

Abstract
A reduced diiron(I) complex reacts with CO2 to give two iron-containing products. One product has a carbonate bridge, which isomerizes rapidly at −70 °C and may be derived from an oxodiiron intermediate. Formation of this product releases free CO, which leads to a four-coordinate iron dicarbonyl complex. This product is the first crystallographically characterized example of a four-coordinate iron dicarbonyl species, a moiety that may be present in the active site of Hmd (“iron-sulfur cluster free”) hydrogenase.
Carbon dioxide plays an important role in synthetic and biological chemistry as a one-carbon building block.1 Despite the large bond enthalpy of the C=O double bond in CO2 (532 kJ/mol),2 nature is capable of using late first row transition metal ions to reduce CO2 to CO in the enzymes acetyl-coenzyme A synthase/CO dehydrogenase3 and nitrogenase.4 This is also the reverse of the famous water-gas shift reaction.5 Interest in the reduction of CO2 to CO has led to the investigation of numerous schemes for achieving the reduction of CO2.1 Transition metal promoted reduction of CO2 to give CO and CO32− has been observed in many systems and this type of transformation (eq 1) is known as reductive disproportionation.6
| (1) |
Recently, Peters reported the reductive cleavage of CO2 by a low-coordinate iron(I) system.7 This reaction afforded as the major product a diiron(II) complex with a Fe(μ-CO)(μ-O)Fe core. Herein, we report the reaction of CO2 with another iron(I) system, which gives a different outcome but may use a similar mechanism.
We have described the iron-dinitrogen complex LtBuFeNNFeLtBu (LtBu = 2,2,6,6-tetramethyl-3,5-bis(2,6-diisopropylphenylimino)hept-4-yl).8 Spectroscopic and magnetic studies are consistent with assignment of its iron atoms as iron(II) and the dinitrogen ligand as N22−.9 However, in its reactivity, it generally behaves as a source of the iron(I) fragment LtBuFe.8b,10 For example, it reacts with neutral ligands C6H6 and PR3 to give monomeric iron(I) complexes, and it reductively couples ketones and aldehydes to give diiron(II) pinacolate complexes.8b
Treatment of LtBuFeNNFeLtBu with 2 equiv of dry carbon dioxide in pentane, benzene, or diethyl ether affords a mixture of the dicarbonyliron(I) compound LtBuFe(CO)2 (1) and the bridging carbonatodiiron(II) compound LtBuFe(μ-OCO2)FeLtBu (2). The 1H NMR spectrum of the crude mixture from the reaction of LtBuFeNNFeLtBu and carbon dioxide shows the formation of 2 as 70 ± 5% of the total iron (using an internal integration standard). The theoretical stoichiometry of this reaction is shown in equation 2, and the observed spectroscopic yield of 2 agrees fairly well with the 80% expected. Because 1 is not NMR active (see below), its presence was deduced by the observation of CO stretching vibrations at 1996 and 1917 cm−1 in the solid-state infrared spectrum of the crude product.
| (2) |
In this reaction 8 iron atoms are oxidized from iron(I) to iron(II), concomitant with the reduction of 8 molecules of carbon dioxide. This creates 4 carbonate anions that bind to iron(II), as well as 4 molecules of CO that are trapped by iron(I).
Compound 1 can be synthesized independently by reacting LtBuFeNNFeLtBu with 4 equiv of carbon monoxide in diethyl ether. Using excess CO results in a mixture of LtBuFe(CO)2 and LtBuFe(CO)3, as described in a recent publication.8b In that work, we were frustrated by the cocrystallization of these dicarbonyl and tricarbonyl complexes, and an X-ray crystal structure showed a superposition of these two molecules. Here, use of the appropriate stoichiometry of starting materials gives the pure dicarbonyliron(I) complex, with CO stretching vibrations at 1994 and 1915 cm−1 in the solid-state IR spectrum.11 Complex 1 has a low-spin d7 (S = 1/2) electronic configuration, as shown by EPR spectroscopy in the solution and solid state.8b,11 The crystal structure of 1 is illustrated in Figure 1. The iron atom has a square planar geometry (sum of angles around iron 361.8(1)°). The C-O bond lengths are 1.149(2) and 1.151(2) Å and the Fe-C-O units are slightly bent (172.8(1)° and 172.9(2)°).
Figure 1.
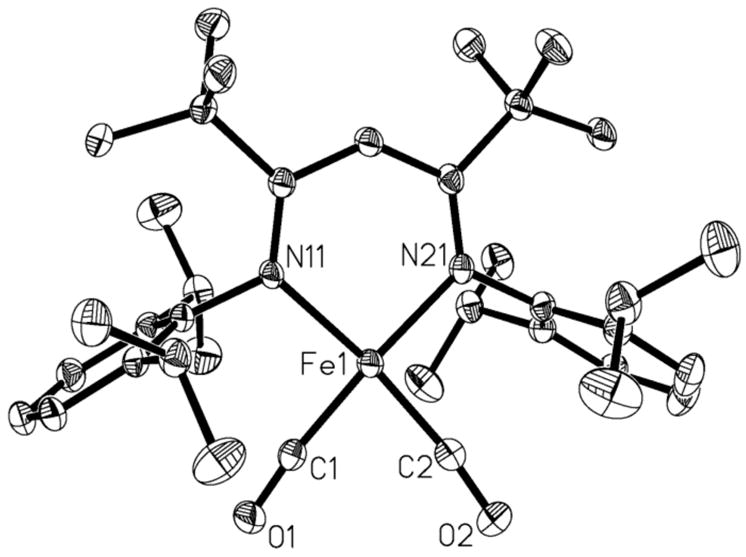
Crystal structure of 1. Thermal ellipsoids shown at 50% probability. Hydrogen atoms have been omitted for clarity. Fe1-C1 1.784(2) Å, Fe1-C2 1.785(2) Å Fe1-N11 1.967(1) Å, C1-O1 1.151(2) Å, C2-O2 1.149(2) Å, N11-Fe1-N21 93.43(5)°, C1-Fe1-C2 81.44(8)°, O1-C1-Fe1 172.8(1)°, O2-C1-Fe1 172.9(2)°.
Compound 1 is interesting because it is the first example of a four-coordinate dicarbonyl complex of iron. This structural feature has been postulated to be present in the active site of the “Fe-S cluster free” hydrogenase Hmd, on the basis of infrared, Mössbauer, and X-ray absorption data.12 Infrared spectra of the enzyme showed two bands of identical intensity, leading to an estimate of the C-Fe-C angle of 86–94° based on the literature relationship tan θ = Iasymm/Isymm.12a,13 However, 1 shows virtually identical intensities for the two CO stretching vibrations despite the C-Fe-C angle of 81.44(8)° in the X-ray crystal structure. On this basis, we suggest that a broader range of CO angles should be considered as possible in the enzyme.
The use of 1 for further comparison to the enzyme is limited because the iron atom in Hmd hydrogenase is diamagnetic, either low-spin iron(II) or low-spin iron(0), in contrast to the iron(I) oxidation state in 1. So, although 1 is not an electronic match for the active site of Hmd, its structure is the best mimic yet known. The redox chemistry of 1 is clearly of interest, and will be reported in the future.
The other product of CO2 reduction, LtBuFe(μ-OCO2)FeLtBu (2), could be isolated in pure form by performing the reaction of LtBuFeNNFeLtBu and carbon dioxide in the presence of Pd/C to trap the carbon monoxide in situ. The crude product of this reaction shows no carbonyl stretching bands in the IR spectrum. The ability to divert the reaction shows that free CO is involved in the formation of complex 1.
Complex 2 has been characterized by 1H NMR and infrared spectroscopies, X-ray diffraction, and elemental analysis. The IR spectrum of 2 shows two strong bands at 1493 and 1481 cm−1 that are characteristic of a bridging carbonate ligand.14 The solution magnetic moment of 6.0(3) BM/molecule in C6D6 suggests that the high-spin iron(II) centers in 2 have very weak magnetic coupling. The solid-state structure of 2 has two different molecules in the unit cell (Figure 2). In one molecule (A) the bridging carbonate is coordinated in a μ-η2:η1 mode and in the other (B) it is coordinated in a μ-η2:η2 mode to the metal centers. The bond lengths and bond angles of these two molecules are compared in Table 1. Structural type A is unprecedented for a carbonate bridge on iron, but has been seen with other metals.15 Structural type B has precedent in a tris(pyrazolyl)boratoiron(II) complex, which was derived from a metal hydroxide and CO2.16
Figure 2.
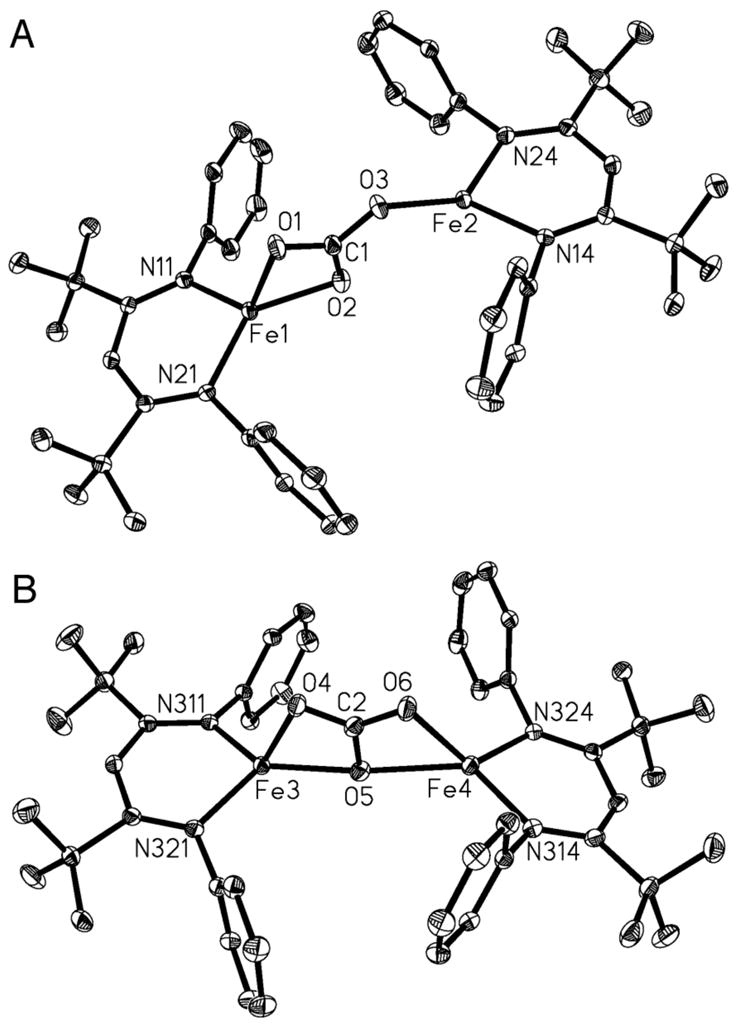
Crystal structure of 2, showing molecules A and B with different bridging modes. Thermal ellipsoids shown at 50% probability. Hydrogen atoms and isopropyl groups have been omitted for clarity. See Table 1 for metrical parameters.
Table 1.
Bond lengths (Å) and bond angles (°) of Fe(μ–CO3)Fe cores in the two independent molecules found in the X-ray crystal structure of 2.
| A | B | |
|---|---|---|
| Fe1-O1 | 2.079(1) | 2.079(1) |
| Fe1-O2 | 2.034(1) | 2.107(1) |
| Fe2-O3 | 1.881(1) | 1.982(1) |
| Fe2-O2 | 3.156(1) | 2.358(1) |
| C1-O1 | 1.270(2) | 1.256(2) |
| C1-O2 | 1.291(2) | 1.323(2) |
| C1-O3 | 1.293(2) | 1.275(2) |
| O1-Fe1-O2 | 64.52(4) | 63.53(4) |
| O2-Fe2-O3 | -- | 60.45(4) |
| O1-C1-O2 | 118.1(1) | 117.4(1) |
| O1-C1-O3 | 120.9(1) | 126.2(1) |
| O2-C1-O3 | 121.0(1) | 116.4(1) |
The 1H NMR spectrum of 2 in C6D6 shows seven peaks, which is characteristic of the protons on β-diketiminate ligands in local C2v symmetry. There is no decoalescence in the temperature range +70 to −70 °C. This implies at least two rapid dynamic processes. First, the bridging carbonate ligand must exchange between binding modes A and B rapidly, making the two metals equivalent on the NMR time scale. Second, the equivalence of the two faces of the β-diketiminate ligand indicates that there is rapid rotation of the iron atom around the Fe-O bond, probably in the iron ion with η1-carbonate binding.
There are multiple possibilities for the mechanism through which 1 and 2 are formed. One is the reductive disproportionation of CO2, a series of one-electron reductions that is well-established in electrochemical and other reactions with reduced transition-metal complexes.1 In other systems, reductive disproportionation is thought to proceed through the intermediacy of the radical [O=C–O–C(=O)–O] −, for which we have no evidence here. Another possibility is the reductive cleavage of CO2 to give CO and CO2−. While this work was in progress, Peters reported an iron(I) complex that reacts with 0.5 equiv of CO2 to give a diiron(II) complex with an Fe(μ-CO)( μ-O)Fe core.7 This outcome corresponds to direct reductive cleavage of the C=O bond. Earlier low-valent iron complexes also give the overall reductive cleavage of CO2.17 In our system, we have reported the isolation of the oxodiiron(II) complex [LtBuFe]2O, which would be the analogous oxo intermediate (Scheme 1).18 This hindered oxo complex binds a number of Lewis bases reversibly, so it is reasonable to assume that it could lose CO, at least transiently. Although we have no direct spectroscopic evidence that [LtBuFe]2O is formed during the course of the reaction of CO2 and LtBuFeNNFeLtBu, treatment of isolated [LtBuFe]2O with CO2 in C6D6 rapidly yields 2 as shown by 1H NMR spectroscopy. Therefore, the oxo complex is kinetically competent to be an intermediate in the formation of 2. As shown above, CO reacts rapidly with LtBuFeNNFeLtBu to give 1 in independent experiments; further, formation of 1 can be suppressed by trapping CO with Pd/C. Therefore, Scheme 1 represents a reasonable mechanism for this interesting new CO2 reduction reaction.
Scheme 1.
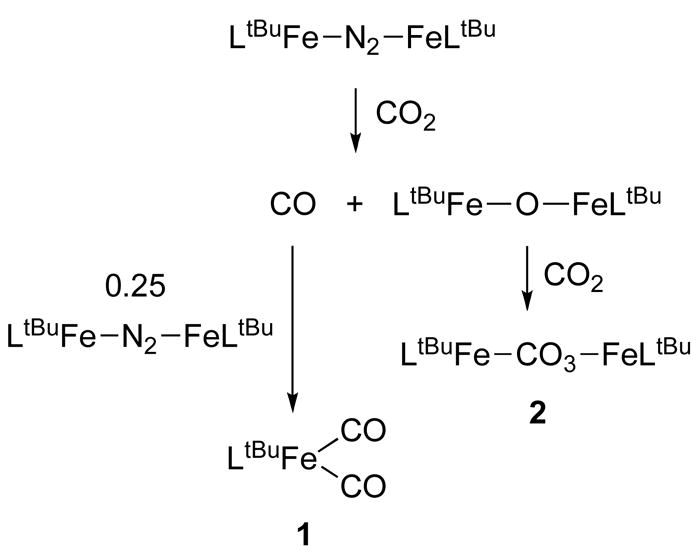
Proposed mechanism for formation of 1 and 2.
In conclusion, CO2 can be reduced cleanly to CO and carbonate at a diiron(I) site. The CO (1) and CO3 2− (2) containing products are notable because of a structural similarity between 1 and a newly discovered hydrogenase enzyme, and an interesting dynamic process in 2. The formation of 2 may proceed through the intermediacy of an oxodiiron(II) complex.
Acknowledgments
This work was supported by the National Institutes of Health (GM065313).
Footnotes
Supporting Information Available: Synthetic, spectroscopic, and crystallographic details. This material is available free of charge via the Internet at http://pubs.acs.org.
References and Notes
- 1.Reviews: Arakawa H, et al. Chem Rev. 2001;101:953–996. doi: 10.1021/cr000018s.Louie J. Curr Org Chem. 2005;9:605–623.Omae I. Catal Today. 2006;115:33–52.Darensbourg DJ. Chem Rev. 2007;107:2388–2410. doi: 10.1021/cr068363q.Aresta M, Dibenedetto A. Dalton Trans. 2007:2975–2992. doi: 10.1039/b700658f.
- 2.Heats of formation taken from: CRC Handbook, 87th edition, http://www.hbcpnetbase.com/
- 3.(a) Ragsdale SW, Kumar M. Chem Rev. 1996;96:2515–2539. doi: 10.1021/cr950058+. [DOI] [PubMed] [Google Scholar]; (b) Drennan CL, Doukov TI, Ragsdale SW. J Biol Inorg Chem. 2004;9:511–515. doi: 10.1007/s00775-004-0563-y. [DOI] [PubMed] [Google Scholar]; (c) Hegg EL. Acc Chem Res. 2004;37:775–783. doi: 10.1021/ar040002e. [DOI] [PubMed] [Google Scholar]
- 4.Rasche ME, Seefeldt LC. Biochemistry. 1997;36:8574–8585. doi: 10.1021/bi970217e. [DOI] [PubMed] [Google Scholar]
- 5.(a) King RB, King AD, Yang DB. ACS Symp Ser. 1981;152:123–132. [Google Scholar]; (b) Ford PC. Electrochem Electrocatal React Carbon Dioxide. 1993:68–93. [Google Scholar]; (c) Kochloefl K. Water Gas Shift and COS Removal. In: Ertl G, et al., editors. Handbook of Heterogeneous Catalysis. Vol. 4. VCH; Weinheim: 1997. pp. 1831–1843. [Google Scholar]
- 6.(a) Fachinetti G, Floriani C, Chiesi-Villa A, Guastini C. J Am Chem Soc. 1979;101:1767–1775. [Google Scholar]; (b) Lee GR, Maher JM, Cooper NJ. J Am Chem Soc. 1987;109:2956–2962. [Google Scholar]; (c) Alvarez R, Atwood JL, Carmona E, Perez PJ, Poveda ML, Rogers RD. Inorg Chem. 1991;30:1493–1499. [Google Scholar]
- 7.Lu CC, Saouma CT, Day MW, Peters JC. J Am Chem Soc. 2007;129:4–5. doi: 10.1021/ja065524z. This paper reports a minor oxalate product; in the work presented here there is no sign of oxalate formation.
- 8.(a) Smith JM, Lachicotte RJ, Pittard KA, Cundari TR, Lukat-Rodgers G, Rodgers KR, Holland PL. J Am Chem Soc. 2001;123:9222–9223. doi: 10.1021/ja016094+. [DOI] [PubMed] [Google Scholar]; (b) Smith JM, Sadique AR, Cundari TR, Rodgers KR, Lukat-Rodgers G, Lachicotte RJ, Flaschenriem CJ, Vela J, Holland PL. J Am Chem Soc. 2006;128:756–769. doi: 10.1021/ja052707x. [DOI] [PubMed] [Google Scholar]
- 9.Stoian SA, Vela J, Smith JM, Sadique AR, Holland PL, Münck E, Bominaar EL. J Am Chem Soc. 2006;128:10181–10192. doi: 10.1021/ja062051n. [DOI] [PubMed] [Google Scholar]
- 10.(a) Vela J, Stoian S, Flaschenriem CJ, Münck E, Holland PL. J Am Chem Soc. 2004;126:4522–4523. doi: 10.1021/ja049417l. [DOI] [PubMed] [Google Scholar]; (b) Yu Y, Smith JM, Flaschenriem CJ, Holland PL. Inorg Chem. 2006;45:5742–5751. doi: 10.1021/ic052136+. [DOI] [PubMed] [Google Scholar]; (c) Sadique AR, Gregory EA, Brennessel WW, Holland PL. J Am Chem Soc. 2007;129:8112–8121. doi: 10.1021/ja069199r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.In ref 8b, we reported bands at 2036, 2000, 1967, 1953, and 1922 cm−1 in a pentane solution of LtBuFe(CO)n (n = 2,3), and at 2042, 1971, and 1960 in a pentane solution of LMeFe(CO)3. This implies that the 2000 and 1922 cm−1 solution bands are from LtBuFe(CO)2, consistent with the solid-state bands at 1994 and 1915 cm−1 in solid 1 as isolated here. The IR spectrum of a solution of 1 in pentane shows that in solution, 1 undergoes partial disproportionation to form some LtBuFe(CO)3. This disproportionation in solution is also evident in Xband EPR spectra at 77 K. See Supporting Information for more detail.
- 12.(a) Lyon EJ, Shima S, Boecher R, Thauer RK, Grevels FW, Bill E, Roseboom W, Albracht SPJ. J Am Chem Soc. 2004;126:14239–14248. doi: 10.1021/ja046818s. [DOI] [PubMed] [Google Scholar]; (b) Shima S, Lyon EJ, Thauer RK, Mienert B, Bill E. J Am Chem Soc. 2005;127:10430–10435. doi: 10.1021/ja051895o. [DOI] [PubMed] [Google Scholar]
- 13.Beck W, Melnikoff A, Stahl R. Angew Chem Int Ed. 1965;4:692–693. [Google Scholar]
- 14.Palmer DA, Van Eldik R. Chem Rev. 1983;83:651–731. [Google Scholar]
- 15.See, for example: Chatt J, Kubota M, Leigh GJ, March FC, Mason R, Yarrow DJ. J Chem Soc Chem Commun. 1974:1033–1034.
- 16.Kitajima N, Hikichi S, Tanaka M, Moro-oka Y. J Am Chem Soc. 1993;115:5496–5508. [Google Scholar]
- 17.Karsch HH. Chem Ber. 1977;110:2213–2221. [Google Scholar]
- 18.Eckert NA, Stoian S, Smith JM, Bominaar EL, Münck E, Holland PL. J Am Chem Soc. 2005;127:9344–9345. doi: 10.1021/ja0436704. [DOI] [PubMed] [Google Scholar]