclusively on their lipid bodies (Fig 3S, panels Q–T in Appendix A, Supplementary data). In any event, AA’s translocating action is not limited to 293 cells and the translocation of cPLA2α fluorescent proteins to lipid bodies occurs in response to diverse stimuli.
4. Discussion
Ca2+ co-ordinates with acidic residues on the membrane-binding face of cPLA2α’s C2 domain; this lowers the face’s electronegativity thereby enabling it to bind electrically neutral membranes, insert hydrophobic residues into these membranes, and form a complex persisting as long as Ca2+ remains available. Since Ca2+-coordinated cPLA2α retains its repulsion for negatively charged membranes [2,4,14,18–22], rises in [Ca2+]i cause cPLA2α to translocate to the perinuclear ER and Golgi, organelles with cytoplasmic leaflets rich in electrically neutral PL, but not to the plasma membrane whose cytoplasmic leaflet is rich in electronegative PLs [4,15,20,22]. The single-leafleted lipid body membrane may originate from the ER [24,25], has a PL content level similar to the ER [26], and may be constitutively associated with numerous lipid mediator-forming enzymes including cPLA2α in some cell types [27–29]. These considerations suggest that the lipid body may be a common target for translocating cPLA2α. Indeed, we found that AA, Io, A23187, GTPγS, and/or S1P did in fact cause 293 and MDA-MB-231 cells to move cPLA2α fluorescent proteins to cytoplasmic organelles identified as lipid bodies in phase-contrast and BODIPY studies (Figs. 1–3, Fig. 3S, Appendix A, Supplementary data). We also found that PC3 and to a lesser extent MBA-MD-231 cells data) localize EYFP-cPLA2 on their lipid bodies in the absence of stimulation (Fig. 2S, Appendix A, Supplementary data). Resting A549 lung [30] and Caco-2 colon [31] cancer cells likewise localize cPLA2α to internal membranes. Since PC3, MDA-MB-231, A549, and Caco-2 cells overproduce eicosanoids or platelet-activating factor and use these products to promote their own growth [27,32–35], constitutive localization of cPLA2α to membranes may underlie the over-production of PL-derived mediators and thereby certain aspects of the malignant phenotype.
AA and the ionophores also caused C2-EYFP to translocate. Like their effects on cPLA2α fluorescent proteins, AA induced slowly evolving responses targeted to the perinuclear ER and lipid bodies but not Golgi (Fig. 5); ionophores induced more rapid responses targeting all three sites (Fig. 5), and neither AA (Fig. 1, panels P–R) nor Io [17] induced EGFP not fused to cPLA2α to translocate. Thus, AA and the ionophores act via the C2 domain to trigger and direct cPLA2α’s movements. In spite of this similarity, however, AA’s ability to translocate cPLA2α and C2 domain fluorescent proteins, unlike those of the ionophores, did not require Ca2+ (Figs. 4, 5, and 1S in Appendix A, Supplementary data). We evaluated three other mechanisms that might explain its action. AA did not operate after being metabolized by AA oxygenases to receptor-interacting products, as found for the C2 domain-dependent translocations of certain protein kinases C [27,36,37]: nordihydroguairetic acid and indomethacin, which block these enzymes, did not inhibit AA; eicosadienoic acid and triacsin C, while not known to be metabolized by the oxygenases, had translocating activity; and pertussis toxin failed to alter AA’s translocating action under conditions where it blocks responses to various AA metabolites (Fig. 6). AA also did not act through GPR40 or GPR120 long-chain fatty acid receptors: these receptors have similar affinities for α-linolenate and AA [27,38–41] whereas AA is 100-fold more potent than α-linolenic acid in translocating EYFP-cPLA2 (Fig 6). Moreover, 293 cells do not express these receptors [27,38,39,42–44]. Finally, AA did not act by being acylated into membrane PLs to form a cPLA2α docking site: triacsin C lacks the carboxy residue needed for this acylation yet has potent translocating activity (Fig. 7). These compounds thus appear to raise the affinity of cPLA2α’s C2 domain for electrically neutral membranes without requiring a metabolic processing step.
The results of our studies lead us to propose that cPLA2α translocates to lipid bodies in cells responding to a wide range of stimuli and that these translocations contribute to the stimulus-induced synthesis of PL-derived mediators. This agrees with prior studies implicating lipid bodies in the production and priming for the production of eicosanoids in stimulated and/or primed leukocytes [45–47]. We further propose that AA is a Ca2+-independent signal for translocating cPLA2α, at least when this occurs in the absence of [Ca2+]i rises. The AA performing this role may arise from Ca2+-independent cPLA2’s, extracellular PLA2’s, other cells by trans-cellular passage, or cPLA2α itself. Relevant to the last point, suboptimal cell stimulation causes fleeting cPLA2α translocations that are not accompanied by AA release or lipid mediator synthesis whereas optimal stimulation causes irreversible cPLA2α translocation, AA release, and eicosanoids synthesis [9,12]. While proposed as resulting from the more prolonged [Ca2+]i rise that occurs under the latter conditions [9], these effects ultimately may result from AA released by translocated cPLA2α. In this scenario, an initial product of cPLA2α’s action sustains the translocation response to convert a non-productive event into one productive of lipid mediators.
Supplementary Material
Supplementary data for this article can be found in its online version.
Acknowledgments
This work was supported by NIH grants PO1CA106742 (J.O., L.D.), BM61754, HL61378, and HL34303.
The abbreviations used are
- cPLA2
cytosolic phospholipase A2
- PL
phospholipids
- AA
arachidonic acid
- [Ca2+]i
cytosolic Ca2+
- EGFP
EYFP, or ECFP enhanced green, yellow, or cyanine fluorescent protein
- Io
ionomycin
- S1P
D-erythro-sphingosine-2-phosphate
- DIC
differential interference contrast
- 293
HEK 293 cells
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2–3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
Reference List
- 1.Kita Y, Ohto T, Uozumi N, Shimizu T. Biochemical properties and pathophysiological roles of cytosolic phospholipase A2s. Biochim Biophys Acta. 2006;1761:1317. doi: 10.1016/j.bbalip.2006.08.001. [DOI] [PubMed] [Google Scholar]
- 2.Cho W. Membrane targeting by C1 and C2 domains. J Biol Chem. 2001;276:32407. doi: 10.1074/jbc.R100007200. [DOI] [PubMed] [Google Scholar]
- 3.Evans JH, Spencer DM, Zweifach A, Leslie CC. Intracellular calcium signals regulating cytosolic phospholipase A2 translocation to internal membranes. J Biol Chem. 2001;276:30150. doi: 10.1074/jbc.M100943200. [DOI] [PubMed] [Google Scholar]
- 4.Evans JH, Leslie CC. The cytosolic phospholipase A2 catalytic domain modulates association and residence time at Golgi membranes. J Biol Chem. 2004;279:6005. doi: 10.1074/jbc.M311246200. [DOI] [PubMed] [Google Scholar]
- 5.Hirabayashi T, Murayama T, Shimizu T. Regulatory mechanism and physiological role of cytosolic phospholipase A2. Biol Pharm Bull. 2004;27:1168. doi: 10.1248/bpb.27.1168. [DOI] [PubMed] [Google Scholar]
- 6.Schievella AR, Regier MK, Smith WL, Lin LL. Calcium-mediated translocation of cytosolic phospholipase A2 to the nuclear envelope and endoplasmic reticulum. J Biol Chem. 1995;270:30749. doi: 10.1074/jbc.270.51.30749. [DOI] [PubMed] [Google Scholar]
- 7.Balsinde J, Balboa MA, Li WH, Llopis J, Dennis EA. Cellular regulation of cytosolic group IV phospholipase A2 by phosphatidylinositol bisphosphate levels. J Immunol. 2000;164:5398. doi: 10.4049/jimmunol.164.10.5398. [DOI] [PubMed] [Google Scholar]
- 8.Bayon Y, Hernandez M, Alonso A, Nunez L, Garcia-Sancho J, Leslie C, Sanchez CM, Nieto ML. Cytosolic phospholipase A2 is coupled to muscarinic receptors in the human astrocytoma cell line 1321N1: characterization of the transducing mechanism. Biochem J. 1997;323(Pt 1):281. doi: 10.1042/bj3230281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Grewal S, Smith J, Ponnambalam S, Walker J. Stimulation-dependent recruitment of cytosolic phospholipase A2-alpha to EA.hy.926 endothelial cell membranes leads to calcium-independent association. Eur J Biochem. 2004;271:69. doi: 10.1046/j.1432-1033.2003.03903.x. [DOI] [PubMed] [Google Scholar]
- 10.Poeckel D, Tausch L, Kather N, Jauch J, Werz O. Boswellic acids stimulate arachidonic acid release and 12-lipoxygenase activity in human platelets independent of Ca2+ and differentially interact with platelet-type 12-lipoxygenase. Mol Pharmacol. 2006;70:1071. doi: 10.1124/mol.106.024836. [DOI] [PubMed] [Google Scholar]
- 11.Qiu ZH, Gijon MA, de Carvalho MS, Spencer DM, Leslie CC. The role of calcium and phosphorylation of cytosolic phospholipase A2 in regulating arachidonic acid release in macrophages. J Biol Chem. 1998;273:8203. doi: 10.1074/jbc.273.14.8203. [DOI] [PubMed] [Google Scholar]
- 12.Hirabayashi T, Kume K, Hirose K, Yokomizo T, Iino M, Itoh H, Shimizu T. Critical duration of intracellular Ca2+ response required for continuous translocation and activation of cytosolic phospholipase A2. J Biol Chem. 1999;274:5163. doi: 10.1074/jbc.274.8.5163. [DOI] [PubMed] [Google Scholar]
- 13.Chang WC, Parekh AB. Close functional coupling between Ca2+ release-activated Ca2+ channels, arachidonic acid release, and leukotriene C4 secretion. J Biol Chem. 2004;279:29994. doi: 10.1074/jbc.M403969200. [DOI] [PubMed] [Google Scholar]
- 14.Six DA, Dennis EA. Essential Ca(2+)-independent role of the group IVA cytosolic phospholipase A(2) C2 domain for interfacial activity. J Biol Chem. 2003;278:23842. doi: 10.1074/jbc.M301386200. [DOI] [PubMed] [Google Scholar]
- 15.Evans JH, Gerber SH, Murray D, Leslie CC. The calcium binding loops of the cytosolic phospholipase A2 C2 domain specify targeting to Golgi and ER in live cells. Mol Biol Cell. 2004;15:371. doi: 10.1091/mbc.E03-05-0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang WC, Nelson C, Parekh AB. Ca2+ influx through CRAC channels activates cytosolic phospholipase A2, leukotriene C4 secretion, and expression of c-fos through ERK-dependent and -independent pathways in mast cells. FASEB J. 2006;20:2381. doi: 10.1096/fj.06-6016fje. [DOI] [PubMed] [Google Scholar]
- 17.O’Flaherty JT, Chadwell BA, Kearns MW, Sergeant S, Daniel LW. Protein kinases C translocation responses to low concentrations of arachidonic acid. J Biol Chem. 2001;276:24743. doi: 10.1074/jbc.M101093200. [DOI] [PubMed] [Google Scholar]
- 18.Gijon MA, Spencer DM, Kaiser AL, Leslie CC. Role of phosphorylation sites and the C2 domain in regulation of cytosolic phospholipase A2. J Cell Biol. 1999;145:1219. doi: 10.1083/jcb.145.6.1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nalefski EA, Wisner MA, Chen JZ, Sprang SR, Fukuda M, Mikoshiba K, Falke JJ. C2 domains from different Ca2+ signaling pathways display functional and mechanistic diversity. Biochemistry. 2001;40:3089. doi: 10.1021/bi001968a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Perisic O, Paterson HF, Mosedale G, Lara-Gonzalez S, Williams RL. Mapping the phospholipid-binding surface and translocation determinants of the C2 domain from cytosolic phospholipase A2. J Biol Chem. 1999;274:14979. doi: 10.1074/jbc.274.21.14979. [DOI] [PubMed] [Google Scholar]
- 21.Stahelin RV, Cho W. Roles of calcium ions in the membrane binding of C2 domains. Biochem J. 2001;359:679. doi: 10.1042/0264-6021:3590679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stahelin RV, Rafter JD, Das S, Cho W. The molecular basis of differential subcellular localization of C2 domains of protein kinase C-alpha and group IVa cytosolic phospholipase A2. J Biol Chem. 2003;278:12452. doi: 10.1074/jbc.M212864200. [DOI] [PubMed] [Google Scholar]
- 23.O’Flaherty JT, Taylor JS, Kuroki M. The coupling of 5-oxo-eicosanoid receptors to heterotrimeric G proteins. J Immunol. 2000;164:3345. doi: 10.4049/jimmunol.164.6.3345. [DOI] [PubMed] [Google Scholar]
- 24.Wan HC, Melo RC, Jin Z, Dvorak AM, Weller PF. Roles and origins of leukocyte lipid bodies: proteomic and ultrastructural studies. FASEB J. 2007;21:167. doi: 10.1096/fj.06-6711com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murphy DJ, Vance J. Mechanisms of lipid-body formation. Trends Biochem Sci. 1999;24:109. doi: 10.1016/s0968-0004(98)01349-8. [DOI] [PubMed] [Google Scholar]
- 26.Tauchi-Sato K, Ozeki S, Houjou T, Taguchi R, Fujimoto T. The surface of lipid droplets is a phospholipid monolayer with a unique Fatty Acid composition. J Biol Chem. 2002;277:44507. doi: 10.1074/jbc.M207712200. [DOI] [PubMed] [Google Scholar]
- 27.Yu W, Bozza PT, Tzizik DM, Gray JP, Cassara J, Dvorak AM, Weller PF. Co-compartmentalization of MAP kinases and cytosolic phospholipase A2 at cytoplasmic arachidonate-rich lipid bodies. Am J Pathol. 1998;152:759. [PMC free article] [PubMed] [Google Scholar]
- 28.Bozza PT, Melo RC, Bandeira-Melo C. Leukocyte lipid bodies regulation and function: contribution to allergy and host defense. Pharmacol Ther. 2007;113:30. doi: 10.1016/j.pharmthera.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 29.Meadows JW, Pitzer B, Brockman DE, Myatt L. Expression and localization of adipophilin and perilipin in human fetal membranes: association with lipid bodies and enzymes involved in prostaglandin synthesis. J Clin Endocrinol Metab. 2005;90:2344. doi: 10.1210/jc.2004-1199. [DOI] [PubMed] [Google Scholar]
- 30.Grewal S, Ponnambalam S, Walker JH. Association of cPLA2-alpha and COX-1 with the Golgi apparatus of A549 human lung epithelial cells. J Cell Sci. 2003;116:2303. doi: 10.1242/jcs.00446. [DOI] [PubMed] [Google Scholar]
- 31.Parhamifar L, Jeppsson B, Sjolander A. Activation of cPLA2 is required for leukotriene D4-induced proliferation in colon cancer cells. Carcinogenesis. 2005;26:1988. doi: 10.1093/carcin/bgi159. [DOI] [PubMed] [Google Scholar]
- 32.Cellai C, Laurenzana A, Vannucchi AM, Caporale R, Paglierani M, Di LS, Pancrazzi A, Paoletti F. Growth inhibition and differentiation of human breast cancer cells by the PAFR antagonist WEB-2086. Br J Cancer. 2006;94:1637. doi: 10.1038/sj.bjc.6603156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghosh J, Myers CE. Inhibition of arachidonate 5-lipoxygenase triggers massive apoptosis in human prostate cancer cells. Proc Natl Acad Sci U S A. 1998;95:13182. doi: 10.1073/pnas.95.22.13182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ihara A, Wada K, Yoneda M, Fujisawa N, Takahashi H, Nakajima A. Blockade of leukotriene B4 signaling pathway induces apoptosis and suppresses cell proliferation in colon cancer. J Pharmacol Sci. 2007;103:24. doi: 10.1254/jphs.fp0060651. [DOI] [PubMed] [Google Scholar]
- 35.Kudryavtsev IA, Gudkova MV, Pavlova OM, Oreshkin AE, Myasishcheva NV. Lipoxygenase pathway of arachidonic acid metabolism in growth control of tumor cells of different type. Biochemistry (Mosc ) 2005;70:1396. doi: 10.1007/s10541-005-0275-0. [DOI] [PubMed] [Google Scholar]
- 36.Alpert SE, Walenga RW, Mandal A, Bourbon N, Kester M. 15-HETE-substituted diglycerides selectively regulate PKC isotypes in human tracheal epithelial cells. Am J Physiol. 1999;277:L457–L464. doi: 10.1152/ajplung.1999.277.3.L457. [DOI] [PubMed] [Google Scholar]
- 37.Sharma GD, Ottino P, Bazan NG, Bazan HE. Epidermal and hepatocyte growth factors, but not keratinocyte growth factor, modulate protein kinase Calpha translocation to the plasma membrane through 15(S)-hydroxyeicosatetraenoic acid synthesis. J Biol Chem. 2005;280:7917. doi: 10.1074/jbc.M408852200. [DOI] [PubMed] [Google Scholar]
- 38.Briscoe CP, Tadayyon M, Andrews JL, Benson WG, Chambers JK, Eilert MM, Ellis C, Elshourbagy NA, Goetz AS, Minnick DT, Murdock PR, Sauls HR, Jr, Shabon U, Spinage LD, Strum JC, Szekeres PG, Tan KB, Way JM, Ignar DM, Wilson S, Muir AI. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J Biol Chem. 2003;278:11303. doi: 10.1074/jbc.M211495200. [DOI] [PubMed] [Google Scholar]
- 39.Hirasawa A, Tsumaya K, Awaji T, Katsuma S, Adachi T, Yamada M, Sugimoto Y, Miyazaki S, Tsujimoto G. Free fatty acids regulate gut incretin glucagon-like peptide-1 secretion through GPR120. Nat Med. 2005;11:90. doi: 10.1038/nm1168. [DOI] [PubMed] [Google Scholar]
- 40.Itoh Y, Kawamata Y, Harada M, Kobayashi M, Fujii R, Fukusumi S, Ogi K, Hosoya M, Tanaka Y, Uejima H, Tanaka H, Maruyama M, Satoh R, Okubo S, Kizawa H, Komatsu H, Matsumura F, Noguchi Y, Shinohara T, Hinuma S, Fujisawa Y, Fujino M. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature. 2003;422:173. doi: 10.1038/nature01478. [DOI] [PubMed] [Google Scholar]
- 41.Katsuma S, Hatae N, Yano T, Ruike Y, Kimura M, Hirasawa A, Tsujimoto G. Free fatty acids inhibit serum deprivation-induced apoptosis through GPR120 in a murine enteroendocrine cell line STC-1. J Biol Chem. 2005;280:19507. doi: 10.1074/jbc.M412385200. [DOI] [PubMed] [Google Scholar]
- 42.Kotarsky K, Nilsson NE, Flodgren E, Owman C, Olde B. A human cell surface receptor activated by free fatty acids and thiazolidinedione drugs. Biochem Biophys Res Commun. 2003;301:406. doi: 10.1016/s0006-291x(02)03064-4. [DOI] [PubMed] [Google Scholar]
- 43.Pacheco P, Bozza FA, Gomes RN, Bozza M, Weller PF, Castro-Faria-Neto HC, Bozza PT. Lipopolysaccharide-induced leukocyte lipid body formation in vivo: innate immunity elicited intracellular Loci involved in eicosanoid metabolism. J Immunol. 2002;169:6498. doi: 10.4049/jimmunol.169.11.6498. [DOI] [PubMed] [Google Scholar]
- 44.Schnell S, Schaefer M, Schofl C. Free fatty acids increase cytosolic free calcium and stimulate insulin secretion from beta-cells through activation of GPR40. Mol Cell Endocrinol. 2007;263:173. doi: 10.1016/j.mce.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 45.Weller PF, Bozza PT, Yu W, Dvorak AM. Cytoplasmic lipid bodies in eosinophils: central roles in eicosanoid generation. Int Arch Allergy Immunol. 1999;118:450. doi: 10.1159/000024161. [DOI] [PubMed] [Google Scholar]
- 46.Bozza PT, Yu W, Penrose JF, Morgan ES, Dvorak AM, Weller PF. Eosinophil lipid bodies: specific, inducible intracellular sites for enhanced eicosanoid formation. J Exp Med. 1997;186:909. doi: 10.1084/jem.186.6.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bozza PT, Payne JL, Morham SG, Langenbach R, Smithies O, Weller PF. Leukocyte lipid body formation and eicosanoid generation: cyclooxygenase-independent inhibition by aspirin. Proc Natl Acad Sci U S A. 1996;93:11091. doi: 10.1073/pnas.93.20.11091. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data for this article can be found in its online version.