

Summary
Objectives
The goal of the present study was to develop a chemical seizure model using the convulsant, 3-mercaptopropionic acid (3-MPA). A pharmacodynamics approach was taken, combining in vivo microdialysis sampling with electrophysiological methods to simultaneously monitor, in real-time, the 3-MPA concentration in the brain and the corresponding electrocorticographic (ECoG) activity.
Methods
The 3-MPA was administered in two doses (50 and 100 mg/kg) in order to study its pharmacokinetics. Microdialysis samples were collected from the striatum, hippocampus, and jugular vein every 5 min. The microdialysates were analyzed using high-performance liquid chromatography with electrochemical detection (HPLC-EC). The ECoG activity was monitored via screws placed onto the cortex. Noncompartmental pharmacokinetics analysis was performed to obtain the elimination constants (Ke), the maximum concentration (Cmax), the time to achieve maximum concentration (Tmax), and the area under the concentration—time curves (AUCinf).
Results
The average brain Ke for the 50 and the 100 mg/kg doses were 0.060 and 0.018 min−1, respectively. The brain AUCinf for the 50 and 100 mg/kg doses were 353 and 2168 mg min−1 mL−1, respectively. This led to a 67-fold increase in the observed number of seizures in the higher dose with the average seizure intensity double that of the smaller dose. These data led to the dosing scheme for the chemical seizure model of administering a 3-MPA loading dose of 60 mg/kg followed by a constant infusion of 50 mg/(kg min−1).
Conclusions
This study describes, to our knowledge, the first successful attempt to combine in vivo microdialysis with electrophysiology to monitor in real-time, the concentration and effects of 3-MPA in the brain. This led to the development of a steady-state chemical seizure model.
Keywords: Epilepsy, Seizure models, 3-Mercaptopropionic acid, Microdialysis, Pharmacokinetics, High-performance liquid chromatography
Introduction
Epilepsy affects approximately 1% of the world population (Fisher and Coyle, 1991) of which an estimated 20% are resistant to current medications (Burnham et al., 2002). This explains the interest in seizure models, as they allow probing into mechanisms of seizure generation, which if fruitful, may translate into more efficacious therapies. Chemical seizure models provide information that is unique to them and easier to obtain, than that provided by other models such as kindling. The mechanisms of action and the seizures induced by 3-mercaptopropionic acid (3-MPA) (Sprince et al., 1969; Sprince et al., 1970; Loscher, 1973) have been well characterized (Skeritt and Johnston, 1983; Mares et al., 1993; Netopilova et al., 1997); 3-MPA decreases γ-aminobutyric acid (GABA) concentrations in the brain (de Lores Arnaiz et al., 1972; de Lores Arnaiz et al., 1973; Fan et al., 1981; Timmerman et al., 1992) by inhibiting glutamatic acid decarboxlylase (GAD) (Lamar, 1970; Tunnicliff, 1990), that converts glutamate (Glu) to GABA. The imbalance between Glu and GABA, the main excitatory and inhibitory neurotransmitters in brain, respectively, manifests in this model as generalized seizures. Quantitative data about the changes in Glu and GABA, as a function of the concentration of 3-MPA in the brain, is lacking. If available, knowledge of the concentration of 3-MPA in the brain would serve as an important independent variable that can be used to investigate the type and degree of correlation between it and changes in the Glu/GABA ratio. Also, a correlation could be formed between the changes in Glu and GABA and the seizure frequency or rate and intensity.
The ability to monitor the concentration of 3-MPA in real-time, using in vivo microdialysis (Tossman and Ungerstedt, 1986; Robinson and Justice, 1991; Davies, 1999; de Lange et al., 2000; Ungerstedt et al., 2000; Weiss et al., 2000) would allow its regulation and achievement and maintenance of a steady-state concentration. This step will enable investigation of seizure frequency or rate and intensity, under conditions that either eliminate or allow precise tracking of fluctuations of the independent variable (convulsant concentration in the brain), thus allowing more accurate interpretation of results and better insight into seizure dynamics. Microdialysis sampling is a well-known technique that can be employed for monitoring in vivo pharmacokinetics. This technique involves implanting a probe containing a semi-permeable membrane into the brain or other organ of interest and collecting samples that will contain the analytes of interest. A solution that is isotonic to that of the cerebral spinal fluid (CSF) is constantly perfused through the implanted probe leading to no net loss of fluid. Microdialysis is an advantageous technique due to the semi-permeable membrane containing a particular molecular weight cutoff, which allows proteins and other large material to be excluded from the sample.
We describe to our knowledge, the first successful attempt to monitor in vivo and in real-time, the concentration and pharmacokinetics of 3-MPA in the brain and a dosing scheme for maintaining steady-state concentration in order to more accurately model the changes in neurotransmitters associated with epileptic seizures and how they may be correlated.
Methods
Animals
Male Wistar rats weighing 300−450 g (Charles River Laboratories, Wilmington, MA) were used. The animals were kept on 12 h light—dark cycles until the beginning of the experiment. Free access to food and water were allowed. The research described in this report was conducted in compliance with all applicable federal statutes and regulations related to animals and experiments involving animals and adheres to the principles stated in the Guide for the Care and Use of Laboratory Animals, NIH publication 86−23, 1996 edition.
Surgical procedure
Brain implantation of cortical electrodes, microdialysis guide cannula and probe
On the day of the experiment, rats were pre-anaesthetized with isoflurane. A subcutaneous injection of 67.5 mg/kg ketamine: 3.4 mg/kg xylazine: 0.67 mg/kg acepromazine was then administered for full anesthesia. Supplemental doses of 100 mg/mL ketamine were given at a rate of 0.2 mL/h to maintain the same plane of anesthesia. The anaesthetized rat was placed on a stereotaxic instrument (Harvard Appartus, Holliston, MA, USA) and then connected to a Homeothermic Blanket Control Unit (Harvard Apparatus, Holliston, MA, USA) where the body temperature was maintained at 37.0 ± 0.3 °C. A midline incision was made on the scalp and the skull was exposed. Four electrodes (1 mm o.d. stainless steel screws (Ace Hardware, Lawrence, KS, USA)) were placed over the cortex for recording of electrical activity. Two of the four electrodes were placed over the right hemisphere 4.2 mm anterior and 5.8 mm posterior and −1.4 mm lateral with respect to bregma; of the remainder electrodes, one was used as a ground electrode on the right hemisphere 5.8 mm posterior and +1.4 mm lateral with respect to bregma, and the other as reference (nasion).
Microdialysis intracerebral guide cannulas (CMA Microdialysis Inc., North Chemlsford, MA, USA) were implanted into the brain with the following coordinates: posterior 0.2 mm, lateral +3.2 mm, ventral 3.5 mm (striatum) and anterior 5.6 mm, lateral +4.8 mm, ventral 3.5 mm (hippocampus) with respect to the bregma (Paxinos and Watson, 1986). The guide cannulas were fixed to the skull surface with Duralay dental cement (Worth, IL, USA). A CMA/12 microdialysis probe with a 4 mm membrane (CMA Microdialysis Inc., North Chemlsford, MA, USA) was then placed through the guide cannula into both the striatum and hippocampus.
Fabrication and implantation of the jugular vein microdialysis probe
Vascular microdialysis probes were manufactured in-house using a modification to a previous concentric probe design (Telting-Diaz et al., 1992). Twelve-inch (probe inlet) and 6 in. (probe outlet) sections of polyimide tubing, 170 μm o.d. and 122 μm i.d. (MicroLumen Inc., Tampa, FL, USA) were UV glued into a 14 mm section of 40 kDa MWCO polyacrylonitrile (AN69HF) hollow fiber microdialysis membrane (Hospal Industries, Lyon, France). The final implantable microdialysis probe had a working semi-permeable membrane 10 mm in length.
A small incision was made in the neck, and the right jugular vein was dissected and ligated. An incision was made in the vein distal to the ligature, and the vascular microdialysis probe was inserted toward the heart and affixed in place with suturing. Tissue staples were used to close the neck incision.
Femoral vein cannulation
A 25 mm length of MRE-033 tubing was inserted in the femoral vein and affixed properly by suturing. The skin incision was closed with tissue staples.
Dosing schemes
Several dosing schemes were tested to firstly profile the kinetics of 3-MPA in the brain and blood, and secondly to find the scheme best suited for maintaining steady-state concentrations of the convulsant. The initial scheme was intraperitoneal (IP) bolus doses. The 3-MPA (Aldrich (Milwaukee, WI, USA)) dose was prepared by combining the proper volume of 3-MPA per rat body weight with sterile 0.9% saline. The next dosing scheme tested was multiple dosing regimens in which 3-MPA was administered as a loading dose of a pharmacokinetically determined concentration followed by booster doses of the convulsant. The third dosing scheme tested was the administration of a loading dose of 3-MPA followed by an intravenous (IV) constant infusion.
Experimental design
Following the implantation of the electrodes and the brain and vascular microdialysis probes, the rat was connected to a CMA/100 microinjection pump (CMA Microdialysis Inc., North Chelmsford, MA, USA). Both microdialysis probes were first perfused with a solution of 2.65 μg/mL 3-MPA at a rate of 1 μL/min for 3 h to allow for microdialysis probe calibration. Then both probes were flushed with artificial cerebrospinal fluid (aCSF) for microdialysis [145 mM NaCl, 2.7 mM KCl, 1.0 mM MgCl2, 1.2 mM CaCl2, 0.45 mM NaH2PO4, and 2.33 mM Na2HPO4, pH 7.4], at a rate of 1 μL/min for 3 h to allow for cleansing and stabilization of the probe environment. NANOpure water (18.2 Ω, Labconco, USA) was used in preparing all solutions.
The rats were divided into five groups. Group 1 acted as a control and received a bolus dose of sterile 0.9% saline. Group 2 received a bolus dose of 3-MPA in order to study the pharmacokinetics of 3-MPA. Groups 3 and 4 received either multiple doses of 3-MPA or a loading dose of 3-MPA followed by a constant infusion in order to determine which scheme will allow the attainment of a steady-state concentration for longer periods of time. Group 5 received the dosing scheme determined to provide the steady-state concentration most effectively simultaneously to two different brain regions, the striatum and hippocampus, in order to compare the method in different areas of the brain. Following the administration of 3-MPA, microdialysate samples were collected every 5 min until the termination of the experiment. The calculated lag time for the brain microdialysis probe to flush the exit tubing was 1 min and for the vascular microdialysis probe was 2 min. These lag times were taken into account when analyzing the data. The microdialysis samples were analyzed the day of the experiment.
Microdialysis probe calibration
In vitro vascular microdialysis probe
The in vitro vascular microdialysis probe calibration was determined by placing the semi-permeable membrane of the probe into a bath of aCSF that was stirred and heated to 37 °C. A solution of 2.65 μg/mL 3-MPA was delivered through the probe at a flow rate of 1 μL/min and the dialysate was collected and analyzed. The following equation was used to calculate the percent delivery of 3-MPA through the microdialysis probe:
| (1) |
where Cp is the concentration of 3-MPA in the perfusate and Cd is the concentration of 3-MPA in the dialysate.
In vivo vascular and brain microdialysis probes
The in vivo vascular and brain microdialysis probes were calibrated using the same procedure described above only post-implantation. Eq. (1) was used to again calculate the percent delivery of 3-MPA through the microdialysis probe. The concentration of 3-MPA in the dialysate was obtained using this probe correction factor.
Electrocorticographical (ECoG) recording
ECoG recordings were obtained beginning 30 min prior to dosing with 3-MPA (baseline). The signals were acquired through a Biomedical amplifier (SA Instruments, San Diego, CA, USA) using a 0.1 Hz high pass filter, a 6000 Hz low pass filter, a sampling rate of 15 kHz and a NIDAQ PCI 6731 data acquisition card (National Instruments, Austin, TX). The data were stored in the computer hard drive and analyzed, using custom built software (Osorio et al., 1998, 2001). The total number of seizures, their duration and intensity were determined using an algorithm developed by Osorio et al. (1998, 2002). Seizures are detected by filtering the ictal component from the raw ECoG using a wavelet spectral filter that enhances frequencies between 8 and 42 Hz, with a peak at 25 Hz and computing in real-time the median power of the filtered foreground (2 s), and dividing it by the median power of the filtered background (30 min). The resulting ratio is an estimate of the instantaneous power in the high frequency bands (15−30 Hz). For this study, seizures were defined as any automated detection with a maximal ratio that reached a threshold of 22.
Microdialysis sample analysis
In total, 2 μL of microdialysis sample was directly injected onto a Synergi 4 μ Hydro-RP column (150 mm × 2.0 mm, Phenomenex, Torrance, CA, USA). A liquid chromatographic system with electrochemical detection was used to analyze the samples. The liquid chromatographic system consisted of a Shimadzu LC-20AD pump, and a Rheodyne 9725i PEEK sample injector valve connected to a Phenomenex C18 guard column. The mobile phase consisted of 25 mM monobasic sodium phosphate and 0.5 mM disodium EDTA with the pH adjusted to 2.5 using 85% o-phosphoric acid. Methanol (Fisher, Pittsburgh, PA, USA) was then added making the final composition 80% phosphate buffer:20% methanol (v:v). The electrochemical detector consisted of a thin-layer gold mercury amalgam electrode. Preparation of the gold electrode was as described by Allison and Shoup (1983). In brief, a 3 mm gold electrode embedded in a PEEK block (Bioanalytical Systems, West Lafayette, IN, USA) was polished with 0.3 μm alumina powder, then rinsed with 100% methanol followed by copious amounts of water. Triple-distilled mercury (Bethlehem Apparatus Company (Hellertown, PA, USA) was placed onto the surface of the electrode and allowed to remain for 5 min before removal with the edge of a notecard. The amalgamation process was allowed to occur overnight before placement into the electrochemical flowcell. The electrode was set at a potential of +100 mV versus a Ag/AgCl reference electrode. The optimal detection potential was determined from a hydrodyanamic voltammagram of 3-MPA. This potential was controlled by a LC-4C potentiostat (Bioanalytical Systems, West Lafayette, IN, USA). The data were collected at 10 Hz and processed using a Chrom&Spec Chromatography Data System (Ampersand International, Beach-wood, OH, USA).
Pharmacokinetics analysis
Noncompartmental PK analysis was performed on the data using WinNonlin (Pharsight Corporation, Mountain View, CA, USA). The parameters of interest obtained from the blood and brain microdialysates were the elimination constant (Ke), time to achieve maximum concentration (Tmax), maximum concentration (Cmax), the area under the curve (AUCinf), and the area under the curve per dose administered (AUCinf/dose). The paired t-test was used to study statistical significance of the data.
Results
Control experiments
A bolus dose of 0.9% saline was administered to the rat while recording the ECoG response. No changes were observed from basal ECoG activity during these control injections (data not shown).
Bolus dosing
The 3-MPA was administered as an IP bolus dose at two different concentrations, 50 mg/kg (n = 5) and 100 mg/kg (n = 5). The resulting pharmacokinetics curves are shown in Fig. 1. Semi-logarithmic concentration versus time plots are shown for 3-MPA in the blood and in the brain. The slopes of the concentration—time curves appear parallel for the blood and brain in the 50 mg/kg 3-MPA dose. This is not the case, however, for the 100 mg/kg 3-MPA dose. The slopes of the blood and brain concentration—time curves for the higher dose appear to converge onto each other, and if extrapolated, appear that they may cross paths. It is inferred from the pharmacokinetics curves and the ECoG data that, in the brain, the minimum effective concentration (MEC) of 3-MPA needed to induce seizure activity is 2.65 μg/mL and the minimum toxic concentration (MTC) for the convulsant is 22.2 μg/mL. These observations may be explained by the pharmacokinetics parameters shown in Table 1A. There are significant differences in the pharmacokinetics data for both the 50 and 100 mg/kg doses. The Ke values are very similar in both the blood and the brain for the 50 mg/kg dose as was depicted on the semi-log concentration—time curves (Fig. 1A). At the 100 mg/kg dose where the slopes appear to converge upon one another, the Ke values for the blood and brain differ (p < 0.05). The Tmax differs significantly (p < 0.10) between the blood and brain for the 100 mg/kg dose. The AUCinf/dose (p < 0.01) values differ significantly between the blood and the brain for both doses administered. The Cmax values differ between the blood and the brain for the 50 mg/kg dose (p < 0.01) whereas a lesser difference lays between the blood and the brain the 100 mg/kg dose (p < 0.05).
Fig. 1.
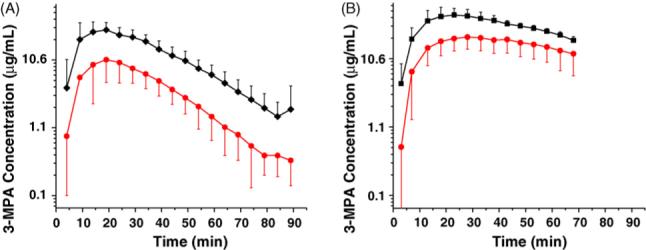
Semi-log pharmacokinetics curves from blood and brain microdialysates for 3-MPA bolus dosing (n = 5): (A) 50 mg/kg IP bolus dose; (B) 100 mg/kg IP bolus dose [blood microdialysate (◆); brain microdialysate (•)]. 3-MPA was administered at t = 0 min on the x-axis.
Table 1.
Pharmacokinetics parameters for 50 and 100 mg/kg 3-MPA bolus doses
| Dose | Sample | Ke (min−1) | Tmax (min) | Cmax (μg/mL) | AUCinf (mg min μL−1) | AUCinf/dose (min μL−1) |
|---|---|---|---|---|---|---|
| A | ||||||
| 50 mg/kga | Blood | 0.052 ± 0.012 | 15.0 ± 4.5 | 32.8 ± 12.1d | 1125 ± 387 | 23 ± 8d |
| Brain | 0.060 ± 0.016 | 18.0 ± 2.2 | 10.8 ± 5.9 | 353 ± 178 | 7 ± 4 | |
| 100 mg/kga | Blood | 0.026 ± 0.014b | 21.0 ± 2.7b | 47.6 ± 13.0c | 3171 ± 641 | 32 ± 6d |
| Brain | 0.018 ± 0.014 | 31.0 ± 8.4 | 22.4 ± 7.4 | 2168 ± 973 | 22 ± 10 | |
| B | ||||||
| 50 mg/kg | Blood/brain | 0.89 ± 0.14b | 0.82 ± 0.18 | 3.36 ± 1.06 | 3.54 ± 1.27c | |
| 100 mg/kg | Blood/brain | 1.52 ± 0.47 | 0.72 ± 0.22 | 2.64 ± 1.97 | 1.59 ± 0.40 |
A: average data from the blood and brain microdialysates; B: blood/brain ratios for each parameter.
n = 5 rats.
p < 0.10.
p < 0.05.
p < 0.01.
The blood and brain pharmacokinetics across doses have significant differences as well. The Ke values for the blood and brain both differ (p < 0.01) between the 50 and 100 mg/kg doses. There is also a difference (p < 0.05) for the Tmax for both the blood and the brain across doses. The Cmax for the brain differs significantly (p < 0.05) between the two doses, whereas the blood Cmax values are not. The AUCinf/dose differs (p < 0.05) for both the brain and the blood pharmacokinetics.
The blood/brain data are shown in Table 1B. For the 50 mg/kg dose, the Ke is around 1. There is a noticeable difference (p < 0.05) in the Ke blood/brain values between the two doses, however. The other significant difference arises with the AUCinf blood/brain values for each dose (p < 0.05). The AUCinf in the brain for the smaller dose is 31% of that in the blood while at the higher dose it is 68%. The average maximum concentration of 3-MPA in the brain is 33% of that in the blood for the 50 mg/kg dose, whereas for the 100 mg/kg dose, it is 47%. These values do not differ significantly.
A typical raw electrocortigram displaying 3-MPA seizure activity using single channel continuous recording is shown in Fig. 2. These raw data are filtered through a seizure detection algorithm (Osorio et al., 1998, 2002) to provide pertinent information regarding the dosing scheme applied. A typical ECoG recording after manipulation via the seizure detection algorithm is shown in Fig. 3A and B for the administration of a 50 and 100 mg/kg bolus doses, respectively. Seizure detections are denoted by increases in the algorithm ratio to or above 22. After administration of the 50 mg/kg dose (Fig. 3A) short-lived seizure activity occurred on average within 11.3 min. For the 100 mg/kg dose (Fig. 3B), seizure activity occurred on average within 8.5 min and was much longer lived. These data are summarized in Table 2. There is a statistically significant difference (p < 0.01) in the number of seizures detected between the 50 and 100 mg/kg doses. The maximum seizure intensity (Rmax) is on average double for the 100 mg/kg dose versus the 50 mg/kg dose, but is not significantly different over all of the experiments.
Fig. 2.
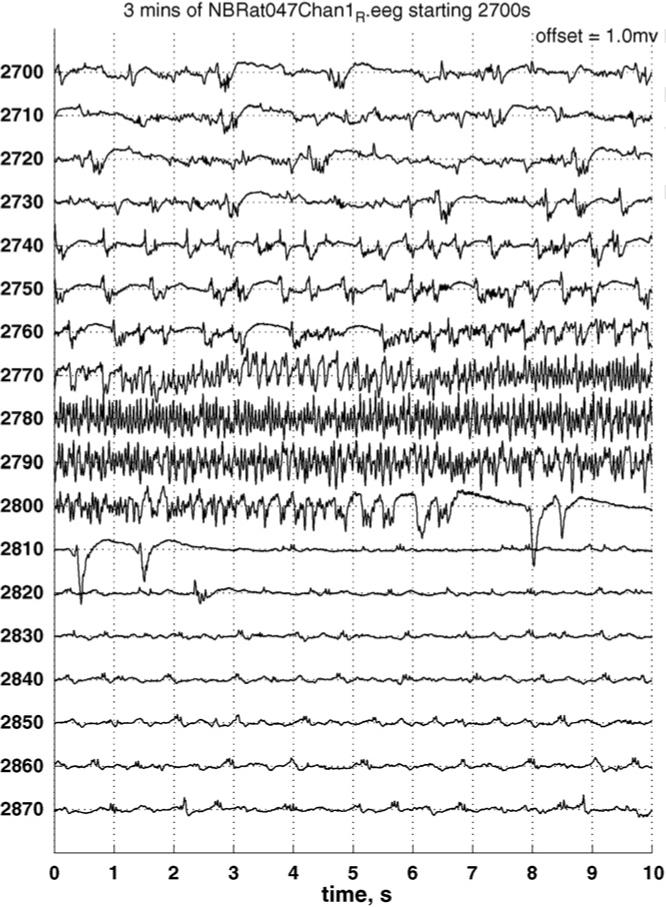
Electrocortigram showing a typical 3-MPA seizure with onset at approximately 2765 s and termination at 2808 s and “interictal” epileptiform discharges, recorded from two subdural electrodes placed symmetrically on each side of bregma.
Fig. 3.
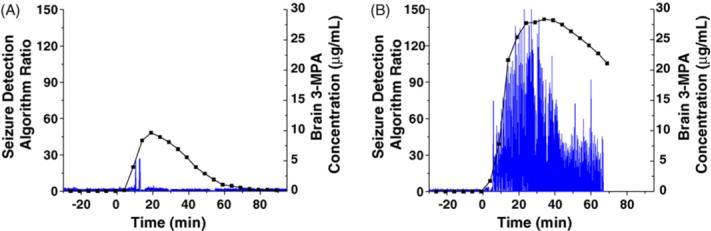
Example seizure detection algorithm ratio plots: (A) representative data for brain 3-MPA concentration superimposed onto the ECoG recording for a 50 mg/kg bolus dose; (B) representative data for brain 3-MPA concentration superimposed onto the ECoG recording for a 100 mg/kg bolus dose. The x-axis denotes the time (t = 0 corresponds to the 3-MPA administration), and the y-axis denotes an estimate of the seizure content (intensity) of the raw ECoG signal. Increases in the algorithm ratio to 22 or above correspond to seizures.
Table 2.
ECoG data for bolus dosing and steady-state seizure model with the correlation of 3-MPA concentration in the brain
| Dose | Latency to seizure onset (s) | Brain [3-MPA] at seizure onset (μg/mL) | Number of seizures detectedc | Average seizure durationd (s) | Rmax |
|---|---|---|---|---|---|
| 50 mg/kga | 680.7 ± 123.5 | 6.11 ± 3.95 | 9 ± 2 | 1.72 ± 4.00 | 61.7 ± 38.1 |
| 100 mg/kga | 510.2 ± 305.2 | 9.75 ± 3.00 | 603 ± 85 | 1.03 ± 2.11 | 123.8 ± 69.5 |
| 60 mg/kg bolus + 50 mg/(kg min−1) infusionb | 363.2 ± 148.8 | 4.80 ± 2.03 | 592 ± 187 | 0.87 ± 1.78 | 71.8 ± 22.2 |
n = 4.
n = 6.
p < 0.01.
Represents the average of the average seizure duration for each experiment.
Multiple dosing regimens
In the next portion of the study, two multiple dosing regimens were employed in an attempt to obtain a steady concentration of 3-MPA in the brain. Based on the single dose pharmacokinetics the first dosing regimen consisted of administering an IP loading dose of 50 mg/kg followed by two IP maintenance doses of 46.7 mg/kg at 30 min intervals. These values were arrived at by using the maintenance dosing equation shown below (Schoenwald, 2001):
| (2) |
A τ of 30 min represented approximately one elimination half-life for the 50 mg/kg dose. The second dosing regimen consisted of administering an IP loading dose of 50 mg/kg followed by two IP maintenance doses of 42.3 mg/kg at 45 min intervals. The τ of 45 min represented approximately two elimination half-lives for the loading dose.
Fig. 4A and B depicts the concentration—time curves for the two models. For the first model, τ = 30 min, the maximum concentration in the brain of 32.9 μg/mL occurred just before the rat died. When maintenance dosing was extended to every 45 min, the maximum concentration in the brain was 18.6 μg/mL. Only one experiment of each dosing regimen was carried out due to the lethal effect of the first dosing regimen and not achieving a steady 3-MPA concentration in the brain with either regimen. These results are consistent with the bolus dosing experiments in which the hypothesized MTC in the brain is 22.2 μg/mL.
Fig. 4.
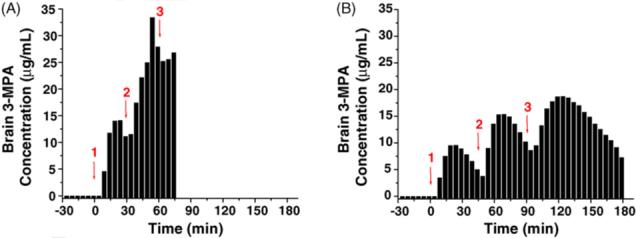
Concentration—time curves for different multiple dosing schemes of 3-MPA: (A) 50 mg/kg IP bolus dose administered at t = 0 min on the x-axis with booster doses of 46.7 mg/kg administered on 30 min intervals (n = 1 rat); (B) 50 mg/kg IP bolus dose administered at t = 0 min on the x-axis with booster doses of 42.3 mg/kg administered on 45 min intervals (n = 1 rat) (1 = administration of loading dose; 2, 3 = administration of booster doses).
Constant infusion dosing
The next set of experiments involved using a loading dose followed by a constant infusion in an attempt to attain and maintain a steady-state concentration of 3-MPA in the brain. An IV loading dose of 60 mg/kg 3-MPA was chosen, then immediately followed by a femoral infusion of 50 mg/(kg min−1). The infusion was stopped 50 min after the initial administration. Fig. 5A and B shows the concentration—time curves using this constant infusion regimen for the blood and the brain. Fig. 5C displays a typical experimental data set with the 3-MPA concentration—time plot superimposed onto the corresponding EcoG recording. The time interval between 20 and 50 min, denoted in a box on each plot, is the time during which a steady brain concentration of 3-MPA is achieved. Table 2 displays the ECoG data for this dosing scheme. Seizure activity using the constant infusion dosing was obtained on average within 6.1 min, and the seizures were long-lived resembling those of the 100 mg/kg bolus dose. The average number of seizures obtained with the constant infusion dosing (592) differed significantly (p < 0.05) with the average number of seizures obtained with the 50 mg/kg bolus dose (nine), but not with the 100 mg/kg dose (603). The average seizure intensity for this scheme, while more similar in value to the 50 mg/kg bolus dose, was not significantly different from either of the intensities arising from the administered bolus doses.
Fig. 5.

Blood and brain concentration profiles for constant infusion dosing of 3-MPA: (A) average blood 3-MPA concentration (n = 9 rats); (B) average brain 3-MPA concentration in striatum (n = 16 rats); (C) representative plot displaying ECoG recording with the corresponding brain 3-MPA concentration superimposed. The 3-MPA was administered at t = 0 min. The boxes in each plot (t = 20−50 min) represent the time when a steady-state concentration of 3-MPA was achieved in the brain. The arrow represents the time in which the infusion was stopped.
The elimination pharmacokinetics for the infusion model (Table 3A) for the blood and brain are significantly different (p < 0.01) compared to that of the bolus dose of 50 mg/kg 3-MPA (data from Table 1). The infusion model elimination pharmacokinetics, however, are more similar to the 100 mg/kg bolus dose of 3-MPA differing significantly from the blood and the brain (p < 0.05 and 0.10, respectively).
Table 3.
Elimination pharmacokinetic parameters for constant infusion model
| Dosing scheme | Sample | Ke (min−1) |
|---|---|---|
| Constant infusion | Blood | 0.034 ± 0.019 |
| Brain | 0.030 ± 0.011 | |
| Constant infusion | Blood/brain | 1.34 ± 0.48 |
A: average data from the blood (n = 9 rats) and brain (n = 16 rats); B: blood/brain ratios for elimination parameters.
The blood/brain Ke data for the infusion model are significantly different from the 50 mg/kg dose (p < 0.01), whereas there is no difference between this value and the 100 mg/kg dose.
Comparison of striatum versus hippocampus dosing
The final set of experiments involved comparing two different brain regions, the striatum and the hippocampus, employing the constant infusion dosing scheme. Again, an IV loading dose of 60 mg/kg 3-MPA was administered immediately followed by an IV infusion of 50 mg/(kg min−1). The infusion was ceased at 50 min post-introduction. Microdialysis samples were collected every 5 min from the striatum and hippocampus. Fig. 6A and B details brain concentration—time plots for the constant infusion dosing regimen. The average concentration of 3-MPA achieved within the hippocampus is approximately 31% higher than that obtained in the striatum. Table 4 details these observations. The Cmax differs significantly (p < 0.05) between the striatum and hippocampus during the steady-state phase of the infusion model. The Ke does not differ significantly between the striatum and hippocampus. The 3-MPA eliminates from the hippocampus at a comparable rate to that of the striatum.
Fig. 6.
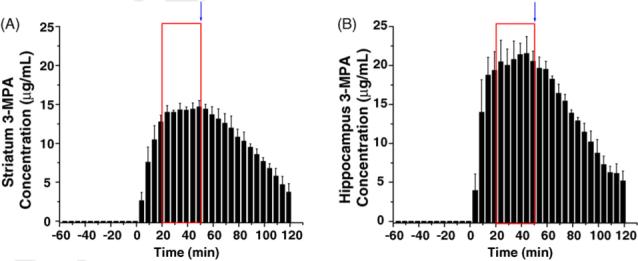
Brain concentration vs. time profiles for constant infusion dosing of 3-MPA measured from the striatum and hippocampus simultaneously (n = 3 rats). The 3-MPA was administered at t = 0 min. Boxes represent the time in which a steady-state concentration of 3-MPA was achieved in each region. The arrow represents the time in which the infusion was stopped.
Table 4.
Comparison of constant infusion dosing model in striatum and hippocampus (n = 3 rats)
| Cmax (μg/mL)a | Ke (min−1) | |
|---|---|---|
| Striatum | 14.9 ± 0.6 | 0.020 ± 0.007 |
| Hippocampus | 21.7 ± 1.9 | 0.023 ± 0.003 |
p < 0.05.
Discussion
The major findings in this study concern the in vivo pharmacokinetics of 3-MPA and the ability to track in real-time the convulsant's concentration in the brain and the development of a scheme to control the concentration of 3-MPA in the brain.
3-MPA pharmacokinetics
Two doses (50 and 100 mg/kg) were used to evaluate the pharmacokinetics of 3-MPA. From these two concentrations and the ECoG recordings, it is hypothesized that the MEC and MTC for 3-MPA in the rat brain are 2.65 and 22.2 μg/mL, respectively. One major observation of these experiments was that the rats dosed with the higher concentration of 3-MPA did not survive as long as those dosed with the lower concentration. At the conclusion of the experiments where the rats were dosed with 50 mg/kg, the average concentration of 3-MPA remaining in the brain was approximately 0.53 μg/mL whereas for the rats dosed with 100 mg/kg, the final brain concentration measured was approximately 12.7 μg/mL. This shows that a saturation is occurring in the brain with the higher dose of 3-MPA. The pharmacokinetics parameters show this effect also. The Ke for the 100 mg kg−1 dose is three times smaller than that of the 50 mg/kg dose. The Cmax for the brain was significantly different (p < 0.05) between the two doses. This is a much smaller difference than that observed for the blood Cmax values.
Another interesting observation was the blood/brain data for each dose. At the lower dose, the slopes of the blood and brain elimination curves appear parallel. This led to approximately a 33% ratio of brain 3-MPA to blood 3-MPA for both the Cmax and the AUCinf. However, at the higher dose these parameters are much different. The Cmax for the brain is 47% that of the blood while the AUCinf in the brain is 68% that of the blood. These data again point to a saturation effect occurring in the brain upon administration of higher doses of 3-MPA. The AUCinf/dose blood/brain data differs between doses (p < 0.05). At the 50 mg/kg dose, the AUCinf of 3-MPA in the brain is on average 3.5 times less than the 3-MPA in the blood. However, at the 100 mg/kg dose, the brain 3-MPA is only 1.5 times less than that in the blood. This again shows the saturation effect that is dose dependent in the rat.
The ECoG data from the bolus dosing is displayed in Table 2. The latency period for the onset of seizures after administration of the 100 mg/kg bolus dose is about 3 min shorter than that of the 50 mg/kg bolus dose. This corresponds to a slightly higher average concentration in the brain at onset (9.75 μg/mL versus 6.11 μg/mL, respectively). This represents a statistically significant difference in the concentrations at seizure onset (p < 0.05). There is a 67-fold increase in the average number of seizures detected for the 100 mg/kg dose compared to the 50 mg/kg dose (p < 0.01). The corresponding seizure duration times show that the average seizure lasts longer for the 50 mg/kg dose (1.72 s) than for the 100 mg/kg dose (1.03 s). This is directly related to the total number of seizures observed for each experiment. For the 100 mg/kg dose, there are more total seizures observed per experiment and their average duration is shorter than the lower dose administered. The range for the seizure durations for the 100 mg/kg dose is also less variable than the 50 mg/kg dose as shown by the average standard deviation. The average Rmax is higher for the corresponding higher bolus dose, but not statistically different from that of the lower dose.
Dosing regimen for steady 3-MPA in the brain
The first attempt to achieve a steady concentration of 3-MPA in the brain was by multiple dosing. Due to the accumulation of 3-MPA in the brain with the dose of 100 mg/kg, the lower dose of 50 mg/kg was used as a loading dose. When a maintenance dose was administered every 30 min (Fig. 4A), an accumulation of 3-MPA is observed in the brain which led to a toxic effect with a maximum concentration in the brain of 32.9 μg/mL. This is well above the noted MTC of 22.2 μg/mL. The timing of the maintenance dose was extended to 45 min (Fig. 4B) allowing more of the 3-MPA to eliminate from the brain before additional dosing. In this case, the rat fell out of seizure before each successive dose (data not shown). Due to not being able to achieve a steady 3-MPA concentration in the brain with this method, it was abandoned for one using constant infusion.
The second attempt to achieve a steady-state concentration of 3-MPA in the brain was by administering a constant infusion. An IV loading dose of 60 mg/kg was administered, and then immediately followed by a femoral infusion of 50 mg/(kg min−1). The infusion was continued for 50 min following its onset before being removed. The loading dose was chosen to be higher than the CD50 of 50 mg/kg as reported by Mares et al. (1993) in order to assure seizure activity would occur before the infusion took effect. Fig. 5B shows success 20 min after the loading dose/start of infusion with a steady 3-MPA concentration in the brain. In these experiments, the concentration of 3-MPA was held steady in the brain for a time period of 30 min (as shown boxed in). This time frame is a parameter that could be shortened or broken into segments to study differences in seizure activities due to 3-MPA. It was found that 30 min of steady-state convulsant in the brain was the longest time acceptable for inducing seizure activity and still obtain meaningful data after cessation of the 3-MPA infusion.
To further examine the constant infusion dosing method, experiments were conducted in which both the striatum and hippocampus were examined by microdialysis for 3-MPA content. Fig. 6B displays again success in obtaining a steady-state of 3-MPA concentration in the hippocampus with approximately 20 min after the start of dosing. This is comparable with that of the striatum. The one noticeable difference with the hippocampus in the Cmax obtained with the constant infusion dosing. The Cmax in the hippocampus (21.7 μg/mL) is significantly higher than that obtained in the striatum (14.9 μg/mL). While still below the previously noted MTC of 22.2 μg/mL for the striatum, the concentration in the hippocampus is slowly approaching this value. This would then further the finding that 30 min of steady-state convulsant in the brain is an acceptable timeframe to induce seizures without the possibility of crossing the MTC threshold.
The elimination pharmacokinetics parameters are significantly different from that of the administration of both a bolus dose of 50 and 100 mg/kg. The infusion model elimination kinetics are more similar to those of the 100 mg/kg bolus dose, however, where saturation within the brain is observed. This accumulative effect within the brain during the infusion model can be discussed comparatively to that of the 100 mg/kg bolus dose. The infusion model elimination kinetics (Table 3) show that these data do not differ significantly when compared against the 100 mg/kg dose. These data point to more 3-MPA residing within the brain when the infusion occurs. This may explain why all of the 3-MPA is not eliminated before the experiment is finished. The rats only survive approximately 70 min after the removal of the infusion of 3-MPA and the average concentration in the brain upon death was 1.06 μg/mL. The slow elimination kinetics are believed to play a role in this effect. The elimination kinetics are also found to be similar when the striatum is compared with the hippocampus (Table 4). While it appears from Fig. 6 that the 3-MPA eliminates more rapidly from the hippocampus than the striatum, there is no statistical difference between the two brain regions.
The ECoG data for the infusion model are shown in Table 2. The latency to seizure onset for this model is significantly different from the 50 mg/kg bolus dose (p < 0.05), but not different from the 100 mg/kg dose. Interestingly, this difference in latency to first seizure is not explained by the 3-MPA concentration, since the infusion model's does not differ statistically from that of the 50 mg/kg dose, but it does differ from the 100 mg/kg dose (p < 0.05). This could possibly be explained by the dosing routes. The bolus dosing was administered IP, while the loading dose for the infusion model was administered IV. The 3-MPA concentration at the time of seizure onset could be closer to the 50 mg/kg dose due to the smaller amount of the convulsant introduced during the loading dose followed by the infusion with a small steady amount of convulsant.
Conclusion
This is the first study to our knowledge that reports on the pharmacokinetics of the known convulsant, 3-MPA. We were successful in monitoring in vivo and in real-time, the concentration of 3-MPA in the brain (both striatum and hippocampus), and were able to maintain a steady concentration for a given time period. This chemical seizure model will allow for the future analysis of neurochemical events as they are related to the convulsant concentration in the brain. Having this independent variable accessible for future experiments will allow for a better correlation of the neurochemical changes that occur due to the administration of 3-MPA.
Acknowledgements
The funding for this project was provided by the NIH (R01 HL069014) and the Alliance for Epilepsy Research (Kansas City, Kansas). The authors would like to thank Dr. Jinping Qiao for valuable assistance in surgical procedures and sample collection and Dr. Mark Frei for the ECoG analyses and insightful discussions of the data.
Footnotes
Publisher's Disclaimer: This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2−3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
References
- Allison LA, Shoup RE. Dual electrode liquid chromatography detector for thiols and disulfides. Anal. Chem. 1983;55:8–12. [Google Scholar]
- Burnham WM, Carlen PL, Hwang PA. Intractable Seizures: Diagnosis, Treatment, and Prevention. Kluwer Academic/Plenum; New York: 2002. [Google Scholar]
- Davies MI. A review of microdialysis sampling for pharmacokinetic applications. Anal. Chim. Acta. 1999;379:227–249. [Google Scholar]
- de Lange ECM, de Boer AG, Breimer DD. Methodological issues in microdialysis sampling for pharmacokinetic studies. Adv. Drug Deliv. Rev. 2000;45:125–148. doi: 10.1016/s0169-409x(00)00107-1. [DOI] [PubMed] [Google Scholar]
- de Lores Arnaiz GR, de Canal MA, De Robertis E. Alteration of GABA system and Purkinje cells in rat cerebellum by the convulsant 3-mercaptopropionic acid. J. Neurochem. 1972;19:1379–1385. doi: 10.1111/j.1471-4159.1972.tb01462.x. [DOI] [PubMed] [Google Scholar]
- de Lores Arnaiz GR, de Canal MA, Robiolo B, de Pacheco MM. The effect of the convulsant 3-mercaptopropionic acid on enzymes of the γ-aminobutyrate system in the rat cerebral cortex. J. Neurochem. 1973;21:615–623. doi: 10.1111/j.1471-4159.1973.tb06006.x. [DOI] [PubMed] [Google Scholar]
- Fan SG, Wusteman M, Iverson LL. 3-Mercaptopropionic acid inhibits GABA release from rat brain slices in vitro. Brain Res. 1981;229:379–387. doi: 10.1016/0006-8993(81)91001-5. [DOI] [PubMed] [Google Scholar]
- Fisher RS, Coyle JT. Neurotransmitters and Epilepsy. Wiley–Liss Inc.; New York: 1991. [Google Scholar]
- Lamar C. Mercaptopropionic acid: A convulsant that inhibits glutamate decarboxylase. J. Neurochem. 1970;17:165–170. doi: 10.1111/j.1471-4159.1970.tb02197.x. [DOI] [PubMed] [Google Scholar]
- Loscher W. 3-Mercaptopropionic acid: convulsant properties, effects on enzymes of the gamma-aminobutyric acid system in mouse brain and antagonism by certain anticonvulsant drugs, aminooxyacetic acid and gabaculine. Biochem. Pharmacol. 1973;28:1397–1407. doi: 10.1016/0006-2952(79)90443-x. [DOI] [PubMed] [Google Scholar]
- Mares P, Kubova H, Zouhar A, Folbergrova J, Koryntova H, Stankova L. Motor and electrocorticographic epileptic activity induced by 3-mercaptopropionic acid in immature rats. Epilepsy Res. 1993;16:11–18. doi: 10.1016/0920-1211(93)90034-5. [DOI] [PubMed] [Google Scholar]
- Netopilova M, Drsata J, Haugvicova R, Kubova H, Mares P. Inhibition of glutamate decarboxylase activity by 3-mercaptopropionic acid has different time course in the immature and adult rat brains. Neurosci. Lett. 1997;226:68–70. doi: 10.1016/s0304-3940(97)00241-3. [DOI] [PubMed] [Google Scholar]
- Osorio I, Frei MG, Giftakis J, Peters T, Ingram J, Turn-bull M, Herzog M, Rise MT, Schaffner S, Wennberg RA, Walczak TS, Risinger MW, Ajmone-Marsan C. Performance reassessment of a real-time seizure-detection algorithm on long ECoG series. Epilepsia. 2002;43:1522–1535. doi: 10.1046/j.1528-1157.2002.11102.x. [DOI] [PubMed] [Google Scholar]
- Osorio I, Frei MG, Manly BFJ, Sunderam S, Bhavaraju NC, Wilkinson SB. An introduction to contingent (closed-loop) brain electrical stimulation for seizure blockage, to ultra-short-term clinical trials, and to multidimensional statistical analysis of therapeutic efficacy. J. Clin. Neurophysiol. 2001;18:533–544. doi: 10.1097/00004691-200111000-00003. [DOI] [PubMed] [Google Scholar]
- Osorio I, Frei MG, Wilkinson SB. Real-time automated detection and quantitative analysis of seizures and short-term prediction of clinical onset. Epilepsia. 1998;39:615–627. doi: 10.1111/j.1528-1157.1998.tb01430.x. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Justice JB. Microdialysis in the Neurosciences. Elsevier Science Publishers BV; Amsterdam: 1991. [Google Scholar]
- Schoenwald RD. Pharamacokinetic Principles of Dosing Adjustments: Understanding the Basics. Technomic; Pennsylvania: 2001. p. 258. [Google Scholar]
- Skeritt JH, Johnston GAR. Enhancement of excitant amino acid release from rat brain slices by the convulsant 3-mercaptopropionic acid. Brain Res. 1983;258:165–169. doi: 10.1016/0006-8993(83)91245-3. [DOI] [PubMed] [Google Scholar]
- Sprince H, Parker CM, Josephs JA, Magazino J. Convulsant activity of homocysteine and other short-chain mercaptoacids: protection therefrom. Ann. NY Acad. Sci. 1969;166:323–325. doi: 10.1111/j.1749-6632.1969.tb46402.x. [DOI] [PubMed] [Google Scholar]
- Sprince H, Parker CM, Smith GG. 3-Mercaptopropionic acid: convulsant and lethal properties compared with other sulfur-convulsants; protection therefrom. Agents Actions. 1970;1:231–233. doi: 10.1007/BF01968695. [DOI] [PubMed] [Google Scholar]
- Telting-Diaz M, Scott DO, Lunte CE. Intravenous micro-dialysis sampling in awake, freely-moving rats. Anal. Chem. 1992;64:806–810. doi: 10.1021/ac00031a019. [DOI] [PubMed] [Google Scholar]
- Timmerman W, Zwaveling J, Westerink BHC. Characterization of extracellular GABA in the substantia nigra reticulata by means of brain microdialysis. Naunyn-Schmiedeberg's Arch. Pharmacol. 1992;345:661–665. doi: 10.1007/BF00164580. [DOI] [PubMed] [Google Scholar]
- Tossman U, Ungerstedt U. Microdialysis in the study of extracellular levels of amino acids in the rat brain. Acta Physiol. Scand. 1986;128:9–14. doi: 10.1111/j.1748-1716.1986.tb07943.x. [DOI] [PubMed] [Google Scholar]
- Tunnicliff G. Action of inhibitors on brain glutamate decarboxylase. Int. J. Biochem. 1990;22:1235–1241. doi: 10.1016/0020-711x(90)90304-l. [DOI] [PubMed] [Google Scholar]
- Ungerstedt U, Bellander B-M, Nordstrom C-H. [February 2003];2000 :1–8. Microdialysis in neuromonitoring: Principles, procedures, and interpretations. http://www.microdialysis.com/handbook/
- Weiss DJ, Lunte CE, Lunte SM. In vivo microdialysis as a tool for monitoring pharmacokinetics. Trends Anal. Chem. 2000;19:606–616. [Google Scholar]