

Abstract
Multiple myeloma is a fatal hematologic malignancy associated with clonal expansion of malignant plasma cells within the bone marrow and the development of a destructive osteolytic bone disease. The principal cellular mechanisms involved in the development of myeloma bone disease are an increase in osteoclastic bone resorption, and a reduction in bone formation. Myeloma cells are found in close association with sites of active bone resorption, and the interactions between myeloma cells, and other cells within the specialized bone marrow microenvironment are essential, both for tumor growth and the development of myeloma bone disease. This review discusses the many different factors which have been implicated in myeloma bone disease, including the evidence for their role in myeloma and subsequent therapeutic implications.
INTRODUCTION
Multiple myeloma is a hematologic malignancy characterized by the development of a progressive and destructive osteolytic bone disease, associated with severe bone pain, pathological fractures, osteoporosis, hypercalcemia and spinal cord compression. Despite many significant advances in the understanding of the biology of multiple myeloma, it remains an incurable malignancy, and the destructive osteolytic bone disease is a major cause of morbidity in patients with multiple myeloma.
Myeloma cells are found in close association with sites of active bone resorption, and the interactions between myeloma cells, osteoclasts, stromal cells, myeloma-associated fibroblasts and osteoblasts are crucial both for the development of the osteolytic bone disease and for myeloma cell growth and survival in the bone marrow (1, 2). The destructive nature of myeloma bone disease is increased by the vicious cycle that develops between myeloma cells and the bone marrow microenvironment. Although the precise molecular mechanisms responsible for the bone destruction in multiple myeloma are not completely understood, however it is known that the bone destruction is primarily mediated by osteoclasts, and that this destruction is exacerbated by a reduction in osteoblastic bone formation. Patients with multiple myeloma have abnormal bone remodeling, where resorption and formation become uncoupled, with the end result being an increase in bone resorption and a decrease in bone formation. Histomorphometric studies have demonstrated that bone resorption is increased in patients with multiple myeloma, with an increase in both the number and activity of osteoclasts (3–5). The increase in resorption is thought to be an early event in the development of myeloma bone disease (5). Although early stages of multiple myeloma have been associated with an increase in osteoblast recruitment, a very marked impairment of bone formation due to reduced osteoblast number and activity is a common feature in the later stages of the osteolytic bone disease (5–7). This has been confirmed in studies which demonstrate that markers of bone formation are decreased in patients with multiple myeloma (8, 9).
Over recent years, our understanding of the osteoclastogenic and osteoblastic factors involved in the development of myeloma bone disease has improved. The location of myeloma cells adjacent to sites of active bone resorption suggests that the mechanism for osteoclastic bone destruction in myeloma bone disease is locally mediated. In contrast, the generalized osteoporosis associated with multiple myeloma suggests that the reduction in bone formation may be due, at least in part, to systemic regulation. Factors which have been implicated include RANKL, OPG, MIP-1α, SDF-1, TGF-β, Dkk1 and sFRP-2 (Figure 1). Many of these factors have been demonstrated to be dysregulated in patients with multiple myeloma, and all may play a role in either bone resorption or bone formation, either alone or in combination. Thus, these factors represent potential therapeutic targets for the treatment of myeloma bone disease.
Figure 1. Factors implicated in myeloma bone disease.
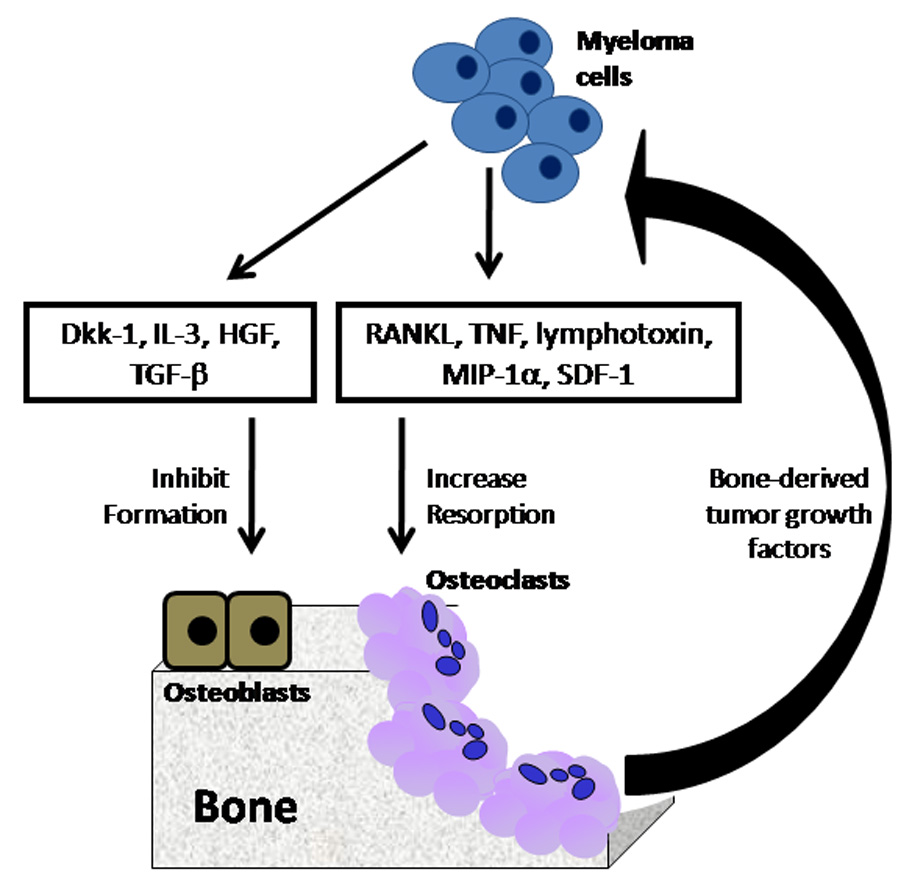
Myeloma bone disease is associated with an increase in osteoclastic bone resorption and a reduction in osteoblastic bone formation. A number of key factors have been identified which play a role in either bone resorption or bone formation.
PATHOPHYSIOLOGY OF INCREASED OSTEOCLASTIC BONE RESORPTION
Myeloma cells secrete a number of osteoclast activating factors which can promote osteoclast formation and activation, including lymphotoxin, TNF-α, IL-1, IL-3 and IL-6 (10–13). While in vitro studies have demonstrated a role for these factors in the development of myeloma bone disease, their role in vivo remains unclear. Cell-cell contact between myeloma cells and bone marrow stromal cells is critically important in the development of myeloma bone disease and plays a key role in the abnormal regulation of many factors implicated in myeloma bone disease. Although it has been known for more than 30 years that osteoclasts are hyperstimulated by cytokines in myeloma, there are still unresolved questions as to the nature of the local mediators responsible for the increased osteoclast recruitment and activation.
The RANK/RANKL/OPG System
The identification of the TNF and TNF receptor superfamily members RANK, RANKL and OPG, and their key role in osteoclast formation and activity has markedly increased our understanding of the mechanisms involved in myeloma bone disease. RANKL is expressed on the surface of osteoblasts and bone marrow stromal cells and the interaction between RANKL and its signalling receptor RANK, found on the surface of osteoclast precursor cells, stimulates osteoclast formation and bone resorption (14, 15) (15). OPG is a soluble decoy receptor which is secreted by bone marrow stromal cells and osteoblasts, and binds to RANKL, preventing its interaction with its cognate receptor RANK (16). The interaction between RANKL and RANK is critical in the formation and activation of osteoclasts, and inhibition by OPG results in inhibition of bone resorption. The critical role of these TNF family members is highlighted by in vivo studies where mice deficient in RANK or RANKL have extensive osteopetrosis due to a lack of functional osteoclasts (17, 18), whereas OPG knock out mice have severe osteoporosis (19).
In patients with multiple myeloma, the ratio of RANKL and OPG has been shown to be markedly dysregulated, with an increase in RANKL expression, and a concomitant decrease in OPG expression and serum concentration (20–24) An increase in the serum RANKL:OPG ratio has been demonstrated in patients with multiple myeloma, which correlates with both bone disease and survival (25). There is evidence to support both an increase in RANKL expression by bone marrow stromal cells and osteoblasts(20–22), and also for expression of RANKL by myeloma cells (26, 27). Indeed, in vitro studies have demonstrated that RANKL expression by myeloma cells can promote osteoclastic bone resorption (26). Human myeloma cells do not express or release OPG. However, myeloma cells can interact with bone marrow stromal cells and osteoblasts to reduce the concentration of OPG released from these cells, which is consistent with the hypothesis that decreased OPG may play a role in the pathogenesis of myeloma bone disease (20, 21, 28).
The key role of the RANK/RANKL/OPG system in the pathogenesis of myeloma bone disease is demonstrated by preclinical in vivo studies which target this system using either a recombinant OPG construct (Fc.OPG) or a soluble form of RANK (RANK.Fc). Administration of Fc.OPG to mice with established myeloma prevented the development of osteolytic bone lesions and was associated with a decrease in osteoclast number and bone mineral density (29). In a separate study, preventative treatment (from the time of tumour cell inoculation) of the 5T33 murine model of myeloma with recombinant Fc.OPG resulted in a significant decrease in serum paraprotein and tumour burden and increase in time to morbidity (30). This effect was attributed to the inhibitory effect of OPG on myeloma bone disease altering the bone marrow microenvironment and thus indirectly inhibiting myeloma growth. RANK.Fc functions as a RANKL antagonist which, in contrast to Fc.OPG, does not bind to TRAIL. Therefore the effects of RANK.Fc reflect specific inhibition of RANKL. RANK.Fc was shown to prevent the development of myeloma bone disease, and to reduce tumour burden in the 5TGM1 murine model of myeloma (31) and in the SCID-hu model of human myeloma (20, 32). Taken together, the use of preclinical models has both demonstrated the potential of targeting the RANK/RANKL/OPG system in treating myeloma bone disease, and raised the possibility that targeting this system may also influence myeloma growth and survival in the bone marrow.
MIP-1α
Macrophage inflammatory protein-1α (MIP-1α) is a chemokine that is produced by myeloma cells and induces osteoclast formation, thus making it a good candidate for a therapeutic target for the treatment of myeloma bone disease. In patients with multiple myeloma, serum levels of MIP-1α have been shown to correlate both with severity of bone disease and survival (33). Choi et al. identified MIP-1α as an osteoclastogenic factor in multiple myeloma, demonstrating both an increase in the concentration of MIP-1α in bone marrow plasma from patients with multiple myeloma, and that the osteoclastogenic activity of this plasma was inhibited by the addition of a neutralising antibody to MIP-1α (34). MIP-1α interacts with several chemokine receptors, and recent evidence suggests that its osteoclastogenic effect is mediated by the chemokine receptors CCR1 and CCR5, raising the possibility that specific antagonists to these receptors may be of therapeutic value in myeloma bone disease (35). The osteoclastogenic effect of MIP-1α in murine calvariae is not observed in RANK knockout mice, suggesting that the osteoclastogenic effect of this chemokine may be at least partially mediated through RANKL (36).
There have been several preclinical studies targeting MIP-1α in the development of myeloma bone disease. Choi et al. used ARH myeloma cells stably transfected with an anti-sense construct to MIP-1α in SCID mice to study the effect of inhibition of tumour-derived MIP-1α (37). Antisense inhibition of MIP-1α resulted in an inhibition of the development of myeloma bone disease, and also an increase in survival. Oyajobi et al. determined the effect of neutralising monoclonal antibodies to MIP-1α in the 5TGM1 murine model of myeloma (36). The antibodies significantly inhibited the development of myeloma bone disease, reduced tumour load and tumour growth in bone. MIP-1α thus appears to be a major mediator of osteoclastic bone destruction in myeloma bone disease, and may represent a therapeutic target for the treatment of myeloma bone disease.
VEGF
Vascular endothelial growth factor (VEGF) has an important role in the induction of neovascularization in solid tumors. However it was not until 1994 that the microvessel density of bone marrow in myeloma patients was defined to correlate with disease progression and poor prognosis (38). The majority of myeloma cells can secrete VEGF. In addition, VEGF can substitute for M-CSF in the induction of in vitro osteoclast differentiation (39). A recent study has demonstrated that blockade of both VEGF and osteopontin almost completely abrogated angiogenesis and bone resorption enhanced by coculture of myeloma cells and osteoclasts (40), suggesting VEGF may support osteoclastic bone resorption in myeloma.
SDF-1 alpha/CXCR-4
Stromal derived factor-1 alpha (SDF1; CXCL12) is a CXC chemokine. Its receptor is CXCR4 which expressed on hematopoeitic stem cells and lymphocytes, as well as malignant cells and osteoclast precursors (41–43). SDF-1 is expressed by bone marrow stromal cells and endothelial cells. SDF-1/CXCR-4 plays an important role in hematopoietic stem cell homing and tumor migration and proliferation. Recent data showed SDF-1 can not only mediate the migration and homing of myeloma cells (44), but increase osteoclast motility and bone resorbing activity (45). This was associated with an overexpression of osteoclast activation-related genes, including RANKL, RANKL, TRAP, MMP-9, CA-II and Cathepsin K. Osteoclast activation mediated by myeloma cells could be reduced by CXCR-4 specific inhibitor (45). These findings suggest that SDF-1α is an important factor in the pathogenesis of myeloma bone disease.
Cell-cell interactions
Myeloma cells are found in close association with sites of active bone resorption, and direct cell-cell interaction has been demonstrated to play an important role in both tumor growth and the development of myeloma bone disease. The adhesion of myeloma cells to bone marrow stromal cells activates the NF-kB pathway, resulting in the stimulation of myeloma cell growth, survival, drug resistance and migration. Interactions between myeloma cells and bone marrow stromal cells are known to up-regulate a number of factors which promote osteoclastogenesis, including IL-6 and RANKL. Interactions between alpha4-beta1 integrin and vascular cell adhesion molecule 1 (VCAM-1) have been demonstrated to increase the production of osteoclast stimulating activity, which could not be attributed to IL-6, TNF or PTHrP(46). Furthermore, inhibition of this activity, using a monoclonal antibody to alpha4 integrin was found to inhibit the development of multiple myeloma and the associated osteolysis in the 5TGM1 murine model of myeloma (47). Interactions between bone marrow stromal cells and myeloma cells have also been demonstrated to increase the expression of RANKL by the bone marrow stromal cells, with data supporting a role for the VCAM-1-α4b1 interaction (48). Further evidence for the importance of cell-cell interactions to disrupt the RANKL-OPG system was provided by the demonstration that interactions between myeloma cells and osteoblasts or bone marrow stromal cells decrease the production of OPG (21, 28). MIP-1 alpha is a critical factor in myeloma bone disease, and has been demonstrated to increase adhesion of myeloma cells to bone marrow stromal cells by interactions with CCR1 on osteoclasts and CCR5 on myeloma cells, thus potentiating myeloma growth and development of myeloma bone disease (35).
In addition to interacting with bone marrow stromal cells, myeloma cells can also adhere directly to osteoclasts. The direct interactions between myeloma cells and osteoclasts have been shown to both increase myeloma cell proliferation and increase osteoclastic differentiation (49, 50). These effects were only partially blocked by an antibody to IL-6, but were completely abrogated by inhibition of cellular contact, demonstrating the critical role of direct contact between myeloma cells and osteoclasts(50). Interactions between myeloma cells and osteoclasts have also been shown to result in an increase in the secretion of proangiogenic factors, and an increase in vascular tubule formation, suggesting a link between myeloma cells, osteoclasts and angiogenesis (40).
PATHOPHYSIOLOGY OF OSTEOBLAST INHIBITION
For many years, the increase in osteoclastic bone resorption was thought to be the primary mechanism for the development of myeloma bone disease. It is now recognized that a reduction in bone formation plays a critical role in the development of myeloma bone disease, resulting in both a generalized loss of bone and an inability to repair the osteolytic lesions which occur as a result of increased bone resorption. Although the precise cellular and molecular mechanisms which regulate osteoblast function in myeloma remain to be determined, several studies have identified a number of factors which are dysregulated in multiple myeloma and which may play a role in the suppression of osteoblast formation and differentiation, including Dkk1, sFRP-2, IL-3, Runx2 and TGF-β.
Inhibition of Wnt signaling
The Wnt signaling pathway plays a key role in the regulation of bone mass, as demonstrated by human genetic bone diseases and in vivo mouse models. Inactivating mutations in the gene for LRP5 result in osteoporosis-pseudoglioma syndrome in humans, whereas ‘gain of function’ mutations in LRP5 are associated with a syndrome of hereditary high bone density (51, 52). Over-expression of β-catenin in osteoblasts has been demonstrated to induce osteoblast proliferation and a high bone mass phenotype. (53), furthermore, increasing Wnt signaling by inhibition of GSK-3b has been demonstrated to increase bone mass in vivo (54). The key role of Wnt signaling in stimulating bone formation has raised the possibility that this signaling pathway may be important in diseases associated with abnormal bone formation, including multiple myeloma. In support of this, increasing Wnt signaling in the bone microenvironment in multiple myeloma, by inhibition of GSK-3b with LiCl, resulted in the inhibition of myeloma bone disease, using a murine model of myeloma (55).
There are 2 classes of extracellular antagonists of the Wnt signaling pathway, with distinct inhibitory mechanisms, acting either by binding directly to Wnt (secreted FRPs, WIF-1) or by binding to part of the Wnt receptor complex (Dkk family). Recent evidence suggests a role for each of these classes of inhibitors in the development of myeloma bone disease. The soluble, extracellular antagonist of the Wnt signaling pathway, Dkk-1, has been identified as a potential mediator of osteoblast dysfunction in myeloma bone disease (56). Dkk1 is expressed by osteoblasts and bone marrow stromal cells, and has been demonstrated to inhibit bone formation in osteoblasts in vitro (57). In vivo evidence of the critical role of Dkk1 in bone biology is provided by transgenic mice overexpressing the soluble antagonist of Wnt, Dickkopf 1 (Dkk1) in osteoblasts that develop severe osteopenia, whereas deletion of a single allele of Dkk1 resulted in an increase in bone mass (58, 59). Over-expression of Dkk1 in bone marrow aspirates from patients with multiple myeloma was initially identified by cDNA microarray. Dkk1 concentrations were increased in bone marrow plasma and peripheral blood of patients with multiple myeloma, as compared to healthy individuals or to patients with MGUS (56). In addition, osteoblast differentiation was blocked by bone marrow serum from patients with myeloma, and the inhibitory effect was found to be the result of Dkk1 in the serum. The development of myeloma bone disease is dependent upon interactions within the local bone marrow microenvironment and myeloma cells have been demonstrated to interact with osteoblasts and up-regulate expression of Dkk1 by osteoblasts (60). Furthermore, interactions between myeloma cells and mesenchymal stem cells have been demonstrated to promote the growth of myeloma cells, resulting in an increase in Dkk1 and consequent inhibition of mesenchymal stem cell differention into osteoblasts (61). Preclinical studies have demonstrated the potential for targeting Dkk1 in myeloma bone disease. The treatment of myeloma-bearing SCID-rab mice with anti-Dkk1 antibody reduced osteolytic bone lesions, increased osteoblast number and significantly reduced tumor burden (62). Secreted Frizzled related protein-2 (sFRP-2) is a soluble antagonist of Wnt signaling which has been shown to be expressed by human myeloma cells (63). Furthermore, both exogenous sFRP-2 and myeloma-cell derived sFRP-2 have been shown to play a key role in bone formation in vitro, raising the possibility that sFRP-2 may play a role in the development of myeloma bone disease.
Transforming Growth Factor-β
Transforming growth factor-beta is a ubiquitous, multifunctional growth factor that is released from the bone matrix during osteoclastic bone resorption and acts to inhibit osteoblast differentiation. In addition to inhibiting myeloma growth, inhibition of TGF-β signaling has been shown to block the ability of myeloma cells to inhibit osteoblast differentiation in vitro. These results suggest a role for TGF-β in mediating osteoblast dysfunction in myeloma, and the potential for targeting TGF-β in the treatment of myeloma bone disease (64).
Runx2
Runt-related transcription factor 2 (Runx2) is a transcription factor which acts to promote the formation and differentiation of osteoblasts from mesenchymal stem cells. Myeloma cells were found to inhibit Runx2 activity and reduce osteoblast differentiation in osteoprogenitor cells; an effect that was mediated by both cell-cell contact and interleukin 7 (65). In support of a role for Runx2 in the pathophysiology of myeloma bone disease, a significant reduction in the proportion of Runx2 positive osteoblasts was observed in patients with myeloma with evidence of osteolytic bone lesions, compared to those patients with no evidence of bone disease (65).
IL-3
Recent evidence suggests that IL-3 may play a role in regulation of bone formation in myeloma bone disease. (12, 66). While IL-3 had no effect on osteoblast cell lines, IL-3 inhibited basal and BMP-2 stimulated osteoblast formation from primary stromal cells in vitro. The inhibitory effects of IL-3 on osteoblast differentiation were found to be an indirect effect as a result of increasing the number of CD45+ hematopoietic cells in stromal cultures. Furthermore, bone marrow plasma from patients with myeloma was found to inhibit osteoblast differentiation, and this effect could be blocked with a neutralizing antibody to IL-3. These data identify IL-3 as a potential mediator of myeloma bone disease, with a dual effect to directly increase osteoclastic bone resorption, and indirectly inhibit osteoblast differentiation.
HGF
Hepatocyte growth factor (HGF) is produced by myeloma cells and is increased in the serum of patients with multiple myeloma (67) where levels of HGF are correlated with a poor prognosis (68). Furthermore, serum concentrations of HGF were negatively correlated with levels of bone specific alkaline phosphatase in patients with multiple myeloma, suggesting a role for HGF in myeloma bone disease (69). In support of this, HGF was found to inhibit BMP-induced osteoblastogenesis, including expression of the transcription factors Runx2 and Osterix, potentially by inhibiting nuclear translocation of receptor-activated SMADs (69).
Proteasome Inhibitors
Proteasome inhibition is a new therapeutic strategy in the treatment of multiple myeloma, which is based on the exquisite sensitivity of myeloma cells to the activity of the proteasome. Bortezomib (Velcade™; PS-341) is a potent inhibitor of myeloma cell growth and survival in vitro and results indicate striking therapeutic efficacy and acceptable toxicity profile of intravenous bortezomib in myeloma patients when administered alone or in combination with dexamethasone and/or chemotherapeutic agents, after relapse or at first presentation (70–74). In addition to myeloma cells, osteoblasts are also sensitive to proteasome inhibitors, and we have recently shown that structurally-unrelated inhibitors of the ubiquitin-proteasome pathway (including bortezomib) have beneficial anabolic effects on the skeleton in vivo (60, 75). Furthermore, proteasome inhibitors have also recently been demonstrated to inhibit osteoclast formation and bone resorption (76). These studies suggest that the effects of bortezomib in myeloma may be due, at least in part, to the direct effects of proteasome inhibitors to prevent myeloma bone disease. In support of these observations, Bortezemib has been demonstrated to reduce tumor burden and increase BMD in myeloma-bearing mice using the SCID-rab model of myeloma, an effect associated with an increase in osteoblasts and a reduction in osteoclasts (77). Clinical studies have demonstrated significant increases in markers of bone formation, including alkaline phosphatase and osteocalcin in those patients who responded to bortezomib treatment (78–81). In addition to changes in markers of bone formation, bortezomib treatment has also been shown to result in a reduction in serum Dkk1 and RANKL (82). These studies also suggest that the effect of bortezomib on bone formation is specific to proteasome inhibition, and not a result of reducing tumor burden. Thus, bortezomib may be effective both for the treatment of myeloma and of the associated bone disease.
THERAPEUTIC APPROACHES
Current therapeutic approaches for the treatment of myeloma bone disease include the use of bisphosphonates, radiation therapy, surgery, kyphoplasty and vertebroplasty. Bisphosphonates are effective in the treatment of myeloma bone disease, however are limited by their inability to promote new bone formation. Furthermore, bisphosphonates have recently been associated with the development of osteonecrosis of the jaw, identified in 4–6% of myeloma patients receiving intravenous bisphosphonates. As our understanding of the mechanisms involved in the development of myeloma bone disease increases, this identifies novel potential therapeutic approaches. Dysregulation of the RANKL-OPG system is a critical mechanism in the development of myeloma bone disease. Initial attempts to target this system used a recombinant OPG construct (AMGN-0007), which was found to be well tolerated and caused a rapid and sustained dose-dependent decrease of bone resorption marker levels, comparable to pamidronate (83). However, concerns about the development of antibodies to OPG prevented further development of this drug. More recently, a humanized monoclonal antibody to RANKL has been developed (Denosumab, AMG 162). This antibody is specific for RANKL, and does not bind to any other members of the TNF superfamily. In a recent clinical trial, the effect of Denosumab on safety and efficacy was investigated in patients with breast cancer or multiple myeloma with radiologically confirmed bone lesions. (84) A single subcutaneous dose of Denosumab was well tolerated and caused a sustained reduction in markers of bone resorption. Preclinical trials in murine models of myeloma suggest that targeting osteoblastic bone formation in multiple myeloma may be an effective therapeutic approach, however, with the exception of proteasome inhibitors, as described previously, these approaches have not yet reached clinical trial.
SUMMARY
The dramatic and destructive bone loss associated with myeloma bone disease is the result of the powerful combination of an increase in osteoclastic bone resorption and a decrease in osteoblastic bone formation. As our understanding of these cellular and molecular mechanisms increases, potential therapeutic targets are identified which may be effective in the treatment of myeloma bone disease (Figure 3). Furthermore, there is increasing evidence to demonstrate the efficacy of reducing tumor burden to indirectly reduce myeloma bone disease. Current therapeutic approaches include the use of bisphosphonates, which, while effective inhibitors of osteoclastic bone resorption, are limited by their inability to stimulate new bone formation and repair existing osteolytic bone lesions. Based on our increasing knowledge, the most effective therapeutic approach may be to target both the osteoclastic and the osteoblastic components of myeloma bone disease, to not only prevent further bone loss but to repair bone already damaged during the development of multiple myeloma.
Figure 3. Therapeutic approaches in myeloma bone disease.
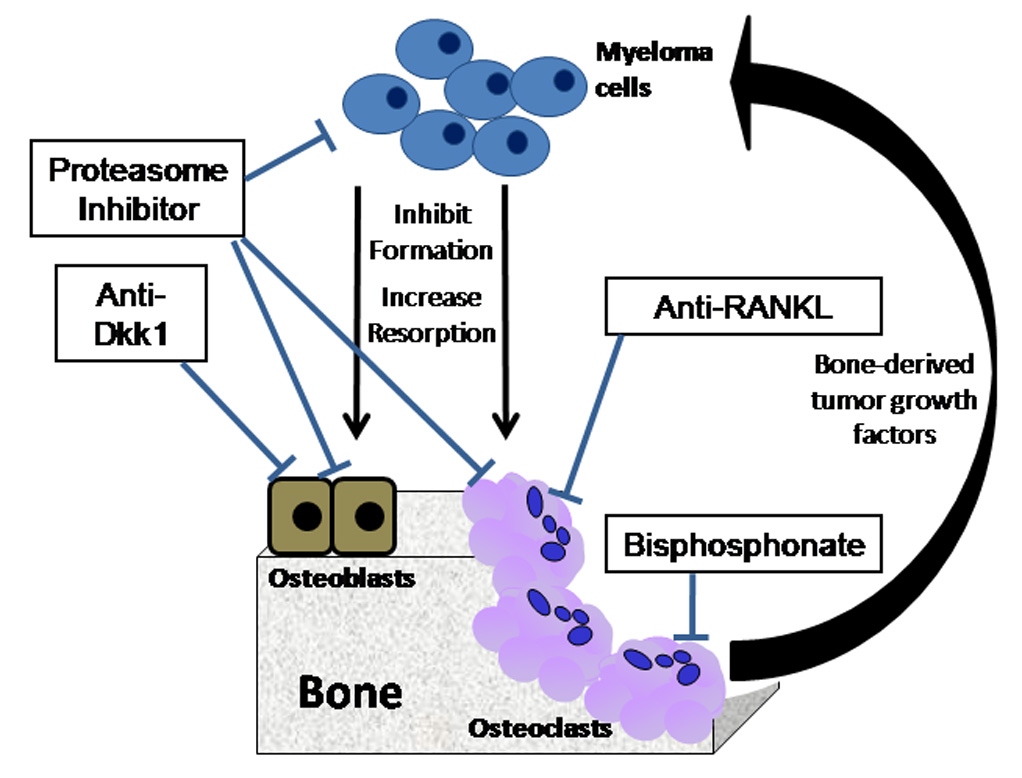
The osteoclast and osteoblast are potential therapeutic targets in the treatment of myeloma bone disease. Proteasome inhibitors and inhibitors of Dkk1 increase osteoblastic bone formation, whereas bisphosphonates and anti-RANKL inhibit osteoclastic bone resorption.
Figure 2. Cell-cell interactions in myeloma bone disease.
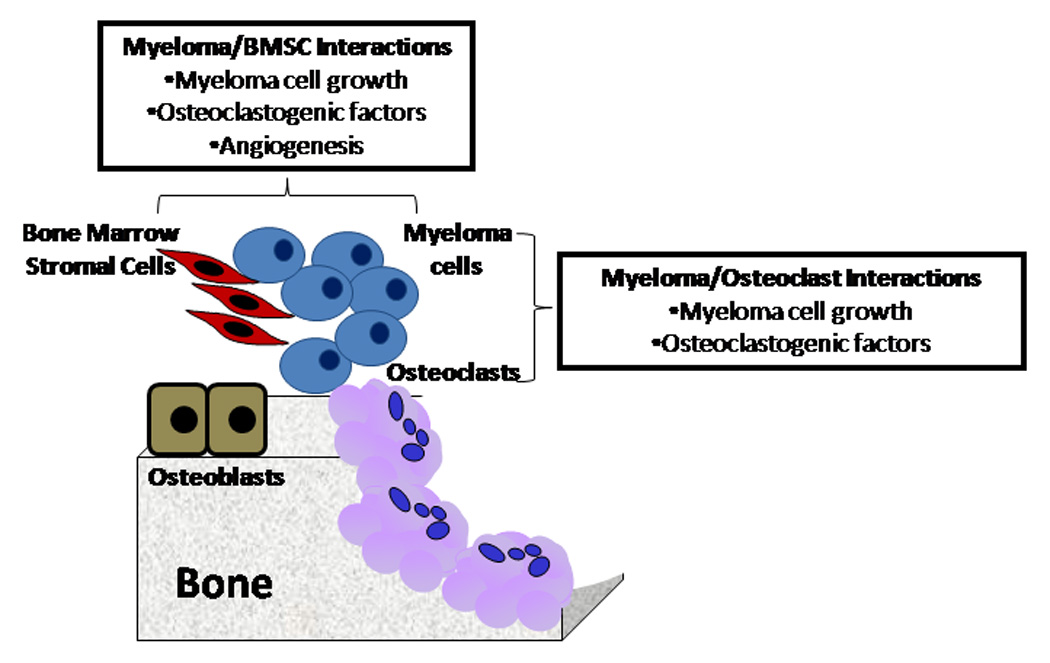
Direct Interactions between myeloma cells and bone marrow stromal cells or osteoclasts play a critical role in the development of myeloma bone disease.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Mundy GR. Myeloma bone disease. European Journal of Cancer. 1998;34:246–251. doi: 10.1016/s0959-8049(97)10133-2. [DOI] [PubMed] [Google Scholar]
- 2.Mundy GR, Raisz LG, Cooper RA, Schechter GP, Salmon S. Evidence for the secretion of an osteoclast stimulating factor in myeloma. New England Journal of Medicine. 1974;291:1041–1046. doi: 10.1056/NEJM197411142912001. [DOI] [PubMed] [Google Scholar]
- 3.Valentin OA, Charhon SA, Meunier PJ, Edouard CM, Arlot ME. Quantitative histology of myeloma-induced bone changes. British Journal of Haematology. 1982;52:601–610. doi: 10.1111/j.1365-2141.1982.tb03936.x. [DOI] [PubMed] [Google Scholar]
- 4.Taube T, Beneton MNC, McCloskey EV, Rogers S, Greaves M, Kanis JA. Abnormal bone remodelling in patients with myelomatosis and normal biochemical indices of bone resorption. European Journal of Haematology. 1992;49:192–198. doi: 10.1111/j.1600-0609.1992.tb00046.x. [DOI] [PubMed] [Google Scholar]
- 5.Bataille R, Chappard D, Marcelli C, Dessauw P, Balset P, Sany J, Alexandra C. Recruitment of new osteoblasts and osteoclasts is the earliest critical event in the pathogenesis of human multiple myeloma. Journal of Clinical Investigation. 1991;88:62–66. doi: 10.1172/JCI115305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Evans CE, Galasko CS, Ward C. Does myeloma secrete an osteoblast inhibiting factor? J Bone Joint Surg Br. 1989;71:288–290. doi: 10.1302/0301-620X.71B2.2925748. [DOI] [PubMed] [Google Scholar]
- 7.Bataille R, Delmas PD, Chappard D, Sany J. Abnormal serum bone Gla protein levels in multiple myeloma. Crucial role of bone formation and prognostic implications. Cancer. 1990;66:167–172. doi: 10.1002/1097-0142(19900701)66:1<167::aid-cncr2820660130>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 8.Abildgaard N, Rungby J, Glerup H, Brixen K, Kassem M, Brincker H, Heickendorff L, Eriksen EF, Nielsen JL. Long-term oral pamidronate treatment inhibits osteoclastic bone resorption and bone turnover without affecting osteoblastic function in multiple myeloma. Eur J Haematol. 1998;61:128–134. doi: 10.1111/j.1600-0609.1998.tb01073.x. [DOI] [PubMed] [Google Scholar]
- 9.Woitge HW, Horn E, Keck AV, Auler B, Seibel MJ, Pecherstorfer M. Biochemical markers of bone formation in patients with plasma cell dyscrasias and benign osteoporosis. Clin Chem. 2001;47:686–693. [PubMed] [Google Scholar]
- 10.Garrett IR, Durie BG, Nedwin GE, Gillespie A, Bringman T, Sabatini M, Bertolini DR, Mundy GR. Production of lymphotoxin, a bone-resorbing cytokine, by cultured human myeloma cells. N Engl J Med. 1987;317:526–532. doi: 10.1056/NEJM198708273170902. [DOI] [PubMed] [Google Scholar]
- 11.Kawano M, Hirano T, Matsuda T, Taga T, Horij Y, Iwato K, Asaoku H, Tang B, Tanabe O, Tanaka H, Kuramoto A, Kishimoto T. Autocrine generation and requirement of BSF-2/IL-6 for human multiple myelomas. Nature. 1988;332:83–85. doi: 10.1038/332083a0. [DOI] [PubMed] [Google Scholar]
- 12.Lee JW, Chung HY, Ehrlich LA, Jelinek DF, Callander NS, Roodman GD, Choi SJ. IL-3 expression by myeloma cells increases both osteoclast formation and growth of myeloma cells. Blood. 2004;103:2308–2315. doi: 10.1182/blood-2003-06-1992. [DOI] [PubMed] [Google Scholar]
- 13.Lichtenstein A, Berenson J, Norman D, Chang MP, Carlile A. Production of cytokines by bone marrow cells obtained from patients with multiple myeloma. Blood. 1989;74:1266–1273. [PubMed] [Google Scholar]
- 14.Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, Teepe MC, DuBose RF, Cosman D, Galibert L. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390:175–179. doi: 10.1038/36593. [DOI] [PubMed] [Google Scholar]
- 15.Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Butgess T, Elliott R, Colombero A, Elliott G, Scully S, Hsu H, Sullivan J, Hawkins N, Davy E, Capparelli C, Eli A, Qian YX, Kaufman S, Sorosi I, Shalhoub V, Senaldi G, Guo J, Delaney J, Boyle W. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 16.Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Lüthy R, Nguyen HQ, Shimamoto G, DeRose M, Elliott R, Sullivan J, Day E, Bucay N, Renshaw-Gegg L, Hughes TM, Hill D, Pattison W, Campbell P, Sander S, Van G, Tarpley J, Derby P, Lee R, Boyle WJ. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 17.Kong Y-Y, Yoshida H, Sarosi I, Tan H-L, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, Khoo W, Wakeham A, Dunstan CR, Lacey DL, Mak TW, Penninger JM. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 18.Li J, Sarosi I, Yan XQ, Morony S, Capparelli C, Tan HL, McCabe S, Elliott R, Scully S, Van G, Kaufman S, Juan SC, Sun Y, Tarpley J, Martin L, Christensen K, McCabe J, Kostenuik P, Hsu H, Fletcher F, Dunstan CR, Lacey DL, Boyle WJ. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc Natl Acad Sci U S A. 2000;97:1566–1571. doi: 10.1073/pnas.97.4.1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mizuno A, Amizuka N, Irie K, Murakami A, Fujise N, Kanno T, Sato Y, Nakagawa N, Yasuda H, Mochizuki S, Goibuchi T, Yano K, Shima N, Washida N, Tsuda E, Morinaga T, Higashio K, Ozawa H. Severe osteoporosis in mice lacking osteoclastogenesis inhibitory factor/osteoprotegerin. Biochemical and Biophysical Research Communications. 1998;247:610–615. doi: 10.1006/bbrc.1998.8697. [DOI] [PubMed] [Google Scholar]
- 20.Pearse RN, Sordillo EM, Yaccoby S, Wong BR, Liau DF, Colman N, Michaeli J, Epstein J, Choi Y. Multiple myeloma disrupts the TRANCE/osteoprotegerin cytokine axis to trigger bone destruction and promote tumor progression. PNAS. 2001;98:11581–11586. doi: 10.1073/pnas.201394498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Giuliani N, Bataille R, Mancini C, Lazzaretti M, Barillé S. Myeloma cells induce imbalance in the osteoprotegerin/osteoprotegerin ligand system in the human bone marrow environment. Blood. 2001;98:3527–3533. doi: 10.1182/blood.v98.13.3527. [DOI] [PubMed] [Google Scholar]
- 22.Roux S, Amazit L, Meduri G, Guiochon-Mantel A, Milgrom E, Mariette X. RANK (receptor activator of nuclear factor kappa B) and RANK ligand are expressed in giant cell tumors of bone. Am J Clin Pathol. 2002;117:210–216. doi: 10.1309/BPET-F2PE-P2BD-J3P3. [DOI] [PubMed] [Google Scholar]
- 23.Seidel C, Hjertner Ø, Abildgaard N, Heickendorff L, Hjorth M, Westin J, Nielsen JL, Hjorth-Hansen H, Waage A, Sundan A, Børset M. Serum osteoprotegerin levels are reduced in patients with multiple myeloma with lytic bone disease. Blood. 2001;98:2269–2271. doi: 10.1182/blood.v98.7.2269. [DOI] [PubMed] [Google Scholar]
- 24.Politou M, Terpos E, Anagnostopoulos A, Szydlo R, Laffan M, Layton M, Apperley JF, Dimopoulos MA, Rahemtulla A. Role of receptor activator of nuclear factor-kappa B ligand (RANKL), osteoprotegerin and macrophage protein 1-alpha (MIP-1α) in monoclonal gammopathy of undetermined significance (MGUS) Br J Haematol. 2004;126:686–689. doi: 10.1111/j.1365-2141.2004.05092.x. [DOI] [PubMed] [Google Scholar]
- 25.Terpos E, Szydlo R, Apperley JF, Hatjiharissi E, Politou M, Meletis J, Viniou N, Yataganas X, Goldman JM, Rahemtulla A. Soluble receptor activator of nuclear factor kappaB ligand-osteoprotegerin ratio predicts survival in multiple myeloma: proposal for a novel prognostic index. Blood. 2003;102:1064–1069. doi: 10.1182/blood-2003-02-0380. [DOI] [PubMed] [Google Scholar]
- 26.Farrugia AN, Atkins GJ, To LB, Pan B, Horvath N, Kostakis P, Findlay DM, Bardy P, Zannettino AC. Receptor activator of nuclear factor-kappaB ligand expression by human myeloma cells mediates osteoclast formation in vitro and correlates with bone destruction in vivo. Cancer Res. 2003;63:5438–5445. [PubMed] [Google Scholar]
- 27.Shipman CM, Holen I, Lippitt JM, Vandeberghe E, Croucher PI. Tumour cells isolated from patients with multiple myeloma express the critical osteoclastogenic factor, RANKL. Blood. 2000;96:360a. [Google Scholar]
- 28.Shipman CM, Croucher PI. Osteoprotegerin is a soluble decoy receptor for tumor necrosis factor-related apoptosis-inducing ligand/Apo2 ligand and can function as a paracrine survival factor for human myeloma cells. Cancer Research. 2003;63:912–916. [PubMed] [Google Scholar]
- 29.Croucher PI, Shipman CM, Lippitt J, Perry M, Asosingh K, Hijzen A, Brabbs AC, van Beek EJ, Holen I, Skerry TM, Dunstan CR, Russell GR, Van Camp B, Vanderkerken K. Osteoprotegerin inhibits the development of osteolytic bone disease in multiple myeloma. Blood. 2001;98:3534–3540. doi: 10.1182/blood.v98.13.3534. [DOI] [PubMed] [Google Scholar]
- 30.Vanderkerken K, De Leenheer E, Shipman C, Asosingh K, Willems A, Van Camp B, Croucher P. Recombinant osteoprotegerin decreases tumor burden and increases survival in a murine model of multiple myeloma. Cancer Res. 2003;63:287–289. [PubMed] [Google Scholar]
- 31.Oyajobi BO, Mundy GR. Receptor activator of NF-kappaB ligand, macrophage inflammatory protein-1alpha, and the proteasome: novel therapeutic targets in myeloma. Cancer. 2003;97:813–817. doi: 10.1002/cncr.11133. [DOI] [PubMed] [Google Scholar]
- 32.Yaccoby S, Pearse RN, Johnson RN, Johnson CL, Barologie B, Choi Y, Epstein J. Myeloma interacts with the bone marrow microenvironment to induce osteoclastogenesis and is dependent on ostoeclast activity. British Journal of Haematology. 2002;116:278–290. doi: 10.1046/j.1365-2141.2002.03257.x. [DOI] [PubMed] [Google Scholar]
- 33.Terpos E, Politou M, Szydlo R, Goldman JM, Apperley JF, Rahemtulla A. Serum levels of macrophage inflammatory protein-1 alpha (MIP-1alpha) correlate with the extent of bone disease and survival in patients with multiple myeloma. Br J Haematol. 2003;123:106–109. doi: 10.1046/j.1365-2141.2003.04561.x. [DOI] [PubMed] [Google Scholar]
- 34.Choi SJ, Cruz JC, Craig F, Chung H, Devlin RD, Roodman GD, Alsina M. Macrophage inflammatory protein 1-alpha is a potential osteoclast stimulatory factor in multiple myeloma. Blood. 2000;96:671–675. [PubMed] [Google Scholar]
- 35.Oba Y, Lee JW, Ehrlich LA, Chung HY, Jelinek DF, Callander NS, Horuk R, Choi SJ, Roodman GD. MIP-1alpha utilizes both CCR1 and CCR5 to induce osteoclast formation and increase adhesion of myeloma cells to marrow stromal cells. Exp Hematol. 2005;33:272–278. doi: 10.1016/j.exphem.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 36.Oyajobi BO, Franchin G, Williams PJ, Pulkrabek D, Gupta A, Munoz S, Grubbs B, Zhao M, Chen D, Sherry B, Mundy GR. Dual effects of macrophage inflammatory protein-1alpha on osteolysis and tumor burden in the murine 5TGM1 model of myeloma bone disease. Blood. 2003;102:311–319. doi: 10.1182/blood-2002-12-3905. [DOI] [PubMed] [Google Scholar]
- 37.Choi SJ, Oba Y, Gazitt Y, Alsina M, Cruz J, Anderson J, Roodman GD. Antisense inhibition of macrophage inflammatory protein 1-alpha blocks bone destruction in a model of myeloma bone disease. J Clin Invest. 2001;108:1833–1841. doi: 10.1172/JCI13116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vacca A, Ribatti D, Roncali L, Ranieri G, Serio G, Silvestris F, Dammacco F. Bone marrow angiogenesis and progression in multiple myeloma. Br J Haematol. 1994;87:503–508. doi: 10.1111/j.1365-2141.1994.tb08304.x. [DOI] [PubMed] [Google Scholar]
- 39.Niida S, Kaku M, Amano H, Yoshida H, Kataoka H, Nishikawa S, Tanne K, Maeda N, Nishikawa S, Kodama H. Vascular endothelial growth factor can substitute for macrophage colony-stimulating factor in the support of osteoclastic bone resorption. J Exp Med. 1999;190:293–298. doi: 10.1084/jem.190.2.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tanaka Y, Abe M, Hiasa M, Oda A, Amou H, Nakano A, Takeuchi K, Kitazoe K, Kido S, Inoue D, Moriyama K, Hashimoto T, Ozaki S, Matsumoto T. Myeloma cell-osteoclast interaction enhances angiogenesis together with bone resorption: a role for vascular endothelial cell growth factor and osteopontin. Clin Cancer Res. 2007;13:816–823. doi: 10.1158/1078-0432.CCR-06-2258. [DOI] [PubMed] [Google Scholar]
- 41.Juarez J, Bendall L. SDF-1 and CXCR4 in normal and malignant hematopoiesis. Histol Histopathol. 2004;19:299–309. doi: 10.14670/HH-19.299. [DOI] [PubMed] [Google Scholar]
- 42.Yu X, Huang Y, Collin-Osdoby P, Osdoby P. Stromal cell-derived factor-1 (SDF-1) recruits osteoclast precursors by inducing chemotaxis, matrix metalloproteinase-9 (MMP-9) activity, and collagen transmigration. J Bone Miner Res. 2003;18:1404–1418. doi: 10.1359/jbmr.2003.18.8.1404. [DOI] [PubMed] [Google Scholar]
- 43.Smith MC, Luker KE, Garbow JR, Prior JL, Jackson E, Piwnica-Worms D, Luker GD. CXCR4 regulates growth of both primary and metastatic breast cancer. Cancer Res. 2004;64:8604–8612. doi: 10.1158/0008-5472.CAN-04-1844. [DOI] [PubMed] [Google Scholar]
- 44.Alsayed Y, Ngo H, Runnels J, Leleu X, Singha UK, Pitsillides CM, Spencer JA, Kimlinger T, Ghobrial JM, Jia X, Lu G, Timm M, Kumar A, Cote D, Veilleux I, Hedin KE, Roodman GD, Witzig TE, Kung AL, Hideshima T, Anderson KC, Lin CP, Ghobrial IM. Mechanisms of regulation of CXCR4/SDF-1 (CXCL12)-dependent migration and homing in multiple myeloma. Blood. 2007;109:2708–2717. doi: 10.1182/blood-2006-07-035857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zannettino AC, Farrugia AN, Kortesidis A, Manavis J, To LB, Martin SK, Diamond P, Tamamura H, Lapidot T, Fujii N, Gronthos S. Elevated serum levels of stromal-derived factor-1alpha are associated with increased osteoclast activity and osteolytic bone disease in multiple myeloma patients. Cancer Res. 2005;65:1700–1709. doi: 10.1158/0008-5472.CAN-04-1687. [DOI] [PubMed] [Google Scholar]
- 46.Michigami T, Shimizu N, Williams PJ, Niewolna M, Dallas SL, Mundy GR, Yoneda T. Cell-cell contact between marrow stromal cells and myeloma cells via VCAM-1 and α4β1-integrin enhances production of osteoclast-stimulating activity. Blood. 2000;96:1953–1960. [PubMed] [Google Scholar]
- 47.Mori Y, Shimizu N, Dallas M, Niewolna M, Story B, Williams PJ, Mundy GR, Yoneda T. Anti-alpha4 integrin antibody suppresses the development of multiple myeloma and associated osteoclastic osteolysis. Blood. 2004;104:2149–2154. doi: 10.1182/blood-2004-01-0236. [DOI] [PubMed] [Google Scholar]
- 48.Oyayobi BO, Ytaianedes K, Yoneda T, Mundy GR. Expression of RANK ligand (RANKL) by myeloma cells requires binding to bone marrow stromal cells via an α4β1-VCAM-1 interaction. Bone. 1998;23:S180. [Google Scholar]
- 49.Yaccoby S, Wezeman MJ, Henderson A, Cottler-Fox M, Yi Q, Barlogie B, Epstein J. Cancer and the microenvironment:Myeloma-osteoclast interactions as a model. Cancer Research. 2004;64:2016–2023. doi: 10.1158/0008-5472.can-03-1131. [DOI] [PubMed] [Google Scholar]
- 50.Abe M, Hiura K, Wilde J, Shioyasono A, Moriyama K, Hashimoto T, Kido S, Oshima T, Shibata H, Ozaki S, Inoue D, Matsumoto T. Osteoclasts enhance myeloma cell growth and survival via cell-cell contact: a vicious cycle between bone destruction and myeloma expansion. Blood. 2004;104:2484–2491. doi: 10.1182/blood-2003-11-3839. [DOI] [PubMed] [Google Scholar]
- 51.Gong Y, Slee RB, Fukai N, Rawadi G, Roman-Roman S, Reginato AM, Wang H, Cundy T, Glorieux FH, Lev D, Zacharin M, Oexle K, Marceline J. LDL receptor-related protein 5 (LRP5) affects bone accrual and eye development. Cell. 2001;107:513–523. doi: 10.1016/s0092-8674(01)00571-2. [DOI] [PubMed] [Google Scholar]
- 52.Boyden LM, Mao J, Belsky J, Mitzner L, Farhi A, Mitnick MA, Wu D, Insogna K, Lifton RP. High bone density due to a mutation in LDL-receptor-related protein 5. N Engl J Med. 2002;346:1513–1521. doi: 10.1056/NEJMoa013444. [DOI] [PubMed] [Google Scholar]
- 53.Kato M, Patel M, Levasseur R, Lobov I, Chang BH, Glass DA, Hartmann C, Li L, Hwang TH, Brayton CF, Lang RA, Karsenty G, Cha L. Cbfa1-independent decrease in osteoblast proliferation, osteopenia, and persistant embryonic eye vascularization in mice deficient in LRP5, a Wnt coreceptor. Journal of Cell Biology. 2002;157:303–314. doi: 10.1083/jcb.200201089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Clement-Lacroix P, Ai M, Morvan F, Roman-Roman S, Vayssiere B, Belleville C, Estrera K, Warman ML, Baron R, Rawadi G. Lrp5-independent activation of Wnt signaling by lithium chloride increases bone formation and bone mass in mice. Proc Natl Acad Sci U S A. 2005;102:17406–17411. doi: 10.1073/pnas.0505259102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Edwards CM, Edwards JR, Esparza J, Oyajobi BO, McCluskey B, Munoz S, Grubbs B, Mundy GR. Lithium inhibits the development of myeloma bone disease in vivo. Journal of Bone and Mineral Research. 2006;21:S82. (abstract) [Google Scholar]
- 56.Tian E, Zhan F, Walker R, Rasmussen E, Ma Y, Barlogie B, Shaughnessy JD. The role of the Wnt-signaling antagonist in the development of osteolytic lesions in multiple myeloma. The New England Journal of Medicine. 2003;349:2483–2494. doi: 10.1056/NEJMoa030847. [DOI] [PubMed] [Google Scholar]
- 57.Rawadi G, Vayssiere B, Dunn F, Baron R, Roman-Roman S. BMP-2 controls alkaline phosphatase expression and osteoblast mineralization by a Wnt autocrine loop. Journal of Bone and Mineral Research. 2003;18:1842–1853. doi: 10.1359/jbmr.2003.18.10.1842. [DOI] [PubMed] [Google Scholar]
- 58.Li J, Sarosi I, Cattley RC, Pretorius J, Asuncion F, Grisanti M, Morony S, Adamu S, Geng Z, Qiu W, Kostenuik P, Lacey DL, Simonet WS, Bolon B, Qian X, Shalhoub V, Ominsky MS, Zhu Ke H, Li X, Richards WG. Dkk1-mediated inhibition of Wnt signaling in bone results in osteopenia. Bone. 2006;39:754–766. doi: 10.1016/j.bone.2006.03.017. [DOI] [PubMed] [Google Scholar]
- 59.Morvan F, Boulukos K, Clement-Lacroix P, Roman Roman S, Suc-Royer I, Vayssiere B, Ammann P, Martin P, Pinho S, Pognonec P, Mollat P, Niehrs C, Baron R, Rawadi G. Deletion of a single allele of the Dkk1 gene leads to an increase in bone formation and bone mass. J Bone Miner Res. 2006;21:934–945. doi: 10.1359/jbmr.060311. [DOI] [PubMed] [Google Scholar]
- 60.Oyajobi BO, Garrett IR, Gupta A, Banerjee M, Esparza X, Flores A, Sterling J, Rossinni G, Zhao M, Mundy GR. Role of Dickkopf 1(Dkk) in myeloma bone disease and modulation by the proteasome inhibitor velcade. Journal of Bone and Mineral Research. 2004;19:1011. [Google Scholar]
- 61.Gunn WG, Conley A, Deininger L, Olson SD, Prockop DJ, Gregory CA. A crosstalk between myeloma cells and marrow stromal cells stimulates production of DKK1 and interleukin-6: a potential role in the development of lytic bone disease and tumor progression in multiple myeloma. Stem Cells. 2006;24:986–991. doi: 10.1634/stemcells.2005-0220. [DOI] [PubMed] [Google Scholar]
- 62.Yaccoby S, Ling W, Zhan F, Walker R, Barlogie B, Shaughnessy JD., Jr Antibody-based inhibition of DKK1 suppresses tumor-induced bone resorption and multiple myeloma growth in vivo. Blood. 2007;109:2106–2111. doi: 10.1182/blood-2006-09-047712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Oshima T, Abe M, Asano J, Hara T, Kitazoe K, Sekimoto E, Tanaka Y, Shibata H, Hashimoto T, Ozaki S, Kido S, Inoue D, Matsumoto T. Myeloma cells suppress bone formation by secreting a soluble Wnt inhibitor, sFRP-2. Blood. 2005 doi: 10.1182/blood-2004-12-4940. [DOI] [PubMed] [Google Scholar]
- 64.Takeuchi K, Abe M, Oda A, Amou H, Hiasa M, Asano J, Kitazoe K, Kido S, Inoue D, Hashimoto T, Ozaki S, Matsumoto T. Enhancement of osteoblast differentiation by inhibition of TGF-beta signaling suppresses myeloma cell growth and protects from destructive bone lesions. Journal of Bone and Mineral Research. 2006;21:1101. [Google Scholar]
- 65.Giuliani N, Colla S, Morandi F, Lazzaretti M, Sala R, Bonomini S, Grano M, Colucci S, Svaldi M, Rizzoli V. Myeloma cells block RUNX2/CBFA1 activity in human bone marrow osteoblast progenitors and inhibit osteoblast formation and differentiation. Blood. 2005;106:2472–2483. doi: 10.1182/blood-2004-12-4986. [DOI] [PubMed] [Google Scholar]
- 66.Ehrlich LA, Chung HY, Ghobrial I, Choi SJ, Morandi F, Colla S, Rizzoli V, Roodman GD, Giuliani N. IL-3 is a potential inhibitor of osteoblast differentiation in multiple myeloma. Blood. 2005;106:1407–1414. doi: 10.1182/blood-2005-03-1080. [DOI] [PubMed] [Google Scholar]
- 67.Seidel C, Berset M, Turesson I, Abildgaard N, Sundan A, Waage A. Elevated serum concentrations of hepatocyte growth factor in patients with multiple myeloma. Blood. 1998;91:806–812. [PubMed] [Google Scholar]
- 68.Andersen NF, Standal T, Nielsen JL, Heickendorff L, Borset M, Sorensen FB, Abildgaard N. Syndecan-1 and angiogenic cytokines in multiple myeloma: correlation with bone marrow angiogenesis and survival. Br J Haematol. 2005;128:210–217. doi: 10.1111/j.1365-2141.2004.05299.x. [DOI] [PubMed] [Google Scholar]
- 69.Standal T, Abildgaard N, Fagerli UM, Stordal B, Hjertner O, Borset M, Sundan A. HGF inhibits BMP-induced osteoblastogenesis: possible implications for the bone disease of multiple myeloma. Blood. 2006 doi: 10.1182/blood-2006-07-034884. [DOI] [PubMed] [Google Scholar]
- 70.Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, Rajkumar SV, Hideshima T, Xiao H, Esseltine D, Schenkein D, Anderson KC. Clinical factors predictive of outcome with bortezomib in patients with relapsed, refractory multiple myeloma. Blood. 2005 doi: 10.1182/blood-2005-02-0691. [DOI] [PubMed] [Google Scholar]
- 71.Yang HH, Vescio R, Schenkein D, Berenson JR. A prospective, open-label safety and efficacy study of combination treatment with bortezomib (PS-341, velcade and melphalan in patients with relapsed or refractory multiple myeloma. Clin Lymphoma. 2003;4:119–122. doi: 10.3816/clm.2003.n.022. [DOI] [PubMed] [Google Scholar]
- 72.Jagannath S, Barlogie B, Berenson JR, Singhal S, Alexanian R, Srkalovic G, Orlowski RZ, Richardson PG, Anderson J, Nix D, Esseltine DL, Anderson KC. Bortezomib in recurrent and/or refractory multiple myeloma. Initial clinical experience in patients with impared renal function. Cancer. 2005;103:1195–1200. doi: 10.1002/cncr.20888. [DOI] [PubMed] [Google Scholar]
- 73.Jagannath S, Durie BG, Wolf J, Camacho E, Irwin D, Lutzky J, McKinley M, Gabayan E, Mazumder A, Schenkein D, Crowley J. Bortezomib therapy alone and in combination with dexamethasone for previously untreated symptomatic multiple myeloma. Br J Haematol. 2005;129:776–783. doi: 10.1111/j.1365-2141.2005.05540.x. [DOI] [PubMed] [Google Scholar]
- 74.Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina M, Alexanian R, Siegel D, Orlowski RZ, Kuter D, Limentani SA, Lee S, Hideshima T, Esseltine DL, Kauffman M, Adams J, Schenkein DP, Anderson KC. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003;348:2609–2617. doi: 10.1056/NEJMoa030288. [DOI] [PubMed] [Google Scholar]
- 75.Garrett IR, Chen D, Gutierrez G, Zhao M, Escobedo A, Rossini G, Harris SE, Gallwitz W, Kim KB, Hu S, Crews CM, Mundy GR. Selective inhibitors of the osteoblast proteasome stimulate bone formation in vivo and in vitro. J Clin Invest. 2003;111:1771–1782. doi: 10.1172/JCI16198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zavrski I, Krebbel H, Wildemann B, Heider U, Kaiser M, Possinger K, Sezer O. Proteasome inhibitors abrogate osteoclast differentiation and osteoclast function. Biochem Biophys Res Commun. 2005;333:200–205. doi: 10.1016/j.bbrc.2005.05.098. [DOI] [PubMed] [Google Scholar]
- 77.Pennisi A, Ling W, Perkins P, Saha R, Barlogie B, Tricot G, Zangari M, Yaccoby S. PTH and Bortezomib suppresses growth of primary human myeloma thorugh increased bone formation in vivo. Blood. 2006;108:154a. [Google Scholar]
- 78.Zangari M, Esseltine D, Lee CK, Barlogie B, Elice F, Burns MJ, Kang SH, Yaccoby S, Najarian K, Richardson P, Sonneveld P, Tricot G. Response to bortezomib is associated to osteoblastic activation in patients with multiple myeloma. Br J Haematol. 2005;131:71–73. doi: 10.1111/j.1365-2141.2005.05733.x. [DOI] [PubMed] [Google Scholar]
- 79.Shimazaki C, Uchida R, Nakano S, Namura K, Fuchida SI, Okano A, Okamoto M, Inaba T. High serum bone-specific alkaline phosphatase level after bortezomib-combined therapy in refractory multiple myeloma: possible role of bortezomib on osteoblast differentiation. Leukemia. 2005;19:1102–1103. doi: 10.1038/sj.leu.2403758. [DOI] [PubMed] [Google Scholar]
- 80.Zangari M, Yaccoby S, Cavallo F, Esseltine D, Tricot G. Response to bortezomib and activation of osteoblasts in multiple myeloma. Clin Lymphoma Myeloma. 2006;7:109–114. doi: 10.3816/CLM.2006.n.047. [DOI] [PubMed] [Google Scholar]
- 81.Giuliani N, Morandi F, Tagliaferri S, Lazzaretti M, Bonomini S, Crugnola M, Petro D, Mancini C, Martella E, Rizzoli V. The proteasome inhibitor Bortezomib affects osteoblastogenesis and bone formation in vitro and in vivo in multiple myeloma patients. Blood. 2006;108:508. doi: 10.1182/blood-2006-11-059188. [DOI] [PubMed] [Google Scholar]
- 82.Terpos E, Heath DJ, Rahemtulla A, Zervas K, Chantry A, Anagnostopoulos A, Pouli A, Katodritou E, Verrou E, Vervessou EC, Dimopoulos MA, Croucher PI. Bortezomib reduces serum dickkopf-1 and receptor activator of nuclear factor-kappaB ligand concentrations and normalises indices of bone remodelling in patients with relapsed multiple myeloma. Br J Haematol. 2006;135:688–692. doi: 10.1111/j.1365-2141.2006.06356.x. [DOI] [PubMed] [Google Scholar]
- 83.Body JJ, Greipp P, Coleman RE, Facon T, Geurs F, Fermand JP, Harousseau JL, Lipton A, Mariette X, Williams CD, Nakanishi A, Holloway D, Martin SW, Dunstan CR, Bekker PJ. A phase I study of AMGN-0007, a recombinant osteoprotegerin construct, in patients with multiple myeloma or breast carcinoma related bone metastases. Cancer. 2003;97:887–892. doi: 10.1002/cncr.11138. [DOI] [PubMed] [Google Scholar]
- 84.Body JJ, Facon T, Coleman RE, Lipton A, Geurs F, Fan M, Holloway D, Peterson MC, Bekker PJ. A study of the biological receptor activator of nuclear factor-kappaB ligand inhibitor, denosumab, in patients with multiple myeloma or bone metastases from breast cancer. Clin Cancer Res. 2006;12:1221–1228. doi: 10.1158/1078-0432.CCR-05-1933. [DOI] [PubMed] [Google Scholar]