

Summary
The molecular mechanisms linking the stress of unfolded proteins in the endoplasmic reticulum (ER stress) to glucose intolerance in obese animals are poorly understood. In this study enforced expression of a translation initiation 2α(eIF2α)-specific phosphatase, GADD34, was used to selectively compromise signaling in the eIF2(αP)-dependent arm of the ER unfolded protein response in liver of transgenic mice. The transgene resulted in lower liver glycogen levels and susceptibility to fasting hypoglycemia in lean mice and glucose tolerance and diminished hepato-steatosis in animals fed a high fat diet. Attenuated eIF2(αP) correlated with lower expression of the adipogenic nuclear receptor PPARγ and its upstream regulators, the transcription factors C/EBPα and C/EBPβ, in transgenic mouse liver, whereas eIF2α phosphorylation promoted C/EBP translation in cultured cells and primary hepatocytes. These observations suggest that eIF2(αP)-mediated translation of key hepatic transcriptional regulators of intermediary metabolism contributes to the detrimental consequences of nutrient excess.
Keywords: Protein folding, translation, transgenic mice, diabetes mellitus
Introduction
Eukaryotic cells respond to fluctuations in the load of unfolded proteins in the endoplasmic reticulum by an unfolded protein response (UPR). The oldest arm of the UPR, which is conserved in all eukaryotes, is mediated by IRE1, an ER stress regulated kinase-endoribonuclease that signals through a transcription factor, XBP-1 (Hac1p, in yeast) to activate UPR target genes. In animals XBP-1 is joined by ATF6, a transcription factor that senses ER stress directly, to activate genes that enhance cells’ ability to cope with the load of unfolded proteins facing their ER (reviewed in: Schroder and Kaufman, 2005; Bernales et al., 2006). These transcriptional events are complemented by a third arm of the UPR mediated by PERK, an ER-localized stress-activated kinase whose only known substrate is the alpha subunit of translation initiation factor 2 (eIF2α). Phosphorylation of eIF2α on serine 51 inhibits the guanine nucleotide exchange factor for eIF2 and reduces rates of translation initiation. The consequent repression of protein synthesis diminishes the load of unfolded proteins entering the ER and conserves ATP and amino acids in ER stressed cells. eIF2(αP) also activates gene expression, which is accounted for, in part, by the translational up-regulation of the transcription factor ATF4 (reviewed in: Ron and Harding, 2007).
Earlier studies emphasized the UPR’s role in maintaining homeostasis of the protein-folding environment in the ER lumen and its physiological significance was sought in the context of cellular adaptation to the stress posed by unfolded and misfolded ER proteins. The phenotype of mutations in components of the UPR is certainly consistent with this notion – knockout of IRE1, XBP-1, ATF6 and PERK all reduce the ability of cells to cope with ER stress (Delepine et al., 2000; Harding et al., 2001; Shen et al., 2001; Zhang et al., 2002; Lee et al., 2005; Wu et al., 2007; Yamamoto et al., 2007). However, accrued evidence now suggests that ER stress and the response to it modulate mammalian physiology in ways that cannot be explained simply by the aforementioned cell autonomous processes.
In addition to imparting hypersensitivity to ER stress, a homozygous Eif2aS51A mutation (that abolishes regulatory phosphorylation of eIF2α) blocks hepatic glucose production in neonatal mice (Scheuner et al., 2001). By contrast, heterozygosity for the same Eif2aS51A mutation disposes adult mice to obesity, insulin resistance and glucose intolerance (Scheuner et al., 2005). Obesity promotes ER stress, presumably by increasing the load of unfolded proteins in the ER, which is detected as enhanced UPR signaling in liver and fat (Ozcan et al., 2004; Nakatani et al., 2005). Importantly, compromised signaling in the UPR by a partial loss-of-function mutation in XBP-1 (which increases further the level of ER stress) or a mutation in the ER chaperone ORP150, increased insulin resistance in obese mice (Ozcan et al., 2004; Nakatani et al., 2005) whereas chemical chaperones or ORP150 over-expression that reduced the level of ER stress substantially reversed the insulin resistance and glucose intolerance of obese mice (Ozawa et al., 2005; Ozcan et al., 2006). These studies imply that ER stress, or aspects of the response to it, modulate intermediary metabolism in a manner that cannot be simply attributed to variation in survival of secretory cells.
Several kinases phosphorylate eIF2α to activate a downstream gene expression program that we refer to as the integrated stress response (ISR) (Harding et al., 2003). In yeast, a GCN2-mediated eIF2(αP)-dependent transcriptional program responds to diverse metabolic perturbations (Hinnebusch and Natarajan, 2002) and the homologous mammalian eIF2α kinase is also implicated in metabolic regulation (Hao et al., 2005; Maurin et al., 2005; Guo and Cavener, 2007). These observations suggest that links between metabolic regulation and eIF2α phosphorylation are conserved and might contribute to the physiological response to ER stress in mammals. Here we have focused on eIF2(αP) signaling in the liver, a key organ for intermediary metabolism, and present evidence that the hepatic ISR contributes to the metabolic syndrome of obesity and insulin resistance by translational upregulation of transcription factors involved in carbohydrate and lipid metabolism.
RESULTS
The major determinant of ISR activity in liver is the ER stress-activated kinase PERK, whose transient activation is difficult to detect (Harding et al., 2001). To determine if the ISR is modulated by normal fluctuations in nutrient intake, we first monitored two perdurable markers, BiP and XBP-1 mRNA, as surrogates for UPR (and PERK activity). Feeding coincided with a clear peak of these marker mRNAs (Figure S1A), suggesting that physiological ER stress is induced by feeding, as predicted by previous studies (Dhahbi et al., 1997). To gage the transient phosphorylation of eIF2α we fasted animals for 18 hours and re-fed them normal (low fat) and high fat chow. Levels of eIF2(αP) were barely detectable in liver after a 18 hour fast, but increased 4 hours after feeding ordinary chow and were higher yet in animals provided high fat chow (Figure S1B). These observations confirmed the previously noted (Ozcan et al., 2004) correlation between physiological nutritional fluctuations and the eIF2(αP)-dependent ISR in liver.
Selective attenuation of the ISR in liver of transgenic mice
To study the potential significance of physiological levels of eIF2(αP) in mouse liver, we sought to selectively interfere with this phosphorylation event. Gadd34 (PPP1R15a) encodes a substrate-specific regulatory subunit of a phosphatase that selectively de-phosphorylates eIF2α phospho-serine 51 (Novoa et al., 2001). Gadd34 activity is tightly regulated at the transcriptional level and the gene is normally turned on by the ISR as part of a negative feed-back loop that terminates signaling (Novoa et al., 2003), however enforced expression of an active C-terminal fragment of GADD34 is sufficient to dephosphorylate eIF2(αP) and inhibit the ISR (Novoa et al., 2001). We exploited this feature by expressing a GADD34 C-terminal fragment from a liver-specific albumin promoter in transgenic mice. The GADD34 C-terminal active fragment (GC) was detected by immunoblot in liver lysates of transgenic Alb::GC mice (Figure 1A). Expression of single copy of the transgene attenuated feeding-induced eIF2α phosphorylation and two copies of the transgene blocked phosphorylation even during severe ER stress in mice injected with tunicamycin (Figure 1A). We conclude that the Alb::GC transgene interferes with eIF2α phosphorylation in the liver.
Figure 1. Enforced de-phosphorylation of eIF2α blocks the integrated stress response in the liver of transgenic mice.
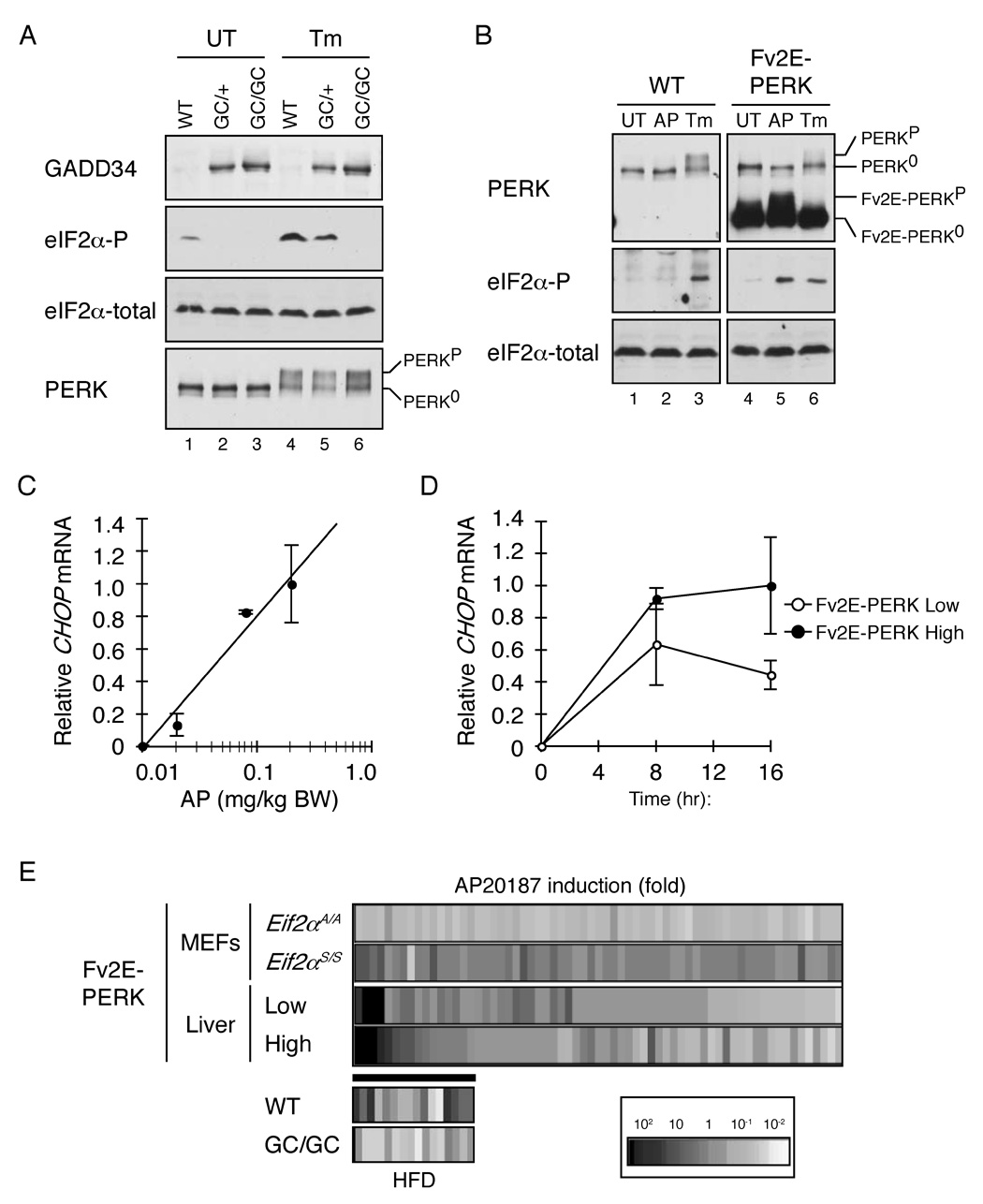
A. Immunoblot of immunopurified GADD34 C-terminal fragment, phosphorylated eIF2α, total eIF2α and immunopurified PERK from liver lysates of untreated (UT) and tunicamycin injected (Tm) non-transgenic (WT), heterozygous (GC/+) and homozygous Alb::GC (GC/GC) transgenic mice. The migration of inactive (PERK0) and active (PERKP) is indicated.
B. Immunoblot of immunopurified PERK, phosphorylated eIF2α and total eIF2α from liver lysates of untreated (UT), AP20187 injected (AP) and tunicamycin injected (Tm) non-transgenic (WT) and Ttr::Fv2E-PERK low-expressing transgenic (Fv2E-PERK) mice. The inactive and active endogenous and transgenic Fv2E-PERK proteins are labeled.
C. Relative levels of Chop mRNA in liver of Ttr::Fv2E-PERK low-expressing transgenic mice 8 hours after injected with AP20187. Shown are mean ± SEM of a representative experiment (n=3). Linear regression analysis shows correlation factor r2=0.969).
D. Relative levels of Chop mRNA in liver after injection with 0.2 mg/Kg AP20187. Shown are mean ± SEM (n=4) of a representative experiment performed in low (p=0.047, one-way ANOVA) and high level expressing lines of Ttr::Fv2E-PERK transgenic mice (p=0.006, one-way ANOVA).
E. Expression profiling of ISR target genes revealed by AP20187 treatment of Fv2E-PERK transgenic fibroblasts with wild-type (Eif2aS/S) and mutant (Eif2aA/A) genotypes (“MEFs”) (from Lu et al., 2004b). The induction profile of the same genes in liver of AP20187 injected Ttr::Fv2E-PERK transgenic mice expressing low (n=2) and high (n=4) levels of the transgene is shown below that (data in table S1). The inset at the bottom depicts the expression level of a subset of these validated hepatic ISR target genes (that are induced more that two fold by AP20187 expression in both Ttr::Fv2E-PERK transgenic lines) in high fat diet fed non-transgenic (WT, n=2) and Alb::GC transgenic (n=2) animals (data in table S2).
To estimate the consequences of the Alb::GC transgene on the eIF2(αP)-mediated ISR, and to generate hypothesis for possible mechanisms, we established a reference data-base for the activity of this pathway in the liver. PERK’s kinase activity can be uncoupled from ER stress by fusion of the cytosolic PERK kinase domain to an artificial dimerization domain (Fv2E) which subordinates eIF2α phosphorylation to a soluble, otherwise inert ligand, AP20187 (Lu et al., 2004b) (figure S2A). In the absence of ligand the Fv2E-PERK chimera, expressed in the liver of transgenic mice (Ttr::Fv2E-Perk) is inert. However, following intraperitoneal injection of AP20187 the chimeric protein is activated (reflected by a shift in its mobility on SDS-PAGE) and phosphorylates its substrate (Figure 1B). The Chop marker gene was used to confirm the ligand and gene dose-dependent activation of the ISR in the liver of Ttr::Fv2E-Perk mice (figure 1C, 1D and S2B) and a profile of genes thereby induced was assembled using oligonucleotide hybridization microarrays of liver mRNA. About half of the genes previously identified as ISR targets in fibroblasts (Harding et al., 2003; Lu et al., 2004b) were also induced in the liver of the transgenic mice following injection of AP20187 ligand (Figure 1E and supplemental table 1).
Next we sought to determine if the Alb::GC transgene, which blocked eIF2α phosphorylation, interfered with expression of this hypothesized set of ISR genes in liver. As feeding of a high fat diet promotes eIF2(αP) in the liver, we compared the profile of genes expressed in the liver of wildtype and Alb::GC mice fed a high or low fat chow. As expected, the genes constituting the hepatic ISR were expressed at higher levels in the liver of high fat diet fed wildtype mice. These dietary-induced differences in expression were attenuated by the Alb::GC transgene (Figure 1E bottom and same data in table S2), suggesting that the latter blocks signaling in a dietary-induced ISR and that such mice might constitute a useful tool for studying the role of this pathway in metabolism.
Metabolic profile of the ISR-defective Alb::GC mice
Apart from slight gene-dosage dependent reduced body mass (Figure 2A) adult Alb::GC mice are superficially indistinguishable from wildtype littermates when fed a normal diet. However, they are markedly impaired in defending blood glucose during a fast (Figure 2B). This functional defect correlated with diminished hepatic glycogen reserves (Figure 2C & 2D) and attenuated glucose production in response to pyruvate loading (a measure of gluconeogenesis) in fasted Alb::GC mice, compared with wildtype (Figure 2E).
Figure 2. Fasting hypoglycemia, enhanced insulin sensitivity and reduced glycogen stores in the liver of ISR-defective Alb::GC transgenic mice.
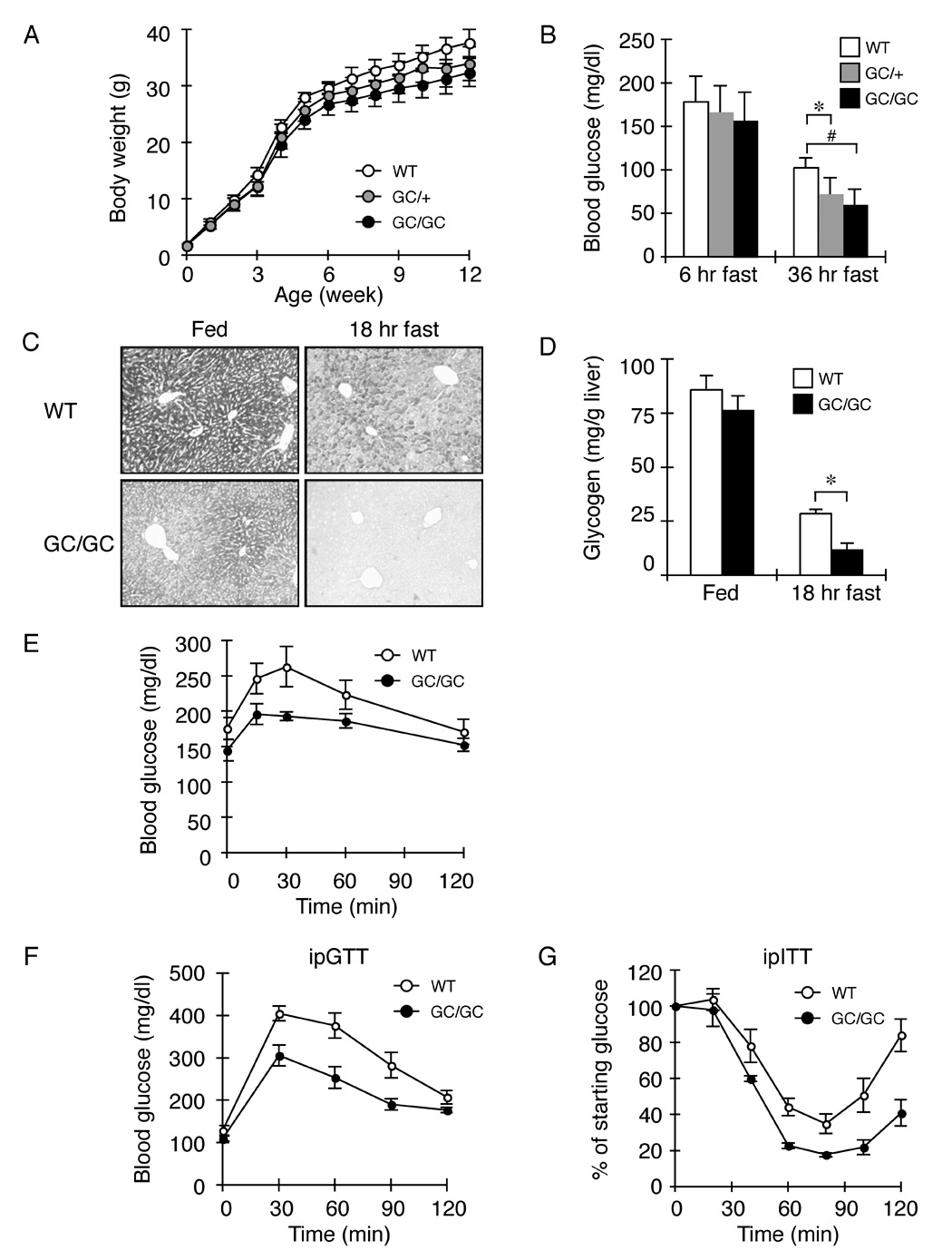
A. Body weight (mean ± SEM, n=20) of non-transgenic (WT), heterozygous (GC/+) and homozygous Alb::GC (GC/GC) transgenic male mice as function of age.
B. Blood glucose after fasting of adult male mice of indicated genotype (mean ± SEM, n=4, *p<0.05, #p<0.01).
C. PAS stain (glycogen) of representative liver sections of fed and fasted mice of the indicated genotype.
D. Glycogen content of liver of fed and fasted (18 hr) mice of the indicated genotype (mean ± SEM, WT n=3, Alb::GC n=5, *p<0.05)
E. Blood glucose levels as a function of time after pyruvate loading in mice of the indicated genotype (mean ± SEM, n=5, p<0.001 two-way ANOVA).
F. Blood glucose as a function of time after intra-peritoneal injection of glucose in mice of the indicated genotype (mean ± SEM, n=3, p<0.001 versus WT by two-way ANOVA)
G. Blood glucose as a function of time (expressed as a percent of level at t=0) after intra-peritoneal injection of insulin in mice of the indicated genotype (mean ± SEM, n=4, p=0.005 versus WT by two-way ANOVA)
A tendency towards fasting hypoglycemia may also explain the high rates of perinatal attrition of Alb::GC transgenic mice (21 of 58 pups found dead at postnatal day one in Alb::GC transgenic litters compared with 1 of 41 in the wildtype FVB/n control, P<0.05 by Xi2 test); a defect they share with homozygous Eif2aS51A mice (Scheuner et al., 2001). Consistent with these observations, Alb::GC mice also had enhanced glucose tolerance in experimental surrogates of the fed state, as reflected in lower serum glucose levels following intra-peritoneal injection of glucose (ipGTT, figure 2F) and enhanced sensitivity to the hypoglycemic effects of injected insulin (ipITT, figure 2G).
To analyze the ISR’s role in the context of nutrient excess we combined dietary manipulation with aurothioglucose injection, thus overcoming the known resistance of FVB/n mice (the background for the wildtype and transgenic mice studied here) to dietary-induced obesity (Hu et al., 2004). Between 6 and 21 weeks of age, the body weight of wildtype mice increased 2.43 ± 0.16-fold and that of the Alb::GC transgenic mice by 1.96 ± 0.08 fold (mean ± SEM n=12 p<0.05) (Figure 3A). The resulting glucose intolerance and insulin resistance were also less in the Alb::GC mice compared to the wildtype (Figure 3B–D). Hepatic steatosis, a predictable feature of obese, wildtype, male animals was also significantly lower in ISR defective mice fed a high fat diet, as reflected in a lower histochemical steatosis index of 0.8 ± 0.2 (mean ± SEM) in Alb::GC versus 6.4 ± 0.2 in wildtype (n=5, p<0.005 and Figure 3E) (Kleiner et al., 2005) and in lower liver tissue triglyceride content: 10.72 ± 1.69 mg/gm in Alb::GC versus 21.34 ± 4.11 mg/gm in the wildtype (mean ± SEM, n=5, p<0.05) (Figure 3F).
Figure 3. Sustained insulin sensitivity and reduced hepato-steatosis in ISR-defective Alb::GC transgenic mice on a high fat diet.
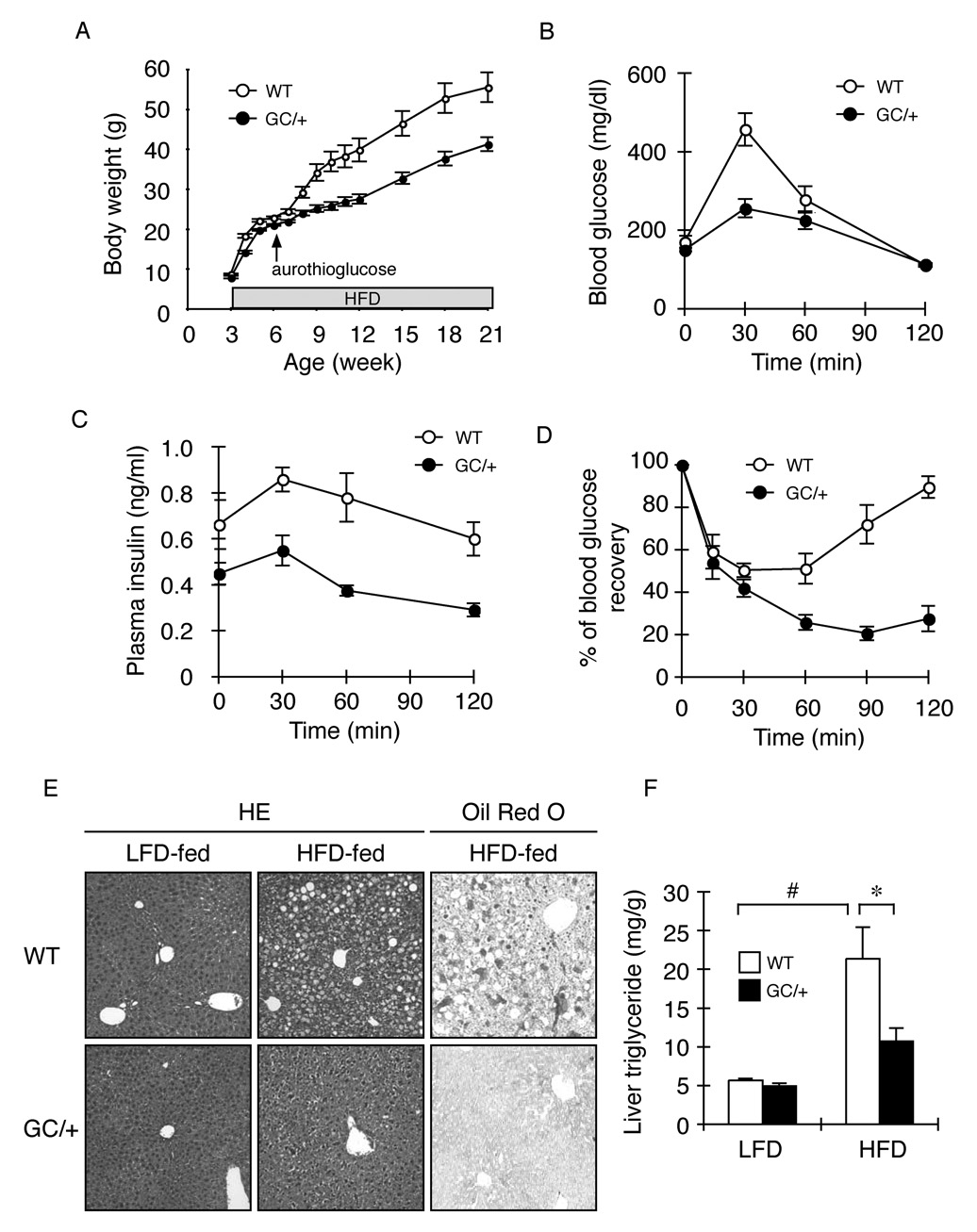
A. Body weight of a cohort of 12 non-transgenic (WT) and 12 Alb::GC transgenic mice over time (mean ± SEM, p<0.001 versus WT by two-way ANOVA). High fat diet (HFD) was instituted at weaning (3 weeks) whereas the aurothioglucose injection was introduced at 6 weeks of age.
B. Blood glucose as a function of time after intra-peritoneal injection of glucose in obese (high fat diet fed) mice of the indicated genotype (mean ± SEM, n=5, p<0.001 versus WT by two-way ANOVA)
C. Plasma insulin of the samples from “B” (mean ± SEM, n=5, p<0.001 versus WT by two-way ANOVA).
D. Blood glucose as a function of time after intra-peritoneal injection of insulin in obese mice of the indicated genotype (mean ± SEM, n=5, p<0.001 versus WT by two-way ANOVA).
E. Hematoxylin and Eosin (HE) and Oil Red O staining of representative liver sections of mice of the indicated genotype fed normal rodent chow (LFD) or high fat diet (HFD).
F. Triglyceride content of liver from wildtype and Alb::GC transgenic mice fed a low (LFD) and high fat diet (HFD) (mean ± SEM, n=5, *p<0.05)
The ISR regulates genes involved in intermediary metabolism
Two transcription factors are known to be activated by eIF2(αP): ATF4, whose translation is paradoxically stimulated (Harding et al., 2000; Lu et al., 2004a; Vattem and Wek, 2004) and NFκB that undergoes de-repression in the ISR (Jiang et al., 2003; Deng et al., 2004). However, defective signaling to their targets seemed unlikely to explain the altered hepatic gene expression program or the metabolic phenotype of the Alb::GC mice (Harding et al., 2003)(and table S3). Therefore, in our search for potential mediators of the metabolic effects of the ISR we focused on other transcription factor encoding genes that were differentially expressed in wildtype and Alb::GC mice. At the top of this list was peroxisome proliferator-activated receptor gamma (PPARγ) (table 1).
Table 1.
List of genes induced ≥2-fold by HFD feeding in wild-type mice and reduced by at least 25% in HFD-fed Alb::GC transgenic mice compared to high fat diet (HFD)-fed wildtype (WT). "Induction" is the ratio of the hybridization signals in LFD-fed versus HFD-fed mice. "% of GC in WT" is defined as the percentage of the signal of HFD-fed Alb::GC in HFD-fed WT mice. The mean ± the average deviation of the expression level (see methods) in WT and Alb::GC from two independent mice is shown. (A) A list of genes encoding transcription factors that satisfy the above criteria but are also induced ≥ 2-fold in AP20187-injected (0.2µg/gm body weight) Ttr::Fv2E-PERK high expressing (line #30) transgenic mice. (B) A list of genes involved in carbohydrate, lipid and amino acid metabolism that satisfy the above criteria.
| A | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Expression level |
HFD induction (fold) |
||||||||
| Entrez | WT | Alb::GC | WT | Alb::GC | % of GC in WT | ||||
| Name | Description | Gene | LFD | HFD | LFD | HFD | HFD/LFD | HFD/LFD | HFD |
| Transcription factor | |||||||||
| Pparg | peroxisome proliferator activated receptor gamma | 19016 | 0.97 ± 0.28 | 4.05 ± 0.04 | 0.68 ± 0.71 | 0.25 ± 0.11 | 4.18 ± 1.03 | 0.37 ± 0.13 | 6 ± 2 |
| Crsp6 | cofactor required for Sp1 transcriptional activation, subunit 6 | 234959 | 0.69 ± 0.13 | 2.29 ± 0.51 | 1.02 ± 0.33 | 0.65 ± 0.22 | 3.30 ± 0.09 | 0.63 ± 0.01 | 28 ± 3 |
| Jarid1d | jumonji, AT rich interactive domain 1D (Rbp2 like) | 20592 | 0.49 ± 0.14 | 1.38 ± 1.14 | 1.74 ± 3.07 | 0.45 ± 0.35 | 2.84 ± 1.01 | 0.26 ± 0.12 | 32 ± 1 |
| Klf13 | Kruppel-like factor 13 | 50794 | 0.65 ± 0.27 | 4.20 ± 0.19 | 0.87 ± 0.16 | 1.41 ± 0.63 | 6.44 ± 1.95 | 1.61 ± 0.33 | 33 ± 11 |
| Ppara | peroxisome proliferator activated receptor alpha | 19013 | 0.83 ± 0.13 | 5.41 ± 0.29 | 0.61 ± 0.26 | 1.82 ± 1.35 | 6.51 ± 0.57 | 2.99 ± 0.59 | 34 ± 18 |
| Sox-4 | SRY-box containing gene 4 | 20677 | 0.86 ± 0.45 | 2.93 ± 0.60 | 0.67 ± 0.02 | 1.02 ± 0.53 | 3.41 ± 0.81 | 1.54 ± 0.61 | 35 ± 8 |
| Nfic | nuclear factor I/C | 18029 | 0.47 ± 0.05 | 3.80 ± 0.38 | 0.59 ± 0.03 | 1.67 ± 0.35 | 8.04 ± 0.01 | 2.81 ± 0.40 | 44 ± 4 |
| Cphx | cytoplasmic polyadenylated homeobox | 105594 | 0.28 ± 0.37 | 1.55 ± 0.62 | 0.13 ± 0.04 | 0.69 ± 0.22 | 5.49 ± 2.81 | 5.24 ± 0.12 | 44 ± 3 |
| Akna | AT-hook transcription factor | 100182 | 0.39 ± 0.24 | 1.69 ± 0.48 | 0.60 ± 0.14 | 0.79 ± 0.25 | 4.33 ± 1.79 | 1.31 ± 0.35 | 47 ± 6 |
| Mxd3 | Max dimerization protein 3 | 17121 | 0.29 ± 0.06 | 1.10 ± 0.09 | 0.54 ± 0.23 | 0.56 ± 0.52 | 3.84 ± 0.44 | 1.04 ± 0.32 | 51 ± 31 |
| Mef2a | myocyte enhancer factor 2A | 17258 | 0.27 ± 0.02 | 1.73 ± 0.19 | 0.51 ± 0.81 | 0.96 ± 0.32 | 6.39 ± 0.29 | 1.88 ± 1.33 | 55 ± 10 |
| Klf3 | Kruppel-like factor 3 (basic) | 16599 | 0.74 ± 0.04 | 1.74 ± 0.08 | 0.93 ± 0.04 | 1.04 ± 0.17 | 2.37 ± 0.01 | 1.12 ± 0.12 | 60 ± 6 |
| Nfkb2 | NF-kappaB2/p100 | 18034 | 0.27 ± 0.03 | 3.27 ± 0.10 | 0.32 ± 0.00 | 1.98 ± 0.36 | 11.93 ± 0.81 | 6.27 ± 0.97 | 61 ± 8 |
| Bach2 | BTB and CNC homology 2 | 12014 | 0.52 ± 0.62 | 1.90 ± 0.18 | 0.50 ± 0.11 | 1.16 ± 0.39 | 3.66 ± 2.73 | 2.29 ± 0.23 | 61 ± 12 |
| Zkscan1 | zinc finger with KRAB and SCAN domains 1 | 74570 | 0.73 ± 0.14 | 1.60 ± 0.05 | 0.98 ± 0.04 | 1.11 ± 0.14 | 2.20 ± 0.33 | 1.13 ± 0.09 | 69 ± 6 |
| B | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Expression level |
HFD induction (fold) |
||||||||
| Entrez | WT | Alb::GC | WT | Alb::GC | % of GC in WT | ||||
| Name | Description | Gene | LFD | HFD | LFD | HFD | HFD/LFD | HFD/LFD | HFD |
| Carbonhydrate metabolism | |||||||||
| Pdha2 | pyruvate dehydrogenase E1 alpha 2 | 18598 | 0.16 ± 0.02 | 0.37 ± 0.26 | 0.23 ± 0.14 | 0.14 ± 0.06 | 2.26 ± 1.01 | 0.61 ± 0.07 | 39 ± 7 |
| Pdk3 | pyruvate dehydrogenase kinase, isoenzyme 3 | 236900 | 0.33 ± 0.02 | 0.87 ± 0.09 | 0.38 ± 0.05 | 0.37 ± 0.06 | 2.61 ± 0.14 | 0.98 ± 0.03 | 42 ± 3 |
| Mdh2 | malate dehydrogenase 2, NAD (mitochondrial) | 17448 | 0.75 ± 0.18 | 1.91 ± 0.10 | 0.64 ± 0.04 | 1.11 ± 0.04 | 2.54 ± 0.42 | 1.72 ± 0.04 | 58 ± 1 |
| Agl | amylo-1,6-glucosidase, 4-alpha-glucanotransferase | 77559 | 0.85 ± 0.13 | 1.85 ± 0.07 | 0.71 ± 0.04 | 1.09 ± 0.09 | 2.17 ± 0.23 | 1.54 ± 0.03 | 59 ± 2 |
| Ppp2r5e | protein phosphatase 2, regulatory subunit B (B56), epsilon | 26932 | 0.43 ± 0.03 | 2.57 ± 0.19 | 0.45 ± 0.35 | 1.52 ± 0.40 | 6.00 ± 0.02 | 3.37 ± 1.17 | 59 ± 10 |
| Ppp1r1a | protein phosphatase 1, regulatory (inhibitor) subunit 1A | 58200 | 0.60 ± 0.11 | 1.39 ± 0.04 | 0.79 ± 0.10 | 0.84 ± 0.20 | 2.33 ± 0.32 | 1.07 ± 0.09 | 61 ± 11 |
| Gyg | glycogenin | 27357 | 0.68 ± 0.09 | 1.99 ± 0.12 | 0.31 ± 0.61 | 1.26 ± 0.20 | 2.93 ± 0.18 | 4.11 ± 4.48 | 63 ± 6 |
| Pcx | pyruvate carboxylase | 18563 | 0.72 ± 0.07 | 1.90 ± 0.08 | 0.72 ± 0.11 | 1.19 ± 0.08 | 2.64 ± 0.17 | 1.66 ± 0.20 | 63 ± 2 |
| Pklr | pyruvate kinase liver and red blood cell | 18770 | 0.66 ± 0.09 | 1.67 ± 0.10 | 0.99 ± 0.15 | 1.11 ± 0.06 | 2.53 ± 0.25 | 1.12 ± 0.07 | 66 ± 3 |
| Gbe1 | glucan (1,4-alpha-), branching enzyme 1 | 74185 | 0.60 ± 0.01 | 2.07 ± 0.17 | 0.71 ± 0.01 | 1.40 ± 0.14 | 3.43 ± 0.22 | 1.96 ± 0.15 | 68 ± 1 |
| Suclg2 | succinate-Coenzyme A ligase, GDP-forming, beta subunit | 20917 | 0.73 ± 0.02 | 1.59 ± 0.03 | 0.84 ± 0.06 | 1.13 ± 0.04 | 2.17 ± 0.02 | 1.34 ± 0.05 | 71 ± 1 |
| Lipid metabolism | |||||||||
| Scd1 | stearoyl-Coenzyme A desaturase 1 | 20249 | 0.81 ± 0.19 | 3.43 ± 0.11 | 1.18 ± 0.25 | 0.90 ± 0.10 | 4.24 ± 0.77 | 0.76 ± 0.06 | 26 ± 2 |
| Fasn | fatty acid synthase | 14104 | 0.85 ± 0.25 | 2.26 ± 0.09 | 0.98 ± 0.08 | 0.88 ± 0.06 | 2.66 ± 0.58 | 0.90 ± 0.01 | 39 ± 1 |
| Gpd2 | glycerol phosphate dehydrogenase 2, mitochondrial | 14571 | 0.86 ± 0.29 | 1.86 ± 0.05 | 0.89 ± 0.03 | 0.84 ± 0.24 | 2.17 ± 0.58 | 0.95 ± 0.21 | 45 ± 10 |
| Lpl | lipoprotein lipase | 16956 | 0.66 ± 0.15 | 1.96 ± 0.68 | 0.83 ± 0.36 | 0.93 ± 0.48 | 2.95 ± 0.27 | 1.13 ± 0.06 | 48 ± 6 |
| Olah | oleoyl-ACP hydrolase | 99035 | 0.12 ± 0.00 | 0.34 ± 0.07 | 0.12 ± 0.01 | 0.17 ± 0.05 | 2.82 ± 0.47 | 1.43 ± 0.27 | 50 ± 4 |
| Acaca | acetyl-Coenzyme A carboxylase alpha | 107476 | 0.62 ± 0.43 | 1.82 ± 0.30 | 0.71 ± 0.02 | 1.11 ± 0.19 | 2.94 ± 1.12 | 1.58 ± 0.20 | 61 ± 0 |
| Gyk | glycerol kinase | 14933 | 0.53 ± 0.04 | 1.84 ± 0.06 | 0.69 ± 0.16 | 1.16 ± 0.00 | 3.46 ± 0.16 | 1.68 ± 0.35 | 63 ± 2 |
| Hadh | hydroxyacyl-Coenzyme A dehydrogenase | 15107 | 0.32 ± 0.16 | 2.78 ± 0.02 | 0.25 ± 0.02 | 1.87 ± 0.44 | 8.57 ± 3.53 | 7.45 ± 0.94 | 67 ± 14 |
| Translation, amino acid import and metabolism | |||||||||
| Eif3s6 | eukaryotic translation initiation factor 3, subunit 6 | 16341 | 0.42 ± 0.11 | 1.03 ± 0.18 | 0.81 ± 0.92 | 0.33 ± 0.11 | 2.43 ± 0.14 | 0.41 ± 0.21 | 32 ± 4 |
| Aoc3 | amine oxidase, copper containing 3 | 11754 | 0.21 ± 0.04 | 0.67 ± 1.23 | 0.55 ± 1.35 | 0.19 ± 0.05 | 3.17 ± 3.17 | 0.35 ± 0.42 | 29 ± 26 |
| Pgd | phosphogluconate dehydrogenase | 110208 | 0.91 ± 0.18 | 2.04 ± 0.05 | 1.00 ± 0.11 | 0.82 ± 0.02 | 2.24 ± 0.35 | 0.82 ± 0.06 | 40 ± 0 |
| Rps18 | ribosomal protein S18 | 20084 | 0.61 ± 0.11 | 2.39 ± 2.01 | 1.03 ± 0.09 | 0.96 ± 0.08 | 3.91 ± 1.77 | 0.93 ± 0.01 | 40 ± 22 |
| RPL39L | ribosomal protein L39-like | 68172 | 0.50 ± 0.20 | 1.32 ± 0.71 | 0.12 ± 0.03 | 0.75 ± 0.03 | 2.65 ± 0.26 | 6.33 ± 1.31 | 57 ± 23 |
| Slc12a1 | solute carrier family 12, member 1 | 20495 | 0.16 ± 0.10 | 1.80 ± 0.07 | 0.57 ± 0.84 | 1.25 ± 1.38 | 11.52 ± 5.41 | 2.22 ± 0.37 | 70 ± 53 |
| Tph1 | tryptophan hydroxylase 1 | 21990 | 0.44 ± 0.14 | 1.40 ± 0.34 | 1.25 ± 0.55 | 0.99 ± 0.43 | 3.22 ± 0.23 | 0.79 ± 0.00 | 71 ± 10 |
Originally identified as an activator of adipocyte differentiation (reviewed in: Rosen et al., 2000; Farmer, 2006), the nuclear receptor PPARγ has also been implicated in hepatic steatosis (Gavrilova et al., 2003; Schadinger et al., 2005). RT-PCR analysis revealed that PPARγ mRNA levels were ~2.7 fold lower in the liver of high fat diet fed Alb::GC mice compared to the wildtype (Figure 4A). Profiling also suggested a corresponding lower expression of PPARγ target genes involved in fatty acid synthesis in the Alb::GC transgenic mice (table 1), which was confirmed by quantitative RT-PCR analysis of fatty acid synthetase, acetyl-CoA carboxylase α and β and stearoyl-CoA desaturase (Figure 4B–E). This analysis places PPARγ downstream of the ISR in the liver and suggests that its lower expression might contribute to reduced hepatic steatosis in the Alb::GC transgenic mice.
Figure 4. Reduced expression of PPARγ and its target lipogenic enzymes in the liver of ISR-defective Alb::GC transgenic mice on a high fat diet.
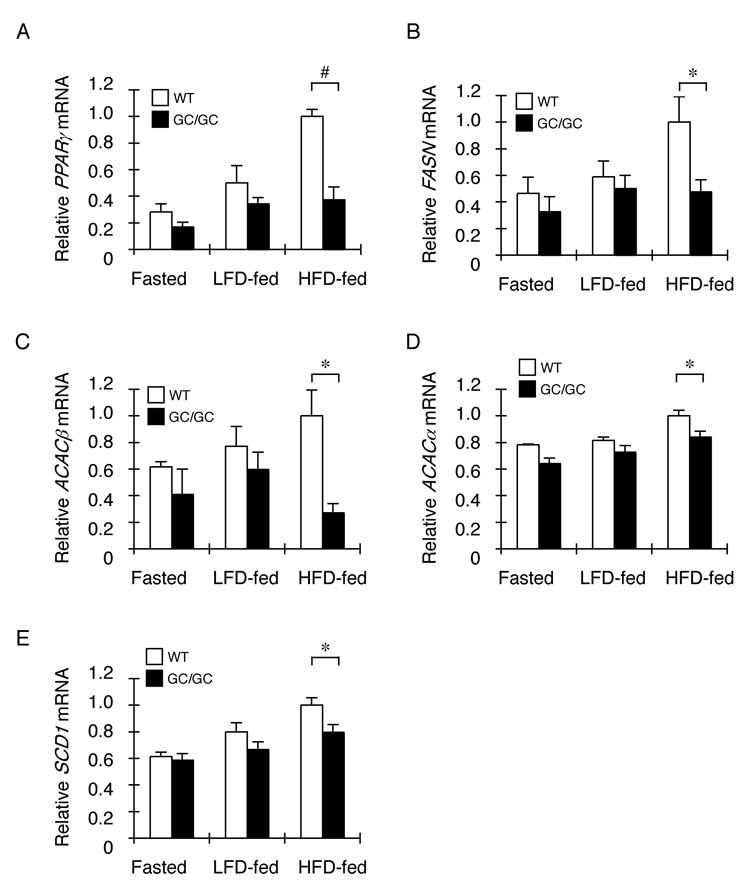
A–E. Relative levels of PPARγ, FASN, ACACβ, ACACα, and SCD1 mRNA in liver of non-transgenic (WT) and Alb::GC transgenic mice in the fasted state or fed a low (LFD and high fat diet) (mean ± SEM, n=3–4, *p<0.05).
Translational activation of C/EBP proteins by the ISR
In adipocytes C/EBPβ and the related C/EBPα isoform, positively regulate PPARγ expression through self-reinforcing feed forward loops (reviewed in: Rosen et al., 2000; Farmer, 2006). Recent data suggests that this relationship extends to the liver, as C/EBPβ deletion reduces levels of PPARγ2 and lipid accumulation in liver of obese mice (Millward et al., 2007; Schroeder-Gloeckler et al., 2007). Furthermore, C/EBP proteins also promote glycogen synthesis and hepatic glucose production (Wang et al., 1995; Liu et al., 1999). These features are shared by the ISR defective, Eif2aS51A mice (Scheuner et al., 2001) and the Alb::GC mice here (Figure 2 and Figure 3), prompting us to probe further the relationship of the ISR to C/EBP expression.
Levels of C/EBPβ protein were more than two-fold lower in nuclei isolated from livers of Alb::GC mice than wildtype and similar differences were observed in the abundance of C/EBPα (Figure 5A and 5B). Given that the ISR regulates its best known target, ATF4, translationally (Harding et al., 2000; Lu et al., 2004a; Vattem and Wek, 2004), and that isoform choice among C/EBP proteins has previously been shown to be regulated translationally by the virally-induced eIF2α kinase, PKR (Calkhoven et al., 2000), we decided to determine if the effects of the ISR on C/EBP protein levels also had a translation component.
Figure 5. Defective expression of C/EBPα and β in the liver of ISR-defective Alb::GC transgenic mice correlates with translational upregulation of C/EBPα and β by the ISR in cultured cells.
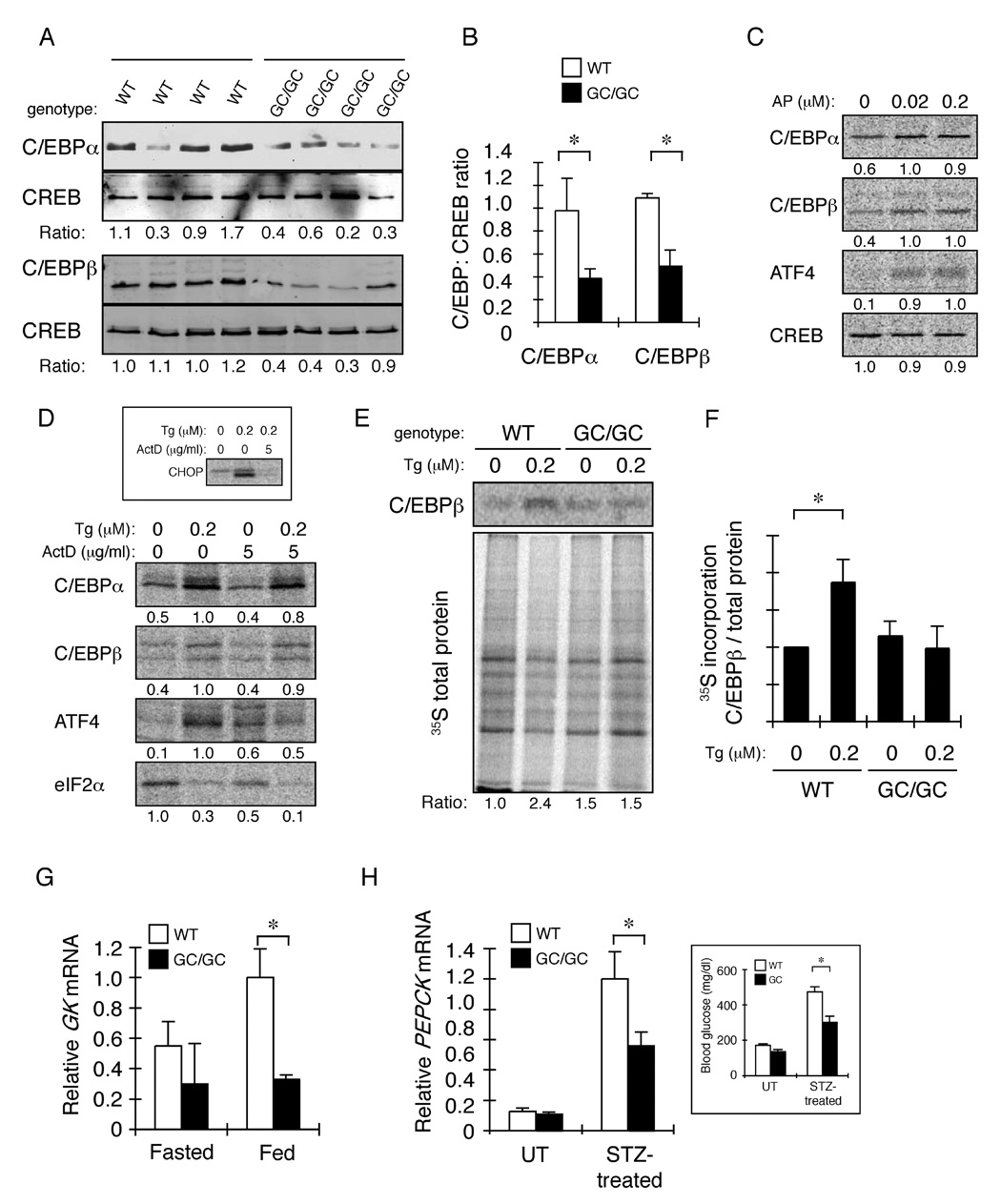
A. Immunoblot of C/EBPα, C/EBPβ and CREB (a loading control) in nuclear extract of livers of individual animals of the indicated genotype. The ratio of C/EBP:CREB signal in each sample is indicated.
B. Graphic presentation of the data in “A” (mean ± SEM, n=4, *p<0.05).
C. Autoradiograph of 35S-met/cys labeled endogenous proteins immunoprecipitated from Fv2E-PERK transgenic CHO cells after a 30 minute labeling pulse in the presence of the indicated concentration of AP20187 (AP). The relative signal level in each sample is indicated below each panel.
D. Autoradiograph of 35S-met/cys labeled endogenous proteins immunoprecipitated from HepG2 cells after a 30 minute labeling pulse in the presence of the indicated concentration of thapsigragin (Tg) and actinomycin D (ActD). The inset is an autoradiogram of CHOP immunoprecipiated from cells exposed to thapsigragin for 6 hours and and actinomycin D for two hours before the labeling pulse.
E Autoradiogram of an experiment identical in design to that shown in “D”, performed on primary hepatocytes obtained from wildtype and Alb::GC mice. The upper panel shows metabolically-labeled immunopurified C/EBPβ and the lower panel metabolically-labeled proteins in the cell lysate. The ratio of label incorporated into C/EBPβ versus total protein, normalized to the untreated WT sample is reported below. Shown is a typical experiment reproduced three times.
F. Plot of the ratio of labeled C/EBPβ versus total protein in all experiments performed on untreated and thapsigargin-treated primary hepatocytes from wildtype and Alb::GC transgenic mice. The ratio in the untreated wildtype cells is arbitrarily set to 1 (mean ± S.E.M., n=3, *p<0.05
G. Relative levels of glucokinase (GK) mRNA in liver of fasted and fed non-transgenic (WT) and Alb::GC transgenic mice (mean ± SEM, n=3, *p<0.05).
H. Relative levels of PEPCK mRNA in liver of untreated and streptozotocin injected animals of the indicated genotype. The inset is of morning (non-fasted) blood glucose of the same animals.
The incorporation of 35S-methionine/cysteine into newly synthesized C/EBPα and C/EBPβ was measured by pulse-labeling followed by immunoprecipitation. Activation of the ISR by AP20187 in Fv2E-PERK expressing Chinese Hamster Ovary cells led to a ~2-fold increase in label incorporated into newly synthesized endogenous proteins (figure 5C). This increase occurred in the face of global repression of protein synthesis (reflected here in the incorporation of label into the transcription factor CREB (figure 5C) and eIF2α (Figure 5D). As activated Fv2E-PERK phosphorylates eIF2α without causing ER stress, these findings suggest that the ISR can enhance C/EBP translation, independently of other signaling pathways.
To extend these observations to a surrogate of the liver ISR, a similar analysis was performed in HepG2 hepatoma cells in which PERK was activated by thapsigargin. Incorporation of label into newly synthesized C/EBPα and C/EBPβ increased ~2 fold within 30’ minutes of treatment. Furthermore, the increase in translation observed over 30 minutes of ISR induction in the thapsigargin-treated HepG2 cells was sustained in the face of transcriptional inhibition by actinomycin D (figure 5D, the effectiveness of which is revealed by the block to activation of a transcriptional target of the ISR, CHOP, figure 5D, inset), attesting to its independence of new mRNA synthesis.
To determine if attenuating the ISR by Alb::GC expression affects translational induction of C/EBP in hepatocytes, we compared the incorporation of labeled methionine/cysteine into C/EBPβ in untreated and thapsigragin-treated primary hepatocytes explanted from wildtype and Alb::GC transgenic mice. Thapsigargin reproducibly enhanced the incorporation of label into immunopurified C/EBPβ in the wildtype, but not the Alb::GC transgenic hepatocytes (Figure 5E and 5F). As expected attenuation of total protein synthesis by thapsigargin was also less conspicuous in the Alb::GC transgenic sample. The rapid de-differentiation of cultured primary hepatocytes likely leads to an under-estimate of the transgene’s effect in vivo. Dedifferentiation may also account for our inability to detect labeled C/EBPα in these samples.
The physiological significance of regulated expression of C/EBP isoforms by the ISR is supported by the finding that genes encoding key enzymes in hepatic glucose metabolism that are known to be regulated by C/EBPs, were reduced in the Alb::GC transgenic mice. This relationship extended across a range of physiological states from normal feeding (Figure 5G) to animals in which diabetes mellitus had been induced by streptozotocin injection (Figure 5H).
Expression profiling revealed that many ISR target genes were induced to lower levels in the livers of transgenic mice from the line with more Fv2E-PERK (Figure 1E and table S1). This finding suggested negative feedback in the ISR. For example, by CHOP, a late downstream transcription factor induced by the ISR (Harding et al., 2000) that inhibits conventional C/EBP target genes (Ron and Habener, 1992) and blocks adipocytic differentiation (Batchvarova et al., 1995) or another transcriptional target of the ISR, ATF3 that inhibits ATF4 (Jiang et al., 2004). To explore this suggestion of biphasic regulation of gene expression by the ISR we injected Ttr::Fv2E-PERK transgenic animals (of the lower-expressing line) with increasing amounts of the AP20187 activator and measured mRNA levels in their liver by RT-PCR. While CHOP, ATF3 and C/EBPβ mRNA levels increased mono-phasically with AP20187 dose, other genes such as, PPARγ, PEPCK and other C/EBP target genes responded bi-phasically (Figure 6A). These observations suggest that at lower levels of signaling the ISR contributes positively to the regulation of the same metabolic pathways that it inhibits at unusually high, possibly pathological, levels of signaling.
Figure 6. Biphasic regulation of genes involved in intermediary metabolism by the ISR.
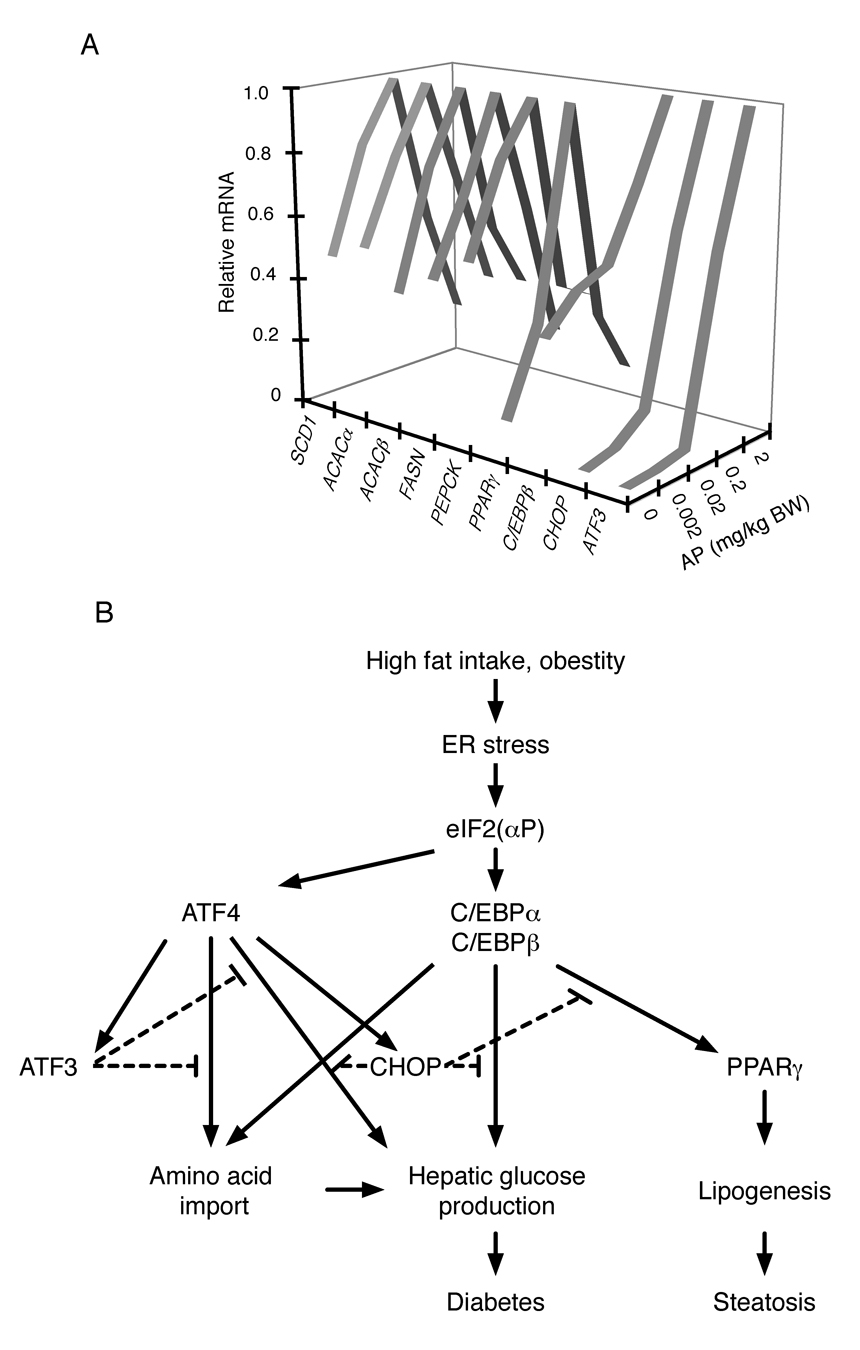
A. Relative levels of the indicated mRNA in liver of Ttr::Fv2E-PERK transgenic mice 8 hours after intra-peritoneal injection with the indicated dose of AP20187 (AP). The complete data set and statistical analysis for this experiment is presented in table S5).
B. Graphic summary of interactions between components of the ISR regulating intermediary metabolism.
DISCUSSION
By focusing on one branch of the UPR - the eIF2(αP)-mediated ISR - and by restricting the genetic manipulation to a single tissue - the liver - this study clarifies earlier work in mice with pervasive alterations in the ER stress response that were effected by germline mutations in UPR pathway genes. Both the hepatic ISR-defective Alb::GC mice described here and the globally ISR-defective homozygous Eif2aS51A mutant mice (Scheuner et al., 2001), share a tendency towards fasting hypoglycemia and reduced hepatic glycogen content. This argues that at least part of the metabolic consequences of a defective ISR are autonomous to the hepatocyte.
The aforementioned hepatic defect is well explained by lower levels of C/EBPα and β protein in the liver of the Alb::GC mice, as both C/EBPα and β are known to positively regulate genes involved in glycogen synthesis and hepatic glucose production (Wang et al., 1995; Liu et al., 1999). Our findings argue that ISR mediated activation of C/EBPα and β can proceed by a translational mechanism that is mobilized within minutes of induction eIF2α phosphorylation and functions independently of new mRNA synthesis. Translational activation is aided by previously-recognized transcriptional upregulation, which likely reinforces further C/EBP expression (Chen et al., 2004). In these respects the C/EBPs resemble the well-validated translational target of the mammalian ISR, ATF4, and its yeast counterpart, GCN4. However, the arrangement of conserved upstream open reading frames in the C/EBPα and β genes suggests important differences between the molecular mechanism by which eIF2(αP) promotes their translation and that of ATF4/GCN4. Two conserved upstream open reading frames specify regulated translational re-initiation at the ATF4 (and GCN4) coding sequence when levels of phosphorylated eIF2α are high (Hinnebusch and Natarajan, 2002; Lu et al., 2004a; Vattem and Wek, 2004). In the case of C/EBPα & β, our observations and those of Calkhoven and colleagues (Calkhoven et al., 2000) are more readily explained by a model whereby eIF2(αP) disfavors initiation at the single inhibitory short open reading frame conserved in their mRNAs and favors initiation at downstream AUG(s). The mechanism linking eIF2(αP) to regulated initiation at two consecutive open reading frames, remains to be resolved, however the phenomena is not restricted to C/EBPα and β, as it is observed on the ATF4 mRNA when the 5’-most of the 2 upstream open reading frames is deleted (Lu et al., 2004a, figure 5B therein).
Our study also implicates the ISR in regulating lipid metabolism in the liver, as the Alb::GC mice accumulated less neutral lipid in their liver when placed on a high fat diet. This alteration, too, appears to be mediated by changes in gene expression, as levels of enzymes involved in fatty acid synthesis were lower in the ISR-defective transgenic mice, compared with the wildtype. Lower levels of PPARγ might explain part of this defect, as a requirement for hepatic PPARγ in the development of hepatic steatosis has been noted recently (Gavrilova et al., 2003). C/EBP proteins positively regulate PPARγ expression (Millward et al., 2007; Rahman et al., 2007), which is consistent with a linear pathway from eIF2(αP) to the C/EBP proteins and from there to PPARγ (Figure 6B). Furthermore, the ISR defective Alb::GC transgenic mice gain significantly less weight, when placed on high fat diet. While a detailed understanding of the physiological mechanisms awaits further studies, the pervasive role of eIF2(αP) in regulating genes involved in glycogenesis and lipid synthesis is consistent with the idea that impaired conversion of ingested nutrients to their storage forms limits weight gain in these ISR-defective animals.
In cultured cells transient activation of the ISR promotes survival and adaptation, whereas unremitting signaling promotes cell death (Rutkowski et al., 2006). The observations made here provide an interesting parallel in terms of intermediary metabolism in the liver: Low level signaling in the ISR (as observed under physiological circumstances) promotes expression of genes involved in glycogen synthesis, gluconeogenesis and fatty acid synthesis, whereas higher levels of signaling repress the expression of the same genes. These findings might be explained by differential responsiveness of the ISR’s effectors to signaling at different intensities, as proposed (Rutkowski et al., 2006). CHOP, ATF3 and C/EBPβ mRNA levels increase monophasically with signal strength, but expression of downstream C/EBP target genes decline at high levels of ISR activity (Figure 6A). CHOP-mediated inhibition of C/EBP proteins (Ron and Habener, 1992) and direct repression of PEPCK by ATF3 (Allen-Jennings et al., 2002) could contribute to the declining limb of the bi-phasic relationship between strength of ISR signal and downstream target gene expression.
Bi-phasic regulation of enzymes involved in fatty acid biosynthesis may also explain an apparent discrepancy between this study, in which signaling in the hepatic ISR is shown to promote steatosis in mice fed a high fat diet, and the observation that global Gcn2 deletion predisposes mice fed a leucine deficient diet to steatosis (Guo and Cavener, 2007). Perhaps leucine deficiency is associated with levels of ISR signaling that repress lipid synthesis in wildtype mice but fail to achieve this level in the Gcn2−/− mice. Alternatively, the lower levels of amino acid transporters noted in ISR defective cells (Harding et al., 2003) may further reduce hepatic uptake of leucine and sensitize amino-acid deprived Gcn2−/− mice to fatty liver by further reducing the building blocks for lipoprotein synthesis and thereby lipid export.
It has previously been reported that a partial compromise in downstream signaling in the IRE1 branch of the UPR (effected by haploid insufficiency for XBP-1) accentuates insulin resistance and promotes glucose intolerance in obese mice (Ozcan et al., 2004) and that protein and chemical chaperones that reduce ER stress in insulin target tissues ameliorate that phenotype (Ozawa et al., 2005; Ozcan et al., 2006). Our study suggests a parallel process operating in hepatocytes, whereby heightened activity of the ISR and its downstream target genes contributes to the link between (physiological) ER stress and the metabolic syndrome of obesity and diabetes. In regard to intermediary metabolism, signaling by IRE1 and the ISR proceed in parallel and neither seems to dominate the metabolic phenotype. This is exemplified by the observations that insulin signaling to its proximal targets is not obviously affected by the ISR perturbation (data not shown); whereas such a defect is predicted in a system dominated by IRE1 signaling. Though loss of translational control in the ISR(−) state leads to more IRE1 signaling, which impairs insulin signaling, our study suggests that the net effect of a compromised ISR is to ameliorate the metabolic phenotype in mice exposed to nutrient excess.
It is interesting to speculate on the evolutionary origins of the link between the ISR and intermediary metabolism. Gene knockout experiments show that in most mammalian tissues the ER stress inducible kinase PERK dominates ISR activity (Harding et al., 2001). However, the GCN2-posessing ancestor in which PERK first evolved already had in place a gene expression program responsive to eIF2(αP) that modulated intermediary metabolism (Hinnebusch and Natarajan, 2002). We propose that this pre-existing link was co-opted by PERK to coordinate intermediary metabolism with nutritionally entrained variation in load of unfolded client proteins that enter the ER of insulin responsive tissues. This model is supported by the observation that yeasts, that lack PERK, also use ER activity as a proxy for nutrient availability, but effect this by IRE1 signaling (Schroder et al., 2000; Patil et al., 2004). Presumably, under conditions of limited nutrient availability this fine tuning of intermediary metabolism by physiological levels of ER stress (activity) is adaptive, however in the presence of excess nutrients the ISR’s contribution to lipid synthesis and hepatic glucose production are counterproductive and promote glucose intolerance and liver steatosis. Time will tell if this example of failure of homeostasis can be exploited therapeutically, by targeting distinct facets of the ISR with inhibitors.
Experimental Procedures
Transgenic mice
A 2.3Kb mouse albumin enhancer/promoter fragment was used to drive expression of Hamster GADD34 (aa 292–590) in the Alb::GC transgene. Two transgenic lines were established in the FvB/n strain (#14 & #16). They exhibited similar sensitivity to fasting hypoglycemia and the higher expressing line, #14, was used in subsequent studies.
As we were unable to obtain lines expressing Fv2E-PERK using the Alb promoter, the transthyretin (Ttr) promoter was used instead to produce six Ttr::Fv2E-Perk transgenic lines in FvB/n, two of which, that express the protein at different levels (#30 “high” and #58 “low”, Figure S2B), were selected for further study. For the experiment reported on in table S3, the low expressing Ttr::Fv2E-Perk transgene (#58) was bred into the Atf4 knockout strain and the derivative compound heterozygous mice (in the mixed FvB/n; Swiss Webster background) were backcrossed to the Atf4+/− parental stock and Ttr::Fv2E-PERK positive siblings with Atf4+/+ and Atf4−/− genetypes were analyzed.
Animal Experiments
All experiments in mice were approved by the Institutional Animal Care and Use Committee. Because of their enhanced susceptibility to the metabolic consequences of nutrient excess, male mice were used. These were maintained on a LFD containing 12% fat or a HFD containing 60% fat (Research Diets #D12492) with 12 hr light and dark cycles.
To induce hypo-insulinemic diabetes, streptozotocin (STZ, 100 mg/kg, Sigma), freshly dissolved in citrate buffer [pH 4.5] was injected intraperitoneally into 10–12 weeks old mice on 2 consecutive days.
For diet-induced obesity, aurothioglucose (0.5 mg/g) (Schering-Plough) was injected intraperitoneally as a single dose into 6 weeks old mice.
For the glucose tolerance test, mice were fasted overnight (14 hr) and injected intraperitoneally with a glucose solution (2 g/kg). For the insulin tolerance test, mice were fasted for 4 hr and human insulin (0.75 mU/kg) (Eli Lilly) was injected intraperitoneally. For pyruvate loading, mice were fasted for 16 hours and injected intraperitoneally with 1.5 g/Kg sodium pyruvate. Blood glucose and plasma insulin concentrations were measured from tail blood using a OneTouch Ultra glucometer (Johnson and Johnson) and a rat/mouse insulin ELISA kit (Linco Research), respectively.
To determine liver glycogen content, the livers (100–150 mg) were digested in 0.3 ml of 30% KOH for 30 min at 100°C, followed by addition of 0.1 ml of 20% NaSO4 and 0.8 ml of ethanol. Macromolecules containing glycogen were precipitated by centrifugation (20,000× g, 10 min), and washed with 70% ethanol. The pellets were hydrolyzed in 0.5 ml of 4 N H2SO4 for 10 min at 100°C, and neutralized by 0.5 ml of 4 N NaOH. To measure the glucose concentration, 5 µl of the sample supernatant was added in a Glucose (HK) assay reagent (Sigma). The liver glycogen concentration was determined by comparison with a standard curve constructed by glycogen from oyster type II (Sigma).
Triglyceride content of liver tissue was measured enzymatically (L-type TG H test, Wako pure chemical, Japan) on a lipids extracted from tissue samples with chloroform:methanol (2:1 mix).
Histological Analysis
Livers from animals perfused with 10% paraformaldehyde were fixed in the same, paraffin embedded, sectioned in 5 µm, and stained with hematoxylin and eosin or periodic acid-Schiff (PAS). Oil red O was used to stain neutral lipids in frozen liver sections. Hepatic steatosis was assessed in three categories: Grade; Location, Microvesicularity and a composite score was obtained (based on Kleiner et al., 2005).
RNA Analysis
Livers were snap-frozen in liquid nitrogen and total RNA was isolated using RNA-STAT60 (Tel-Test) and RNeasy kit (Qiagen). For Gene expression profiling, total RNA was fluorescently labeled and hybridized to Affymetrix mouse genome 430A 2.0 GeneChip or Affymetrix murine genome U74Av2 GeneChip under standard conditions. Primary image analysis of the arrays was performed using the Genechip 3.2 software (Affymetrix). The raw data from the hybridization experiments were analyzed by GeneSpring GX (Agilent Technologies). The raw signal from each gene was normalized to the mean strength of all genes from the same chip to obtain the normalized signal strength. Then, to allow visualization of all data on the same scale for subsequent analysis, the normalized signal strength of each gene was divided by the median signal strength for that gene among all samples to obtain the normalized expression level. The complete data set has been submitted to the NCBI GEO database (GSE11116 and GSE11210).
Quantitative RT-PCR was performed with iScript one-step RT-PCR kit using the MyIQ single-color real-time PCR detection system (Bio-Rad Laboratories). All PCR reactions were performed in duplicate or triplicate and PCR products were subjected to a melting curve analysis. The abundance of specific mRNAs was determined by comparison with a standard curve constructed by serial dilution of the sample and normalized to β-actin. Primers used for PCR reaction are listed in table S4.
Cell Culture
The HepG2 cells were cultured in regular DMEM supplemented with 10% FetalClone II serum (Hyclone), and penicillin-streptomycin. CHO-KI cells were grown in Hams F12 supplemented with 10% FetalClone II serum and penicillin-streptomycin.
Primary hepatocytes were isolated from wildtype and Alb::GC mice, cultured on type I collagen-coated plates in Waymouth’s medium supplemented with 0.1 nM insulin and metabolically labeled as previously described (Oyadomari et al., 2006).
Protein Analysis
PERK and GADD34 were detected in liver lysates by immunoprecipitation followed by immunoblotting as described or by direct immunoblotting of the lysate to detect phosphorylated and total eIF2α as described (Harding et al., 2001). C/EBPα, C/EBPβ and CREB were blotted in a nuclear extract prepared from liver. Antibodies for immunoprecipitation for GADD34 and PERK and immunoblotting procedures for eIF2α, GADD34, PERK, C/EBPα, C/EBPβ and CREB have previously been described (Batchvarova et al., 1995; Harding et al., 2001; Novoa et al., 2001). Phosphorylated eIF2α was detected using the BioSource Ab #44-728G lot #0201.
The translation of C/EBPα, C/EBPβ, ATF4, CREB and eIF2α was measured by pulse-labeling and immunoprecipitation in Fv2E-PERK expressing CHO cells (Lu et al., 2004a), HepG2 cells and primary hepatocytes. For metabolic labeling, cells were switched to methionine and cystine minus DMEM with 10% dialyzed fetal calf serum 5 min before addition of -TRAN35S-LABEL (MP Biomedicals) at 400 µCi/ml for 30 min.
Statistical analysis
All results are expressed as means ± SEM. Unpaired two-tailed Student’s t tests were performed to determine p values for paired samples and two-way ANOVA with repeat measurements was performed to analyze measurements obtained by time course‥
Supplementary Material
ACKNOWLEDGMENTS
We thank Richard Palmiter (University of Washington) for the pBS-Albe/p plasmid, Domenico Accili (Columbia University) for the pTTR-ExV3 plasmid, and ARIAD Inc. for the Fv2E dimerization system and the AP20187 compound. Supported by NIH grant DK47119 to D.R. and fellowships from the Uehara Memorial Foundation and The Naito Foundation to S.O.
Footnotes
Institute for Genome Research, The University of Tokushima
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Allen-Jennings AE, Hartman MG, Kociba GJ, Hai T. The roles of ATF3 in liver dysfunction and the regulation of phosphoenolpyruvate carboxykinase gene expression. J Biol Chem. 2002;277:20020–20025. doi: 10.1074/jbc.M200727200. [DOI] [PubMed] [Google Scholar]
- Batchvarova N, Wang X-Z, Ron D. Inhibition of adipogenesis by the stress-induced protein CHOP (GADD153) EMBO J. 1995;14:4654–4661. doi: 10.1002/j.1460-2075.1995.tb00147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernales S, Papa FR, Walter P. Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol. 2006;22:487–508. doi: 10.1146/annurev.cellbio.21.122303.120200. [DOI] [PubMed] [Google Scholar]
- Calkhoven CF, Muller C, Leutz A. Translational control of C/EBPalpha and C/EBPbeta isoform expression. Genes Dev. 2000;14:1920–1932. [PMC free article] [PubMed] [Google Scholar]
- Chen C, Dudenhausen EE, Pan YX, Zhong C, Kilberg MS. Human CCAAT/enhancer-binding protein beta gene expression is activated by endoplasmic reticulum stress through an unfolded protein response element downstream of the protein coding sequence. J Biol Chem. 2004;279:27948–27956. doi: 10.1074/jbc.M313920200. [DOI] [PubMed] [Google Scholar]
- Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with Wolcott-Rallison syndrome. Nat Genet. 2000;25:406–409. doi: 10.1038/78085. [DOI] [PubMed] [Google Scholar]
- Deng J, Lu PD, Zhang Y, Scheuner D, Kaufman RJ, Sonenberg N, Harding HP, Ron D. Translational repression mediates activation of Nuclear Factor kappa B by phosphorylated translation initiation factor 2. Molecular and Cellular Biology. 2004;24:10161–10168. doi: 10.1128/MCB.24.23.10161-10168.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer SR. Transcriptional control of adipocyte formation. Cell Metab. 2006;4:263–273. doi: 10.1016/j.cmet.2006.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavrilova O, Haluzik M, Matsusue K, Cutson JJ, Johnson L, Dietz KR, Nicol CJ, Vinson C, Gonzalez FJ, Reitman ML. Liver peroxisome proliferator-activated receptor gamma contributes to hepatic steatosis, triglyceride clearance, and regulation of body fat mass. J Biol Chem. 2003;278:34268–34276. doi: 10.1074/jbc.M300043200. [DOI] [PubMed] [Google Scholar]
- Guo F, Cavener DR. The GCN2 eIF2alpha Kinase Regulates Fatty-Acid Homeostasis in the Liver during Deprivation of an Essential Amino Acid. Cell Metab. 2007;5:103–114. doi: 10.1016/j.cmet.2007.01.001. [DOI] [PubMed] [Google Scholar]
- Hao S, Sharp JW, Ross-Inta CM, McDaniel BJ, Anthony TG, Wek RC, Cavener DR, McGrath BC, Rudell JB, Koehnle TJ, Gietzen DW. Uncharged tRNA and sensing of amino acid deficiency in mammalian piriform cortex. Science. 2005;307:1776–1778. doi: 10.1126/science.1104882. [DOI] [PubMed] [Google Scholar]
- Harding H, Novoa I, Zhang Y, Zeng H, Wek RC, Schapira M, Ron D. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6:1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- Harding H, Zeng H, Zhang Y, Jungreis R, Chung P, Plesken H, Sabatini D, Ron D. Diabetes Mellitus and exocrine pancreatic dysfunction in Perk−/− mice reveals a role for translational control in survival of secretory cells. Mol Cell. 2001;7:1153–1163. doi: 10.1016/s1097-2765(01)00264-7. [DOI] [PubMed] [Google Scholar]
- Harding H, Zhang Y, Zeng H, Novoa I, Lu P, Calfon M, Sadri N, Yun C, Popko B, Paules R, Stojdl D, Bell J, Hettmann T, Leiden J, Ron D. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11:619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- Hinnebusch AG, Natarajan K. Gcn4p, a master regulator of gene expression, is controlled at multiple levels by diverse signals of starvation and stress. Eukaryot Cell. 2002;1:22–32. doi: 10.1128/EC.01.1.22-32.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu CC, Qing K, Chen Y. Diet-induced changes in stearoyl-CoA desaturase 1 expression in obesity-prone and -resistant mice. Obes Res. 2004;12:1264–1270. doi: 10.1038/oby.2004.160. [DOI] [PubMed] [Google Scholar]
- Jiang HY, Wek SA, McGrath BC, Scheuner D, Kaufman RJ, Cavener DR, Wek RC. Phosphorylation of the alpha subunit of eukaryotic initiation factor 2 is required for activation of NF-kappaB in response to diverse cellular stresses. Mol Cell Biol. 2003;23:5651–5663. doi: 10.1128/MCB.23.16.5651-5663.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang HY, Wek SA, McGrath BC, Lu D, Hai T, Harding HP, Wang X, Ron D, Cavener DR, Wek RC. Activating Transcription Factor 3 Is Integral to the Eukaryotic Initiation Factor 2 Kinase Stress Response. Mol Cell Biol. 2004;24:1365–1377. doi: 10.1128/MCB.24.3.1365-1377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, Yeh M, McCullough AJ, Sanyal AJ. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology. 2005;41:1313–1321. doi: 10.1002/hep.20701. [DOI] [PubMed] [Google Scholar]
- Lee AH, Chu GC, Iwakoshi NN, Glimcher LH. XBP-1 is required for biogenesis of cellular secretory machinery of exocrine glands. Embo J. 2005;24:4368–4380. doi: 10.1038/sj.emboj.7600903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Croniger C, Arizmendi C, Harada-Shiba M, Ren J, Poli V, Hanson RW, Friedman JE. Hypoglycemia and impaired hepatic glucose production in mice with a deletion of the C/EBPbeta gene. J Clin Invest. 1999;103:207–213. doi: 10.1172/JCI4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu PD, Harding HP, Ron D. Translation re-initiation at alternative open reading frames regulates gene expression in an integrated stress response. Journal Cell Biology. 2004a;167:27–33. doi: 10.1083/jcb.200408003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu PD, Jousse C, Marciniak SJ, Zhang Y, Novoa I, Scheuner D, Kaufman RJ, Ron D, Harding HP. Cytoprotection by pre-emptive conditional phosphorylation of translation initiation factor 2. Embo J. 2004b;23:169–179. doi: 10.1038/sj.emboj.7600030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurin A, Jousse C, Averous J, Parry LAB, Cherasse Y, Zeng H, Zhang Y, Harding H, Ron D, Fafournoux P. The GCN2 kinase biases feeding behavior to maintain amino-acid homeostasis in omnivores. Cell Metabolism. 2005;1:273–277. doi: 10.1016/j.cmet.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Millward CA, Heaney JD, Sinasac DS, Chu EC, Bederman IR, Gilge DA, Previs SF, Croniger CM. Mice with a deletion in the gene for CCAAT/enhancer-binding protein beta are protected against diet-induced obesity. Diabetes. 2007;56:161–167. doi: 10.2337/db06-0310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani Y, Kaneto H, Kawamori D, Yoshiuchi K, Hatazaki M, Matsuoka TA, Ozawa K, Ogawa S, Hori M, Yamasaki Y, Matsuhisa M. Involvement of endoplasmic reticulum stress in insulin resistance and diabetes. J Biol Chem. 2005;280:847–851. doi: 10.1074/jbc.M411860200. [DOI] [PubMed] [Google Scholar]
- Novoa I, Zeng H, Harding H, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2α. J Cell Biol. 2001;153:1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa I, Zhang Y, Zeng H, Jungreis R, Harding HP, Ron D. Stress-induced gene expression requires programmed recovery from translational repression. Embo J. 2003;22:1180–1187. doi: 10.1093/emboj/cdg112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyadomari S, Yun C, Fisher EA, Kreglinger N, Kreibich G, Oyadomari M, Harding HP, Goodman AG, Harant H, Garrison JL, Taunton J, Katze MG, Ron D. Co-Translocational degradation protects the stressed endoplasmic reticulum from protein overload. Cell. 2006;126:727–739. doi: 10.1016/j.cell.2006.06.051. [DOI] [PubMed] [Google Scholar]
- Ozawa K, Miyazaki M, Matsuhisa M, Takano K, Nakatani Y, Hatazaki M, Tamatani T, Yamagata K, Miyagawa J, Kitao Y, Hori O, Yamasaki Y, Ogawa S. The endoplasmic reticulum chaperone improves insulin resistance in type 2 diabetes. Diabetes. 2005;54:657–663. doi: 10.2337/diabetes.54.3.657. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Gorgun CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science. 2006;313:1137–1140. doi: 10.1126/science.1128294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil CK, Li H, Walter P. Gcn4p and novel upstream activating sequences regulate targets of the unfolded protein response. PLoS Biol. 2004;2:E246. doi: 10.1371/journal.pbio.0020246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman SM, Schroeder-Gloeckler JM, Janssen RC, Jiang H, Qadri I, Maclean KN, Friedman JE. CCAAT/enhancing binding protein beta deletion in mice attenuates inflammation, endoplasmic reticulum stress, and lipid accumulation in diet-induced nonalcoholic steatohepatitis. Hepatology. 2007;45:1108–1117. doi: 10.1002/hep.21614. [DOI] [PubMed] [Google Scholar]
- Ron D, Habener JF. CHOP, a novel developmentally regulated nuclear protein that dimerizes with transcription factors C/EBP and LAP and functions as a dominant negative inhibitor of gene transcription. Genes & Dev. 1992;6:439–453. doi: 10.1101/gad.6.3.439. [DOI] [PubMed] [Google Scholar]
- Ron D, Harding H. eIF2a phosphorylation in cellular stress responses and disease. In: Sonenberg N, Hershey J, Mathews M, editors. Translational Control. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 2007. pp. 345–368. [Google Scholar]
- Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM. Transcriptional regulation of adipogenesis. Genes Dev. 2000;14:1293–1307. [PubMed] [Google Scholar]
- Rutkowski DT, Arnold SM, Miller CN, Wu J, Li J, Gunnison KM, Mori K, Sadighi Akha AA, Raden D, Kaufman RJ. Adaptation to ER stress is mediated by differential stabilities of pro-survival and pro-apoptotic mRNAs and proteins. PLoS Biol. 2006;4:e374. doi: 10.1371/journal.pbio.0040374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadinger SE, Bucher NL, Schreiber BM, Farmer SR. PPARgamma2 regulates lipogenesis and lipid accumulation in steatotic hepatocytes. Am J Physiol Endocrinol Metab. 2005;288:E1195–E1205. doi: 10.1152/ajpendo.00513.2004. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Song B, McEwen E, Gillespie P, Saunders T, Bonner-Weir S, Kaufman RJ. Translational control is required for the unfolded protein response and in-vivo glucose homeostasis. Mol Cell. 2001;7:1165–1176. doi: 10.1016/s1097-2765(01)00265-9. [DOI] [PubMed] [Google Scholar]
- Scheuner D, Mierde DV, Song B, Flamez D, Creemers JW, Tsukamoto K, Ribick M, Schuit FC, Kaufman RJ. Control of mRNA translation preserves endoplasmic reticulum function in beta cells and maintains glucose homeostasis. Nat Med. 2005;11:757–764. doi: 10.1038/nm1259. [DOI] [PubMed] [Google Scholar]
- Schroder M, Chang JS, Kaufman RJ. The unfolded protein response represses nitrogen-starvation induced developmental differentiation in yeast. Genes Dev. 2000;14:2962–2975. doi: 10.1101/gad.852300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- Schroeder-Gloeckler JM, Rahman SM, Janssen RC, Qiao L, Shao J, Roper M, Fischer SJ, Lowe E, Orlicky DJ, McManaman JL, Palmer C, Gitomer WL, Huang W, O'Doherty RM, Becker TC, Klemm DJ, Jensen DR, Pulawa LK, Eckel RH, Friedman JE. CCAAT/enhancer-binding protein beta deletion reduces adiposity, hepatic steatosis, and diabetes in Lepr(db/db) mice. J Biol Chem. 2007;282:15717–15729. doi: 10.1074/jbc.M701329200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Ellis RE, Lee K, Liu C-Y, Yang K, Solomon A, Yoshida H, Morimoto R, Kurnit DM, Mori K, Kaufman RJ. Complementary signaling pathways regulate the unfolded protein response and are required for C. elegans development. Cell. 2001;107:893–903. doi: 10.1016/s0092-8674(01)00612-2. [DOI] [PubMed] [Google Scholar]
- Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci U S A. 2004;101:11269–11274. doi: 10.1073/pnas.0400541101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N-d, Finegold M, Bradley A, Ou C, Abdelsayed S, Wilde M, Taylor L, Wilson D, Darlington G. Impaired energy homeostasis in C/EBPα knockout mice. Science. 1995;269:1108–1112. doi: 10.1126/science.7652557. [DOI] [PubMed] [Google Scholar]
- Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T, Wang J, Song B, Yau GD, Kaufman RJ. ATF6alpha Optimizes Long-Term Endoplasmic Reticulum Function to Protect Cells from Chronic Stress. Dev Cell. 2007;13:351–364. doi: 10.1016/j.devcel.2007.07.005. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H, Harada A, Mori K. Transcriptional Induction of Mammalian ER Quality Control Proteins Is Mediated by Single or Combined Action of ATF6alpha and XBP1. Dev Cell. 2007;13:365–376. doi: 10.1016/j.devcel.2007.07.018. [DOI] [PubMed] [Google Scholar]
- Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, Gannon M, Ma K, McNaughton K, Cavener DR. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol. 2002;22:3864–3874. doi: 10.1128/MCB.22.11.3864-3874.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.