

Abstract
Residual tissue androgens are consistently detected within the prostate tumors of castrate individuals and are thought to play a critical role in facilitating the androgen-receptor-mediated signaling pathways leading to disease progression. The source of residual tumor androgens is attributed in part to the uptake and conversion of circulating adrenal androgens. Whether the de novo biosynthesis of androgens from cholesterol or earlier precursors occurs within prostatic tumors is not known, but it has significant implications for treatment strategies targeting sources of androgens exogenous to the prostate versus ‘intracrine’ sources within the prostatic tumor. Moreover, increased expression of androgen-metabolizing genes within castration-resistant metastases suggests that up-regulated activity of endogenous steroidogenic pathways may contribute to the outgrowth of ‘castration-adapted’ tumors. These observations suggest that a multi-targeted treatment approach designed to simultaneously ablate testicular, adrenal and intracrine contributions to the tumor androgen signaling axis will be required to achieve optimal therapeutic efficacy.
Keywords: prostate cancer, hormone therapy, castration resistant, metastatic, androgen metabolism, intracrinology, steroidogenesis
Androgen-deprivation therapy has been the primary treatment for patients with metastatic prostate cancer since the seminal recognition of the disease as androgen-sensitive by Huggins and Hodges in 1941.[1] Although initially effective, hormonal therapy for metastatic cancer is marked by progression to castration-resistant disease over a period of 18–20 months, with an ensuing median survival of 1–2 years. Importantly, substantial data indicate that in the setting of ‘castrate’ serum testosterone levels, prostatic androgen concentrations remain at approximately 10–25% of the levels found in untreated patients,[2–4] well within the range capable of mediating continued androgen-receptor (AR) signaling and gene expression.[5] Moreover, residual intra-prostatic androgens are implicated in nearly every mechanism whereby AR-mediated signaling leads to the development of castration-resistant disease.[6]
The source of residual androgens within the prostate tumors of castrate men has not been fully elucidated, but has been attributed to the uptake and conversion of circulating adrenal androgens.[7,8] Whether the de novo biosynthesis of androgens from cholesterol or earlier precursors occurs within castration-resistant metastases is not known[9] but has significant implications for treatment strategies targeting sources of androgens exogenous to the prostate versus ‘intracrine’ sources active within the actual tumor tissue.
In this review we examine the data demonstrating the presence of residual tissue androgens despite castration, as well as studies suggesting that residual tumoral androgens are active in prostate cancer progression. Mechanisms by which tumoral androgen levels are maintained despite castration are discussed, including the contribution of circulating adrenal androgens and the potential contribution of de novo steroidogenesis. Finally, we discuss the therapeutic implications of these observations for the optimal treatment of prostate cancer, including data regarding novel steroidogenic enzyme inhibitors under development, and the need for combinatorial treatment strategies targeting all intra-tumoral ligand sources as well as AR-mediated contributions to the tumoral androgen axis.
INTRA-PROSTATIC ANDROGEN LEVELS AFTER CASTRATION
Geller and colleagues examined prostatic levels of dihydrotestosterone (DHT) in men with localized prostate cancer and made several seminal observations: (1) castration by orchiectomy or megace plus diethylstilbestrol (DES) reduced prostatic DHT levels by 75–80% in some but not all patients; (2) protein synthesis in epithelial and stromal cells was strongly correlated with tissue DHT levels; and (3) prostatic DHT levels were further reduced when castration was combined with adrenal androgen blockade using ketoconazole.[5,10] Geller concluded that even small amounts of residual DHT may be sufficient to stimulate tumor growth, and that the goal of therapy for prostate cancer should be to decrease prostatic DHT levels to as low as possible, a concept similarly framed in early studies by Labrie et al.[11]
Subsequent studies in men with normal prostate histology, men with benign prostatic hypertrophy (BPH), and men with localized cancer have consistently demonstrated that castration results in a 70–75% decrease in tissue testosterone levels and an 80–90% decrease in tissue DHT.[2,4,7,12,13] Interestingly, tumor grade was associated with the degree of change in tissue DHT in one report, with an 85% decrease observed in Gleason 6 tumors, but only a 60% decrease in Gleason 7–10 tumors,[14] suggesting that tumor-specific alterations in tissue androgen metabolism may influence the response to castration. In castrate patients with locally recurrent prostate cancer, prostatic DHT levels were decreased approximately 80% compared with untreated BPH tissues; however, testosterone levels were actually equivalent to BPH tissues from untreated patients.[3] Furthermore, in a study evaluating metastatic tumors obtained via rapid autopsy from anorchid men with castration-resistant prostate cancer (CRPC), testosterone levels within metastases from the castrate men were approximately three-fold higher than levels within primary prostate tumors from untreated (eugonadal) patients.[15]
These data clearly demonstrate that achieving castrate levels of circulating testosterone does not eliminate androgens from the prostate tumor microenvironment. Moreover, the persistent and even elevated tumoral testosterone levels observed in castration-resistant tumors, along with the recently reported association between tumor grade and the response to castration,[14] suggest that alterations in androgen metabolism within the tumoral tissue may contribute to residual tissue androgen levels observed in the castrate setting.
EVIDENCE THAT RESIDUAL TISSUE ANDROGENS ARE PHYSIOLOGICALLY ACTIVE
In-vitro and in-vivo data indicate that DHT levels in the 0.5–1.0 ng/g range observed in the prostatic tissue of castrate patients are adequate to activate the AR, stimulate expression of androgen-regulated genes, and support tumor-cell growth and survival.[16] In mice bearing human prostate cancer xenografts, peripheral testosterone supplementation to maintain prostate-tissue DHT levels in the 3–4 pmol/g range (~0.9 ng/g) stabilized tumor xenograft volumes, with higher tissue DHT levels stimulating tumor growth.[17]
The activity of residual tissue androgens is also supported by the observation that AR-mediated transcription is frequently detected in castration resistant-prostate tumors.[9] While this observation has been attributed in part to the development of ligand-independent AR-activation mechanisms,[6] the most parsimonious explanation for persistent AR signaling in the setting of anorchid serum testosterone levels is the continued presence of intracellular androgens at levels adequate to activate wild-type AR. In this regard, residual androgens and persistent expression of AR target genes have been demonstrated in benign prostate tissue after short-term castration, as well as in localized prostate tumors after neoadjuvant androgen suppression,[18,19] tissues in which ligand-independent signaling mechanisms are unlikely to have had the opportunity to develop through clonal selection of advantageous genomic alterations.
THE SIGNIFICANCE OF RESIDUAL ANDROGENS IN CASTRATION-RESISTANT PROSTATE CANCER
Nearly every mechanism whereby persistent AR signaling is proposed to confer a castration-resistant phenotype either still requires or is enhanced by the presence of residual AR ligands. Mechanisms leading to AR activation in the castrate setting include AR over-expression, AR mutations that broaden ligand specificity, alterations in AR co-regulator that modulate AR stability and ligand sensitivity, and activation of the AR or downstream regulatory molecules by ‘cross-talk’ with other signaling pathways.[6] Restoration of AR expression in a xenograft model was instrumental in the progression from androgen-dependent to castration-resistant growth: AR up-regulation allowed tumor-cell proliferation in 80% lower androgen concentrations, and shortened the latency of xenograft tumor formation by >50%.[20] Importantly, ligand binding was required for hormone-refractory growth, and modest increases in AR expression were sufficient to support signaling in a low-androgen environment.
The physiologic role of tumor androgens in disease progression is further substantiated by the observation that suppressing residual androgens with either secondary hormonal maneuvers or combined androgen blockade (CAB) has clinical efficacy in treating advanced and/or recurrent tumors. Nearly 30% of patients with recurrent prostate tumors demonstrate at least transient clinical responses to secondary or tertiary hormonal manipulation, suggesting that these tumors are not truly androgen-independent, but have maintained a clinically relevant degree of androgen sensitivity.[21] Initial studies of CAB suggested significant improvements in survival compared to historical controls.[11] Although subsequent randomized trials have not all been conclusive, meta-analyses have consistently suggested a statistically significant improvement in 5-year survival, on the order of 5%, in favor of CAB over castration alone,[22,23] suggesting that more effective suppression of the androgen axis would improve the degree of benefit observed in patients with advanced metastatic disease.
Thus, a substantial body of evidence suggests that castration-resistant tumors are not in fact androgen-independent, but develop in a setting of continued AR-mediated signaling that is driven by the presence of residual tumoral androgens derived from exogenous sources, endogenous synthesis, or a combination of both.[6,11] Accordingly, therapeutic strategies designed to more effectively ablate tumoral androgen activity are likely to have improved clinically efficacy in both treating hormone-naïve disease and in preventing disease progression.
THE SOURCE OF RESIDUAL TISSUE ANDROGENS AFTER CASTRATION
The intra-prostatic uptake and conversion of adrenally derived androgens is thought to be the most significant source of residual tissue androgens in anorchid patients.[24] While the requisite metabolic enzymes involved in the conversion of adrenal androgens to testosterone and DHT have been demonstrated in benign and neoplastic prostate tissue, an unresolved question is whether intra-tumoral androgens are synthesized de novo from progesterone, cholesterol or earlier precursors (Figure 1).
Figure 1.
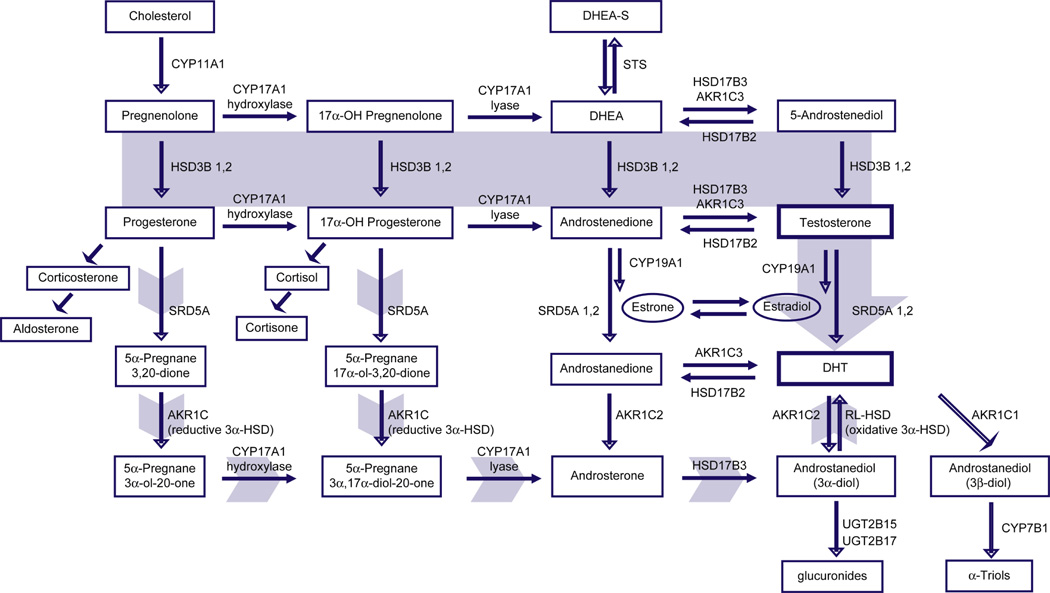
The classical and back-door pathways of androgen biosynthesis. In the classical pathway (solid gray arrow), C21 precursors (pregnenolone and progesterone) are converted to the C19 adrenal androgens dihydroepiandrosterone (DHEA) and androstenedione (AED) by the sequential hydroxylase and lyase activities of CYP17A1. Circulating adrenal androgens (including the sulfated form of DHEA, DHEA-S), enter the prostate and can be converted to testosterone by a series of reactions involving the activity of HSD3B, HSD17B and AKR1C enzymes. Testosterone is then converted to the potent androgen DHT by the activity of SRD5A. In the back-door pathway to DHT synthesis (short gray arrows), C21 precursors are first acted upon by SRD5A and the reductive 3α-hydroxysteroid dehydrogenase (3α-HSD) activity of AKR1C family members, followed by conversion to C19 androgens via the lyase activity of CYP17A, and subsequently to dihydrotestosterone (DHT) by the action of HSD17B3 and an oxidative 3α-HSD enzyme.
Prostatic utilization of adrenal androgens
In a seminal study designed to evaluate the prostatic uptake of circulating androgens, Bruchovsky administered radioactively labeled androgens – including testosterone (T), DHT, and the adrenal androgens dihydroepiandrosterone (DHEA) and androstenedione (AED) – to castrate male rats, and evaluated prostatic metabolites at 60 minutes after injection.[25] Following administration of DHEA, approximately 1% and 8% of the recovered radioactivity was found in T and DHT respectively, with 2% and 12% found after AED injection, compared to 37% conversion of exogenously administered T into DHT. In the Dunning R3327 prostate carcinoma model, administration of adrenal androgens to castrate male rats increased tumor DHT levels and stimulated tumor growth to the level of that in intact controls.[26]
In a remarkable report paralleling Bruchovsky’s animal studies, Harper et al evaluated prostate androgen metabolism by infusing eugonadal men with 3H-T, 3H-AED or 3H-DHEA-sulfate (DHEA-S) 30 minutes before performing radical prostatectomy for BPH.[27] The major metabolite present in prostate tissue after 3H-T infusion was DHT (about 65% conversion). Infusion of 3H-AED resulted in approximately 7–10% radioactivity associated with either T or DHT. 3H-DHEA-S was primarily converted to DHEA (70–90%), with 1–3% conversion to T, DHT and AED. Klein et al evaluated the presence of adrenal androgens and steroid metabolizing activity ex vivo in hormone-naive tumors and lymph-node metastases. Although malignant tissue had a subtotal loss of SRD5A activity, primary tumors and metastases possessed the capacity to metabolize adrenal androgen precursors along the pathway to DHT.[28]
Importantly, intra-prostatic adrenal androgens have been detected at significant levels in patients treated to suppress testosterone.[3,7,13] Levels of DHEA, DHEA-S and AED were decreased by about 50% in tumor tissue from castrate patients with recurrent prostate cancer, and far exceeded the values of testosterone and DHT in the recurrent tumor tissue. Notably, no decrease in tissue levels of 5-androstenediol (a primary metabolite of DHEA and a direct precursor of testosterone) was observed in another study of prostate tissues after castration,[13] which is of particular significance as this androgen has been shown to bind the wild-type AR without being inhibited by flutamide or bicalutamide.[29]
The de-novo synthesis of tumoral androgens
Up-regulated expression of several genes involved in the rate-limiting steps of steroid precursor biosynthesis have been reported in castration-resistant versus primary prostate tumors.[9] Acevedo et al investigated the metabolism of 14C-progesterone in primary prostate cancer tissues, but did not observe significant metabolic conversion beyond the formation of immediate progesterone derivatives.[30] Conversely, Montgomery et al recently reported up-regulated expression of CYP17A (a critical enzyme early in steroidogenesis) in castration-resistant tumor metastases, suggesting that castration-resistant tumors may have the ability to utilize progesterone as androgenic precursors.[15] Furthermore, a novel inhibitor of CYP17A has recently demonstrated relatively high clinical efficacy compared to historical studies of adrenalectomy,[31] suggesting that its activity may be mediated via inhibition of tumoral as well as adrenal CYP17A activity.
An alternative mechanism for prostate tumor androgen production is the de-novo synthesis of androgens via the ‘backdoor pathway’, wherein steroid flux to DHT bypasses the conventional intermediates of AED and testosterone (Figure 1). In the classical pathway, C21 steroids such as pregnenolone and progesterone are first converted to the C19 steroids DHEA and AED via the sequential hydroxylase and lyase activity of CYP17A, followed by the activity of HSD17B and SRD5A. In steroidogenic tissues in which both CYP17A and SRD5A are expressed, an alternate route to DHT is possible, wherein C21 steroids are first acted upon by SRD5A, followed by CYP17A and HSD17B.[32] Interestingly, the production of DHT in the mouse testis via this mechanism is mediated specifically by the SRD5A1 and not the SRD5A2 isoform, which is of relevance in that a clear shift from SRD5A2 to SRD5A1 expression occurs in the transition from benign to neoplastic prostate tissue (see below). Moreover, recombinant human CYP17A displays markedly more robust lyase activity for the 5α-reduced progesterone intermediates than for the classical substrates 17α-OH pregnenolone or 17α-OH progesterone, such that the combination of increased SRD5A1 activity in conjunction with the proposed expression of CYP17A in prostate cancer tissue might actually favor de novo synthesis via the backdoor pathway over the classical pathway, although this hypothesis remains to be tested.
These studies clearly demonstrate the uptake and conversion of adrenal androgens within prostatic tumor tissue. Although Harper et al found only a 5–15% rate of adrenal androgen metabolism to testosterone and DHT within prostatic tissue, intra-prostatic concentrations of adrenal androgens substantially exceed those of testosterone and DHT, and therefore provide a significant reservoir of substrate for conversion. Whether the de novo synthesis of tumoral androgens from cholesterol or earlier precursors occurs to a substantial and/or clinically relevant degree within prostate tumors requires further evaluation.
ALTERATIONS IN INTRACRINE ANDROGEN METABOLISM IN NEOPLASTIC PROSTATE TISSUES
Alterations in a number of critical enzymes responsible for DHT synthesis and catabolism have been identified in primary and castration–resistant prostate tumors, providing mechanistic support for the role of intracrine steroid metabolism in modulating the tumor androgen microenvironment. A subtotal loss of tumoral SRD5A activity, the primary isoform in benign prostate tissue, has been consistently observed in neoplastic prostate tissues,[28] with a relative shift to SRD5A1 in primary and recurrent prostate tumors;[8,33,34] however, some studies have shown Gleason-grade-related increases in both SRD5A1 and SRD5A2.[35] These data indicate that treatment strategies designed to inhibit DHT production must adequately suppress the activity of both isoforms.
Differential expression of several 17β-hydroxysteroid dehydrogenase family members (HSD17B) has been observed in prostate cancer, with increased tumoral expression of reductive enzymes catalyzing conversion of precursors to active androgens (HSD17B3 and HSD17B5 – also known as aldo-keto reductase AKR1C3), and decreased expression of oxidative enzymes catalyzing the reverse reaction (HSD17B2),[8,36,37] suggesting a shift in tumoral androgen metabolism to the formation of testosterone and DHT. Interestingly, increased prostate tumor expression of the oxidative family member HSD17B4 has also been observed; however, this isoform has a unique peroxisomal targeting sequence and acts primarily in peroxisomal β-chain oxidation of fatty acids.[38] Differential expression of AKR1C1 and AKR1C2, which mediate the catabolism of DHT,[39] has also been observed in prostate tumors. Ji et al have demonstrated a selective loss of AKR1C2 and AKR1C1 in primary prostate tumors, accompanied by a reduced capacity to catabolize DHT and increased tumoral DHT levels.[40] As these investigators also noted a decrease in SRD5A2 expression, the elevated tissue DHT levels underscore how effectively the decrease in DHT catabolizing activity can maintain tumoral DHT despite a relative decrease in the conversion of testosterone to DHT.
In summary, androgen metabolism within primary prostate tumors is characterized by steroid enzyme alterations which may potentiate the intracrine conversion of adrenal androgens to testosterone and DHT and/or inhibit the conversion of DHT to inactive metabolites. These observations suggest that tissue-based alterations in steroid metabolism may facilitate prostatic tumor development and underscore these metabolic pathways as critical therapeutic targets.
NOVEL AGENTS FOR THE INHIBITION OF RESIDUAL TUMORAL ANDROGENS
Historically, ketoconazole and glutethimide – or, more rarely, surgical approaches such as adrenalectomy and hypophysectomy – have been utilized for suppression of residual tissue androgens. Due to limited efficacy and significant treatment-related side-effects, only ketoconazole (a weak inhibitor of CYP11A and CYP17A) remains in use in routine clinical practice. However, the limited efficacy of ketoconazole in castration-resistant prostate cancer has prompted the development of more potent inhibitors of androgen biosynthesis.
Inhibitors of CYP17A
CYP17A is a single enzyme which catalyzes sequential steps in the conversion of C21 precursors to the C19 adrenal androgens DHEA and AED. A number of CYP17A inhibitors have been reported,[41] including a series of novel agents exhibiting both CYP17A inhibition and anti-androgen activity.[42] The most potent of these, VN/124-1, effectively inhibited the binding of the synthetic androgen R1881 to either mutant or wild-type AR, acted as a pure AR antagonist in vitro, and was more effective than castration in suppressing growth of the androgen-dependent LAPC prostate tumor xenograft in vivo.
To date, abiraterone is the only CYP17A inhibitor to have been evaluated in phase-I studies, and it shows promising efficacy in reducing circulating androgen levels and achieving PSA responses. In dose-finding studies, castrate men treated with a single 500-mg dose (n = 6) demonstrated suppression of testosterone levels by ≥75% within 48 hours; this was maintained for 2–5 days, and was accompanied by a moderate decrease in serum AED levels. In eugonadal men treated with the same 500-mg dose (n = 3), testosterone levels were suppressed by >50% within 48 hours, and recovered to baseline in 6–9 days, accompanied by a corresponding rise and then decline in luteinizing hormone (LH) levels. Baseline cortisol levels remained normal, although all patients treated in a 12-day continuous dosing regimen developed an abnormal response to adrenocorticotropic hormone (ACTH) provocation.[43] In a small study reported by Ryan et al, 16 patients with CRPC (nine previously treated with ketoconazole) were treated with abiraterone 500 mg per day orally for 28 days. Five of the nine ketoconazole-treated patients experienced a >50% decline in PSA. A rise in mineralocorticoids and a decline in adrenal androgens was observed, with four patients developing grade-1 hypertension and two patients requiring steroid replacement for mild symptoms related to adrenal insufficiency.[44]
Attard et al have reported a phase-I continuous dosing study of abiraterone in men with metastatic CRPC treated for up to 14 months. Doses from 300 to 1000 mg orally per day suppressed DHEA levels by approximately 75%, and suppressed AED and testosterone to undetectable levels. Durable PSA declines ≥50% were observed in 21/34 patients (60%), with PSA declines ≥90% in 11/34 (32%). As observed in studies of ketoconazole,[45] patients achieving ≥50% PSA declines had higher baseline DHEA-S, DHEA and AED levels compared to patients in whom PSA declines did not occur. Of patients evaluable by response evaluation criteria in solid tumors (RECIST) criteria, 12/21 had radiological partial responses and 8/21 had stable disease. Of patients evaluable for toxicity, side-effects were generally mild and primarily related to symptoms of mineralocorticoid excess, including grade-1 and -2 hypertension (11/38), hypokalemia (31/38), edema (10/38) and fatigue (14/38).[31]
Abiraterone resulted in the expected increases in C21 steroids upstream of CYP17A – including deoxycorticosterone (up ten-fold) and corticosterone (up 40-fold) – and the expected decreases in C19 steroids downstream of CYP17A – including DHEA, AED and testosterone. However, a significant decrease in 17-OH pregnenolone (derived from the 17α-hydroxylase activity of CYP17A) was not observed, and cortisol levels (downstream of 17-OH pregnenolone) were decreased only about two-fold, accompanied by a five-fold increase in ACTH levels. These observations suggest that cortisol-driven feedback mechanisms may compensate for the inhibition of 17α-hydroxylase activity caused by abiraterone. As the two reactions catalyzed by CYP17A are independently regulated, an ACTH-mediated feedback mechanism specifically leading to increased CYP17A hydroxylase (but not lyase) activity could maintain 17-OH pregnenolone levels without concomitant elevation of C19 adrenal androgens.
These observations are of relevance in that progestins and corticosteroids have been reported to act as non-canonical AR ligands,[46] consistent with the preliminary observation of Attard et al that the addition of dexamethasone at 0.5 mg per day (in eight patients with disease progression while on abiraterone) suppressed ACTH, deoxycorticosterone, and corticosterone levels and was associated with PSA responses. Furthermore, in contrast to studies with ketoconazole, in which disease progression has been accompanied by a rise in circulating adrenal androgen levels[47] – no increase in testosterone, AED or DHEA levels has been observed at progression in abiraterone-treated patients, further suggesting non-canonical AR activation in disease progression. In this respect, clinical development of CYP17A inhibitors such as VN/124-1, which additionally display AR binding and antagonism, is of significant interest.
Inhibitors of HSD17B3 and AKR1C3
HSD17B3 and AKR1C3 catalyze reduction of 17-ketosteroids, including AED and androstanedione, to the more active 17-OH steroids, testosterone and DHT. Increased expression of these enzymes in prostate tumors suggests they may be important targets for inhibition. Poirier and colleagues recently reported a novel hybrid strategy in which an adenosine moiety (to interact with the cofactor-binding site) is linked via an alkyl spacer to AED in order to obtain a bi-substrate inhibitor of HSD17B3, yielding several compounds with micromolar inhibitory potency in cell-free assays.[48] Lorenzi et al have identified several small-molecule inhibitors of HSD17B3 with nanomolar cellular potency, able to decrease testosterone levels in intact rats by 70% within 3 hours, although with subsequent recovery through unidentified mechanisms.[49]
Interestingly, the AKR1C family members are inhibited by pharmacologic doses of non-steroidal anti-inflammatory drugs (NSAIDs), in rank order of their anti-inflammatory potency, and the COX-2 selective inhibitor, celecoxib.[50] The two most potent classes of NSAIDs targeting the AKR1C and COX isozymes are the indoleacetic acids (e.g. indomethacin) and the N-phenylanthranilic acids. Penning and colleagues generated a series of N-phenylanthranilic acid analogs that retained specificity for inhibition of AKR1C enzymes without inhibiting COX-1 or COX-2, and identified key substituents required for selective inhibition of AKR1C isoforms (which share 86% homology).[50] More recently, an indomethacin analog, N-(4-chlorobenzoyl)-melatonin (CBM) – predicted to prevent inhibition of AKR1C1, AKR1C2 and the COX isoenzymes while retaining the preferential inhibition of AKR1C3 observed with indomethacin – has been described.[51] A number of additional agents with inhibitory activity against AKR1C3 has been reported, including phytoestrogens and cinnamic acid derivatives, but specificity with respect to other AKR1C family members and COX enzymes has not been described.[52,53]
Inhibitors of HSD3B and SRD5A
Isoform-specific differences in tissue expression and/or pharmacogenetic activity of HSD3B and SRD5A have yielded important insight into factors relevant to the effective development of steroidogenic enzyme inhibitors. HSD3B is a dimeric bifunctional enzyme required for the biosynthesis of all classes of steroid hormones. The two human isoforms, encoded by distinct genes, share 93% homology and have an isoform-specific tissue distribution, with HSD3B1 expressed in the placenta and peripheral tissues such as skin, breast and prostate, and HSD3B2 almost exclusively expressed in the adrenal, testis and ovary.[54] Despite differing by only one residue in the catalytic site, HSD3B1 has 14–16-fold higher affinity for the dehydrogenase substrate and three-fold higher affinity for the isomerase substrate versus 3BHSD2. Reciprocal residues in the dimer subunit interface responsible for the differential activity of the two isoforms have been identified, and selective inhibition of HSD3B1 without inhibition of HSD3B2 has been experimentally demonstrated.[55] The isoform-specific differences in tissue distribution and catalytic efficiency of the HSD3B isoforms are particularly relevant to designing HSD3B inhibitors for prostate cancer therapy, as either or (most likely) both sites of androgen synthesis, the adrenal and the prostate, are thought to be important targets for inhibition.
SRD5A1 and SRD5A2, encoded by distinct genes, share 50% amino acid identity and catalyze the 5α-reduction of testosterone to DHT with different pH optima and kinetics.[34] Finasteride (an inhibitor of SRD5A2) and dutasteride (a dual SRD5A inhibitor) are 4-azasteroids used extensively in clinical prostate studies. Significant genetic and pharmacogenetic variation in the SRD5A2 locus has been reported, with several SRD5A2 mutations resulting in increased enzyme activity and reduced inhibition by finasteride. Dutasteride displays significantly less pharmacogenetic variation than finasteride in its inhibitory activity, as well as significantly lower inhibition constants for nearly all SRD5A2 variants examined.[56] These observations underscore the importance of tissue-specific isoform expression and pharmacogenetic variability in enzyme activity in the effective application of steroidogenic enzyme inhibitors. Recently, a third SRD5A isoenzyme has been reported,[57] although the clinical significance of this isoform is unknown.
Inhibitors of steroid sulfatase
Steroid sulfatase (STS) hydrolyzes inactive sulfates of estrogen and DHEA to biologically active steroids. Although not yet tested in prostate cancer, STS inhibitors have been evaluated in patients with breast cancer,[58] and may have efficacy in preventing prostatic utilization of the adrenal androgen DHEA, which circulates primarily as the inactive sulfate, DHEA-S. STS activity has been demonstrated in prostate cancer cell lines and in prostate tissue homogenates,[59] and immunohistochemical expression of STS in 85% of prostatic tumors examined (n = 52) was recently reported.[60] Given the proposed importance of adrenal androgens in maintaining prostate tumor androgen metabolism in anorchid patients, the clinical efficacy of preventing tumoral DHEA-S utilization by STS inhibitors warrants examination.
In summary, agents designed to inhibit critical enzymes in the androgen metabolic pathway are currently under development. Further study is required to delineate which steroidogenic pathways represent the most critical targets for inhibition, as well as elucidating the relative contribution of adrenal versus tumoral sites of intracrine androgen production. In this regard, the relatively high clinical efficacy observed in initial studies of the CYP17A inhibitor abiraterone, compared to historical studies of medical and surgical adrenalectomy, suggests that the clinical activity of this agent may be derived from suppressing tumoral as well as adrenal sources of CYP17A activity.
THE IMPLICATIONS OF INTRACRINE ANDROGEN METABOLISM FOR PROSTATE CANCER THERAPY
Residual androgens are consistently detected within the prostate tumors of castrate individuals and may play a critical role in facilitating the AR-mediated signaling pathways leading to disease progression. Moreover, substantial data demonstrate that multiple and parallel intracrine metabolic pathways are active within prostate tumors and could serve as a primary source of residual tissue androgens in the castrate microenvironment. The increased expression of androgen-metabolism genes within castration-resistant metastatic tumors[8,9,15] strongly suggests that up-regulated activity of endogenous steroidogenic pathways is driving the outgrowth of ‘castration-adapted’ tumors. Furthermore, recent data emphasize the molecular heterogeneity exhibited by both early- and advanced-stage prostate tumors in the response to castration.[14,18,61] These observations suggest that a multi-targeted treatment approach designed to maximally ablate all contributions to the AR signaling axis within the prostate tumor will be required to achieve optimal anti-tumor efficacy.
Important questions requiring further study include determining: (1) which steroidogenic enzymes represent the most critical targets for inhibition; (2) whether these targets are the same or different in different patients or at different time points in therapy; and (3) whether the most relevant sites of extra-testicular steroidogenic inhibition are within the adrenal gland, or are more proximal, within the locally active metabolic pathways of the tumor tissue itself. Currently, the parallel nature of the steroidogenic pathways active within prostate tumors, as well as the underlying heterogeneity observed among individuals in the response to castration, indicate that a combined-treatment approach targeting multiple contributions to the androgen axis is most likely to be successful in achieving a uniform and sustained ablation of the tumoral androgen axis.[62] Such a strategy would include targeting all potential contributors to the prostatic androgen milieu, including testicular, adrenal and intra-prostatic sources, as well as novel agents targeting the stability, degradation and activity of the AR.[6,62]
The data also suggest that initiating therapy with a simultaneous combinatorial approach, rather than the sequentially additive approach more commonly practiced today, will be most effective in preventing castration-adapted disease progression. The minor clinical benefit observed in historical studies of combined androgen blockade versus monotherapy is clearly attributable to the relatively weak efficacy of standard anti-androgens in stably inhibiting the AR, resulting in suboptimal inhibition of residual androgen activity. The incorporation of potent steroidogenic inhibitors such as abiraterone, or the dual CYP17A/AR inhibitor such as the compound VN/124-1, in combination with novel AR inhibitors (such as Hsp90 and histone deacetylase inhibitors), holds promise for significantly improved therapeutic efficacy. Moreover, in contrast to palliative application in advanced disease, androgen suppression is now being applied with potentially curative benefit in the neoadjuvant and adjuvant setting, raising the question as to whether more potent suppression of the androgen axis could result in more significant survival gains than those already observed.
Importantly, treatment strategies intended to ablate prostate tumor AR activity should ideally be demonstrated to have end-organ tissue and molecular effects before conclusions are drawn regarding clinical efficacy. In this regard, neoadjuvant studies, the development of novel AR-based imaging agents (e.g. fluoro-DHT PET), or protocol designs with biopsies of primary and/or metastatic tissues are of paramount importance in evaluating the pharmacodynamics, molecular activity and anti-tumor efficacy of new agents, as well as in identifying potential serum biomarkers of tissue effect.
Practice points
serum testosterone levels in the castrate range (<50 ng/dL or, ideally, undetectable) should be confirmed prior to a diagnosis of CRPC
secondary hormonal maneuvers are effective in up to one third of patients with CRPC
anorchid patients with higher baseline androgen levels at disease progression (e.g. serum testosterone 30–50 ng/dL; adrenal androgens in the mid to upper tertile) may have a higher likelihood of response to secondary hormonal maneuvers
combinatorial strategies targeting the androgen/AR axis (anti-androgens, metabolic enzyme inhibitors) may have benefits in subpopulations of patients and should be considered at the time of hormone manipulation therapy
Research agenda
non-invasive correlates of tumor androgen ablation (such as AR or angiogenesis-based imaging methods, or novel serum metabolites of tissue androgen metabolism) should be developed
studies should be undertaken to determine: (1) which steroidogenic enzymes represent the most critical targets for inhibition; (2) whether these targets are the same or different in different patients or at different time points in therapy; and (3) whether the most relevant sites of extra-testicular steroidogenic inhibition are within the adrenal gland, or are more proximal, within the locally active metabolic pathways of the tumor tissue itself
the efficacy of novel steroidogenic inhibitors should be evaluated in clinical studies employing biopsies of primary and/or metastatic tissues in order to confirm anticipated alterations in tissue androgen levels and metabolism
the efficacy of novel steroidogenic inhibitors should be evaluated in clinical studies employing biopsies of primary and/or metastatic tissues in order to confirm anticipated alterations in tissue androgen levels and metabolism
longitudinal studies evaluating tissue markers or surrogate assays of intracrine androgen metabolism should be established to determine mechanisms by which disease progression occurs in response to hormonal therapy
inter-patient variations in intracrine metabolic pathways (due to tumor-specific metabolic features or inherited variations in steroidogenic enzyme activity) should be identified as potential targets for individualized therapy
SUMMARY
Data regarding the molecular response of prostate cancer to hormone therapy continues to emerge, providing critical insight into growth and signaling pathways that may be exploited as therapeutic targets. Accumulated data emphasize the presence of residual androgens and persistent activation of the AR signaling axis in advanced prostate tumors despite castration. The activity of critical enzymes responsible for DHT synthesis and catabolism has been identified in prostate tumors, providing mechanistic support for intracrine steroid metabolism as a primary source of residual tissue androgens in the castrate microenvironment. Moreover, increased expression of steroidogenic enzymes within castration-resistant metastases suggests that up-regulated activity of endogenous androgen metabolic pathways may contribute to the outgrowth of ‘castration-adapted’ tumors, further underscoring the importance of these enzymes as treatment targets. Together, the basic, translational and clinical studies reviewed in this report indicate that the androgen axis remains an important therapeutic pathway for drug development in advanced prostate cancer.
ACKNOWLEDGEMENTS
This work was supported by a Career Development Award from the Prostate Cancer Foundation, a Young Investigator Award from the American Society of Clinical Oncology, and NIH grant 5K23 CA122820-02 (all to E.A.M.), and the NIH/NCI Pacific Northwest Prostate Cancer SPORE grant P50CA97186 (P.S.N).
Footnotes
CONFLICT OF INTEREST STATEMENT, Dr Nelson has served as an advisor to GlaxoSmithKline and Solvay Pharmaceuticals, and has received research funding from GlaxoSmithKline.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Huggins C, Hodges CV. Studies on prostatic cancer. I. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J Clin. 1972;22(4):232–240. doi: 10.3322/canjclin.22.4.232. [DOI] [PubMed] [Google Scholar]
- 2.Forti G, Salerno R, Moneti G, et al. Three-month treatment with a long-acting gonadotropin-releasing hormone agonist of patients with benign prostatic hyperplasia: Effects on tissue androgen concentration, 5 alpha-reductase activity and androgen receptor content. J Clin Endocrinol Metab. 1989;68(2):461–468. doi: 10.1210/jcem-68-2-461. [DOI] [PubMed] [Google Scholar]
- *3.Mohler JL, Gregory CW, Ford OH, 3rd, et al. The androgen axis in recurrent prostate cancer. Clin Cancer Res. 2004;10(2):440–448. doi: 10.1158/1078-0432.ccr-1146-03. [DOI] [PubMed] [Google Scholar]
- 4.Nishiyama T, Hashimoto Y, Takahashi K. The influence of androgen deprivation therapy on dihydrotestosterone levels in the prostatic tissue of patients with prostate cancer. Clin Cancer Res. 2004;10(21):7121–7126. doi: 10.1158/1078-0432.CCR-04-0913. [DOI] [PubMed] [Google Scholar]
- 5.Geller J, Liu J, Albert J, et al. Relationship between human prostatic epithelial cell protein synthesis and tissue dihydrotestosterone level. Clin Endocrinol (Oxf) 1987;26(2):155–161. doi: 10.1111/j.1365-2265.1987.tb00771.x. [DOI] [PubMed] [Google Scholar]
- 6.Scher HI, Buchanan G, Gerald W, et al. Targeting the androgen receptor: Improving outcomes for castration-resistant prostate cancer. Endocr Relat Cancer. 2004;11(3):459–476. doi: 10.1677/erc.1.00525. [DOI] [PubMed] [Google Scholar]
- 7.Belanger B, Belanger A, Labrie F, et al. Comparison of residual C-19 steroids in plasma and prostatic tissue of human, rat and guinea pig after castration: Unique importance of extratesticular androgens in men. J Steroid Biochem. 1989;32(5):695–698. doi: 10.1016/0022-4731(89)90514-1. [DOI] [PubMed] [Google Scholar]
- *8.Stanbrough M, Bubley GJ, Ross K, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66(5):2815–2825. doi: 10.1158/0008-5472.CAN-05-4000. [DOI] [PubMed] [Google Scholar]
- *9.Holzbeierlein J, Lal P, LaTulippe E, et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am J Pathol. 2004;164(1):217–227. doi: 10.1016/S0002-9440(10)63112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu J, Geller J, Albert J, et al. Acute effects of testicular and adrenal cortical blockade on protein synthesis and dihydrotestosterone content of human prostate tissue. J Clin Endocrinol Metab. 1985;61(1):129–133. doi: 10.1210/jcem-61-1-129. [DOI] [PubMed] [Google Scholar]
- 11.Labrie F, Dupont A, Belanger A, et al. Combination therapy with flutamide and castration (LHRH agonist or orchiectomy) in advanced prostate cancer: A marked improvement in response and survival. J Steroid Biochem. 1985;23(5B):833–841. doi: 10.1016/s0022-4731(85)80024-8. [DOI] [PubMed] [Google Scholar]
- 12.Page ST, Lin DW, Mostaghel EA, et al. Persistent intraprostatic androgen concentrations after medical castration in healthy men. J Clin Endocrinol Metab. 2006;91(10):3850–3856. doi: 10.1210/jc.2006-0968. [DOI] [PubMed] [Google Scholar]
- 13.Mizokami A, Koh E, Fujita H, et al. The adrenal androgen androstenediol is present in prostate cancer tissue after androgen deprivation therapy and activates mutated androgen receptor. Cancer Res. 2004;64(2):765–771. doi: 10.1158/0008-5472.can-03-0130. [DOI] [PubMed] [Google Scholar]
- 14.Nishiyama T, Ikarashi T, Hashimoto Y, et al. The change in the dihydrotestosterone level in the prostate before and after androgen deprivation therapy in connection with prostate cancer aggressiveness using the Gleason score. J Urol. 2007;178(4 Pt 1):1282–1288. doi: 10.1016/j.juro.2007.05.138. discussion 1288–1289. [DOI] [PubMed] [Google Scholar]
- 15.Montgomery B, Mostaghel E, Vessella R, et al. Androgen synthesis in castration-adapted metastatic prostate cancer. Journal of Clinical Oncology, 2007 ASCO Annual Meeting Proceedings. 2007;Part 1, Vol 25(No 18S):98. [Google Scholar]
- 16.Gregory CW, Johnson RT, Jr, Mohler JL, et al. Androgen receptor stabilization in recurrent prostate cancer is associated with hypersensitivity to low androgen. Cancer Res. 2001;61(7):2892–2898. [PubMed] [Google Scholar]
- 17.van Weerden WM, van Steenbrugge GJ, van Kreuningen A, et al. Effects of low testosterone levels and of adrenal androgens on growth of prostate tumor models in nude mice. J Steroid Biochem Mol Biol. 1990;37(6):903–907. doi: 10.1016/0960-0760(90)90441-m. [DOI] [PubMed] [Google Scholar]
- *18.Mostaghel EA, Page ST, Lin DW, et al. Intraprostatic androgens and androgen-regulated gene expression persist after testosterone suppression: Therapeutic implications for castration-resistant prostate cancer. Cancer Res. 2007;67(10):5033–5041. doi: 10.1158/0008-5472.CAN-06-3332. [DOI] [PubMed] [Google Scholar]
- 19.Ryan CJ, Smith A, Lal P, et al. Persistent prostate-specific antigen expression after neoadjuvant androgen depletion: An early predictor of relapse or incomplete androgen suppression. Urology. 2006;68(4):834–839. doi: 10.1016/j.urology.2006.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *20.Chen CD, Welsbie DS, Tran C, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10(1):33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 21.Small EJ, Ryan CJ. The case for secondary hormonal therapies in the chemotherapy age. The Journal of Urology. 2006;176(6):S66–S71. doi: 10.1016/j.juro.2006.06.071. [DOI] [PubMed] [Google Scholar]
- 22.Samson DJ, Seidenfeld J, Schmitt B, et al. Systematic review and meta-analysis of monotherapy compared with combined androgen blockade for patients with advanced prostate carcinoma. Cancer. 2002;95(2):361–376. doi: 10.1002/cncr.10647. [DOI] [PubMed] [Google Scholar]
- 23.Klotz L, Schellhammer P. Combined androgen blockade: The case for bicalutamide. Clin Prostate Cancer. 2005;3(4):215–219. doi: 10.3816/cgc.2005.n.002. [DOI] [PubMed] [Google Scholar]
- *24.Labrie F. Adrenal androgens and intracrinology. Semin Reprod Med. 2004;22(4):299–309. doi: 10.1055/s-2004-861547. [DOI] [PubMed] [Google Scholar]
- *25.Bruchovsky N. Comparison of the metabolites formed in rat prostate following the in vivo administration of seven natural androgens. Endocrinology. 1971;89(5):1212–1222. doi: 10.1210/endo-89-5-1212. [DOI] [PubMed] [Google Scholar]
- 26.Schiller CD, Schneider MR, Hartmann H, et al. Growth-stimulating effect of adrenal androgens on the R3327 dunning prostatic carcinoma. Urol Res. 1991;19(1):7–13. doi: 10.1007/BF00294013. [DOI] [PubMed] [Google Scholar]
- *27.Harper ME, Pike A, Peeling WB, et al. Steroids of adrenal origin metabolized by human prostatic tissue both in vivo and in vitro. J Endocrinol. 1974;60(1):117–125. doi: 10.1677/joe.0.0600117. [DOI] [PubMed] [Google Scholar]
- 28.Klein H, Bressel M, Kastendieck H, et al. Androgens, adrenal androgen precursors, and their metabolism in untreated primary tumors and lymph node metastases of human prostatic cancer. Am J Clin Oncol. 1988;11 Suppl 2:S30–S36. doi: 10.1097/00000421-198801102-00008. [DOI] [PubMed] [Google Scholar]
- 29.Miyamoto H, Yeh S, Lardy H, et al. Delta5-androstenediol is a natural hormone with androgenic activity in human prostate cancer cells. Proc Natl Acad Sci U S A. 1998;95(19):11083–11088. doi: 10.1073/pnas.95.19.11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Acevedo HF, Goldzieher JW. The metabolism of [14C] progesterone by hypertrophic and carcinomatous human prostate tissue. Biochim Biophys Acta. 1965;111(1):294–298. doi: 10.1016/0304-4165(65)90495-2. [DOI] [PubMed] [Google Scholar]
- 31.Attard G, A Y, H RA, et al. Phase I study of continuous oral dosing of an irreversible CYP17 inhibitor, abiraterone (a), in castration refractory prostate cancer (CRPC) patients (p) incorporating the evaluation of androgens and steroid metabolites in plasma and tumor. Journal of Clinical Oncology, 2007 ASCO Annual Meeting Proceedings. 2007;Part 1, Vol 25(No 18S):5063. [Google Scholar]
- 32.Auchus RJ. The backdoor pathway to dihydrotestosterone. Trends Endocrinol Metab. 2004;15(9):432–438. doi: 10.1016/j.tem.2004.09.004. [DOI] [PubMed] [Google Scholar]
- 33.Luo J, Dunn TA, Ewing CM, et al. Decreased gene expression of steroid 5 alpha-reductase 2 in human prostate cancer: Implications for finasteride therapy of prostate carcinoma. Prostate. 2003;57(2):134–139. doi: 10.1002/pros.10284. [DOI] [PubMed] [Google Scholar]
- 34.Titus MA, Gregory CW, Ford OH, III, et al. Steroid 5{alpha}-reductase isozymes I and II in recurrent prostate cancer. Clin Cancer Res. 2005;11(12):4365–4371. doi: 10.1158/1078-0432.CCR-04-0738. [DOI] [PubMed] [Google Scholar]
- 35.Thomas LN, Douglas RC, Lazier CB, et al. Levels of 5[alpha]-reductase type 1 and type 2 are increased in localized high grade compared to low grade prostate cancer. The Journal of Urology. 2008;179(1):147–151. doi: 10.1016/j.juro.2007.08.155. [DOI] [PubMed] [Google Scholar]
- 36.Koh E, Noda T, Kanaya J, et al. Differential expression of 17beta-hydroxysteroid dehydrogenase isozyme genes in prostate cancer and noncancer tissues. Prostate. 2002;53(2):154–159. doi: 10.1002/pros.10139. [DOI] [PubMed] [Google Scholar]
- 37.Fung KM, Samara EN, Wong C, et al. Increased expression of type 2 3alpha-hydroxysteroid dehydrogenase/type 5 17beta-hydroxysteroid dehydrogenase (AKR1C3) and its relationship with androgen receptor in prostate carcinoma. Endocr Relat Cancer. 2006;13(1):169–180. doi: 10.1677/erc.1.01048. [DOI] [PubMed] [Google Scholar]
- 38.Zha S, Ferdinandusse S, Hicks JL, et al. Peroxisomal branched chain fatty acid beta-oxidation pathway is upregulated in prostate cancer. Prostate. 2005;63(4):316–323. doi: 10.1002/pros.20177. [DOI] [PubMed] [Google Scholar]
- 39.Penning TM, Bauman DR, Jin Y, et al. Identification of the molecular switch that regulates access of 5[alpha]-DHT to the androgen receptor. Molecular and Cellular EndocrinologyAdrenal/Molecular Steroidogenesis Conference 2006. 2007;265–266:77–82. doi: 10.1016/j.mce.2006.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ji Q, Chang L, Stanczyk FZ, et al. Impaired dihydrotestosterone catabolism in human prostate cancer: Critical role of AKR1C2 as a pre-receptor regulator of androgen receptor signaling. Cancer Res. 2007;67(3):1361–1369. doi: 10.1158/0008-5472.CAN-06-1593. [DOI] [PubMed] [Google Scholar]
- 41.Attard G, Belldegrun AS, de Bono JS. Selective blockade of androgenic steroid synthesis by novel lyase inhibitors as a therapeutic strategy for treating metastatic prostate cancer. BJU Int. 2005;96(9):1241–1246. doi: 10.1111/j.1464-410X.2005.05821.x. [DOI] [PubMed] [Google Scholar]
- ****42.Handratta VD, Vasaitis TS, Njar VCO, et al. Novel C-17-heteroaryl steroidal CYP17 inhibitors/antiandrogens: Synthesis, in vitro biological activity, pharmacokinetics, and antitumor activity in the LAPC4 human prostate cancer xenograft model. J Med Chem. 2005;48(8):2972–2984. doi: 10.1021/jm040202w. [DOI] [PubMed] [Google Scholar]
- ****43.O'Donnell A, Judson I, Dowsett M, et al. Hormonal impact of the 17alpha-hydroxylase/C(17,20)-lyase inhibitor abiraterone acetate (CB7630) in patients with prostate cancer. Br J Cancer. 2004;90(12):2317–2325. doi: 10.1038/sj.bjc.6601879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ryan CJ, Rosenberg J, Lin A, et al. Phase I evaluation of abiraterone acetate (CB7630), a 17 alpha hydroxylase C17,20-lyase inhibitor in androgen-independent prostate cancer (AIPC) Journal of Clinical Oncology, 2007 ASCO Annual Meeting Proceedings. 2007;Part 1, Vol 25(No 18S):5064. [Google Scholar]
- 45.Ryan CJ, Halabi S, Ou SS, et al. Adrenal androgen levels as predictors of outcome in prostate cancer patients treated with ketoconazole plus antiandrogen withdrawal: Results from a Cancer and Leukemia Group B study. Clin Cancer Res. 2007;13(7):2030–2037. doi: 10.1158/1078-0432.CCR-06-2344. [DOI] [PubMed] [Google Scholar]
- 46.Culig Z, Hobisch A, Cronauer MV, et al. Mutant androgen receptor detected in an advanced-stage prostatic carcinoma is activated by adrenal androgens and progesterone. Mol Endocrinol. 1993;7(12):1541–1550. doi: 10.1210/mend.7.12.8145761. [DOI] [PubMed] [Google Scholar]
- 47.Small EJ, Halabi S, Dawson NA, et al. Antiandrogen withdrawal alone or in combination with ketoconazole in androgen-independent prostate cancer patients: A phase III trial (calgb 9583) J Clin Oncol. 2004;22(6):1025–1033. doi: 10.1200/JCO.2004.06.037. [DOI] [PubMed] [Google Scholar]
- 48.Berube M, Laplante Y, Poirier D. Design, synthesis and in vitro evaluation of 4-androstene-3,17-dione/adenosine hybrid compounds as bisubstrate inhibitors of type 3 17beta-hydroxysteroid dehydrogenase. Med Chem. 2006;2(4):329–347. doi: 10.2174/157340606777724086. [DOI] [PubMed] [Google Scholar]
- 49.Lorenzi MV, Rizzo CA, You D, et al. In vivo suppression of testosterone biosynthesis with a novel series of non-steroidal 17{beta}-hydroxysteroid dehydrogenase type III (17{beta}-HSD3) inhibitors. AACR Meeting Abstracts. 2005;2005(1):605-a. [Google Scholar]
- 50.Bauman DR, Rudnick SI, Szewczuk LM, et al. Development of nonsteroidal anti-inflammatory drug analogs and steroid carboxylates selective for human aldo-keto reductase isoforms: Potential antineoplastic agents that work independently of cyclooxygenase isozymes. Mol Pharmacol. 2005;67(1):60–68. doi: 10.1124/mol.104.006569. [DOI] [PubMed] [Google Scholar]
- 51.Byrns MC, Steckelbroeck S, Penning TM. An indomethacin analogue, n-(4-chlorobenzoyl)-melatonin, is a selective inhibitor of aldo-keto reductase 1C3 (type 2 3alpha-HSD, type 5 17beta-HSD, and prostaglandin F synthase), a potential target for the treatment of hormone dependent and hormone independent malignancies. Biochem Pharmacol. 2008;75(2):484–493. doi: 10.1016/j.bcp.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krazeisen A, Breitling R, Moller G, et al. Phytoestrogens inhibit human 17[beta]-hydroxysteroid dehydrogenase type 5. Molecular and Cellular Endocrinology. 2001;171(1–2):151–162. doi: 10.1016/s0303-7207(00)00422-6. [DOI] [PubMed] [Google Scholar]
- 53.Brozic P, Golob B, Gomboc N, et al. Cinnamic acids as new inhibitors of 17beta-hydroxysteroid dehydrogenase type 5 (akr1c3) Mol Cell Endocrinol. 2006;248(1–2):233–235. doi: 10.1016/j.mce.2005.10.020. [DOI] [PubMed] [Google Scholar]
- 54.Simard J, Ricketts M-L, Gingras S, et al. Molecular biology of the 3{beta}-hydroxysteroid dehydrogenase/{delta}5-{delta}4 isomerase gene family 10.1210/er.2002-0050. Endocr Rev. 2005;26(4):525–582. doi: 10.1210/er.2002-0050. [DOI] [PubMed] [Google Scholar]
- 55.Thomas JL, Boswell EL, Scaccia LA, et al. Identification of key amino acids responsible for the substantially higher affinities of human type 1 3{beta}-hydroxysteroid dehydrogenase/isomerase (3{beta}-HSD1) for substrates, coenzymes, and inhibitors relative to human 3{beta}-HSD2. J Biol Chem. 2005;280(22):21321–21328. doi: 10.1074/jbc.M501269200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Makridakis N, Reichardt JK. Pharmacogenetic analysis of human steroid 5 alpha reductase type II: Comparison of finasteride and dutasteride. J Mol Endocrinol. 2005;34(3):617–623. doi: 10.1677/jme.1.01725. [DOI] [PubMed] [Google Scholar]
- 57.Titus M, Li Y, Kawinski E, et al. Biochemical and pharmacological evidence for a third isozyme of steroid 5a-reductase in prostate cancer. American Urological Association, Anaheim (CA) 2007:268. [Google Scholar]
- 58.Stanway SJ, Purohit A, Woo LW, et al. Phase I study of STX 64 (667 coumate) in breast cancer patients: The first study of a steroid sulfatase inhibitor. Clin Cancer Res. 2006;12(5):1585–1592. doi: 10.1158/1078-0432.CCR-05-1996. [DOI] [PubMed] [Google Scholar]
- 59.Klein H, Molwitz T, Bartsch W. Steroid sulfate sulfatase in human benign prostatic hyperplasia: Characterization and quantification of the enzyme in epithelium and stroma. J Steroid Biochem. 1989;33(2):195–200. doi: 10.1016/0022-4731(89)90294-x. [DOI] [PubMed] [Google Scholar]
- 60.Nakamura Y, Suzuki T, Fukuda T, et al. Steroid sulfatase and estrogen sulfotransferase in human prostate cancer. Prostate. 2006;66(9):1005–1012. doi: 10.1002/pros.20426. [DOI] [PubMed] [Google Scholar]
- 61.Shah RB, Mehra R, Chinnaiyan AM, et al. Androgen-independent prostate cancer is a heterogeneous group of diseases: Lessons from a rapid autopsy program. Cancer Res. 2004;64(24):9209–9216. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 62.Singh P, Uzgare A, Litvinov I, et al. Combinatorial androgen receptor targeted therapy for prostate cancer. Endocr Relat Cancer. 2006;13(3):653–666. doi: 10.1677/erc.1.00797. [DOI] [PubMed] [Google Scholar]