

Abstract
We report a survey of the reactivity of the first isolable iron-hydride complexes with a coordination number less than five. The high-spin iron(II) complexes [(β-diketiminate)Fe(μ-H)]2 react rapidly with representative cyanide, isocyanide, alkyne, N2, alkene, diazene, azide, CO2, carbodiimide and Brønsted acid containing substrates. The reaction outcomes fall into three categories: (1) addition of Fe-H across a multiple bond of the substrate, (2) reductive elimination of H2 to form iron(I) products, and (3) protonation of the hydride to form iron(II) products. The products include imide, isocyanide, vinyl, alkyl, azide, triazenido, benzo[c]cinnoline, amidinate, formate, and hydroxo complexes. These results expand the range of known bond transformations at iron complexes. Additionally, they give insight into the elementary transformations that may be possible at the iron-molybdenum cofactor of nitrogenases, which may have hydride ligands on high-spin, low coordinate metal atoms.
Keywords: iron(II), hydride, nitrogenase, insertion
Introduction
Organometallic chemists have long appreciated the many reactions of transition-metal hydride complexes, especially reductions.1 Recently, hydride chemistry has become established in bioinorganic systems.2 Nitrogenases are prodigious reductants that cleave double and triple bonds in N2, CO2, N2O,N3−, and CN− at iron-sulfur clusters (FeMoco in the molybdenum-iron nitrogenases, FeVco in the vanadium-iron nitrogenases, and FeFeco in the iron-only nitrogenases).3 Because these substrates are reduced by multiples of two electrons accompanied by the addition of protons, chemists have often speculated about the potential role of hydrides.4
Recently, the first direct evidence for hydride intermediates in nitrogenase emerged from ENDOR (Electron-Nuclear Double Resonance) studies of nitrogenase mutants freeze-trapped during substrate turnover. For example, a Val70Ile mutant of A. vinelandii molybdenum-iron nitrogenase freeze-trapped during proton reduction shows two hydrogen nuclei with very strong coupling to the S = ½ iron-sulfur cluster, strongly suggesting the presence of Fe-H bonds.5 This frozen species loses two molecules of H2 upon annealing to −20 °C, raising the possibility that these two hydride ligands can combine with nearby protons to release H2.6 This species is thought to be in the redox state that reacts directly with N2 in the wild-type MoFe nitrogenase,7 underscoring the importance of hydride-containing intermediates in enabling the nitrogen reduction activity characteristic of this enzyme.
From the perspective of the coordination chemist, there are a number of ways that potential FeMoco-bound hydrides differ from the majority of known synthetic transition-metal hydride complexes. First, FeMoco hydride adducts could achieve a number of different oxidation levels. One-electron redox changes undoubtedly occur in the FeMoco because electrons are supplied to the FeMoco one at a time by the Fe protein, which dissociates and reassociates before each one-electron reduction of the FeMoco.3 However, most synthetic hydride complexes have diamagnetic metal centers, and little one-electron chemistry has been reported.9,10 Second, the iron ions in the FeMoco are expected to be high-spin, based on ligand field considerations (weak-field sulfide donor set, coordination number less than five) and the results of computational studies.11 Synthetic hydride complexes, on the other hand, typically have strong-field organometallic or phosphine co-ligands, which enforce a low-spin electronic configuration.12,13 These fundamental differences motivate synthetic research aimed at the creation of iron-hydride complexes with weak-field ligands and low coordination number, to determine their characteristic reactivity patterns and reaction mechanisms, which can in turn be correlated with nitrogenase reactions.
Often the design of functional models of an enzyme is driven by an attempt to achieve structural similarity to the enzyme’s active site.14 In the case of nitrogenase, an accurate structural mimic of the FeMoco hydride species is elusive for several reasons. First, the FeMoco of iron-molybdenum nitrogenase (Figure 1) has eight transition metals in a cluster type (M8S9C or M8S9N or M8S9O) that is unknown in synthetic chemistry.15,16 Second, the crystallographically characterized form of the FeMoco (MN) does not have hydrides, as shown by ENDOR: hydrides are only incorporated concurrent with catalytic turnover.5 In any case, X-ray crystal structures of proteins do not have sufficient resolution to distinguish hydrogen atoms. Third, the N2-binding form of the FeMoco is reduced by 3-4 electrons from MN, and the FeMoco may undergo structural rearrangements upon reduction.7,17 Peters and Holland have independently proposed that dissociation of X is important in enabling substrate binding to iron, based on the binding of N2 to synthetic iron complexes.18 Computational studies come to a variety of conclusions regarding the structural flexibility of the FeMoco during catalysis.11 In short, there are many questions about the structure of the iron-hydride nitrogenase intermediates, and the available data are not sufficient to provide guesses about their structure(s).
Figure 1.

The iron-molybdenum cofactor (“FeMoco”) of iron-molybdenum nitrogenase in the isolated MN form.8 This form is reduced (with incorporation of hydrides in an undisclosed location) to give the intermediate that reacts with N2 and other nitrogenase substrates. X is C4−, N3−, or O2−.
For these reasons, we have created one- and two-iron compounds that focus on two key features of the nitrogenase-hydride intermediates: the weak-field ligands and the coordination number less than five.18a Extremely bulky bidentate β-diketiminate ligands (Figure 2) have π-donating nitrogen atoms that lead to a weak ligand field.19 The bulky diisopropylphenyl groups maintain a low coordination number that mimics the ligand-poor environment of the iron atoms in the FeMoco. Using these ligands, we have isolated the only examples of iron-hydride complexes with a coordination number less than five (Figure 2).20,21
Figure 2.

Bulky β-diketiminate iron complexes used in this work. The ligands are abbreviated LR, where R is the group on the diketiminate backbone. Both complexes have roughly tetrahedral geometry at each iron atom.20,21
Our studies on low-coordinate iron are complemented by those of Peters and coworkers, who use tridentate, strong-field tris(phosphino)borate (BP3) supporting ligands that contain “soft” phosphine donors, and more often give low-spin electronic configurations.22 In the BP3 systems, it has not yet been possible to isolate a low-coordinate iron hydride, but the presence of a terminal hydride in four coordinate complexes is strongly implied by the isolation of products that result from activation of solvent or the supporting ligand.23 Bridging hydrides have recently been observed in a few electronically unsaturated dinuclear complexes,24,25 and borohydrides have been studied on iron-sulfur clusters.26
Recent contributions from our group have described the reaction of [LtBuFeH]2 with azobenzene to cleave the N=N bond,20,27 and the reaction of [LMeFeH]2 with boranes to cleave B-C bonds.28 This manuscript reports a wider variety of reactions with unsaturated small molecules that are nitrogenase substrates or organic-soluble mimics of these substrates. By examining the reactions with representative compounds, one may begin to understand some of the unusual reaction patterns available to hydride complexes with a high-spin electronic configuration. In addition, a number of the products have novel structural and/or electronic features that are interesting from a fundamental perspective.
Results
Iron-Hydride Starting Materials
The synthesis and characterization of [LMeFeH]2 (1a) and [LtBuFeH]2 (1b) have been presented previously.20,21,27,28 They are each synthesized from iron(II) chloride complexes ([LMeFeCl]2 or LtBuFeCl) by reaction with potassium triethylborohydride for 0.5 h in toluene, followed by promptly removing solvent and BEt3, then extracting with pentane and crystallizing to give brown powders or crystals. Crude reaction mixtures are contaminated with the chloride starting material and the dihydridoborate complex LRFe(μ-H)2BEt2.28 Multiple crystallizations are typically necessary to remove these impurities, and 1H NMR spectroscopy is used to judge purity of the hydride complexes. The good yields of many of the products derived therefrom support the purity of 1a and 1b.
The 1H NMR spectrum of 1a consists of seven resonances; the number of signals and their integrations are characteristic of the diketiminate ligand in a local C2v symmetry environment. It has no unusual temperature- or concentration-dependent changes in its 1H NMR or UV-vis spectra, suggesting that the dimeric structure in the solid state is always maintained. To learn more about the electronic structure of 1a, we examined a solid sample using Mössbauer spectroscopy. The zero-field Mössbauer spectrum at 80 K (Figure S-6) exhibits a superposition of two quadrupole doublets with 72% and 28% relative intensities. The major component has δ = 0.70(2) mm/s and ΔEQ= 0.86(2) mm/s, whereas the minor component shows δ = 0.49(2) mm/s and ΔEQ = 2.06(2) mm/s. The values for the minor component are relatively similar to those of a high-spin Fe(I) diketiminate complex (ΔEQ = 2.02 mm/s, δ = 0.41 mm/s at 180 K).19c Therefore, we assign the component to contamination from an unidentified Fe(I) impurity. In contrast, the Mössbauer parameters of the major component, particularly the isomer shift, are in the range of high-spin iron(II) diketiminate complexes (δ = 0.62 – 0.86 mm/s), and so it is assigned to 1a.19,29 Magnetic Mössbauer spectra measured at 4.2 K with applied fields up to 7 T show that the major component is diamagnetic, suggesting antiferromagnetic coupling between the two iron(II) ions. In sum, the spectroscopic data support the formulation of 1a as an exchange-coupled pair of high-spin iron(II) ions with a ground state of S = 0.
The 1H NMR spectrum of 1b has at least 17 overlapping peaks, because of extreme steric crowding in the dimer that presumably renders some ligand bond rotations slow on the NMR time scale.27 Upon heating a solution of 1b in C6D6, there is growth of a simple seven-line 1H NMR spectrum with chemical shifts much like trigonal-planar alkyl complexes LtBuFeR.20 This behavior is ascribable to equilibration between dimeric [LtBuFeH]2 and monomeric LtBuFeH that is slow on the NMR time scale (ms) but rapid on the time scale of the equilibration (min). Therefore, in the discussion below it should be understood that 1b does not refer specifically to the monomer or dimer, but the equilibrating mixture of the two.30
Reactions With C≡N Triple Bonds, and with N2 and CO
Carbon monoxide (CO) is an inhibitor of N2 reduction by nitrogenase.3 Methyl isocyanide (CH3NC) and cyanide (CN−) are also inhibitors but are substrates as well: CN− is reduced to CH3NH2, NH3, and CH4, while CH3NC is reduced to CH3NH2, (CH3)2NH, and CH4.31 Therefore, it is of interest to examine the reactivity of small molecules containing CO and CN triple bonds with synthetic hydride complexes that mimic potential activated forms of the FeMoco. We use alkyl cyanides as surrogates for CN− and tert-butyl isocyanide in place of CH3NC.
The addition of CO or isocyanide to 1a results in the rapid release of H2. Adding an excess of CO to 1a gives LMeFe(CO)3 through formal reduction of the iron. Because LMeFe(CO)3 has been characterized previously, the reader is referred to the earlier paper for its properties.47
Addition of a large excess of tert-butyl isocyanide gives an intractable mixture, but treatment of a solution of 1a in pentane or toluene with 4 equiv (per dimer) of tert-butyl isocyanide results in the formation of a mononuclear iron(I) complex, LMeFe(CNtBu)2 (2a). Integration of the 1H NMR spectrum against an internal standard (LtBuFeCl in a capillary) indicated 62% conversion to 2a, amongst other unidentified products. The production of H2 was quantified by GC to be ca. 0.2 equiv per mole of 1a. The low yield of dihydrogen may be due to hydrogen incorporation into some of the unidentified products. Because of the low conversion, samples of 2a for further spectroscopic study were generated through an alternative method, by adding 4 equiv of tBuNC to LMeFeNNFeLMe. The 1H NMR and X-band EPR spectra of samples prepared in this way were identical to the spectra of material generated from 1a.
Solutions of the isocyanide complex 2a decompose over several hours in solution at room temperature, making the results of bulk magnetic studies unreliable, but EPR spectra of frozen mixtures of LMeFeNNFeLMe and 1–4 equiv of tBuNC in toluene show a rhombic signal with g = 2.45, 2.24, 1.98, suggesting that 2a has an S = ½ ground state.32,33 In the solid-state infrared spectrum of 2a, four stretching bands are observed at 2122, 2050, 1969, and 1948 cm−1. Some bands almost certainly derive from decomposition products, because bands at 2050, 1969 and 1948 cm−1 are observed in the IR spectrum of a sample from allowing 2a to completely decompose. Therefore, only the peak at 2122 cm−1 can be assigned confidently as a C≡N stretching vibration of 2a. Computations (see below for details) predict the second stretching vibration at 2050 cm−1 after correction for anharmonicity, so it is possible that the peak at 2050 cm−1 derives from 2a as well as a decomposition product.
The product has also been characterized by X-ray diffraction of a crystal grown at low temperature. The solid-state structure of 2a is illustrated in Figure 2. The iron atom has a square planar geometry, with the sum of angles 360.4(5)°. The two isocyanides are not identical: one CNC angle is almost linear (170.1(2)°) while the other is bent. The bent isocyanide ligand is disordered; two positions of the tBu group were observed, and the ratio of components is exactly 50:50 due to the presence of a crystallographic inversion center. Both refined ligand positions show bent isocyanide ligands (CNC angles of 164.0(7)° and 148.3(7)°). The refinement model required that geometrical restraints be placed on the NtBu portions of the two ligand disorder components, specifically that the N–C(tBu) bond lengths and angles in both components be similar. The result is a disorder model that confirms the CNC bending, but that should be used with caution with regard to the exact values of the angles.
The ambiguous spectroscopic and crystallographic data left unanswered questions about 2a. Therefore, hybrid quantum mechanics/molecular mechanics (QM/MM) computations were performed to study complete models of 2a. The optimized geometry of the doublet state (Erelative = 0) is square planar about iron, but the higher energy quartet state (Erelative = 3.9 kcal/mol) has a tetrahedral coordination geometry upon QM/MM geometry optimization. The calculated metrical parameters for doublet LMeFe(CNtBu)2 (compared to experimental ones in parentheses) are Fe-C = 1.83 Å (1.82 Å); CN = 1.18 Å (1.20, 1.17 Å). Both the lower calculated energy and the geometrical similarity to the experimental structure support the contention that 2a has an S = ½ ground state.
The computations also address the unusual difference between the CNC angles of the two isocyanide ligands (we denote the difference between these CNC angles as θ). The optimized geometry had CNC angles of 171° (close to the crystallographic angle of 170°) and 162° (between the observed 164° and 148° disorder components). A search of the Cambridge Structural Database for neutral, monometallic complexes with at least two tBuNC ligands indicated that the average θ is 6.9 ± 10.1° (170 examples) with a median value of 2.9°.34 Hence, our computed value of θ = 9°, while somewhat large, is not outside experimental norms. Computations indicate that bending the CNC angle from 180° to 150° requires 4 kcal/mol in free tBuNC and only 0.4 kcal/mol in the QM/MM model of 2a.35 Considering the softness of this bending distortion, it is not surprising that the isocyanide is unusually flexible, and can exist in multiple geometries in the solid state.
In the next section, we turn to reactions with nitriles, mimics of cyanide with substituents on the carbon. Scheme 1 shows the reduction products obtained from CH3CN and tBuCN. Heating 1a with 2 equiv of CH3CN in toluene at 45 °C for 18 h gives an orange-yellow precipitate in 88% yield. The X-ray crystal structure shows that the product is LMeFe(μ-N=CHCH3)2FeLMe (3a) (Figure 4, top), with a crystallographic inversion center relating the two halves of the molecule. Each nitrile molecule has been reduced to a bridging N=CHCH3 ligand with an N=C distance of 1.258(2) Å. The iron centers in 3a have roughly tetrahedral geometries. The 1H NMR spectrum of 3a in toluene-d8 has peaks from 50 ppm to −40 ppm, and the number and integrations are consistent with the C2h symmetry in the crystal structure.
Scheme 1.

Figure 4.
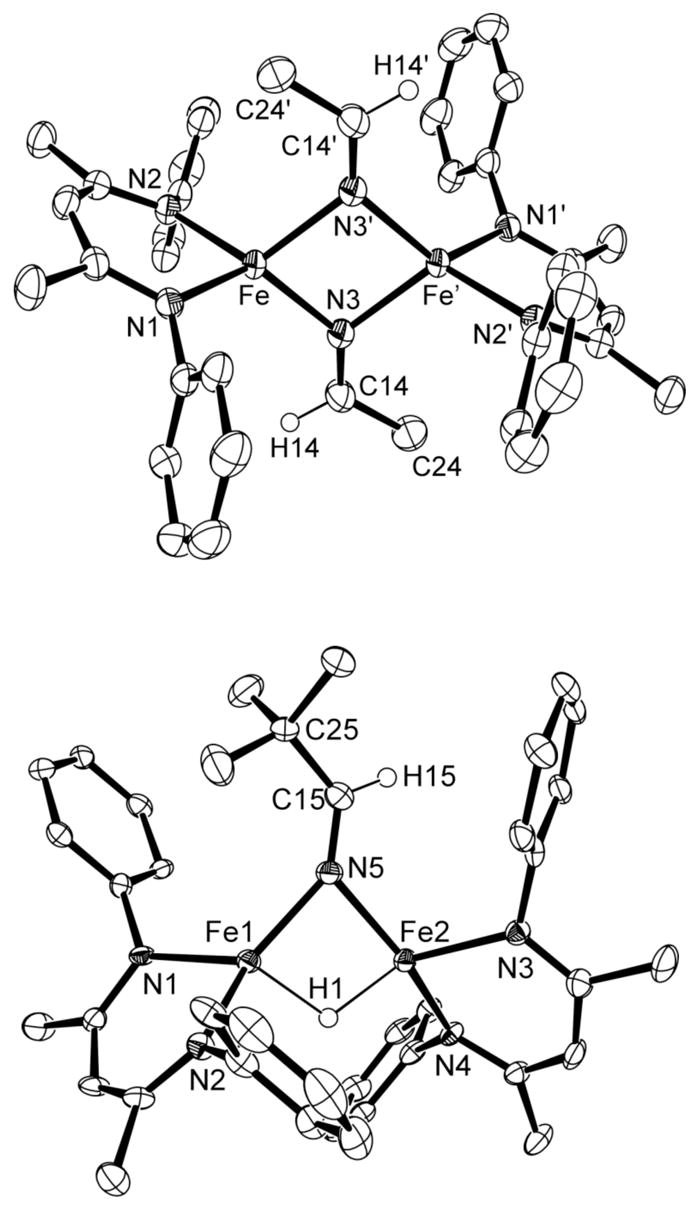
Molecular structures of 3a (top) and 4a (bottom), showing 50% thermal ellipsoids. The aryl isopropyl groups are omitted for clarity. Selected bond distances (Å) and angles (°) for 3a: Fe-N3 2.041(2), Fe-N3′ 2.079(2), N3-C14 1.258(2), Fe-N3-Fe′ 96.72(6), N3-C14-C24 125.4 (2). For 4a: Fe1-Fe2 2.7816(8), Fe1-N5 2.000(3), Fe2-N5 2.016(3), Fe1-H1 1.69(3), Fe2-H1 1.67(3), N5-C15 1.258(4), Fe1-N5-Fe2 87.7(1), N5-C15-C25 129.3(3).
When 1a is treated with the bulkier nitrile tBuCN, brown LMeFe(μ-N=CHtBu)( μ-H)FeLMe (4a) is obtained in 85% yield. The diiron complex incorporates only one equivalent of the nitrile, even in the presence of excess tBuCN. Addition across an N≡C bond has formed a bridging N=CHtBu ligand, while the other bridging hydride remains untouched. The crystal structure of 4a (Figure 4, bottom) indicates that the large tBu group pushes the two β-diketiminate ligands toward the bridging hydride, which significantly reduces the dihedral angle between the two β-diketiminate planes to around 81° in each of the two independent molecules in the crystal structure. This structural distortion makes the 1H NMR spectrum more complicated than in 3a. At ambient temperature, approximately 16 peaks are observed. Severe overlap between peaks has made attempts to assign peaks and measure the solution magnetic moment unsuccessful. At −20 °C, these ~16 peaks split into ~25 peaks, consistent with a reduction of symmetry from Cs (in a conformation like that in the crystal structure, but with the two β-diketiminate ligand planes coplanar on average) to C1 (the symmetry in the solid state).
Another kind of insertion product is observed when 1b is treated with tBuCN. In this case, the orange product is monomeric LtBuFe(N=CHtBu) (5b) in 67% yield. The geometry around the iron atom is trigonal planar (Figure 5). A C=N double bond is indicated by the N14-C14 bond length of 1.256(3) Å and the C=N stretching band at 1687 cm−1. The Fe-N(imide) bond length is 1.857(2) Å, substantially shorter than the Fe-N distances in the bridging imides of 3a and 4a.
Figure 5.

Molecular structure of 5b, showing 50% thermal ellipsoids. Most hydrogen atoms have been omitted for clarity. Selected bond distances (Å) and angles (°): Fe-N14 1.857(2), Fe-N11 1.977(2), Fe-N21 1.966(2), N14-C14 1.256(3), N11-Fe-N21 94.69(6), N11-Fe-N14 126.53(7), N21-Fe-N14 138.70(7), Fe-N14-C14 143.0(2).
Some triple bonds do not insert into the Fe-H bond. There is no evidence for thermal reaction of hydride complexes 1a and 1b with N2: use of high pressures of dinitrogen (up to 800 psi at 60 °C) gave LtBuFeOFeLtBu, the product of reaction with moisture, as the only product observable by 1H NMR spectroscopy.29b However, irradiation of each hydride complex under 1 atm of N2 with a high-pressure mercury lamp yields quantitative conversion to LRFeNNFeLR over the course of 18 h (1a) or 3 d (1b) as shown by 1H NMR spectroscopy. These dinitrogen complexes have a formal oxidation state of +1 at each iron atom, and therefore result from reduction.36 Therefore, it is possible to induce these hydride complexes to reductively eliminate H2 and bind N2, but this transformation requires photolysis.
Reactions With C≡C Triple Bonds
Acetylene is the most commonly studied unnatural substrate of nitrogenases, and it is reduced to ethylene.37 Mutant nitrogenase enzymes can also reduce longer-chain alkynes.38,39 Here, we examine the reactivity of the low-coordinate iron hydride complexes with alkynes diphenylacetylene and 3-hexyne.
Complex 1a inserts into the C≡C triple bond in PhCCPh to form LMeFeC(Ph)=C(H)Ph (6a) in 75% yield. This vinyl product is analogous to LtBuFeC(Et)=C(H)Et (6b), which was previously reported from the reaction of 1b with 3-hexyne.20 Figure 6 presents the crystal structure of 6a. The C-C distance (1.347(2) Å) and C-C-C angles (129.0(2)°, 121.6(2)°) in the vinyl ligand confirm that both carbons of the C=C double bond have sp2 hybridization. The 1H NMR spectrum of 6a shows seven peaks for the β-diketiminate protons, indicating averaged C2v symmetry from rapid rotation around the Fe-C14 bond on the NMR time scale.
Figure 6.

Molecular structure of LMeFeCPh=C(H)Ph (6a), showing 50% thermal ellipsoids. Selected bond distances (Å) and angles (°): Fe-C14 2.017(2), C14-C24 1.347(2), C14-C24-C34 129.0(2), C24-C14-C94 121.6(2).
Monitoring the reaction of 1a with PhC≡CPh, or the reaction of 1b with EtC≡CEt, shows no dependence of the reaction rate on [PhC≡CPh] (Table 2). Therefore, the rate laws are rate = k[1] with first-order rate constants 1.7(2)×10−3 s−1 (1a/PhCCPh at 31 °C) and 5.0(5)×10−4 s−1 (1b/EtCCEt at 15 °C). The first-order rate law is inconsistent with the interaction of alkyne with 1a or 1b during or prior to the rate-limiting step of the reaction. Two mechanistic possibilities consistent with the rate law are shown in Scheme 2. In pathway A, the opening of a single Fe-H bond is the rate determining step. We consider this Fe-H bond opening pathway to be more likely for the reaction of alkyne with 1a because (1) there is no other evidence for any monomer form of 1a by NMR or UV-vis spectroscopy; (2) in the reaction of 1a with boranes, the rate law was first-order in [1a] and first-order in [BEt3] but independent of the steric demands of the borane or iron complex.28 These data were inconsistent with dissociation of 1a into monomers (which predicts a half-order dependence on [1a]) and most consistent with single Fe-H opening, as in the bottom reaction pathway here. In pathway B, dimer cleavage is the rate determining step, as proposed for the reaction of 1b with 3-hexyne.20 This pathway is reasonable for 1b, because monomer is rapidly accessed at room temperature (see above). The rate of the reaction is consistent with the rapid equilibrium of monomer and dimer observed previously.20 So, although the evidence is not definitive, the kinetic data here and elsewhere are most indicative of the pathway A in the reactions of alkynes with 1a, but pathway B for alkyne reactions with 1b. This difference is consistent with the greater steric demands of the diketiminate ligands in 1b than 1a.
Table 2.
| [PhC≡CPh] (mM) | [PhC≡CPh]/[1a] | kobs (s−1) |
|---|---|---|
| 184 | 10.5 | 1.7(1)×10−3 |
| 362 | 20.6 | 1.6(1)×10−3 |
| 725 | 41.2 | 1.7(2)×10−3 |
[1a] = 17.6 mM in C6D6 at 30.8 °C.
Details of the reaction of 1b with 3-hexyne are given in ref 20.
Scheme 2.

Reactions with C=C bonds
We previously reported that LMeFe(alkyl) complexes with β-hydrogens can act as sources of transient hydride species LMeFe(H)(alkene). These react with alkenes to give insertion products, and complexes LMeFeR′ (R′ = ethyl, 1-propyl, 2-propyl, 1-butyl, 2-butyl, cyclohexyl, 2-phenethyl) were fully characterized.30 We have also characterized numerous alkyl complexes of iron with the larger LtBu ligand.40 With isolated iron hydride complexes in hand, we verified that addition of alkenes to isolated 1a or 1b gives alkyl complexes of the same type through [1,2] addition across the double bond. For example, reaction of 1a with 1-hexene quantitatively gives a 1-hexyliron complex, as judged by 1H NMR spectroscopy. It is worth noting that the transient iron hydride LMeFe(H)(alkene) also adds across the C=N bonds of imines (forming LMeFeNR′CHR2 from R2C=NR′) and the C=O bonds of ketones (forming LMeFeOCHR2 from R2C=O).30 Because these reactions have already been characterized starting from the alkyl complexes,30 the reactions with isolated hydride complexes were not investigated further.
Reactions With Azides
The azide ion (N3−) is transformed by nitrogenase into N2, NH3, and N2H4.41 Here, substituted azides AdN3 and Me3SiN3 are used as models. The reactions of organic azides with transition-metal complexes have been reviewed recently.42
Reactions of complex 1a with Me3SiN3 give mixtures, as judged from 1H NMR spectra. However, complex 1b, when treated with 2 equiv of trimethylsilyl azide in diethyl ether, gives a dimeric azide complex, LtBuFe(μ-N3)2FeLtBu (7b) in 79% yield (Scheme 3). Compound 7b can be prepared independently by reacting LtBuFeCl43 with sodium azide.
Scheme 3.
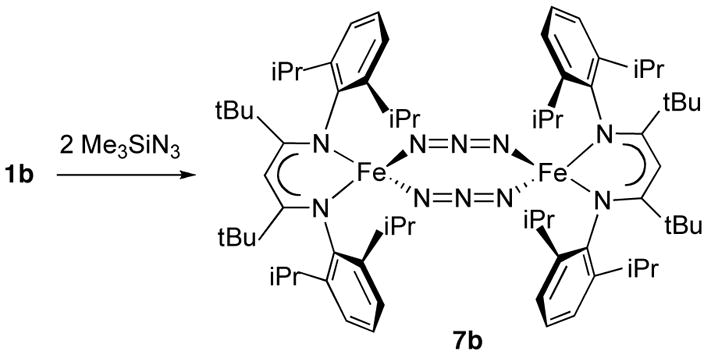
The X-ray crystal structure of 7b (Figure 7) shows that the compound is dinuclear in the solid state with an Fe···Fe distance of 5.0966(4) Å. Each iron atom is coordinated to two azide ions, which bridge the iron atoms in a μ1,3-end-to-end fashion with inequivalent iron-nitrogen bond lengths of 2.061(1) and 2.025(1) Å. The eight atoms of the Fe2N6 core are coplanar, in contrast to the only other crystallographically characterized end-to-end bridged iron azide complex, [(PhBP3)Fe(μ-1,3-N3)]2 (PhBP3 = [PhB(CH2PPh2)3]−), which has a chair conformation.44 The dihedral angle between the Fe2N6 plane and the Fe2N2(diketiminate) plane is 87.41(4)°. The azide ligands are almost linear (N-N-N angle of 177.0(1)°) and the N-N bond lengths within the azide groups (1.167(2), 1.176(2) Å) show double bond character similar to N-N distances in other bridging azide complexes.34 The solid-state IR spectrum of 7b shows characteristic azide bands at 2129 and 2081 cm−1.45
Figure 7.

Molecular structure of LtBuFe(μ-N3)2FeLtBu (7b), showing 50% thermal ellipsoids. Selected bond distances (Å) and angles (°): Fe-N15 2.061(1), Fe-N35 2.025(1), Fe-N11 1.988 (1), Fe-N21 1.989(1), N15-N25 1.167(2), N25-N35 1.176(2), N11-Fe-N21 97.10(4), N15-Fe-N35 95.17(4), N15-N25-N35 177.0(1).
29Si{1H} NMR analysis of the volatile products in the reaction mixture revealed a resonance at −22 ppm for Me3SiSiMe3, but none for Me3SiH. The production of Me3SiSiMe3 implies that two hydrogen atoms were lost. However, H2 was not detected in the headspace by gas chromatography, so the fate of the hydrogen atoms is unclear. The coupling of two trimethylsilyl units strongly implies the intermediacy of Me3Si· radicals. These in turn might derive from the attack of an iron radical on trimethylsilyl azide.46 In order to test this idea, the iron(I) sources LtBuFeNNFeLtBu 47 and LtBuFeClK(solvent)x 47 were each treated separately with Me3SiN3. In each case, [LtBuFeN3]2 was formed. Therefore, iron(I) species of this type are reasonable intermediates that could be formed by homolysis of Fe-H bonds. Homolysis of Fe-H bonds does not occur spontaneously in 1b at room temperature, and therefore we infer that coordination of Me3SiN3 weakens the Fe-H bond and brings about Fe-H homolysis.
Compound 1b undergoes an insertion reaction with AdN3 to give 67% yield of a triazenido complex, LtBuFe(η2-HNNNAd) (8b), which is shown in Scheme 4 and Figure 8. The bidentate triazenide and β-diketiminate ligands are perpendicular to each other, which gives a distorted tetrahedral geometry around the metal center. The presence of an N-H bond in 8b is confirmed by the observation of νN-H at 3371 cm−1 in the infrared spectrum. The 1H NMR spectrum of 8b is indicative of C2v local symmetry at temperatures from room temperature to −75 °C, despite the Cs idealized symmetry of the molecule in the solid state (each face of the diketiminate ligand should be inequivalent). This observation implies that there is rapid exchange between the two possible orientations of the triazenido ligand. Considering the stability of three-coordinate complexes LtBuFeX and the large size of the adamantyl group, it seems most reasonable to attribute this behavior to a rapid, reversible isomerization of the triazenido ligand from η2 to η1, followed by rotation and re-coordination.
Scheme 4.
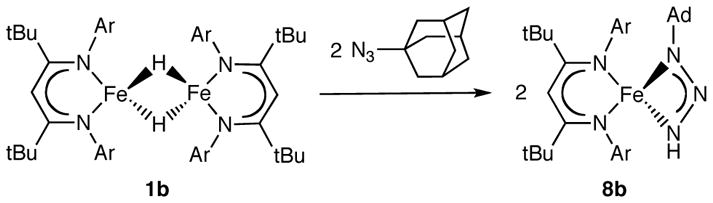
Figure 8.

Molecular structure of triazenido complex 8b, showing 50% thermal ellipsoids. Selected bond distances (Å) and angles (°): Fe-N1 2.081(2), Fe-N3 2.078(2), Fe-N11 1.878(4), Fe-N21 2.117(4), N1-N2 1.310(3), N2-N3 1.285(2), N3-C1 1.463(7), N11-Fe-N21 98.32(9), N1-Fe-N3 60.46(7), N1-N2-N3 107.6(2).
Although disubstituted triazenido complexes are well-known in the literature, there are few examples of triazenido complexes bearing H as one substituent; they come from insertion of azide into a hydride complex,48,49 and from protonation of an azide complex.50,51 Free H2NNNR compounds are extremely unstable with respect to loss of N2. Although compound 8b is thermally stable for hours in solution at room temperature, it eventually decomposes to the Fe(II) amido complex LtBuFeNHAd with loss of N2. (For identification, LtBuFeNHAd was synthesized independently from LtBuFeCl and LiNHAd by analogy to other known iron(II) amido complexes.52) Quantitative transformation from triazenido to amido was observed by 1H NMR spectroscopy after heating a sample of 8b in C6D6 at 80 °C for 5 h.
Reactions With N=N Double Bonds
Diazene (HN=NH) is a possible intermediate of N2 fixation by nitrogenase. Consistent with this idea, both NH=NH and CH3N=NH are nitrogenase substrates,53 and CH3-N=N-CH3 is reduced by nitrogenase to give ammonia, methane and methylamine.54 Very recently, hydrazine (N2H4), methyldiazene (HN=N-CH3) and N2 derived nitrogenase intermediates have been freeze-trapped.55 Free diazenes bearing hydrogen substituents are extremely difficult to handle, because they decompose in seconds or minutes. As an alternative approach, we have used the stable diazenes azobenzene (PhN=NPh) and benzo[c]cinnoline.
We recently reported a detailed study of the the reaction of 1b with PhN=NPh, which leads first to the [1,2]-addition product LtBuFeNPhNHPh, and subsequently to the amido complex LtBuFeNHPh.27 Mechanistic studies were most consistent with a radical chain mechanism, mediated by an iron(I) carrier. 27 Reaction of the smaller 1a with PhN=NPh at ambient temperature gives a mixture of products as judged by the 1H NMR spectrum. It has not been possible to purify and isolate these compounds, but they may be tentatively assigned as LMeFeNPhNHPh and LMeFeNHPh based on the similarity of their 1H NMR spectra with the LtBu analogues.27 Longer reaction time or heating does not drive the reaction mixture to LMeFeNHPh. Instead decomposition occurs, probably due to the instability of LMeFeNHPh.52
We also investigated the reaction of 1a with 2 equiv of benzo[c]cinnoline, which gave 90% conversion to deep green 9a. The detection of 1.07(3) equivalents of H2 by GC is consistent with the reaction stoichiometry shown in Scheme 5. In this case, there is no addition across the N=N double bond: rather, H2 is lost and the benzo[c]cinnoline coordinates to iron. Alternatively, 9a could be isolated in 91% yield from the reaction of LMeFeNNFeLMe and benzo[c]cinnoline.
Scheme 5.
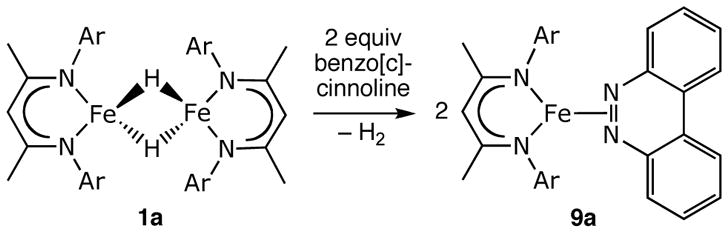
The crystal structure of 9a (Figure 9) shows that benzo[c]cinnoline binds face-on to iron(I) through the N=N π-bond, and the N-N distance is 1.138(5) Å. The dihedral angle between the benzo[c]cinnoline and the β-diketiminate ligand is 55.35(5)°. A side-on interaction between iron and an N=N double bond has been crystallographically confirmed in only two other compounds. In [Fe(NO)2-{PPh2CH2CH2PPh2NNAr}][PF6], the diazene (N=N 1.403(5)) is constrained to bind using a bidentate phosphine;56 and our recently reported LtBuFe(PhNNPh) has an N=N distance of 1.398(2) Å.27
Figure 9.

Two views of the molecular structure of 9a, showing 50% thermal ellipsoids. In the picture on the right, aryl groups are removed for clarity. Selected bond distances (Å) and angles (°): Fe-N2 1.967(1), N2-N2′ 1.385(2), N2-Fe-N2′ 41.22(7). The dihedral angle between the β-diketiminate FeNCCNFe plane and the benzo[c]cinnoline plane is 55.35(5)°.
The peaks in the 1H NMR spectra of C6D6 solutions of 9a are unusually broad. Mixing different ratios of 1a and benzo[c]cinnoline indicates that the broadness of the peaks increases with a greater concentration of benzo[c]cinnoline, and becomes substantially sharper at a 1:1 ratio of 1a to benzo[c]cinnoline (see Supporting Information for spectra). This behavior suggests that free and bound benzo[c]cinnoline exchange at a rate near the NMR time scale (milliseconds). The spectra are substantially sharper in C6D12, indicating that exchange of the aromatic with C6D6 is part of this process. We have previously characterized LMeFe(C6H6), in which the benzene ligand is bound relatively weakly (it is displaced rapidly by phosphines, alkenes, and alkynes).57 Since benzene competes effectively with benzo[c]cinnoline as a ligand, it shows that the heteroaromatic π-ligand is bound weakly as well.
Reactions With Carbon Dioxide and Carbodiimide
After azide, CO2 is another heterocumulene that is a substrate of nitrogenase. Seefeldt and co-workers have shown that CO2 is slowly reduced to CO by the nitrogenase enzyme.58
Complexes 1a and 1b each react with two equivalents of carbon dioxide at room temperature, generating yellow solids that are formulated as formate-bridging diiron complexes (10a, 10b) based on X-ray crystallography (Figure 10). Only a few crystallographically characterized η2-formato bridging iron complexes have been reported in the literature. These complexes were synthesized from iron formate, Fe(O2CH)2·2H2O,59 reaction of iron metal and formic acid,60 or Fe(ClO4)·10H2O with NaO2CH61. Reactions of CO2 with iron hydride complexes are uncommon. 62
Figure 10.
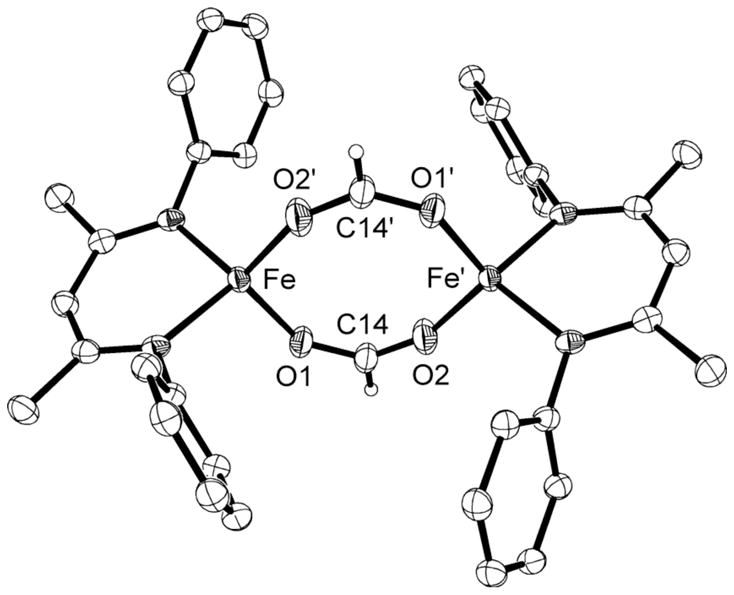
Molecular structure of LMeFe(μ-OCHO)2FeLMe (10a) showing 50% thermal ellipsoids. Isopropyl groups on the aryl rings are omitted for clarity. The structure of 10b is similar, and is shown in the Supporting Information. Selected bond distances (Å) and angles (°) for 10a: Fe-O 1.978(2), 2.006(2), C-O 1.245(3), 1.246(3), O-Fe-O 115.57(7), O-C-O 127.9(2). For 10b: Fe-O 1.967(1), 1.994(1), C-O 1.248(2), 1.245(2), O-Fe-O 106.98(6), O-C-O 128.2(2).
The formate-bridged diiron complexes are insoluble in pentane, and only somewhat soluble in aromatic solvents, a problem that was especially severe for 10b. The solution magnetic moment of 10a is 8.8(2) μB, consistent with two nearly uncoupled high-spin iron(II) ions, and 10b was too insoluble to derive a reliable value. The 1H NMR spectrum of 10a at room temperature has six peaks with chemical shifts that range from 18 ppm to −60 ppm, suggesting idealized D2h symmetry. Although this observation is consistent with either monomer or dimer in solution, no decoalescence is seen in the 1H NMR spectrum of 10a in toluene from −35 °C to 120 °C, indicating that the crystallographically observed dimer is the most likely solution species. In 10b, the 1H NMR spectrum at 85 °C has only eight peaks that integrate as expected for a fully symmetric diketiminate ligand, and the chemical shifts are similar to those in 10a. Interestingly, new peaks appear at lower temperatures. These peaks are consistent with splitting of a isopropyl methine and one isopropyl methyl resonance, which correspond to the protons closest to one another in the dimer. Unfortunately, the poor solubility of 10b, especially at low temperature, prevents us from fully characterizing this phenomenon. The available data are consistent with either a monomer-dimer equilibrium, or a change from slow to rapid flipping of the diketiminate planes to each side of a dimer (change from C2h to effective D2h point group symmetry).
To overcome the issues caused by the low solubility of these products, we investigated the analogous reaction of diisopropyl carbodiimide (iPrN=C=NiPr) with 1b. This reaction proceeded rapidly and quantitatively at room temperature to give the formamidinate complex LtBuFe(iPrNCHNiPr) (11b) in 89% yield. This compound is monomeric, in contrast to the dimeric formate complexes derived from CO2, a result that is most reasonably attributed to the steric hindrance of the isopropyl groups on the carbodiimide. In the solid state structure of 11b (Figure 11) both the diketiminate ligand and the formamidinate ligand are coordinated in an η2 fashion to the Fe atom, giving a distorted tetrahedral geometry around the metal center. The N-Fe-N bite angle of each ligand is typical.34
Figure 11.

Molecular structure of 11b, showing 50% thermal ellipsoids. Selected bond distances (Å) and angles (°): Fe-N1 2.1358(9), Fe-N2 2.0675(8), Fe-N11 2.0663(8), Fe-N21 1.9908(8), N1-C1 1.318 (1), N2-C1 1.323(1), N1-Fe-N2 64.50(3), N11-Fe-N21 97.44(3), N1-C1-N2 116.27(8).
Reactions With Brønsted Acids
As described above, EPR investigations suggest that an FeMoco species with two hydrides can lose two equivalents of H2 to return to the hydride-free resting state.6 This suggests that some low-coordinate iron hydride species could be protonated by nearby Brønsted acids to give H2. To investigate the susceptibility of synthetic Fe-H compounds to protonation of the hydride, we explored the reactions of 1a and 1b with weak acids.
In the presence of excess water, the low-coordinate iron hydride complexes decompose to intractable mixtures that contain free β-diketimine, suggesting that the acid protonates the α position of the β-diketiminate ligand. Reaction with smaller amounts of water often gives mixtures as well. In a few cases, using solutions of H2O in tetrahydrofuran gives mixtures in which one compound predominates, enabling isolation. For example, we reported that addition of 1 equiv of H2O to 1b (0.5 equiv per iron) gives the unique oxodiiron(II) complex LtBuFeOFeLtBu in 71% yield.29b In another reaction, the addition of 2 equiv of water to 1a gives a green iron complex, LMeFe(μ-OH)2FeLMe (12a) in 67% yield (Scheme 6)
Scheme 6.
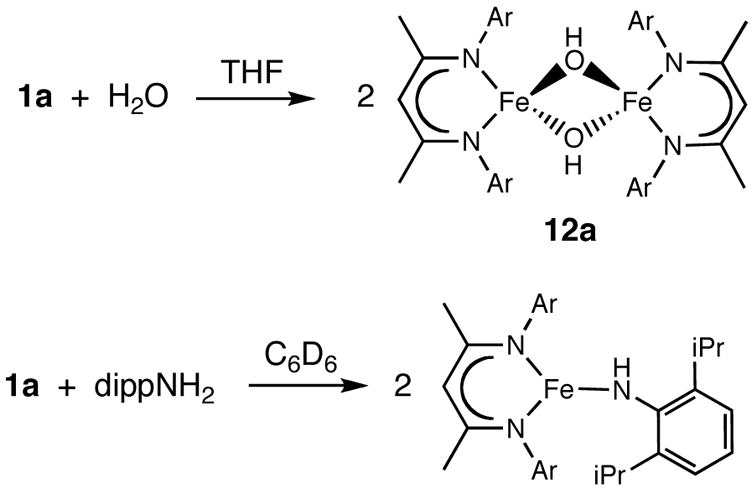
The solid state structure of 12a (Figure 12) has pseudo-D2h symmetry, and half of the molecule is related to the other half by a crystallographic inversion center. The metrical parameters compare well with other complexes containing a Fe2(μ-OH)2 core.34,63 Seven paramagnetically shifted peaks are observed in the 1H NMR spectrum of 12a, consistent with D2h symmetry in solution. No peak clearly corresponding to the bridging OH is observed in the 1H NMR spectrum, but O-H stretching vibrations are seen at 3709 and 3668 cm−1 in the IR spectrum. This binuclear complex has a solution magnetic moment of 7.5(1) μB per dimer, suggesting two uncoupled high-spin Fe(II) centers. Dihydrogen is observed by GC as another product of the reaction. Unfortunately, the THF and H2O peaks overlap with the peak from the internal standard CH4, which prevented quantitative measurement of the H2 produced.
Figure 12.
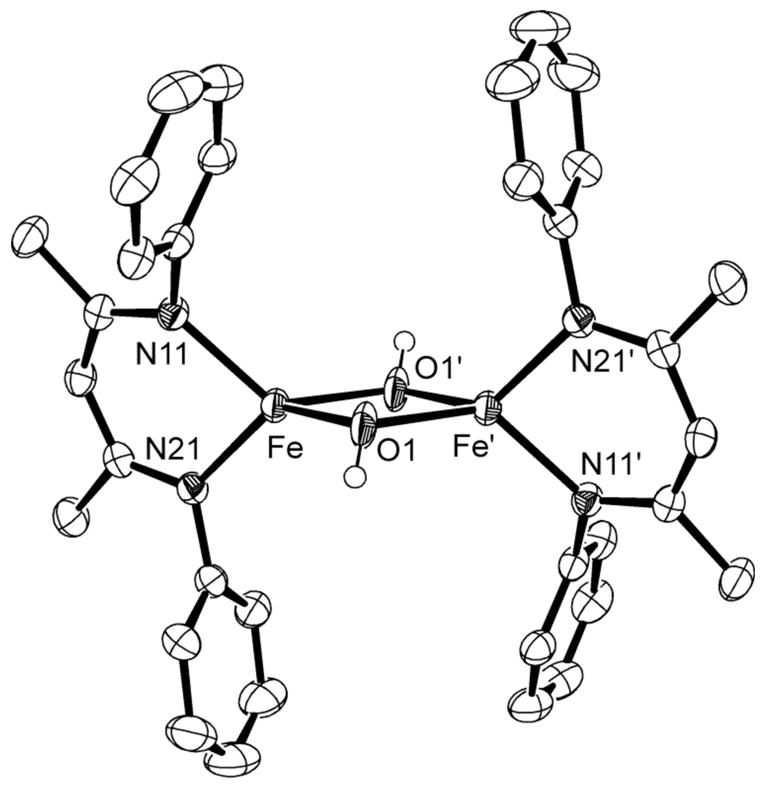
Molecular structure of 12a, showing 50% thermal ellipsoids. Isopropyl groups removed for clarity. Selected bond distances (Å) and angles (°): Fe-O1 2.082(3), 2.059(6), Fe-O1-Fe′ 98.7(2), O1-Fe-O1′ 81.3(2).
Finally, we tested 2,6-diisopropylaniline (dippNH2), which has a similar acidity as water in organic solvents (the pKa values in DMSO are 32 for H2O and 30.6 for PhNH2).64 A mixture of 1a and dippNH2 in C6D6 quantitatively gives the iron amido complex LMeFeNHdipp within a few hours (Scheme 6), as judged by 1H NMR spectroscopy. Dihydrogen (1.6 equiv) is detected from the reaction, close to the 2 equiv expected. This iron amido complex was previously synthesized from [LMeFeCl]2.52
Discussion
Addition of Fe-H Across a Multiple Bond
The low coordination number at the iron atoms in these hydride complexes enables reactions with many unsaturated organic compounds. Rapid [1,2] additions are seen with alkenes, alkynes, imines, ketones, nitriles, carbodiimides, and carbon dioxide. These reactions generally place the iron fragment on the more electronegative atom of the double bond, and the hydrogen on the less electronegative atom. These are not uncommon reactions of transition-metal hydride complexes,1 but they appear to be quite rapid in this system relative to literature iron complexes of higher coordination number. For example, a mixture of trans-[FeHCl(dppe)2] and TlBF4 reacts only with alkynes in which there is an ester functionality that can chelate to the metal.65 The reactions of phosphine-supported octahedral iron hydrides with alkynes often give a variety of products, including acetylide and vinylidene complexes.66 Known saturated iron hydride complexes do not react with alkenes66 except cyclopropene.67 Although there are not many literature examples of heterocumulenes (CO2, CS2, carbodiimides) reacting with iron hydrides, these reactions appear to be facile in one case.68
Cyanide compounds (nitriles) are not usually reactive toward insertion into the Fe-H bonds of iron-hydride complexes, aside from one example of insertion from a dinuclear iron-carbonyl complex.69 Acetonitrile more typically coordinates to iron.70 In the β-diketiminate complexes described here, the iron hydrides reduce RCN to a RC(H)=N− ligand under mild conditions. Interestingly, the outcome of the insertion reactions is dependent on the steric demands of the β-diketiminate ligand and the nitrile. With the bulkiest β-diketiminate (LtBu) and nitrile (tBuCN), the product is a monomeric complex. With the smallest β-diketiminate (LMe) and nitrile (CH3CN), the product is a dimer with two bridging imidoyl ligands. Finally, with tBuCN and the less bulky hydride complex, a single insertion occurs and the steric bulk prevents access of the second nitrile to the remaining bridging hydride.
The reaction of adamantyl azide with 1b gives a triazenido complex that results from formal [1,1]-addition. A few late-metal alkyl complexes have been observed to give [1,1]-addition to azides.71 The lone example of a triazenido ligand on iron is in a dinuclear iron-carbonyl complex, where the triazenide derives from protonation of an anionic azide complex.72 To our knowledge, the only other reaction of an iron hydride complex with an azide involves the addition of sodium azide to L2FeH2 (L = bidentate phosphine ligand) in methanol, which replaces the hydride ligand with azide.73 A search of the CSD shows that LtBuFe(HNNNAd) is the first structurally characterized complex of a triazenido ligand bearing a hydrogen atom.34 Mechanistic and spectroscopic studies indicate the formation of hafnium, rhenium and tungsten triazenido complexes from the reaction of azides with hydride species.51,74 In the hafnium complex, mechanistic studies indicate that the HNNN(aryl)− ligand decomposes to NH(aryl)− through a shift of the hydrogen atom to the carbon-bound nitrogen atom, followed by extrusion of N2. In the iron system studied here, a similar mechanism can explain the conversion of the triazenido complex to LtBuFeNHAd.
Recent reports of other low-coordinate iron systems are also indicative of high reactivity, although hydride species were not isolable in these systems. For example, Chirik has used pyridinediimine-iron and α-diimine-iron complexes as precatalysts for catalytic alkene hydrogenations, though hydride complexes were not isolated.75 Peters has proposed a transient four-coordinate iron hydride complex that adds across an aromatic C=C bond of benzene,76 and Ohki and Tatsumi have proposed the intermediacy of Cp*FeH in the cleavage of N=N bonds.77 These precedents imply that the high activity of the isolable low-coordinate iron-hydride complexes described here is likely to have broader applicability with other supporting ligands.
Reductive Elimination of Dihydrogen
Numerous polyhydride complexes, both monometallic and multimetallic, are capable of formal reduction through loss of H2 when a ligand is added.78 Some synthetic hydride complexes have been observed to lose H2 with addition of N2.79,80 Specifically with iron, a few polyhydride complexes reductively eliminate H2 with ligand binding.25,81 Iron-dihydrogen complexes have been studied in octahedral systems.82 The reactions typically involve reduction of iron(II) to iron(0), not to iron(I) as observed here. In a recent paper, Peters has presented evidence supporting reversible metallation of a ligand alkyl group in a low-coordinate iron(I) complex.23b
We see no evidence of stable H2 complexes in thermal or photochemical elimination reactions of the diketiminate-supported iron hydride complexes, because any putative transient “Lfe” or “LFe(H2)” is trapped by N2 (in the photochemical reaction) or the added ligand (thermal reactions with CO or CNR). The thermal reactions must proceed through a mechanism in which substrate binding precedes H2 loss, because 1a and 1b are stable at room temperature in solution and under vacuum. We infer that interaction with ligands activates the iron hydride species toward H2 loss. As described above in the context of alkene/alkyne insertion, the evidence described above suggests that [LMeFeH]2 reacts through a low-population isomer in which one iron-hydrogen bond has opened in the Fe2H2 core. The subsequent LMeFe(substrate)(μ-H)Fe(H)LMe complex could be unstable with respect to breaking the remaining Fe-H bond to give an iron(I) product and an unstable iron(III) dihydride complex LMeFeH2, which could lose H2 leading to the remaining iron(I) product. Though other mechanisms are consistent with the data, this is a working model for further studies of these reactions; it also rationalizes why the stronger ligands and those more resistant to insertion reactions (isocyanides, benzo[c]cinnoline, some azides) lead to iron(I) products. In the case of Me3SiN3, the product is not the iron(I) compound itself, but instead the result of reductive coupling of trimethylsilyl groups in Me3SiN3. Because the reductive coupling product also results from treatment of Me3SiN3 with the iron(I) source LFeNNFeL, iron(I) species are likely intermediates in this reaction.
The product of the reaction of 1a with benzo[c]cinnoline is especially interesting. Despite the presence of lone pairs on the two nitrogen atoms of this heterocycle, the iron coordinates instead to the π-system. There are only four previous crystal structures in which benzo[c]cinnoline is a ligand to a transition metal: Ti and Yb complexes with the metal in the plane of the aromatic rings,83 a complex with the diazene bridging the iron atoms in a Fe2(CO)6 fragment,84 and the cobalt(0) complex (PMe3)3Co(benzo[c]cinnoline).85 The latter complex is the most similar to the one shown here, from its formal d7 electronic configuration to the similar angle between the MN2 and aromatic planes (68° vs. 55° here).85 We have previously shown that benzene coordinates in a η6 binding mode in LMeFe(C6H6),47 but the η2 binding of benzo[c]cinnoline is clearly stronger, since the benzocinnoline complex is stable in benzene solution. Why, then, is the binding stronger for the more hindered benzo[c]cinnoline? Previous studies on the binding constants of alkenes, alkynes, and other ligands to the iron(I) LMeFe fragment showed that backbonding dominates the selectivity of binding.57 Because of the greater electronegativity of nitrogen than carbon, the stabilizing shift of electron density from the iron to the unsaturated ligand is more effective in the iron(I) complex of benzo[c]cinnoline.
We also note that strongly hydridic M-H bonds like those in 1a and 1b are characteristic of early transition metal hydrides,86 consistent with the Feδ+–Xδ− character of iron alkyl, amido, and alkoxo complexes demonstrated in our earlier studies,30,52,57 Further research will show whether the reactivity of other low-coordinate late-metal systems will also tend to mimic the behavior of transition metals with a lower d electron count.
Protonation to Give Dihydrogen
Many iron-hydride complexes are protonated by acids to release H2. For example, in iron complexes of a PNP ligand, the necessary pKa of the acid was determined to be less than 10.5 (a coordinated nitrogen was protonated in preference to the hydride ligand).87,88 On the other hand, the diketiminate-bound iron hydrides are protonated by weak acids such as diisopropylaniline and diphenylhydrazine.27 Using literature values in DMSO,64 one can conclude that acids with pKa value up to 32 (and perhaps higher) can protonate 1a and 1b.
Protonation of a hydride ligand may be a key step in nitrogen fixation by nitrogenases. In this hypothesis, one of the metal-bound hydride ligands is protonated and released as dihydrogen to open a coordination site for dinitrogen binding.89 This hypothesis is consistent with Hoffman’s ENDOR-based conclusion that the N2 binding form of the iron-molybdenum cofactor is at the E4H2 state.6 There are a number of acidic Arg residues near the binding face of the iron-molybdenum cofactor.3 This hypothesis finds support here, in that weak acids are capable of protonating the hydride ligand to form H2.
Insight into Potential Nitrogenase Mechanisms
Some nitrogenase researchers have explained the production of H2 by nitrogenase during N2 reduction as the result of ligand-assisted reductive elimination.3 In this model, creation of hydrides provides a way for the FeMoco to store electrons for N2 reduction without reaching an unrealistically low charge and reduction potential. Release of H2 from the FeMoco is thought to be concurrent with N2 binding, explaining why H2 is a competitive inhibitor of N2 reduction.7 Other substrates are not inhibited by H2 because the extra “boost” in reducing power from H2 loss is not necessary. The reactivity of complexes 1a and 1b can be viewed using this model, in that they have sufficient reducing power to react thermally with all nitrogenase substrates except N2. Thus, the reactivity of compounds 1 mimics not the E4 state, but a less reduced FeMoco intermediate (E2 or E3) that reduces alternative substrates like alkynes, CO2, cyanide, and azide. Photolysis of the hydride complexes 1a and 1b provides the “boost” required to bind the weak ligand N2.
Even though 1a and 1b do not reduce N2, their reducing power is superior to octahedral Fe-H complexes, a difference that might arise from the low coordination number, the high-spin electronic configuration, the weak Fe-H bond strength, or (most likely) a combination of these effects. Considering that iron atoms in the FeMoco have a low-coordinate geometry and high-spin electronic configuration, these factors are clearly identified as reasonable targets for evaluation in the enzyme.
It is notable that nitrogenase inhibitors (or analogues thereof) like CO, RNC, and RN3 typically cause the synthetic hydride complexes 1a and 1b to lose H2 and bind the small molecule that was added. Therefore, one may hypothesize that these small molecules might inhibit enzymatic N2 reduction by binding strongly to the FeMoco, causing premature loss of H2. Addition of 1a and 1b to small molecules that are substrates of nitrogenase (or analogues thereof), like RCN, alkynes, RN3, and RN=NR, typically forms a new substrate-H bond, and no H2 is lost. An exception is benzo[c]cinnoline: although it mimics the nitrogenase substrate diazene, it displaces H2 like an inhibitor. This may be attributed to the aromatic nature of benzo[c]cinnoline,90 which perhaps makes it more resistant to reduction than other diazenes. Azides and cyanides are inhibitors and substrates in the enzyme, and in the synthetic compounds they sometimes give H2 and sometimes are reduced. Therefore, the reactions of the synthetic compounds 1a and 1b recall the bifurcated roles of these molecules toward the enzyme.
It is also worth comparing the specific products of substrate reduction by nitrogenase and the products from the synthetic iron complexes. The synthetic compounds typically do not complete the reduction and release the appropriate product, but maintain a bond between the iron atom and the partially reduced substrate. For example, 1a and 1b reduce alkynes to iron-vinyl complexes, but do not release alkene. They reduce one C=O bond of CO2, but do not eliminate water to give CO.91 They reduce C≡N bonds to C=N bonds, but do not continue the reduction to methylamine or methane/ammonia. These outcomes may be attributable to the absence of protons in the synthetic system that would release the anionic ligand, either as a product or as an intermediate that is subsequently reduced to the final product. Future studies will examine the further transformations of these small molecules at iron.
Conclusions
In summary, we have shown that the first isolable low-coordinate iron hydride complexes are highly reactive towards various unsaturated small molecules. The reactions can be categorized into three types: (1) insertions of a multiple bond of the substrate into the Fe-H bond with two-electron reduction of the substrate; (2) reductive elimination of H2 and coordination (or one-electron reduction) of the substrate; (3) loss of H− to an acidic proton of the substrate, releasing H2 and giving an iron complex of the conjugate base. Some of the products isolated have novel structural features, such as a square-planar iron(I) complex, a face-bound complex of benzo[c]cinnoline, and a monoalkylated triazenido complex. The reaction patterns are reminiscent of elementary steps in the proposed nitrogenase catalytic mechanism, and support the idea that hydrides on the low-coordinate, high-spin iron atoms of the FeMoco would be capable of some of the characteristic reactions of the FeMoco. In addition, these studies identify some possible reasons why certain small molecules are substrates of nitrogenases, and others are inhibitors.
Experimental Section
General Procedures
All manipulations were performed under a nitrogen atmosphere by Schlenk techniques or in an M. Braun glove box maintained at or below 1 ppm of O2 and H2O. Glassware was dried at 150 °C overnight. NMR data were recorded on a Bruker Avance 500 spectrometer (500 MHz). All peaks in the NMR spectra are referenced to residual protiated solvents (benzene δ 7.16 ppm; toluene δ 2.08 ppm; cyclohexane δ 1.38 ppm). In parentheses are listed integrations and assignments. Resonances were singlets unless otherwise specified. Infrared spectra (450–4000 cm−1) were recorded on KBr pellet samples in a Shimadzu FTIR spectrophotometer (FTIR-8400S) using 32 scans at 2 cm−1 resolution. UV-vis spectra were measured on a Cary 50 spectrophotometer using screw-cap cuvettes; descriptions of the spectra include extinction coefficients in parentheses. Solution magnetic susceptibilities were determined by the Evans method.92 Elemental analyses were determined by Desert Analytics, Tucson, AZ.
Pentane, tetrahydrofuran (THF), diethyl ether and toluene were purified by passage through activated alumina and “deoxygenizer” columns from Glass Contour Co. (Laguna Beach, CA). Deuterated solvents were first dried over CaH2, then over Na/benzophenone, and then vacuum transferred into a storage container. Before use, an aliquot of each solvent was tested with a drop of sodium benzophenone ketyl in THF solution. Celite was dried overnight at 200 °C under vacuum. CO2 was purchased from Air Products, dried by passing through 4Å molecular sieves, and freed from O2 with three freeze-pump-thaw cycles. H2O, tBuNC, tBuCN, MeCN and 2,6-diisopropylphenylamine were degassed before using. Diphenylacetylene, benno[c]cinnoline, and 1-azidoadamantane (AdN3) were crystallized from pentane in the glovebox. Compounds 1a and 1b were prepared by published procedures.20,21
LMeFe(CNtBu)2 (2a)
Reaction of 1a and tBuNC in C6D6 gives complex 2a, in a crude yield of 62% based on 1H NMR integration with an internal integration standard. However, it can be more conveniently synthesized from LMeFeNNFeLMe and tBuNC as follows. A solution of LMeFeNNFeLMe (205 mg, 0.21 mmol) in pentane (15 mL) was treated with tBuNC (105 μL, 0.93 mmol) with stirring. Immediate bubbling was observed and the solution color changed from deep brown to bright orange. The reaction solution was stirred at ambient temperature for 5 h. Volatile materials were removed under vacuum and the residue was extracted with pentane (20 mL) and filtered through Celite. The solution was concentrated to 3 mL and cooled to −35 °C to give bright orange crystals of 2a (145 mg, 54%). 1H NMR (500 MHz, C6D6): δ9.1 (2H, aryl p-H), 5.8 (4H), 3.5 (12H, i Pr-CH3), 2.5 (18H, tBu), 1.3 (4H), −2.0 (6H, backbone CH3), −5.8 (12H, iPr CH3) ppm. (Peaks integrated as 4H could be aryl m-H or iPr methine CH; the backbone CH was not observed). UV-vis (pentane): 317 (25.3 mM−1cm−1), 447 (3.9 mM−1cm−1), 808 (0.2 mM−1cm−1) nm. IR (KBr pellet): 2122 (vs), 2050 (m), 1969 (m), 1948 (m) cm−1. The X-band EPR spectrum is shown in the Supporting Information. Attempts to measure the solution magnetic moment of 2a were not successful because the internal standard peak was obscured. Complex 2a is thermally unstable (decomposition of a solid sample occurred overnight inside the glove box), and all characterizations were done with fresh samples. The instability prevented elemental analysis measurements.
LMeFe(μ-N=CHMe)2FeLMe (3a)
A stirring solution of [LMeFeH]2 (1a) (146 mg, 0.15 mmol) in toluene (30 mL) was treated with MeCN (18.0 μL, 0.34 mmol). The reaction solution was heated at 45 °C for 18 h and the color changed from deep brown to yellow. All volatile materials were removed under vacuum and the residue was extracted with toluene (30 mL), filtered and concentrated to 5 mL. Cooling to −35 ºC gave orange-yellow crystals of 3a (155 mg, 88%). 1H NMR (500 MHz, C7D8): δ50 (6H, N=CHCH3), 20 (12H), 13 (4H), 12(4H), −2 (12H), −7 (12H), −12 (4H), −16 (4H), −21 (12H), −22 (4H), −31(2H, diketiminate backbone), −40 (12H) ppm. (No peak corresponding to the imine proton was identified. Peaks integrated as 4H could be iPr methine, aryl p-H or aryl o-H; peaks integrated as 12H could be iPr CH3 or backbone CH3.) UV-vis (toluene): 337 (34.2 mM−1cm−1), 392 (4.8 mM−1cm−1) nm. IR (KBr pellet): 1637 cm−1 (C=N). μeff (1% Me3SiOSiMe3 in THF-d8, 25 °C): 5.9(1) μB per dimer. Elem. Anal. Calcd. for C62H90N6Fe2: C, 72.22; H, 8.80; N, 8.15. Found: C, 72.04; H, 8.98; N, 7.72.
LMeFe(η1-N=CHtBu)(μ-H)FeLMe (4a)
A solution of 1a (100 mg, 0.11 mmol) in pentane (10 mL) was treated with tBuCN (12.0 μL, 0.11 mmol) and stirred at room temperature for 2 h. Volatile materials were removed under vacuum and the residue was extracted with toluene (30 mL), filtered and concentrated to 5 mL. Crystallization at −35 ºC gave 4a as a brown powder (97 mg, 85%). 1H NMR (500 MHz, C6D6): The narrow range of the NMR spectrum, the broad peaks, and the low symmetry of the molecule lead to severe overlap between peaks, which made assignment unsuccessful. The spectrum is shown in the Supporting Information. UV-vis (pentane): 332 (30 mM−1cm−1). IR (KBr pellet): 1629 cm−1 (C=N). Elem. Anal. Calcd for C63H93N5Fe2: C, 73.31; H, 9.08; N, 6.79. Found: C, 73.50; H, 9.40; N, 6.46.
LtBuFe(NCHtBu) (5b)
A solution of 1b (91 mg, 81 μmol) in diethyl ether (8 mL) was treated with tBuCN (18 μL, 163 μmol) and stirred at room temperature for 30 min. The volatile materials were removed under vacuum. The orange residue was extracted with toluene, filtered through Celite, concentrated, and stored at −35 °C to afford 5b as orange-red crystals (70 mg, 67%). 1H NMR (500 MHz, C6D6): δ 95 (9H, tBu of imide), 92 (1H, backbone C-H), 36 (18H, tBu of diketiminate), 3 (4H, mAr), −24 (12H, iPr-CH3) −99 (4H, iPr-CH), −103 (12H, iPr-CH3), −108 (2H, p-Ar) ppm. There was no clear peak for NCHtBu. UV-vis (toluene): 335 (25 mM−1cm−1), 397 (6.7 mM−1cm−1), 454 (sh), 515 (sh) nm. μeff (C6D6, 298 K): 4.6 μB. IR (KBr pellet): 3058(w), 2960(vs), 2906(w), 2867(m), 2717(w), 2631(w), 1687(s), 1506(s), 1462(w), 1433(w), 1383(vs), 1362(vs), 1317(s), 1255(w), 1217(w), 1182(w), 1151(w), 1095(m), 1024(w) cm−1. Anal. Cald. for C40H63FeN3: C, 74.86; H, 9.89; N, 6.55. Found: C, 74.52; H, 9.80; N, 6.80.
LMeFe-hexyl
In a J. Young NMR tube, 1a (5.6 mg, 5.9 μmol) and 1-hexene (1.5 μL, 12.1 μmol) were dissolved in 0.5 mL of C6D6. After 1 h, a single paramagnetic species was observed by 1H NMR spectroscopy: δ116 (2H), 77 (1H, backbone CH), 51 (2H), 33 (6H, diketiminate CH3), 32 (3H, hexyl CH3), 15 (4H), −19 (12H, iPr-CH3), −21 (4H), −30 (2H), −44 (2H), −75 (12H, iPr-CH3), −91 (2H) ppm. Peaks integrated as 4H could be aryl m-H or iPr methine CH; peaks integrated as 2H could be aryl p-H, hexyl β-CH2, γ-CH2, δCH2, or ε-CH2. The hexyl α–CH2 protons were not observed in the 1H NMR spectrum. This 1H NMR spectrum is very similar to the spectra of other LMeFe(alkyl) species we have previously reported.30
LMeFe(CPh=CHPh) (6a)
A mixture of 1a (88 mg, 93 μmol) and PhCCPh (33 mg, 187 μmol) in ppentane (15 mL) was stirred at room temperature for 17 h. Volatile materials were removed under vacuum. The product was extracted with pentane (20 mL), filtered through Celite, and concentrated to 2 mL. Cooling to −35 °C gave 6a as a yellow powder (87 mg, 75%). 1H NMR (500 MHz, C6D6): δ123 (1H), 60 (1H), 59 (2H), 57 (6H, diketiminate CH3), 51 (1H), 48 (2H), 33 (2H), −9 (4H), −19 (12H, iPr CH3), −78 (2H), −118 (12H + 4H, iPr CH3 + another peak, possible assignments are listed below) ppm. Peaks integrated as 4H could be aryl m-H or iPr methine CH; peaks integrated as 2H could be aryl p-H, vinyl phenyl m-H (2 peaks for the two phenyl groups), or phenyl o-H; peaks integrated as 1H could be backbone CH, vinyl phenyl p-H (2 peaks for the two phenyl groups). The vinyl C=CH and o-H from the vinyl phenyl closer to the iron were not observed. μeff (C6D6, 298 K) = 5.15(2) μB. UV-vis (pentane): 320 (37.9 mM−1cm−1), 495 (1.5 mM−1cm−1) nm.
LtBuFe(μ-N3)2FeLtBu (7b)
A solution of 1b (71 mg, 64 μmol) in diethyl ether (10 mL) was treated with Me3SiN3 (18 μL, 128 μmol) and stirred at room temperature for 2 h. An orange precipitate formed within 30 minutes. The volatile materials were removed under vacuum. The orange residue was extracted with hot toluene and filtered through Celite while hot to give an orange solution. This solution was concentrated and cooled to −35 °C to afford 7b as orange crystals (70 mg, 79%). 7b is insoluble in benzene, and slightly soluble in toluene, at room temperature. 1H NMR (500 MHz, C7D8, 75 °C): δ15, 1, −3, −24, −46 ppm. The broadness of the peaks prevented accurate integration and assignment of the peaks. μeff (C7D8, 348 K): 4.1 μB. IR (KBr pellet): 2962 (s), 2929 (w), 2869 (m), 2129 (vs), 2081 (w), 1529 (w), 1490 (m), 1463 (w), 1436 (w), 1380 (m), 1357 (s), 1317 (s), 1259 (m), 1215(w), 1190 (w), 1097 (m), 1054 (w), 1024 (m) cm−1. UV-vis (toluene): 337 (15.5 mM−1cm−1) nm. Anal. Cald. for C70H106N10Fe2 C 70.10, H 8.91, N 11.68. Found C, 70.04, H 8.71, N 11.02.
LtBuFe(HNNNAd) (8b)
A solution of 1b (88 mg, 79 μmol) in diethyl ether (8 mL) was treated with a solution of AdN3 (28 mg, 157 μmol) in diethyl ether (5 mL) and stirred at room temperature for 30 min to give an orange-red solution. The volatile materials were removed under vacuum. The residue was extracted with toluene and filtered through Celite. This solution was concentrated and cooled to −35 °C to afford LtBuFe(HNNNAd) as brown crystals (77 mg, 67%). 1H NMR (500 MHz, C6D6): δ 39 (3H, Ad-α, β, or γ), 21 (18H, tBu), 15 (4H, m-Ar), 10 (3H, Ad- α, β, or γ), 8 (3H, Ad-α, β, or γ), 3.7 (3H, Ad-α, β, or γ), −7 (12H, iPr-CH3), −27 (1H, NH), −40 (12H, iPr-CH3), −51 (3H, Ad-α, β, or γ), −61 (2H, p-H) ppm. UV-vis (toluene): 340 (18 mM−1cm−1), 415 (sh), 530 (sh) nm. μeff (C6D6, 298 K) 4.6 μB. IR (KBr pellet): 3371(w), 3059(w), 2960(vs), 2926(s), 2905(vs), 2850(m), 1622(w), 1585(w), 1526(w), 1491(s), 1464(m), 1429(w), 1380(vs), 1356(vs), 1311(m), 1263(w), 1215(w), 1182(w), 1153(w), 1099(m), 1022(m) cm−1. Elemental analysis could not be obtained for this compound as 8b is thermally unstable.
Thermal conversion of 8b to LtBuFeNHAd
A sample of 8b (~5 mg) in C6D6 was heated to 80 °C for 5 h. The 1H NMR spectrum was identical to a sample prepared independently through the following method. A mixture of LtBuFeCl (257 mg, 0.433 mmol), LiNHAd (68 mg, 0.43 mmol), and diethyl ether (10 mL) was stirred at room temperature for 2 h. The yellow-orange solution was filtered through Celite, and the volatile materials were removed under vacuum. The orange residue was dissolved in n-hexane (3 mL), and cooling the solution to −45 ºC afforded LtBuFeNHAd as bright orange crystals in 2 crops (230 mg, 75%). 1H NMR (500 MHz, C6D6): d 95 (6H, Ad-α), 67 (3H, Ad-β or γ), 42 (3H, Ad-β or γ), 37 (18H, tBu), 31 (3H, Ad-β or γ), −1 (4H, m-Ar), −24 (12H, iPr-CH3), −93 (4H, iPr-CH), −102 (2H, p-Ar), −118 (12H, iPr-CH3) ppm. The N-H and backbone C-H protons were not located. μeff (C6D6, 298 K): 4.6(3) μB. IR (KBr pellet): 3277 (vw, νN–H), 3052 (w), 3013 (w), 2959 (vs), 2925 (s), 2903 (vs), 2845 (m), 1534 (m), 1502 (s), 1459 (m), 1443 (m), 1430 (m), 1385 (vs), 1364 (vs), 1319 (s), 1303 (m), 1253 (w), 1218 (m), 1197 (m), 1131 (m), 1095 (s), 1055 (m), 1029 (m), 948 (w), 933 (w), 890 (w) cm−1. UV-Vis (pentane): 340 (14 mM−1cm−1), 510 (sh, ~0.4 mM−1cm−1) nm.
LMeFe(benzo[c]cinnoline) (9a)
Although reaction of 1a and benzo[c]cinnoline gives complex 9a (yield 90% by 1H NMR spectroscopy), it is more easily synthesized from LMeFeNNFeLMe and benzo[c]cinnoline. A vial was loaded with LMeFeNNFeLMe (94 mg, 96 μmol) and benzo[c]cinnoline (35 mg, 190 μmol). Pentane (15 mL) was added, causing an immediate color change from brown to green. The reaction solution was stirred at room temperature for 5 h, filtered through Celite, concentrated to 3 mL, and cooled to −35 °C to give dark green plates of 9a (113 mg, 91%). 1H NMR (500 MHz, C6D12): δ140 (4H), 111 (4H), 17 (12H, iPr-CH3), −17 (6H, backbone CH3), −21 (4H), −52 (12H, iPr-CH3), −78 (2H, p-Ar) ppm (peaks integrated as 4H could be m-Ar, iPr CH, or overlapped peaks for benzo[c]cinnoline ligand). UV-vis: 305 (16.3 mM−1cm−1), 325 (13.1 mM−1cm−1), 388 (9.7 mM−1cm−1), 419 (7.0 mM−1cm−1), 585 (2.3 mM−1cm−1) nm. μeff (C6D6, 25 °C): 3.1(1) μB. Elem. Anal. Calcd for C41H49N4Fe: C, 75.33; H, 7.55; N, 8.57. Found: C, 73.31; H, 7.68; N, 7.98. Repeated attempts at elemental analysis did not give better agreement, indicating a possible small impurity not visible by NMR spectroscopy.
LMeFe(μ-OCHO)2FeLMe (10a)
A resealable flask was loaded with 1a (254 mg, 0.27 mmol) and pentane (15 mL). CO2 (256 mbar, 61.7 mL, 0.64 mmol) was condensed in the reaction flask at 77 K over 30 min. The flask was warmed to room temperature and stirred for 22 h. Volatile materials were removed from the yellow mixture under vacuum and the residue was extracted with toluene (30 mL), filtered and concentrated to 5 mL. Cooling to −35 ºC gave yellow blocks of 10a (138 mg, 50%). 1H NMR (500 MHz, C6D6): δ18 (4H), 5 (12H, iPr CH3), −11 (12H + 4H, iPr CH3 + another 4H; possible assignments for this 4H peak are listed below), −19 (1H, backbone CH), −42 (2H, aryl p-H), −60 (6H, backbone CH3) ppm. (Peaks integrated as 4H could be aryl m-H or iPr methine CH; the two bridging formate H were not observed in the NMR spectrum). UV-vis (pentane): 332 (e = 29.9 mM−1cm−1) nm. IR: 1628 cm−1(C-O). μeff (C6D6, 25 °C): 8.8(1) μB per dimer. Elem. Anal. Calcd for C60H84N4O4Fe2: C, 69.49; H, 8.16; N, 5.40. Found: C, 73.52; H, 9.52; N, 5.62. Repeated attempts at elemental analysis did not give better agreement, indicating a possible small impurity not visible by NMR spectroscopy.
LtBuFe(μ-OCHO)2FeLtBu (10b)
This was identical to the synthesis of 10a, but using 1b (17 mg, 15 μmol), diethyl ether (15 mL), and CO2 (99 mbar; 7.73 mL, 30 μmol). The yield of 10b was 15 mg (82%). 1H NMR (500 MHz, C7D8, 85 °C) δ 30 (1, backbone C-H), 19 (18H, tBu), 16 (4H, iPr-CH), −8 (12H, iPr-CH3), −42 (12H, iPr-CH3), −50 (2H, p-H or O2CH), −70 (2H, p-H or O2CH) ppm. A 4H peak for the m-Ar protons was not observed, and is possibly obscured by the solvent. UV-vis (toluene): 339 (43 mM−1cm−1), 393 (6.0 mM−1cm−1), 516 (sh) nm. IR (KBr pellet): 3059(w), 2962(vs), 2929(s), 2869(m), 1626(vs), 1529(w), 1493(s), 1462(m), 1437(m), 1381(vs), 1360(vs), 1317(s), 1259(m), 1215(w), 1190(w), 1157(w), 1099(s), 1024(s) cm−1. Elem. Anal. Calcd. for C72H108Fe2N4O4: C, 71.75; H, 9.03; N, 4.65. Found: 72.79; H, 8.63; N, 4.68.
LtBuFe(iPrNCHNiPr) (11b)
A solution of 1b (57 mg, 51 μmol) in diethyl ether (8 mL) was treated with N,N′-diisopropylcarbodiimide (15.8 μL, 102 μmol) and stirred for 1 h. The reaction mixture turned orange within a couple of minutes. The volatile materials were removed under vacuum. The resultant orange solid was extracted with toluene, filtered through Celite, concentrated, and stored at −35 °C to afford orange-red crystals of 11b (62 mg, 89%). 1H NMR (500 MHz, C6D6): δ 142 (2H, p-Ar), 120 (1H, backbone C-H of diketiminate), 19 (4H, m-Ar or iPr-CH), 10 (18H, tBu), 5 (12H, iPr-CH3), 2.9 (12H, iPr-CH3), 2.0 (4H, m-H or iPr-CH), −9 (12H, iPr-CH3), -36 (2H, iPr-CH of amidinate), −85 (1H, backbone C-H of amidinate) ppm. UV-vis (toluene): 349 (14 mM−1cm−1), 405 (10.2 mM−1cm−1), 475 (sh) nm. μeff (C6D6, 298 K) 4.2(3) μB. IR (KBr pellet): 3057(w), 2962(vs), 2929(w), 2867(m), 1657(w), 1547(s), 1479(m), 1460(m), 1431(w), 1382(vs), 1360(s), 1317(m), 1257(vs), 1191(w), 1097(m), 1020(s) cm−1. Elem. Anal. Cald. for C42H68FeN4: C, 73.66; H, 10.01; N, 8.18. Found: C, 73.80; H, 10.42; N, 8.10.
[LMeFe(μ-OH)]2 (12a)
To a solution of 1a (189 mg, 0.20 mmol) in THF (10 mL), a H2O solution of THF (0.28 mM, 1.45 mL, 0.41 mmol) was added dropwise with stirring. Bubbling was observed and the solution turned to green. The solution was stirred at room temperature for 20 h. Volatile materials were removed under vacuum with heating at 120 °C. The residue was extracted with pentane (20 mL), filtered, and concentrated to 5 mL. Cooling to −35 °C gave a green powder, which was further dried under vacuum with heating at 120 °C for 10 h to give dry 12a (131 mg, 67%). 1H NMR (500 MHz, C6D6): δ9 (4H), 4 (12 H, iPr-CH3), −5 (1H, backbone CH), −6 (6H, diketiminate CH3), −8 (4H), −33 (2H, aryl p-H), −38 (12H, iPr-CH3) ppm (peaks integrated as 4H could be iPr CH or aryl o-H; the bridging OH protons are not observed.). UV-vis (pentane): 326 (27.1 mM−1cm−1) nm. IR (KBr pellet): 3709, 3668 cm−1 (O-H). μeff (C6D6, 25 °C): 7.5(1) μB per dimer. Elem. Anal. Calcd for C58H84N4O2Fe2: C, 71.01; H, 8.63; N, 5.71. Found: C, 71.11; H, 8.72; N, 5.53.
Detection of H2 Using Gas Chromatography
28 The reaction solution from a given reaction (3 mL) and a magnetic stir bar were placed in a 25 mL round bottom flask. The flask was capped by an adaptor with a stopcock leading to a rubber septum. Using a syringe, 8 mL of the gas inside was removed and 8 mL CH4 (1043 mbar) was injected into the flask as an internal standard. An aliquot (20 μL) of the gas was withdrawn and injected into a GC (Shimadzu GC-17A) with a 5 Å molecular sieve column (30 m × 0.25 mm) at 26 °C, carrier gas N2, 600 kPa. The ratio of integrated H2:CH4 responses were compared to a calibration plot previously determined28 by injecting known amounts of H2 into the same flask with 3 mL of toluene.
Mössbauer Spectroscopy
Mössbauer data were recorded on a spectrometer with alternating constant acceleration. The minimum experimental line width was 0.24 mm/s (full width at half-height). The sample temperature was maintained constant either in an Oxford Instruments Variox or an Oxford Instruments Mössbauer-Spectromag cryostat. The latter is a split-pair super-conducting magnet system for applied fields up to 8 T where the temperature of the sample can be varied in the range 1.5 K to 250 K. The field at the sample is perpendicular to the -beam. The 57Co/Rh source (1.8 GBq) was positioned at room temperature inside the gap of the magnet system at a zero-field position. Isomer shifts are quoted relative to iron metal at 298 K. Magnetic Mössbauer spectra for the paramagnetic contamination of 1a were simulated by using a spin-Hamiltonian description of the electronic ground state:
| (1) |
where St is the total spin of the system, and D and E/D are the axial and rhombic zero-field parameters. The hyperfine interactions for 57Fe were calculated by using the usual nuclear Hamiltonian.93 For 1a only the nuclear Hamiltonian was used.
Computational Methods
The Gaussian 03 package94 was used for all calculations described herein. Hybrid quantum mechanics/molecular mechanics (QM/MM) calculations were used to study full experimental models of LMeFe(CNtBu)2. The QM/MM calculations utilized the ONIOM95 methodology. The MM region was modeled with the Universal Force Field (UFF)96 and included the Ar and Me substituents of LMe,Ar and the methyl groups of tBuNC. The remainder of LMeFe(CNtBu)2 was modeled at the B3LYP/6-311+G(d) level of theory. Geometries were fully optimized using gradient methods unless otherwise noted. The unrestricted Kohn-Sham formalism was used for the description of all open-shell species. The energy Hessian was calculated for all stationary points and thus confirmed the calculated stationary points as minima (no imaginary frequencies). All reported enthalpies are calculated at 1 atm and 298.15 K.
Figure 3.

Molecular structure of LMeFe(CNtBu)2 (2a), showing 50% thermal ellipsoids. There is 50:50 disorder in the position of one isocyanide ligand. Selected bond distances (Å) and angles (°): Fe-C14 1.817(1), Fe-C15 1.821(1), C14-N14 1.222(9), C14-N14′ 1.153(9), C15-N15 1.173(2), C14-N14-C24 148.3(7), C14-N14′-C24′ 164.0(7), C15-N15-C25 170.1(2).
Scheme 7.

Table 1.
Comparisons between C≡N insertion products.
| Property | 3a | 4a | 5b |
|---|---|---|---|
| Symmetry from 1H NMR at RT | C2h | Cs | C2v |
| Fe···Fe distance (Å) | 3.0790(5) | 2.7816(8) | N/A |
| N=C distance (Å) | 1.258(2) | 1.258(4) | 1.256(3) |
| Dihedral angle between β-diketiminate ligand planes (°) | 0a | 81.09(9), 80.6 (1) | N/A |
| N=C stretching frequency (cm−1) | 1637 | 1629 | 1687 |
| N-C-C angle in imide (°) | 125.4(2) | 129.3(3) | 127.2(3) |
| Fe–N(imide) distance (Å) | 2.041(2), 2.079(2) | 2.000(3), 2.016(3) | 1.857(2) |
The two β-diketiminate ligand planes are related by a crystallographic inversion center.
Acknowledgments
This work was supported by grants from the National Institutes of Health (GM065313 to P.L.H.) and the National Science Foundation (CHE-0701247 to T.R.C.), as well as a Sloan Research Fellowship (P.L.H.) and a Weissberger Fellowship (Y.Y.). We thank Bernd Mienert for collection of Mössbauer data. Calculations employed the UNT computational chemistry resource, whose purchase was supported by a NSF CRIF grant (CHE-0342824).
Footnotes
Supporting Information Available. Spectra (PDF) and crystallographic data (CIF). This material is available free of charge via the Internet at http://pubs.acs.org.
Contributor Information
Ying Yu, Department of Chemistry, University of Rochester, Rochester, New York, 14627.
Azwana R. Sadique, Department of Chemistry, University of Rochester, Rochester, New York, 14627
Jeremy M. Smith, Department of Chemistry, University of Rochester, Rochester, New York, 14627
Thomas R. Dugan, Department of Chemistry, University of Rochester, Rochester, New York, 14627
Ryan E. Cowley, Department of Chemistry, University of Rochester, Rochester, New York, 14627
William W. Brennessel, Department of Chemistry, University of Rochester, Rochester, New York, 14627
Christine J. Flaschenriem, Department of Chemistry, University of Rochester, Rochester, New York, 14627
Eckhard Bill, Max-Planck-Institut für Bioanorganische Chemie, D-45470 Mülheim an der Ruhr, Germany.
Thomas R. Cundari, Department of Chemistry and Center for Advanced Scientific Computing and Modeling (CASCaM), University of North Texas, Denton, Texas, 76203
Patrick L. Holland, Department of Chemistry, University of Rochester, Rochester, New York, 14627
References and Notes
- 1.(a) Peruzzini M, Poli R. Recent Advances in Hydride Chemistry. Elsevier; New York: 2001. [Google Scholar]; (b) McGrady GS, Guilera G. Chem Soc Rev. 2003;32:383–392. doi: 10.1039/b207999m. [DOI] [PubMed] [Google Scholar]
- 2.Especially rapid progress has occurred in the area of hydrogenase structure and reactivity: Maroney MJ. J Biol Inorg Chem. 2001;6:452. doi: 10.1007/s007750100225.Nicolet Y, Cavazza C, Fontecilla-Camps JC. J Inorg Biochem. 2002;91:1–8. doi: 10.1016/s0162-0134(02)00392-6.Armstrong FA. Curr Opin Chem Biol. 2004;8:133–140. doi: 10.1016/j.cbpa.2004.02.004.Liu X, Ibrahim SK, Tard C, Pickett CJ. Coord Chem Rev. 2005;249:1641–1652.
- 3.(a) Howard JB, Rees DC. Chem Rev. 1996;96:2965–2982. doi: 10.1021/cr9500545. [DOI] [PubMed] [Google Scholar]; (b) Burgess BK, Lowe DJ. Chem Rev. 1996;96:2983–3011. doi: 10.1021/cr950055x. [DOI] [PubMed] [Google Scholar]; (c) Eady RR. Chem Rev. 1996;96:3013–3030. doi: 10.1021/cr950057h. [DOI] [PubMed] [Google Scholar]; (d) Holland PL. Nitrogen Fixation. In: McCleverty J, Meyer TJ, editors. Comprehensive Coordination Chemistry II. Vol. 8. Elsevier; Oxford: 2004. pp. 569–599. [Google Scholar]
- 4.(a) Thorneley RNF, Eady RR, Lowe DJ. Nature. 1978;272:557–8. [Google Scholar]; (b) Leigh GJ, McMahon CN. J Organomet Chem. 1995;500:219–225. [Google Scholar]; (c) Henderson RA. In: Recent Advances in Hydride Chemistry. Peruzzini M, Poli R, editors. Elsevier; New York: 2001. pp. 463–505. [Google Scholar]; (d) Dance I. Biochemistry. 2006;45:6328–6340. doi: 10.1021/bi052217h. [DOI] [PubMed] [Google Scholar]
- 5.Igarashi RY, Laryukhin M, Dos Santos PC, Lee H-I, Dean DR, Seefeldt LC, Hoffman BM. J Am Chem Soc. 2005;127:6231–6241. doi: 10.1021/ja043596p. [DOI] [PubMed] [Google Scholar]
- 6.Lukoyanov D, Barney BM, Dean DR, Seefeldt LC, Hoffman BM. Proc Natl Acad Sci USA. 2007;104:1451–1455. doi: 10.1073/pnas.0610975104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thorneley RNF, Lowe DJ. Met Ions Biol. 1985;7:221–84. [Google Scholar]
- 8.Einsle O, Tezcan FA, Andrade SLA, Schmid B, Yoshida M, Howard JB, Rees DC. Science. 2002;297:1696–1700. doi: 10.1126/science.1073877. [DOI] [PubMed] [Google Scholar]
- 9.Poli R. Paramagnetic Mono- and Polyhydrides of the Transition Metals. In: Peruzzini M, Poli R, editors. Recent Advances in Hydride Chemistry. Elsevier; New York: 2001. pp. 139–188. [Google Scholar]
- 10.Two examples of paramagnetic iron-hydride complexes: Gargano M, Giannoccaro P, Rossi M, Vasapollo G, Sacco A. J Chem Soc, Dalton Trans. 1975:9–12.Rakowski MC, Busch DH. J Am Chem Soc. 1975;97:2570–2571.
- 11.(a) Dance I. Chem Commun. 2003:324–325. doi: 10.1039/b211036a. [DOI] [PubMed] [Google Scholar]; (b) Hinnemann B, Nørskov JK. J Am Chem Soc. 2003;125:1466–1467. doi: 10.1021/ja029041g. [DOI] [PubMed] [Google Scholar]; (c) Lovell T, Liu T, Case DA, Noodleman L. J Am Chem Soc. 2003;125:8377–8383. doi: 10.1021/ja0301572. [DOI] [PubMed] [Google Scholar]; (d) Schimpl J, Petrilli HM, Blöchl PE. J Am Chem Soc. 2003;125:15772–15778. doi: 10.1021/ja0367997. [DOI] [PubMed] [Google Scholar]; (e) Huniar U, Ahlrichs R, Coucouvanis D. J Am Chem Soc. 2004;126:2588–2601. doi: 10.1021/ja030541z. [DOI] [PubMed] [Google Scholar]; (f) Hinnemann B, Nørskov JK. J Am Chem Soc. 2004;126:3920–3927. doi: 10.1021/ja037792s. [DOI] [PubMed] [Google Scholar]; (g) Kästner J, Hemmen S, Blöchl PE. J Chem Phys. 2005;123:074306. doi: 10.1063/1.2008227. [DOI] [PubMed] [Google Scholar]; (h) Dance I. J Am Chem Soc. 2007;129:1076–1088. doi: 10.1021/ja0644428. [DOI] [PubMed] [Google Scholar]; (i) Kästner J, Blöchl PE. J Am Chem Soc. 2007;129:2998–3006. doi: 10.1021/ja068618h. [DOI] [PubMed] [Google Scholar]; (j) McKee ML. J Comput Chem. 2007;28:1796–1808. doi: 10.1002/jcc.20636. [DOI] [PubMed] [Google Scholar]
- 12.[Cp*Fe(dppe)H]+: Hamon P, Toupet L, Hamon JR, Lapinte C. Organometallics. 1992;11:1429–31.Hamon P, Hamon JR, Lapinte C. J Chem Soc, Chem Commun. 1992:1602–1603.
- 13.The hydride ligand itself is a fairly strong-field ligand: Linn DE, Jr, Gibbins SG. Inorg Chem. 1997;36:3461–3465. doi: 10.1021/ic961300d.
- 14.Ibers JA, Holm RH. Science. 1980;209:223–235. doi: 10.1126/science.7384796. [DOI] [PubMed] [Google Scholar]
- 15.Review: Lee SC, Holm RH. Proc Natl Acad Sci USA. 2003;100:3595–3600. doi: 10.1073/pnas.0630028100. Recent progress: Ohki Y, Ikagawa Y, Tatsumi K. J Am Chem Soc. 2007;129:10457–10465. doi: 10.1021/ja072256b.
- 16.Nitrogenase enzymes without Mo probably have a similarly-shaped clusters, based on X-ray absorption results: Krahn E, Weiss BJR, Kröckel M, Groppe J, Henkel G, Cramer SP, Trautwein AX, Schneider K, Müller A. J Biol Inorg Chem. 2002;7:37–45. doi: 10.1007/s007750100263.
- 17.Extracted FeMoco is structurally intact but inactive. See ref 3b.
- 18.(a) Holland PL. Can J Chem. 2005;83:296–301. [Google Scholar]; (b) Peters JC, Mehn MP. Bio-organometallic approaches to nitrogen fixation chemistry. In: Tolman WB, editor. Activation of Small Molecules. Wiley; Weinheim, Germany: 2006. pp. 81–119. [Google Scholar]
- 19.The electronic structure of trigonal-planar diketiminate-iron complexes: Andres H, Bominaar E, Smith JM, Eckert NA, Holland PL, Münck E. J Am Chem Soc. 2002;124:3012–3025. doi: 10.1021/ja012327l.Holland PL, Cundari TR, Perez LL, Eckert NA, Lachicotte RJ. J Am Chem Soc. 2002;124:14416–14424. doi: 10.1021/ja025583m.Stoian SA, Yu Y, Smith JM, Holland PL, Bominaar EL, Münck E. Inorg Chem. 2005;44:4915–4922. doi: 10.1021/ic050321h.Stoian SA, Vela J, Smith JM, Sadique AR, Holland PL, Münck E, Bominaar EL. J Am Chem Soc. 2006;128:10181–10192. doi: 10.1021/ja062051n.
- 20.Smith JM, Lachicotte RJ, Holland PL. J Am Chem Soc. 2003;125:15752–15753. doi: 10.1021/ja038152s. [DOI] [PubMed] [Google Scholar]
- 21.Vela J, Smith JM, Yu Y, Ketterer NA, Flaschenriem CJ, Lachicotte RJ, Holland PL. J Am Chem Soc. 2005;127:7857–7870. doi: 10.1021/ja042672l. [DOI] [PubMed] [Google Scholar]
- 22.(a) Betley TA, Peters JC. J Am Chem Soc. 2003;125:10782–10783. doi: 10.1021/ja036687f. [DOI] [PubMed] [Google Scholar]; (b) Betley TA, Peters JC. Inorg Chem. 2003;42:5074–5084. doi: 10.1021/ic0343096. [DOI] [PubMed] [Google Scholar]; (c) Jenkins DM, Peters JC. J Am Chem Soc. 2003;125:11162–11163. doi: 10.1021/ja036198f. [DOI] [PubMed] [Google Scholar]; (d) Betley TA, Peters JC. J Am Chem Soc. 2004;126:6252–6254. doi: 10.1021/ja048713v. [DOI] [PubMed] [Google Scholar]; (e) Thomas CM, Peters JC. Inorg Chem. 2004;43:8–10. doi: 10.1021/ic0350234. [DOI] [PubMed] [Google Scholar]; (f) Jenkins DM, Peters JC. J Am Chem Soc. 2005;127:7148–7165. doi: 10.1021/ja045310m. [DOI] [PubMed] [Google Scholar]; (g) Hendrich MP, Gunderson W, Behan RK, Green MT, Mehn MP, Betley TA, Lu CC, Peters JC. Proc Natl Acad Sci USA. 2006;103:17107–17112. doi: 10.1073/pnas.0604402103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.(a) Brown SD, Peters JC. J Am Chem Soc. 2004;126:4538–4539. doi: 10.1021/ja0399122. [DOI] [PubMed] [Google Scholar]; (b) Lu CC, Saouma CT, Day MW, Peters JC. J Am Chem Soc. 2007;129:4–5. doi: 10.1021/ja065524z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown SD, Mehn MP, Peters JC. J Am Chem Soc. 2005;127:13146–13147. doi: 10.1021/ja0544509. [DOI] [PubMed] [Google Scholar]
- 25.(a) Ohki Y, Suzuki H. Angew Chem Int Ed Engl. 2000;39:3120–3122. [PubMed] [Google Scholar]; (b) Nowik I, Herber RH. J Phys Chem Solids. 2003;64:313–317. [Google Scholar]
- 26.(a) Koutmos M, Coucouvanis D. Inorg Chem. 2004;43:6508–6510. doi: 10.1021/ic049275w. [DOI] [PubMed] [Google Scholar]; (b) Koutmos M, Georgakaki IP, Coucouvanis D. Inorg Chem. 2006;45:3648–3656. doi: 10.1021/ic052156b. [DOI] [PubMed] [Google Scholar]
- 27.Sadique AR, Gregory EA, Brennessel WW, Holland PL. J Am Chem Soc. 2007;129:8112–8121. doi: 10.1021/ja069199r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yu Y, Brennessel WW, Holland PL. Organometallics. 2007;26:3217–3226. doi: 10.1021/om7003805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.(a) Vela J, Stoian S, Flaschenriem CJ, Münck E, Holland PL. J Am Chem Soc. 2004;126:4522–4523. doi: 10.1021/ja049417l. [DOI] [PubMed] [Google Scholar]; (b) Eckert NA, Stoian S, Smith JM, Bominaar EL, Münck E, Holland PL. J Am Chem Soc. 2005;127:9344–9345. doi: 10.1021/ja0436704. [DOI] [PubMed] [Google Scholar]
- 30.Differences between the behaviors of 1a and 1b are attributed to steric effects: Vela J, Vaddadi S, Cundari TR, Smith JM, Gregory EA, Lachicotte RJ, Flaschenriem CJ, Holland PL. Organometallics. 2004;23:5226–5239.
- 31.(a) Li JG, Burgess BK, Corbin JL. Biochemistry. 1982;21:4393–4402. doi: 10.1021/bi00261a031. [DOI] [PubMed] [Google Scholar]; (b) Rubinson JF, Corbin JL, Burgess BK. Biochemistry. 1983;22:6260–6268. doi: 10.1021/bi00295a034. [DOI] [PubMed] [Google Scholar]; (c) Lowe DJ, Fisher K, Thorneley RNF, Vaughn S, Burgess BK. Biochemistry. 1989;28:8460–8466. doi: 10.1021/bi00447a028. [DOI] [PubMed] [Google Scholar]; (d) Miller RW, Eady RR. Biochim Biophys Acta. 1988;952:290–296. doi: 10.1016/0167-4838(88)90129-x. [DOI] [PubMed] [Google Scholar]
- 32.The X-band EPR spectrum of the solutions are consistent with a mixture of at least two compounds. In addition to the signal described in the text, there is a less rhombic signal with g = 2.08, 2.06, 2.00 (see Supporting Information). These signals are almost identical to those previously identified as LtBuFe(CO)233 and LtBuFe(CO)347, supporting the assignment of the more rhombic signal as LMeFe(CNtBu)2 and the less rhombic signal as LMeFe(CNtBu)3. The solution behavior of iron(I) isocyanide complexes will be described at length in a future publication.
- 33.Sadique AR, Brennessel WW, Holland PL. Inorg Chem. 2008;47:784–786. doi: 10.1021/ic701914m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cambridge Structural Database, 2006 update: Allen FH. Acta Cryst. 2002;B58:380. We used only high-quality (i.e., R < 10%) non-polymeric structures with no crystallographic disorder
- 35.Similarly soft bending potentials have been seen in metal-imido complexes: Cundari TR, Russo M. J Chem Inf Comput Sci. 2001;41:281–287. doi: 10.1021/ci0000068.
- 36.Loss of H2 to give the dinitrogen complex is formally a reductive elimination, but the iron atoms in LMeFeNNFeLMe are best described as Fe2+-N22−-Fe2+ because of charge transfer to the N2 ligand. See ref 19d.
- 37.Yates MG. Molybdenum-Dependent Nitrogen Fixation. In: Stacey G, Burris RH, Evans HJ, editors. Biological Nitrogen Fixation. Chapman & Hall; New York: 1992. pp. 685–735. [Google Scholar]
- 38.Dos Santos PC, Igarashi RY, Lee H-I, Hoffman BM, Seefeldt LC, Dean DR. Acc Chem Res. 2005;38:208–214. doi: 10.1021/ar040050z. [DOI] [PubMed] [Google Scholar]
- 39.Lee H-I, Igarashi RY, Laryukhin M, Doan PE, Dos Santos PC, Dean DR, Seefeldt LC, Hoffman BM. J Am Chem Soc. 2004;126:9563–9569. doi: 10.1021/ja048714n. [DOI] [PubMed] [Google Scholar]
- 40.Smith JM, Lachicotte RJ, Holland PL. Organometallics. 2002;21:4808–4814. [Google Scholar]
- 41.(a) Schöllhorn R, Burris RH. Proc Natl Acad Sci USA. 1967;57:1317–1323. doi: 10.1073/pnas.57.5.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dilworth MJ, Thorneley RNF. Biochem J. 1981;193:971–83. doi: 10.1042/bj1930971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cenini S, Gallo E, Caselli A, Ragaini F, Fantauzzi S, Piangiolino C. Coord Chem Rev. 2006;250:1234–1253. [Google Scholar]
- 43.(a) Smith JM, Lachicotte RJ, Holland PL. Chem Commun. 2001:1542–1543. doi: 10.1039/b103635c. [DOI] [PubMed] [Google Scholar]; (b) Smith JM, Lachicotte RJ, Holland PL. Organometallics. 2002;21:4808–4814. [Google Scholar]
- 44.Brown SD, Peters JC. J Am Chem Soc. 2005;127:1913–1923. doi: 10.1021/ja0453073. [DOI] [PubMed] [Google Scholar]
- 45.These are similar to the bands observed in literature compounds: Cortes R, Drillon M, Solans X, Lezama L, Rojo T. Inorg Chem. 1997;36:677–683.
- 46.The homolytic Si-N bond energy in Me3SiN3 is 76 kcal/mol: Adeosun SO. Inorg Nucl Chem Lett. 1976;12:301–305.
- 47.Smith JM, Sadique AR, Cundari TR, Rodgers KR, Lukat-Rodgers G, Lachicotte RJ, Flaschenriem CJ, Vela J, Holland PL. J Am Chem Soc. 2006;128:756–769. doi: 10.1021/ja052707x. [DOI] [PubMed] [Google Scholar]
- 48.(a) Burgess K, Johnson BFG, Lewis J, Raithby PR. J Organomet Chem. 1982;224:C40–C44. [Google Scholar]; (b) Johnson BFG, Lewis J, Raithby PR, Sankey SW. J Organomet Chem. 1982;228:135–138. [Google Scholar]
- 49.Hillhouse GL, Bercaw JE. Organometallics. 1982;1:1025–1029. [Google Scholar]
- 50.Wang ZX, Miao SB, Zhou ZY, Zhou XG. J Organomet Chem. 2000;601:87–92. [Google Scholar]
- 51.Hillhouse GL, Haymore BL. J Organomet Chem. 1978;162:C23. [Google Scholar]
- 52.Eckert NA, Smith JM, Lachicotte RJ, Holland PL. Inorg Chem. 2004;43:3306–3321. doi: 10.1021/ic035483x. [DOI] [PubMed] [Google Scholar]
- 53.Barney BM, Lukoyanov D, Yang TC, Dean DR, Hoffman BM, Seefeld LC. Proc Natl Acad Sci U S A. 2006;103:17113–17118. doi: 10.1073/pnas.0602130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.McKenna CE, Simeonov AM, Eran H, Bravo-Leerahhandh M. Biochemistry. 1996;35:4502–4514. doi: 10.1021/bi950964g. [DOI] [PubMed] [Google Scholar]
- 55.Barney BM, Yang T-C, Igarashi RY, Dos Santos PC, Laryukhin M, Lee H-I, Hoffman BM, Dean DR, Seefeldt LC. J Am Chem Soc. 2005;127:14960–14961. doi: 10.1021/ja0539342. [DOI] [PubMed] [Google Scholar]
- 56.Atkinson FL, Connelly NG, Crossley JG, Orpen AG. J Chem Soc, Dalton Trans. 1994:1161–1162. [Google Scholar]
- 57.Yu Y, Smith JM, Flaschenriem CJ, Holland PL. Inorg Chem. 2006;45:5742–5751. doi: 10.1021/ic052136+. [DOI] [PubMed] [Google Scholar]
- 58.Seefeldt LC, Rasche ME, Ensign SA. Biochemistry. 1995;34:5382–5389. doi: 10.1021/bi00016a009. [DOI] [PubMed] [Google Scholar]
- 59.(a) Tolman WB, Bino A, Lippard SJ. J Am Chem Soc. 1989;111:8522–8523. [Google Scholar]; (b) Tolman WB, Liu S, Bentsen JG, Lippard SJ. J Am Chem Soc. 1991;113:152–164. [Google Scholar]; (c) Sessler JL, Hugdahl JD, Lynch V, Davis B. Inorg Chem. 1991;30:334–336. [Google Scholar]
- 60.Overgaard J, Rentschler E, Timco GA, Gerbeleu NV, Arion V, Bousseksou A, Tuchagues JP, Larsen FK. J Chem Soc, Dalton Trans. 2002:2981–2986. [Google Scholar]
- 61.Armstrong WH, Spool A, Papaefthymiou GC, Frankel RB, Lippard SJ. J Am Chem Soc. 1984;106:3653–3667. doi: 10.1021/ja00263a600. [DOI] [PubMed] [Google Scholar]
- 62.(a) Field LD, Lawrenz ET, Shaw WJ, Turner P. Inorg Chem. 2000;39:5632–5638. doi: 10.1021/ic991399z. [DOI] [PubMed] [Google Scholar]; (b) Field LD, Shaw WJ, Turner P. Chem Commun. 2002:46–47. doi: 10.1039/b108492e. [DOI] [PubMed] [Google Scholar]; (c) Lu CC, Saouma CT, Day MW, Peters JC. J Am Chem Soc. 2007;129:4–5. doi: 10.1021/ja065524z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Another coordinatively unsaturated bis(μ-hydroxo)diiron(II) complex has more acute angles at iron, presumably from steric clashes between bulky tris(pyrazolyl)borate ligands: Kisko JL, Hascall T, Parkin G. J Am Chem Soc. 1998;120:10561–10562.
- 64.pKa values from http://www.chem.queensu.ca/programs/ug/2005/chem422/evans_pKa_table1.pdf
- 65.Almeida SSP, Duarte MT, Ribeiro LMD, Gormley F, Galvao AM, Frausto Da Silva JJR, Pombeiro AJL. J Organomet Chem. 1996;524:63–66. [Google Scholar]
- 66.(a) Bianchini C, Meli A, Peruzzini M, Frediani P, Bohanna C, Esteruelas MA, Oro LA. Organometallics. 1992;11:138–145. [Google Scholar]; (b) Hughes DL, Leigh GJ, Jimenez-Tenorio M, Rowley AT. J Chem Soc, Dalton Trans. 1993:75–82. [Google Scholar]
- 67.Hughes DL, Leigh GJ, McMahon CN. J Chem Soc, Dalton Trans. 1999:909–914. [Google Scholar]
- 68.(a) Bianco VD, Doronzo S, Rossi M. J Organomet Chem. 1972;35:337–339. [Google Scholar]; (b) Matsubara T, Hirao K. Organometallics. 2001;20:5759–5768. [Google Scholar]; (c) Field LD, Lawrenz ET, Shaw WJ, Turner P. Inorg Chem. 2000;39:5632–5638. doi: 10.1021/ic991399z. [DOI] [PubMed] [Google Scholar]; (d) Field LD, Shaw WJ, Turner P. Organometallics. 2001;20:3491–3499. [Google Scholar]
- 69.Hogarth G, Lavender MH, Shukri K. Organometallics. 1995;14:2325–2341. [Google Scholar]
- 70.(a) Collman JP, Finke RG, Matlock PL, Wahren R, Brauman JI. J Am Chem Soc. 1976;98:4685–4687. [Google Scholar]; (b) Albertin G, Antoniutti S, Lanfranchi M, Pelizzi G, Bordignon E. Inorg Chem. 1986;25:950–957. [Google Scholar]
- 71.Chiu KW, Wilkinson G, Thornton-Pett M, Hursthouse MB. Polyhedron. 1984;3:79–85. [Google Scholar]
- 72.Wang ZX, Miao SB, Zhou ZY, Zhou XG. J Organomet Chem. 2000;601:87–92. [Google Scholar]
- 73.Field LD, George AV, Pike SR. Polyhedron. 1995;14:3133–3137. [Google Scholar]
- 74.Hillhouse GL, Bercaw JE. Organometallics. 1982;1:1025–1029. [Google Scholar]
- 75.(a) Bart SC, Lobkovsky E, Chirik PJ. J Am Chem Soc. 2004;126:13794–13807. doi: 10.1021/ja046753t. [DOI] [PubMed] [Google Scholar]; (b) Bart SC, Hawrelak EJ, Lobkovsky E, Chirik PJ. Organometallics. 2005;24:5518–5527. [Google Scholar]; (c) Archer AM, Bouwkamp MW, Cortez MP, Lobkovsky E, Chirik PJ. Organometallics. 2006;25:4269–4278. [Google Scholar]
- 76.Brown SD, Peters JC. J Am Chem Soc. 2004;126:4538–4539. doi: 10.1021/ja0399122. [DOI] [PubMed] [Google Scholar]
- 77.Ohki Y, Takikawa Y, Hatanaka T, Tatsumi K. Organometallics. 2006;25:3111–3113. [Google Scholar]
- 78.For an iron-containing example, see: Gao Y, Holah DG, Hughes AN, Spivak GJ, Havighurst MD, Magnuson VR, Polyakov V. Polyhedron. 1997;16:2797–2807.
- 79.(a) MacKay BA, Munha RF, Fryzuk MD. J Am Chem Soc. 2006;128:9472–9483. doi: 10.1021/ja061508q. [DOI] [PubMed] [Google Scholar]; (b) Fryzuk MD, MacKay BA, Patrick BO. J Am Chem Soc. 2003;125:3234–3235. doi: 10.1021/ja034303f. [DOI] [PubMed] [Google Scholar]; (c) Fryzuk MD, Johnson SA, Patrick BO, Albinati A, Mason SA, Koetzle TF. J Am Chem Soc. 2001;123:3969–3937. doi: 10.1021/ja0041371. [DOI] [PubMed] [Google Scholar]
- 80.Fryzuk MD, Love JB, Rettig SJ, Young VG. Science. 1997;275:1445–1447. [Google Scholar]
- 81.Gilbertson JD, Szymczak NK, Crossland JL, Miller WK, Lyon DK, Foxman BM, Davis J, Tyler DR. Inorg Chem. 2007;46:1205–1214. doi: 10.1021/ic061570o. [DOI] [PubMed] [Google Scholar]
- 82.(a) Hills A, Hughes DL, Jimenez-Tenorio M, Leigh GJ. J Organomet Chem. 1990;391:C41–C44. [Google Scholar]; (b) Evans DJ, Jimenez-Tenorio M, Leigh GJ. J Chem Soc, Dalton Trans. 1991:1785–1787. [Google Scholar]; (c) Hills A, Hughes DL, Jimenez-Tenorio M, Leigh GJ, McGeary CA, Rowley AT, Bravo M, McKenna CE, McKenna MC. J Chem Soc, Chem Commun. 1991:522–524. [Google Scholar]; (d) Hills A, Hughes DL, Jimenez-Tenorio M, Leigh GJ, Rowley AT. J Chem Soc, Dalton Trans. 1993:3041–3049. [Google Scholar]; (e) Helleren CA, Henderson RA, Leigh GJ. J Chem Soc, Dalton Trans. 1999:1213–1220. [Google Scholar]; (f) Hughes DL, Leigh GJ, McMahon CN. J Chem Soc, Dalton Trans. 1999:909–914. [Google Scholar]
- 83.(a) Nakayama Y, Nakamura A, Mashima K. Chem Lett. 1997;26:803–804. [Google Scholar]; (b) Hill JE, Fanwick PE, Rothwell IP. Inorg Chem. 1991;30:1143–1144. [Google Scholar]
- 84.Doedens RJ. Inorg Chem. 1970;9:429–436. [Google Scholar]
- 85.Klein HF, Helwig M, Karnop M, Koenig H, Hammerschmitt B, Cordier G, Floerke U, Haupt HJ. Z Naturforsch B. 1993;48:785–93. [Google Scholar]
- 86.Hoskin AJ, Stephan DW. Coord Chem Rev. 2002;233–234:107–129. [Google Scholar]
- 87.(a) Henry RM, Shoemaker RK, Newell RH, Jacobsen GM, DuBois DL, Rakowski DuBois M. Organometallics. 2005;24:2481–2491. [Google Scholar]; (b) Jacobsen GM, Shoemaker RK, Rakowski DuBois M, DuBois DL. Organometallics. 2007;26:4964–4971. [Google Scholar]
- 88.Dihydrogen complexes are often strong acids, for example: Jia G, Morris RH. J Am Chem Soc. 1991;113:875–883.Cappellani EP, Drouin SD, Jia G, Maltby PA, Morris RH, Schweitzer CT. J Am Chem Soc. 1994;116:3375–3388.
- 89.(a) Henderson RA. Chem Commun. 1987:1670–1672. [Google Scholar]; (b) Hlatky GG, Crabtree RH. Coord Chem Rev. 1985;65:1–48. [Google Scholar]; (c) Oglieve KE, Henderson RA. Chem Commun. 1992:441–443. [Google Scholar]
- 90.Husseini A, Akasheh TS. Dirasat - Univ Jordan. 1985;12:65–72. [Google Scholar]
- 91.We are not aware that any researchers have investigated the reduction of formate by nitrogenase. This would be a fruitful area of investigation.
- 92.(a) Schubert EM. J Chem Ed. 1992;69:62. [Google Scholar]; (b) Evans DF. J Chem Soc. 1959:2003–2005. [Google Scholar]
- 93.Trautwein AX, Bill E, Bominaar EL, Winkler H. Struct Bonding. 1991;78:1–95. [Google Scholar]
- 94.Frisch MJ, et al. Gaussian Inc; Pittsburgh, PA: 1998. [Google Scholar]
- 95.Svensson M, Humbel S, Froese RDJ, Matsubara T, Sieber S, Morokuma K. J Phys Chem. 1996;100:19357–19363. [Google Scholar]
- 96.(a) Rappé AK, Casewit CJ, Colwell KS, Goddard WA, Skiff WM. J Am Chem Soc. 1992;114:10024–10035. [Google Scholar]; (b) Casewit CJ, Colwell KS, Rappé AK. J Am Chem Soc. 1992;114:10035–10046. [Google Scholar]