

Abstract
Background
The B-vitamin folic acid (FA) is important to mitochondrial protein and nucleic acid synthesis, is an anti-oxidant, and enhances nitric oxide synthase activity. Here, we tested whether FA reduces myocardial ischemic dysfunction and post-reperfusion injury.
Methods
Wistar rats were pretreated with either FA (10mg/d) or placebo for 1-wk, and then underwent in vivo transient left coronary artery occlusion for 30min with or without 90min reperfusion (total:n=131; sub-groups used for various analyses). FA (4.5•10-6M i.c) pretreatment and global ischemia/reperfusion (30 min/30min), was also performed in vitro (n=28).
Results
After 30min ischemia, global function declined more in controls than FA-pretreated rats (ΔdP/dtmax -878±586 mmHg/s vs. placebo -1956±351 mmHg/s, p=0.03), and regional thickening was better preserved (37.3±5.3% vs. 5.1±0.6%-placebo, p=0.004). Anterior-wall perfusion fell similarly (-78.4±9.3% vs. -71.2±13.8%-placebo at 30 min); yet myocardial high energy phosphates ATP and ADP reduced by ischemia in controls were better preserved by FA-pretreatment (e.g. ATP: control: 2740±58; ischemia: 947±55; ischemia+FA: 1332±101 nmol/g, p=0.02). Basal oxypurines (xanthine, hypoxanthine, and urate) rose with FA-pretreatment, but increased less during ischemia than in controls. Ischemic superoxide generation declined (3124±280 FA vs. 5898±474 placebo, cpm/mg, p=0.001). After reperfusion, FA-treated hearts had smaller infarcts (3.8±1.2% vs 60.3±4.1%-placebo area at risk, p<0.002), less contraction band necrosis, TUNEL-positivity, superoxide, and nitric oxide synthase uncoupling. Infarct size declined similarly with 1 mg/d FA as well.
Conclusion
FA-pretreatment blunts myocardial dysfunction during ischemia, and ameliorates post-reperfusion injury. This is coupled to preservation of high energy phosphates, reducing subsequent ROS-generation, eNOS-uncoupling and post-reperfusion cell death.
Keywords: Ischemia, Folic acid, High Energy Phosphates, Superoxide, eNOS-uncoupling
Introduction
During acute coronary occlusion, myocardial perfusion and oxygen delivery become insufficient to support active muscle contraction. In the ischemic zone, myocardial high energy phosphate content falls, inorganic phosphate rises, and the tissue is rendered acidotic1, resulting in regional dyskinesis and global dysfunction. Coronary reperfusion after ischemia limits tissue damage but confers toxicity from activation of reactive oxygen species (ROS), calcium-dependent proteases such as calpain, myofilament contracture, microvascular dysfunction, and inflammatory cytokines2. ATP depletion during ischemia may also contribute to reperfusion injury3. In addition, ROS generated during both ischemia4 and reperfusion5 is thought to play a central role, and various anti-oxidant strategies have been tested to offset this damage.
Ischemia/reperfusion damage can be diminished by subjecting hearts to brief ischemia prior to more prolonged exposure, a phenomenon termed pre-conditioning. This involves activation of protein kinase C6, mitochondrial KATP channels, and nitric oxide synthesis7, with the latter playing a central role. Infarct size after ischemia-reperfusion is greater in eNOS deficient mice8 and reduced in mice over-expressing eNOS9. This role of NOS has led to efforts to enhance its function, including administration of its obligate cofactor, tetrahydrobiopterin (BH4)10 to help maintain NOS in a functionally coupled (NO synthesis, little ROS generation) state11,12.
An far less costly alternative may be folic acid (FA), a B-vitamin that stabilizes BH4 by augmenting its binding-affinity to eNOS13 and enhances BH4 regeneration from oxidized and inactive BH2. FA or its active metabolite is thought to enhance endothelial function by this mechanism14,15. FA is also essential for normal mitochondrial protein and purine synthesis16 and can enhance total high energy phosphate levels in chronically hypertrophied right ventricles17. In the present study, we tested the hypothesis that high dose FA pre-treatment ameliorates IR injury and explored the mechanisms for such an effect. The data show post-reperfusion benefits but more strikingly reveal a marked and surprising effect of FA-pre-treatment on reducing regional and chamber dysfunction and ROS generation during the period of ischemia itself. This benefit appears linked to alterations in purine catabolism and preservation of high energy phosphate levels during ischemia.
Methods
Experiments were performed in accordance with the Guide for the Care and Use of Laboratory Animals published by the US National Institute of Health (NIH-Publication, N° 85-23) and approved by the Ethical Committees of the University of Antwerp and by the Johns Hopkins Medical Institutions.
In vivo ischemia model
Adult Wistar rats received FA (10 mg/day unless otherwise stated) or placebo by oral gavage for 7-days prior to performing the ischemia/reperfusion (IR) experiment. A total of 131 rats were used reflecting the multiple assays that could not be performed in each one. Animals were anesthetized (pentobarbital 60 mg/kg), intubated via tracheotomy, and ventilated (Harvard Apparatus, MA). ECG was monitored and temperature maintained at 37.5°C. The left anterior artery was exposed through the 4-5th intercostal space and a suture placed around it, and transient coronary artery ligation performed for 30 min with (n=85) or without (n=46) 90 min reperfusion. In one subgroup of reperfused animals (n=9), FA was provided 10 minutes after the onset of LAD occlusion (i.e. 20 minutes before the onset of reperfusion), delivered as an IV-bolus.
In vivo hemodynamics
LV function was assessed in vivo by pressure-volume loops (n=14) during both ischemia and following reperfusion. A 1.4F pressure-volume catheter (SciSense, London-Ontario, Canada) was advanced through the apex, positioned along the longitudinal axis, and attached to a stimulator/analyzer (IOX 1.8.9.19, Emka Tech., Paris). Volume data were calibrated using the hypertonic saline method, assuming a gain=1. Two animals had catheter dislocation during ischemia, and their volume data were not used. Open chest myocardial anterior wall motion was also measured using a Sequoia –Acuson C256 equipped with 15 MHz linear transducer (Sequoia C256 Echocardiography System, Acuson Corporation, Mountain View, CA) at the parasternal view of the left ventricle chamber as described.
Assessment of Redox and Energy Metabolism
Snap frozen samples (n=24) from the anterior wall (±FA pre-treatment; ±30 min ischemia, no reperfusion) were de-proteinized and subjected to HPLC analysis of water-soluble low-molecular weight compounds reflecting tissue oxid-oreductive and energy status. High-energy phosphates, oxypurines (hypoxanthine, xanthine, and uric acid), nucleosides (inosine and adenosine), malondialdehyde, and reduced and oxidized glutathione were measured by ion-pairing HPLC as described18
Myocardial flow measurements
Regional myocardial blood flow was assessed (n=12) by nuclear-activated microspheres (15μM diameter, BioPal, Worcester, MA) injected in the left atrium (0.3ml of 2.5*106 spheres/ml) at baseline and after 5 and 30 min of ischemia19. Total counts per minute (cpm) were normalized to weight and results from the ischemic zone normalized to the remote region to provide relative blood flow before and during ischemia.
In vitro IR model
Adult Wistar rats (n=28) were anesthetized (pentobarbital 60 mg/kg), and hearts rapidly excised and mounted onto a retrograde perfusion system (Emka, Paris, France) using warmed, oxygenated buffered Krebs-Henseleit solution at constant perfusion pressure (75 mmHg). Hearts were paced at 300 bpm and maintained unloaded. Coronary flow was measured by an in-line ultrasonic flow probe (Transonic Systems, Ithaca, NY). After 30 min equilibration, bradykinin or sodium nitroprusside were infused by bolus injection (50 μl, 10-8-10-5.5 M i.c.) and coronary flow reserve assessed at constant perfusion pressure. After re-establishing baseline, hearts received FA (4.5 10-6M i.c.) or vehicle for 30 min, and then subjected to 40-min zero flow-ischemia followed by 40 min reperfusion. Coronary effluent was collected, concentrated (Sartorius-Sipan, Lier, Belgium) and analysed for lactate dehydrogenase (Vitros 950AT, O.C.D., Beerse, Belgium). Coronary vasodilator reserve studies were then repeated.
Infarct size analysis and Histology
Infarct size was assessed (n=31) with area at risk (AAR) determined by Evans blue negative staining, and TTC staining to detect myocardial necrosis. Regions were planimetered and digitized and quantified. AAR was not assessed for the in vitro studies since global ischemia was created. Contraction band necrosis was examined in vivo (n=22) by fixation in Carnoy solution and Masson's trichrome staining. Serial adjacent fields were examined throughout the LV to calculate the percent myocardium with contraction bands present. Similar analysis was performed for TUNEL-positivity (Chemicon Intern, Temecula, CA), also expressed as percent LV area.
ROS determination
Superoxide was assessed by lucigenin (5μM)-enhanced chemiluminescence (Beckman LS6000IC, n=23)20 and fluorescent microtopography (dichlorodihydro-fluorescein diacetate, DCF, n=16) and dihydroethidium, DHE, n=24)18. The direct anti-oxidants effects of folic acid were analysed using an in vitro xanthine/xanthine oxidase system21 and compared with Tempol.
eNOS monomer/dimer formation and enzymatic activity
SDS-resistant eNOS dimers and monomers were assayed on ischemia/reperfusion-tissue (n=16) using low temperature SDS- PAGE as previously described 18. NOS enzymatic activity was assessed by arginine to citrulline conversion assay from extracts obtained from frozen myocardium (n=15)18.
Data analysis
Data are presented as mean ± sem with p-values<0.05 considered statistically significant. Coronary flow-reserve and in vivo hemodynamic data were analyzed by repeated measures ANOVA. Other comparisons used either 1-way ANOVA or Kruskal-Wallis tests to compare between multiple independent groups, with a Bonferroni correction for multiple comparisons. Analysis utilized SPS6 version 11.0 (Chicago, IL).
The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written. None of the authors has conflict of interest disclosures.
Results
Folic acid pre-treatment improves cardiac function with I/R
Figure 1A displays example and summary left ventricular PV data in hearts with or without FA (10mg/d) pre-treatment. Data were measured at initial baseline in open-chest rats, during 30 minutes of coronary occlusion, and after 90 minutes of reperfusion. Control hearts displayed markedly reduced function with a rightward-downshift of PV loops at 30 min ischemia that persisted with reperfusion (upper panel). With FA pre-treatment, cardiac function was better preserved during ischemia and reperfusion (lower panel, Fig 1B). Peak systolic pressure changed little despite LAD occlusion in FA pre-treated rats but fell nearly 25% in controls. Similar disparities were observed in dP/dtmax. Cardiac output and stroke work also declined less in FA-pretreated animals, particularly in late ischemia and reperfusion.
Figure 1. Cardiac function measured by pressure/volume-loop analysis.
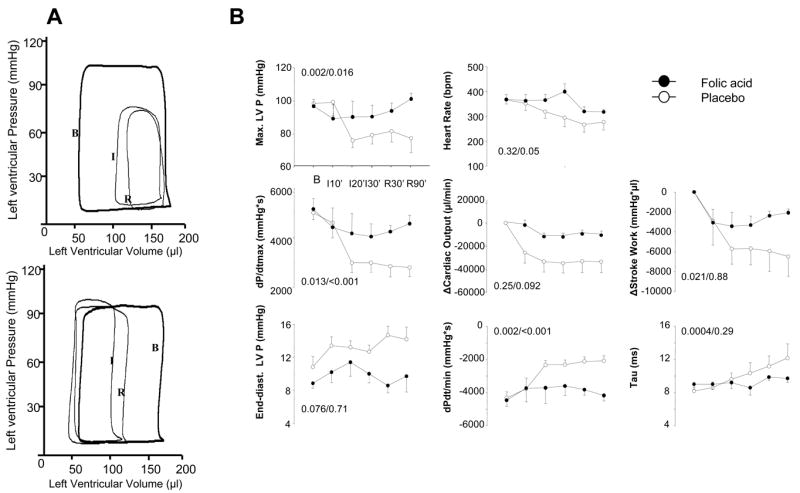
A] Example of pressure-volume loops at baseline (1), end of ischemia (2), and 90 min reperfusion (3) for a placebo (upper) and FA pre-treated (lower) animal. FA pre-treatment improved function both during ischemia and following reperfusion. B] Summary hemodynamics for the full IR protocol shows the time course for systolic and diastolic parameters. (p values are from repeated measures ANOVA, time × treatment interaction/ treatment effect).
The relative preservation of global function during ischemia was somewhat surprising, and suggested less regional dysfunction despite coronary occlusion in FA pre-treated hearts. This was tested by echocardiography measured before and during LAD occlusion (Fig 2). Example M-mode tracings (upper left panel) show marked reductions of anterior wall thickening during ischemia in controls but preserved thickening in FA treated animals (5±0.6% versus 37±5.3%, p=0.004). Ejection fraction was much higher despite ischemia (72.8±1.2% vs. placebo: 27.4±2.2 %, p<0.001 at 30 min), consistent with the PV-loop data.
Figure 2. Open chest echocardiography.
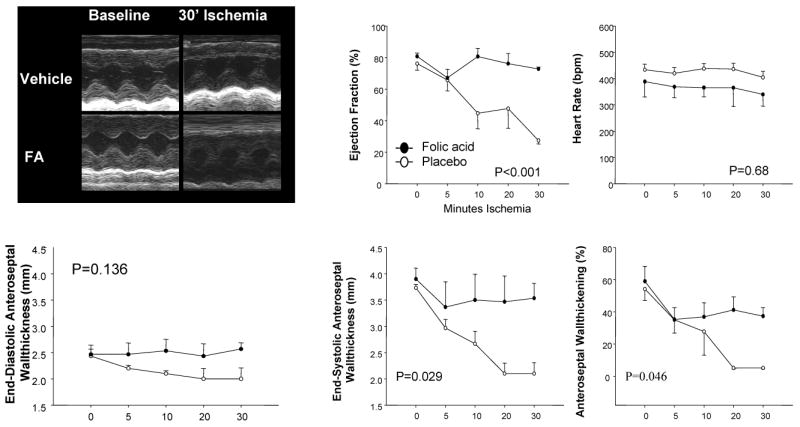
The upper left panel shows examples of M-mode echocardiography at baseline and after 30 min of ischemia in the placebo and FA-treated group. Summary plots show results obtained from these echocardiograms versus time. Ejection fraction and anterior-septal wall-thickening declined in the placebo group but were unchanged in the FA-group (p-values are for treatment effect by RMANOVA).
Folic acid and myocardial flow
Since FA pre-treatment improved both regional and global function during LAD occlusion, we tested whether it enhanced myocardial blood flow to reduce the ischemic insult per se. However, after 5 min of LAD ligation, the ratio of ischemic/remote zone myocardial perfusion obtained by microsphere analysis declined similarly in placebo and FA-pre-treated groups (-73.7±6.0% and -77.7±5.1%, respectively). Flow remained low in both groups at 30 minutes (-78.4±9.3% vs. placebo -71.2±13.8%).
Folic acid preserves myocardial levels of high energy phosphates
Since improved perfusion could not explain the FA-treatment effect, we next tested whether FA altered high energy phosphate (HEP) metabolism at baseline and/or during ischemia. As shown in Figure 3, FA-pretreatment did not alter HEP at baseline, but did elevate levels of inositol monophosphate (IMP) and its catabolites (oxypurines: xanthine, hypoxanthine, uric acid). During ischemia, myocardial ATP and ADP declined more than 66% in controls, consistent with reported data1. However, both were better maintained in FA pre-treated hearts, (p<0.001 for drug-interaction effect). Oxypurines rose markedly during ischemia in controls, consistent with reduced HEP and enhanced AMP catabolism, but changed little or declined in FA-treated hearts. Redox-state indexed by malondialdehyde (a marker of lipid peroxidation) and reduced/oxidized glutathione ratio was little changed by FA pre-treatment with or without myocardial ischemia.
Figure 3. HPLC-analysis of parameters of high energy phosphate (HEP) metabolism and redox-state.

Under baseline conditions, FA pre-treatment did not alter HEP (ATP, ADP, AMP), but did increase inositol monophosphate (IMP) and its catabolites (oxypurines: xanthine, hypoxanthine and uric acid). After 30 minutes of ischemia, the levels of HEP were better maintained in the FA-treated hearts compared with placebo animals. While oxypurines rose markedly after ischemia in placebo-treated animals, this was not observed in the FA-treated rats. P-values are from a 2-way ANOVA, with the first value indicating the effect of ischemia, and the second the interaction between treatment group and ischemia.
Folic acid pre-treatment reduces myocardial infarct size
A potential consequence of improving both function and HEP metabolism during ischemia is reduction of infarct size. Infarct size was 60.3±4.1% of the area of risk in placebo-treated animals versus 3.8±1.2% with FA-pre-treatment (Fig 4A, p<0.002). Similar reduction was observed using 40% or 10% of the FA dose (1 or 4 mg/d), though this is still a fairly high dose compared to that typically used in humans. Contraction band necrosis (CBN) was found in 26.7±2.6% of the LV in controls, versus 4.6%±1.2% in FA treated hearts (p=0.001, Fig. 4B). Similarly, TUNEL-positive myocytes were prevalent (63.0±5.8% of LV fields) in controls but rare (4.3±1.3%) with FA treatment (p=0.001, Fig 4C). Lethal ventricular arrhythmia was fairly common during ischemia in controls but not FA treated rats (36.7% vs 8.3%) and reduced in frequency during reperfusion as well (6.1 vs 0%, p<0.01 for both).
Figure 4. Effect of folic acid on myocardial necrosis.

A] Oral (7 days) FA pre-treatment reduced infarct size in vivo (*: p<0.001; Mann-Whitney). Upper panel: Area at risk (AAR) was comparable between all groups. Middle panel: Infarct size, expressed as % AAR was significantly reduced in all FA-groups. Lower panel: plot-graph of the individual correlation between myocardial necrosis and AAR. B] FA pre-treatment in vivo reduces contraction band necrosis (*p=0.001). C] FA pretreatment in vivo reduces apoptosis (TUNEL-staining) (*p=0.005 and p=0.001). D] FA pre-treatment reduced myocardial necrosis by ∼80% in hearts studied in vitro. (*: p< 0.0001). E] Increased lactate dehydrogenase (LDH) after ischemia-reperfusion peaked at 10 – 20′, whereas hearts receiving FA pre-treatment had minimal LDH release. (p<0.001).
Since post-reperfusion infarct size in vivo is partially related to coupling of function with coronary perfusion, we also tested the impact of FA-pretreatment in isolated hearts. FA reduced infarct size markedly (7.7±2.8% vs. 41.1±4.9%, p<0.0001, Fig 4D) with less lactate dehydrogenase in the coronary effluent (Fig. 4E) consistent with reduced necrosis.
Folate pre-treatment vs. acute folate administration
In a separate group of 9 animals, FA was acutely administered starting after 10 min of coronary occlusion (10 mg i.v.) when functional responses first appeared to diverge (c.f. Fig 1, 2), and continued for the remaining 20 min ischemic period. Infarct size relative to AAR was also reduced (n=5; 3.0±2.2%, p<0.001 vs. placebo) with AAR itself similar to placebo (52.4±5.5%). Histology (n=4) found reduced TUNEL staining (4.3±1.3% LV) and CBN (5.1±0.7% LV; both p<0.001 vs. placebo). Thus, the FA effect on infarct reduction did not appear to be a classic preconditioning effect, as it could be generated by FA administration after ischemia had commenced.
Folate pre-treatment reduces ROS generation
Myocardial superoxide (lucigenin chemiluminescence) declined ∼50% in FA pre-treated animals during ischemia and after 90 minutes reperfusion (Fig 5A). When extracts were pre-incubated with 100 μM BH4, O2- generation declined 90.9±0.7% in vehicle-controls, but less so in FA pre-treated hearts (52.1±11.3%, p<0.03, Fig 5B). This suggested that an anti-oxidant pathway targeted by BH4 (e.g. NOS coupling) was either lacking in FA-pre-treated hearts or already ameliorated by FA therapy. DHE and DCF-stained myocardial slices also showed marked ROS generation in the placebo group that was reduced with FA pre-treatment (Fig 5C-E). To test for direct anti-oxidant effects of FA, we performed an in vitro assay using a xanthine/xanthine oxidase O2- generating system (Fig 5F). FA anti-oxidant effects were substantial in this assay, and similar to the superoxide dismutase mimetic Tempol.
Figure 5. Effects of FA treatment on ROS generation.

A] Lucigenin-enhanced superoxide detection shows marked reduction of ischemia and IR-induced superoxide with FA pre-treatment. B] O2- formation in control-vehicle treated IR hearts was markedly suppressed by acute addition of BH4, whereas this was blunted by nearly half in myocardial extracts obtained from FA-pre-treated hearts. C-D] DHE and E] DCF stained myocardium reveal increased ROS generation in ischemic or IR myocardium that was substantially reduced in heart receiving FA pre-treatment. F] Direct antioxidant effects of FA compared with Tempol. Superoxide was generated by xanthine/xanthine oxidase in vitro, and measured by lucigenin-enhanced chemiluminescence at varying added concentrations of either FA or tempol.
Folate pre-treatment improves eNOS dimerization and activity, and endothelial function
Since FA and its metabolites have been linked to BH4-mediated improvement in NOS coupling and decline in ROS generation14,21, we examined NOS coupling in I/R hearts. Immunoblots showed a decline in the NOS dimer/monomer ratio that was preserved near normal in FA pre-treated hearts (Fig 6A-C). Total eNOS was similar among the conditions (Fig 6B). NOS activity (arginine-citrulline conversion) was borderline improved by FA pre-treatment (p=0.08, data not shown).
Figure 6. Effect of FA pre-treatment on NO-pathway.

A] SDS-Page gel shows increased eNOS monomer in IR myocardium that was reduced towards control levels by FA pre-treatment. B-C] Summary densitometry data of gel analysis as shown in panel A. FA pre-treatment reduced the monomer/dimmer ratio, but had no effect on total eNOS protein expression. D] Bradykinin administered to hearts in vitro before versus after IR showed a marked decline in endothelium-dependent flow response. This was restored to a normal response by pre-treating with FA. E] FA treatment did not influence the flow-response to sodium nitroprusside, supporting that the disparity observed with bradykinin was endothelium-dependent.
Coronary endothelial function also improved with FA pre-treatment. Bradykinin induced a maximal 108.3±9.2% rise in coronary flow at baseline but 67.1±8.1% after IR, (p<0.001 Fig 6D). This decline was absent in FA pre-treated hearts (122.0±11.3%). Basal flow before (placebo: 10.2±0.7 ml/min, FA: 11.4±13.8 ml/min, p=0.5) or after ischemia (placebo: 9.8±0.7 ml/min FA:8.9±1.4 ml/min, p=0.5) was similar between groups. Coronary flow rose similarly pre- and post-ischemia with sodium nitroprusside (Fig 6E), supporting endothelium dependence of the prior effect.
Discussion
This study demonstrates that pretreatment with high doses of oral folic acid markedly reduces ischemic dysfunction during coronary occlusion, and enhances function and diminishes infarct size following reperfusion. This was associated with better preserved high energy phosphates during ischemia despite flow reduction, sustaining regional function and reducing oxidative stress, with preserved eNOS coupling after reperfusion. Preserved function during ischemia is unusual and quite different from the influence of anti-oxidants and classical preconditioning agents which typically benefit the heart only after post-reperfusion. For that matter, the benefit of FA on reducing infarct size even when delivered after ischemia had commenced implies a different mechanism.
During normoxia, ATP synthesis is highly regulated and levels maintained. With ischemia, ATP supply from anaerobic glycolysis and HEP reserves such as phosphocreatine are insufficient to meet demand, and net ATP level fall. Theoretically, interventions that could maintain ATP even in ischemic myocardium should delay the advent of lethal injury because low tissue ATP-levels (< 5 μmoles/g dry weight) are associated with depressed high-energy phosphate re-synthesis, failure of cell volume regulation and irreversible myocardial injury. FA is a known to regulate mitochondrial function plays an important role as a methylation co-factor for the synthesis of thymidylate, purines, and methionine. With regard to purine synthesis, the metabolite N10- formyl tetrahydrofolate contributes two carbons (C2, and C8) to the ring structure, with the primary end-point being formation of IMP. IMP is then further metabolized to generate AMP or GMP, or catabolized to oxypurines.
The present results support a role of FA in HEP generation. The increased basal level of oxypurines suggests that high dose of FA drove their synthesis (via IMP) by mass-action, but as HEP was adequate, this enhanced IMP catabolites with AMP levels unchanged. During ischemia, however, ATP and ADP levels fell, leading in controls to increased AMP and AMP catabolism (IMP, oxypurinols). In FA treated hearts, however, purine synthesis would be primed to help re-derive HEP (FA treated hearts had virtually no decline in ADP during ischemia, and ATP levels decline was about half). Actual ATP generation was probably higher since in FA treated hearts, since the anterior wall still actively contracted and was therefore utilizing more ATP despite LAD occlusion versus the controls. The concept of FA-enhanced HEP is supported by a recent study by Lamberts et al22 who found chronic high dose FA restored total HEP (ATP+ADP+AMP) in hypertrophied rat hearts accompanied by improved diastolic function. Our finding that FA administered i.v. 10 minutes into the ischemic period still diminished infarct size could also be explained by such a mass-action effect as this may occur quickly. This remains to be confirmed, but has intriguing therapeutic implications.
High dose FA may have potent anti-oxidant effects both directly and via weak but competitive inhibition of xanthine oxidase13, and this could also benefit the heart during ischemic and post-ischemic periods. While our in vitro data showed similar antioxidant effects between FA and Tempol, in vivo, scavenging effects of FA are thought to be modest even at high doses14. Importantly, reduced oxidant stress per se has not been previously shown to enhance HEP, rather, a decline in HEP is linked to mitochondrial damage and ROS generation23. Klawitter 4 administered 1,2-dihydroxybenzene-3,5-disulfonate (Tiron), a superoxide scavenger, or N-acetyl-L-cysteine before ischemia, and found no change in energetic recovery as compared with untreated hearts. Tempol reduces post-reperfusion infarct size in vivo in rat and rabbit by ∼50 and 33%, respectively24, but only modestly improves LV pressure recovery post reperfusion25, and similar results are reported with other free radical scavengers26. Thus, it seems unlikely that the sole or primary mechanism for FA benefit was its antioxidant capacity.
Another potential mechanism is the re-coupling of NOS. NOS uncoupling is thought to contribute to the pathophysiology of diseases such as hypertension, atherosclerosis, and cardiac hypertrophy18,27-29. Uncoupling involves a decline in the normal electron transfer from the reductase to oxidase domains, leading to reduced NO synthesis and greater ROS generation by NOS30. BH4 plays a key role in maintaining normal eNOS coupling, and FA (or 5-MTHF) can increase BH4 levels by facilitating enzymatic reduction of its oxidized forms31, and are associated with improved consequentially improved endothelial function14. The current study supports this mechanism based on the diminished eNOS uncoupling post-reperfusion, and in vitro analysis showing that addition of BH4 reduced ROS generation far more in untreated versus FA-treated myocardium. Improved NOS coupling could also contribute to improved HEP generation, as endogenous NO generation reduces oxygen consumption and improves energetic efficiency32 partly by enhancing HEP synthesis itself.
This is the first study to test FA effects on in vivo ischemia and IR injury. Several recent studies have found FA and/or its active metabolite 5-methyltetrahydrofolate (5-MTHF) improves endothelial function21,33, though the dose required has been somewhat controversial. Tawakol et al34 used high dose folic acid (30mg p.o.) in patients with coronary artery disease and found it increased both adenosine-stimulated myocardial blood flow and flow reserve in segments with impaired dilator reserve, but also acutely lowered arterial pressure. The anti-necrotic effect of FA observed in the current study is consistent with earlier in vitro data in which high dose folate (>1 mM) suppressed apoptosis from oxidant injury in U937 cells35. The beneficial effect of FA on in vivo ischemia and reperfusion-induced arrhythmias is similar to the reduction of in vitro reperfusion-induced arrhythmias by FA described by Manning36. One prior study10 has examined BH4 effects on post-IR infarct size, and found more modest changes than we observed with folate. This can be due to dose differences, but also to specific influences that folate has unrelated to BH4.
FA has been studied in clinical trials, particularly to test its potential to lower cardiovascular risk in patients with myocardial vascular disease. For example, Oster37 demonstrated that long-term folic acid treatment (∼10 years) at a dose far higher than typically used (40-80 mg/day), reduced the incidence of myocardial infarction, angina pectoris and requirement for nitroglycerin in patients with coronary artery disease. This observation was not confirmed by a placebo controlled trial nor was the mechanism explored. Other studies focused on the ability of FA to lower homocysteine, and found a 3 μmol/l decrease in serum homocysteine (achievable with 0.8mg/d). Despite these positive results, meta-analyses of multiple FA trials for use in cardiovascular prevention have been unimpressive38,39 and it remains unclear if dose, study duration, target population, or other factors explain this. The doses of FA used in the present study are far above those employed in prior cardiovascular interventional studies (5-25mg/70kg/d) or prevention trials (500 μg-1mg). Even taking into account that rodents have a higher metabolic turn-over, the present dose would likely correlate with between 0.25-2.5 g/70kg/d in a human. While long-term treatment at this level may have adverse off-target effects, short term dosing in a peri-infarct period should be well tolerated. Admittedly, this remains to be determined.
A few limitations to our study should be noted. While FA pre-treatment improved HEP balance and function during a 30 minute coronary occlusion, it might become less effective if the ischemic period were more prolonged. It is also unknown how long after an occlusion can might still intervene with FA and see a benefit. Lastly, our data support but do not prove that FA directly helps re-derive HEP by mass action effects on purine synthesis. This will require future studies employing 14C-labeled folate where the carbons can then be tracked and direct involvement tested.
In conclusion, we have found marked and novel evidence for improved ischemic and post-ischemic/reperfusion myocardial function and reduced infarction by pre-treatment with high dose FA. The relation to preservation of myocardial HEP during the ischemic period is intriguing, and suggests that FA can alter the manner by which HEP are synthesized despite a decline in myocardial perfusion. This may relate to changes in purine synthesis, enhanced NOS coupling modulated by folate/BH4 interactions, and/or by effects of FA on improving mitochondrial function during ischemia. Given that FA is inexpensive, safe even at high doses, and readily available, it is possible that such treatment in high risk patients could provide a novel means to limit ischemic damage.
Acknowledgments
The authors thank Prof. dr. C. Van Campenhout for help with the LDH measurements on the coronary effluent, F. Terloo, R. Spillemaeckers and D. Vindevogel for technical assistance, J. Van Daele and D. De Rijck for graphic assistance.
Supported by: Research fund of the University of Antwerp, the Belgian-American Education Foundation (Collen)-Grant, and an American Heart Association Post-doctoral Fellowship Grant (ALM), the Fund for Scientific Research FWO-Flanders, n° 2204/4951 (CJV/ JPT), NHLBI grants HL31069, HL43023 and HL66331 (MSW), American Heart Association Scientist Development Grant (HCC), and, NIH grants RO1-AG18324, HL-47511, and PO1-HL59408 (DAK).
Reference List
- 1.Jennings RB, Steenbergen C., Jr Nucleotide metabolism and cellular damage in myocardial ischemia. Annu Rev Physiol. 1985;47:727–749. doi: 10.1146/annurev.ph.47.030185.003455. [DOI] [PubMed] [Google Scholar]
- 2.Gross ER, Gross GJ. Ligand triggers of classical preconditioning and postconditioning. Cardiovasc Res. 2006;70:212–221. doi: 10.1016/j.cardiores.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 3.Gunduz D, Kasseckert SA, Hartel FV, Aslam M, Abdallah Y, Schafer M, Piper HM, Noll T, Schafer C. Accumulation of extracellular ATP protects against acute reperfusion injury in rat heart endothelial cells. Cardiovasc Res. 2006;71:764–773. doi: 10.1016/j.cardiores.2006.06.011. [DOI] [PubMed] [Google Scholar]
- 4.Klawitter PF, Murray HN, Clanton TL, Angelos MG. Reactive oxygen species generated during myocardial ischemia enable energetic recovery during reperfusion. Am J Physiol Heart Circ Physiol. 2002;283:H1656–H1661. doi: 10.1152/ajpheart.00041.2002. [DOI] [PubMed] [Google Scholar]
- 5.Zweier JL, Talukder MA. The role of oxidants and free radicals in reperfusion injury. Cardiovasc Res. 2006;70:181–190. doi: 10.1016/j.cardiores.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 6.Yamamura K, Steenbergen C, Murphy E. Protein kinase C and preconditioning: role of the sarcoplasmic reticulum. Am J Physiol Heart Circ Physiol. 2005;289:H2484–H2490. doi: 10.1152/ajpheart.00590.2005. [DOI] [PubMed] [Google Scholar]
- 7.Jones SP, Bolli R. The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol. 2006;40:16–23. doi: 10.1016/j.yjmcc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 8.Jones SP, Girod WG, Palazzo AJ, Granger DN, Grisham MB, Jourd'Heuil D, Huang PL, Lefer DJ. Myocardial ischemia-reperfusion injury is exacerbated in absence of endothelial cell nitric oxide synthase. Am J Physiol. 1999;276:H1567–H1573. doi: 10.1152/ajpheart.1999.276.5.H1567. [DOI] [PubMed] [Google Scholar]
- 9.Jones SP, Greer JJ, Kakkar AK, Ware PD, Turnage RH, Hicks M, van Haperen R, de Crom R, Kawashima S, Yokoyama M, Lefer DJ. Endothelial nitric oxide synthase overexpression attenuates myocardial reperfusion injury. Am J Physiol Heart Circ Physiol. 2004;286:H276–H282. doi: 10.1152/ajpheart.00129.2003. [DOI] [PubMed] [Google Scholar]
- 10.Wajima T, Shimizu S, Hiroi T, Ishii M, Kiuchi Y. Reduction of myocardial infarct size by tetrahydrobiopterin: possible involvement of mitochondrial KATP channels activation through nitric oxide production. J Cardiovasc Pharmacol. 2006;47:243–249. doi: 10.1097/01.fjc.0000201360.71813.8a. [DOI] [PubMed] [Google Scholar]
- 11.Hevel JM, Marletta MA. Macrophage nitric oxide synthase: relationship between enzyme-bound tetrahydrobiopterin and synthase activity. Biochemistry. 1992;31:7160–7165. doi: 10.1021/bi00146a019. [DOI] [PubMed] [Google Scholar]
- 12.Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA., Jr Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc Natl Acad Sci U S A. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stroes ES, van Faassen EE, Yo M, Martasek P, Boer P, Govers R, Rabelink TJ. Folic acid reverts dysfunction of endothelial nitric oxide synthase. Circ Res. 2000;86:1129–1134. doi: 10.1161/01.res.86.11.1129. [DOI] [PubMed] [Google Scholar]
- 14.Shirodaria C, Antoniades C, Lee J, Jackson CE, Robson MD, Francis JM, Moat SJ, Ratnatunga C, Pillai R, Refsum H, Neubauer S, Channon KM. Global improvement of vascular function and redox state with low-dose folic acid: implications for folate therapy in patients with coronary artery disease. Circulation. 2007;115:2262–2270. doi: 10.1161/CIRCULATIONAHA.106.679084. [DOI] [PubMed] [Google Scholar]
- 15.Moat SJ, Clarke ZL, Madhavan AK, Lewis MJ, Lang D. Folic acid reverses endothelial dysfunction induced by inhibition of tetrahydrobiopterin biosynthesis. Eur J Pharmacol. 2006;530:250–258. doi: 10.1016/j.ejphar.2005.11.047. [DOI] [PubMed] [Google Scholar]
- 16.Depeint F, Bruce WR, Shangari N, Mehta R, O'Brien PJ. Mitochondrial function and toxicity: role of B vitamins on the one-carbon transfer pathways. Chem Biol Interact. 2006;163:113–132. doi: 10.1016/j.cbi.2006.05.010. [DOI] [PubMed] [Google Scholar]
- 17.Lamberts RR, Caldenhoven E, Lansink M, Witte G, Vaessen RJ, St Cyr JA, Stienen GJ. Preservation of diastolic function in monocrotaline-induced right ventricular hypertrophy in rats. Am J Physiol Heart Circ Physiol. 2007;293:H1869–H1876. doi: 10.1152/ajpheart.00294.2007. [DOI] [PubMed] [Google Scholar]
- 18.Takimoto E, Champion HC, Li M, Ren S, Rodriguez ER, Tavazzi B, Lazzarino G, Paolocci N, Gabrielson KL, Wang Y, Kass DA. Oxidant stress from nitric oxide synthase-3 uncoupling stimulates cardiac pathologic remodeling from chronic pressure load. J Clin Invest. 2005;115:1221–1231. doi: 10.1172/JCI21968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Reinhardt CP, Dalhberg S, Tries MA, Marcel R, Leppo JA. Stable labeled microspheres to measure perfusion: validation of a neutron activation assay technique. Am J Physiol Heart Circ Physiol. 2001;280:H108–H116. doi: 10.1152/ajpheart.2001.280.1.H108. [DOI] [PubMed] [Google Scholar]
- 20.Gupte SA, Kaminski PM, Floyd B, Agarwal R, Ali N, Ahmad M, Edwards J, Wolin MS. Cytosolic NADPH may regulate differences in basal Nox oxidase-derived superoxide generation in bovine coronary and pulmonary arteries. Am J Physiol Heart Circ Physiol. 2005;288:H13–H21. doi: 10.1152/ajpheart.00629.2004. [DOI] [PubMed] [Google Scholar]
- 21.Antoniades C, Shirodaria C, Warrick N, Cai S, de Bono J, Lee J, Leeson P, Neubauer S, Ratnatunga C, Pillai R, Refsum H, Channon KM. 5-methyltetrahydrofolate rapidly improves endothelial function and decreases superoxide production in human vessels: effects on vascular tetrahydrobiopterin availability and endothelial nitric oxide synthase coupling. Circulation. 2006;114:1193–1201. doi: 10.1161/CIRCULATIONAHA.106.612325. [DOI] [PubMed] [Google Scholar]
- 22.Lamberts RR, Caldenhoven E, Lansink M, Witte G, Vaessen RJ, St Cyr JA, Stienen GJ. Preservation of diastolic function in monocrotaline-induced right ventricular hypertrophy in rats. Am J Physiol Heart Circ Physiol. 2007;293:H1869–H1876. doi: 10.1152/ajpheart.00294.2007. [DOI] [PubMed] [Google Scholar]
- 23.Hyslop PA, Hinshaw DB, Halsey WA, Jr, Schraufstatter IU, Sauerheber RD, Spragg RG, Jackson JH, Cochrane CG. Mechanisms of oxidant-mediated cell injury. The glycolytic and mitochondrial pathways of ADP phosphorylation are major intracellular targets inactivated by hydrogen peroxide. J Biol Chem. 1988;263:1665–1675. [PubMed] [Google Scholar]
- 24.McDonald MC, Zacharowski K, Bowes J, Cuzzocrea S, Thiemermann C. Tempol reduces infarct size in rodent models of regional myocardial ischemia and reperfusion. Free Radic Biol Med. 1999;27:493–503. doi: 10.1016/s0891-5849(99)00100-8. [DOI] [PubMed] [Google Scholar]
- 25.Kutala VK, Khan M, Mandal R, Potaraju V, Colantuono G, Kumbala D, Kuppusamy P. Prevention of postischemic myocardial reperfusion injury by the combined treatment of NCX-4016 and Tempol. J Cardiovasc Pharmacol. 2006;48:79–87. doi: 10.1097/01.fjc.0000242050.16790.65. [DOI] [PubMed] [Google Scholar]
- 26.Penumathsa SV, Thirunavukkarasu M, Koneru S, Juhasz B, Zhan L, Pant R, Menon VP, Otani H, Maulik N. Statin and resveratrol in combination induces cardioprotection against myocardial infarction in hypercholesterolemic rat. J Mol Cell Cardiol. 2007;42:508–516. doi: 10.1016/j.yjmcc.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Alp NJ, McAteer MA, Khoo J, Choudhury RP, Channon KM. Increased endothelial tetrahydrobiopterin synthesis by targeted transgenic GTP-cyclohydrolase I overexpression reduces endothelial dysfunction and atherosclerosis in ApoE-knockout mice. Arterioscler Thromb Vasc Biol. 2004;24:445–450. doi: 10.1161/01.ATV.0000115637.48689.77. [DOI] [PubMed] [Google Scholar]
- 28.Bendall JK, Alp NJ, Warrick N, Cai S, Adlam D, Rockett K, Yokoyama M, Kawashima S, Channon KM. Stoichiometric relationships between endothelial tetrahydrobiopterin, endothelial NO synthase (eNOS) activity, and eNOS coupling in vivo: insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ Res. 2005;97:864–871. doi: 10.1161/01.RES.0000187447.03525.72. [DOI] [PubMed] [Google Scholar]
- 29.Adlam D, Bendall JK, De Bono JP, Alp NJ, Khoo J, Nicoli T, Yokoyama M, Kawashima S, Channon KM. Relationships between nitric oxide-mediated endothelial function, eNOS coupling and blood pressure revealed by eNOS-GTP cyclohydrolase 1 double transgenic mice. Exp Physiol. 2007;92:119–126. doi: 10.1113/expphysiol.2006.035113. [DOI] [PubMed] [Google Scholar]
- 30.Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–1714. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- 31.Hyndman ME, Verma S, Rosenfeld RJ, Anderson TJ, Parsons HG. Interaction of 5-methyltetrahydrofolate and tetrahydrobiopterin on endothelial function. Am J Physiol Heart Circ Physiol. 2002;282:H2167–H2172. doi: 10.1152/ajpheart.00935.2001. [DOI] [PubMed] [Google Scholar]
- 32.Loke KE, Laycock SK, Mital S, Wolin MS, Bernstein R, Oz M, Addonizio L, Kaley G, Hintze TH. Nitric oxide modulates mitochondrial respiration in failing human heart. Circulation. 1999;100:1291–1297. doi: 10.1161/01.cir.100.12.1291. [DOI] [PubMed] [Google Scholar]
- 33.Moat SJ, Madhavan A, Taylor SY, Payne N, Allen RH, Stabler SP, Goodfellow J, McDowell IF, Lewis MJ, Lang D. High- but not low-dose folic acid improves endothelial function in coronary artery disease. Eur J Clin Invest. 2006;36:850–859. doi: 10.1111/j.1365-2362.2006.01739.x. [DOI] [PubMed] [Google Scholar]
- 34.Tawakol A, Migrino RQ, Aziz KS, Waitkowska J, Holmvang G, Alpert NM, Muller JE, Fischman AJ, Gewirtz H. High-dose folic acid acutely improves coronary vasodilator function in patients with coronary artery disease. J Am Coll Cardiol. 2005;45:1580–1584. doi: 10.1016/j.jacc.2005.02.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang RF, Yaong HC, Chen SC, Lu YF. In vitro folate supplementation alleviates oxidative stress, mitochondria-associated death signalling and apoptosis induced by 7-ketocholesterol. Br J Nutr. 2004;92:887–894. doi: 10.1079/bjn20041259. [DOI] [PubMed] [Google Scholar]
- 36.Manning A, Bernier M, Crome R, Little S, Hearse D. Reperfusion-induced arrhythmias: a study of the role of xanthine oxidase-derived free radicals in the rat heart. J Mol Cell Cardiol. 1988;20:35–45. doi: 10.1016/s0022-2828(88)80177-9. [DOI] [PubMed] [Google Scholar]
- 37.Oster K. Atherosclerosis treated with folic acid. FASEB J. 1981;40 [Google Scholar]
- 38.Wang X, Qin X, Demirtas H, Li J, Mao G, Huo Y, Sun N, Liu L, Xu X. Efficacy of folic acid supplementation in stroke prevention: a meta-analysis. Lancet. 2007;369:1876–1882. doi: 10.1016/S0140-6736(07)60854-X. [DOI] [PubMed] [Google Scholar]
- 39.Wald DS, Wald NJ, Morris JK, Law M. Folic acid, homocysteine, and cardiovascular disease: judging causality in the face of inconclusive trial evidence. BMJ. 2006;333:1114–1117. doi: 10.1136/bmj.39000.486701.68. [DOI] [PMC free article] [PubMed] [Google Scholar]