

Abstract
Efficacious delivery of drugs and genes to the heart is an important goal. Here, a radiolabeled peptide-targeted liposome was engineered to bind to the heart, and the biodistribution and pharmacokinetics were determined by dynamic positron emission tomography in the FVB mouse. Efficient targeting occurred only with an exposed ligand and a dense concentration of peptide (6000 peptides/particle). Liposomes targeted with CRPPR or other arginine-rich peptides with an exposed guanidine moiety bound within 100 seconds after intravenous injection and remained stably bound. With CRPPR-targeted particles, the radioisotope density in the heart averaged 44 ± 9% injected dose per gram of tissue, more than 30 fold higher than in skeletal muscle. The rapid and efficient targeting of these particles can be exploited in drug and gene delivery systems and with dynamic positron emission tomography provides a model system to optimize targeting of engineered particles.
Keywords: endothelium, heart, liposome, peptides, positron emission tomography (PET), molecular imaging, pharmacokinetics
1 Introduction
The vascular endothelium has been recognized as an important target for therapeutic interventions which use liposomes and nanoparticles to carry drugs and genes [1, 2]. Specific adhesion to normal or pathological organs has been reported using ligands tailored to vascular zip codes or luminally-expressed pathological targets [3–5]. While many previous studies have targeted particles to tumor vessels and tumor cells, effectively combining the enhanced permeability and retention effect within tumors with the effect of the targeting ligand [1, 6], few studies have evaluated the pharmacokinetics of particles in targeting the intact vessel wall [7–9]. For a vascular target, binding of particles to the target site can be very rapid, with a time constant on the order of 30 seconds[8]. Here for the first time, we use dynamic positron emission tomography (PET) to elucidate the dynamics of cardiac targeting in real time while varying the peptide coating and surface architecture of the particle.
The short linear peptide CRPPR has previously been reported to specifically bind to the heart endothelium [4]. Other short linear arginine-containing peptides were similarly identified by phage display and demonstrated substantial but less specific cardiac targeting [4]. Arginine-rich peptides have been widely shown to enhance the cellular internalization of drugs, genes and particles [10], as most cationic cell penetrating peptides contain at least one residue of arginine. Alternatively, RGD peptides targeted to platelets or endothelial integrins have been commonly used to target long-circulating particles [11, 12]. Here, we use c(RGDY(OMe)KE)[13] as a neutrally-charged non-binding control. We evaluate cardiac targeting of intravenously-administered particles, incorporating CRPPR as well as scrambled controls (CPPRR and CRRPP) and peptides with greater and lesser charge (CRRRR and c(RGDY(OMe)KE)), to determine whether the lipid particles retain the organ specificity of the peptide and to characterize the kinetics of particles targeted by arginine-containing peptides.
Among various drug-gene delivery vectors, targeted phospholipid-based liposomes have been widely studied but have yet had limited clinical impact. In limited pre-clinical studies, antibody targeting of liposomes to intravascular targets has shown impressive localization of delivery vehicles and subsequent therapeutic effect [14, 15]. However, a major drawback of antibody targeted liposomes is low tissue penetration and high molecular weight. In contrast, small peptides and small molecules that selectively recognize cell surface markers can be employed to target vehicles, decreasing the immunogenic effect and expanding the potential applications [6, 16–18]. While the effect of the tether length, peptide concentration, and multivalency have been intensively studied in vitro in recent years, quantitative data on the biodistribution and pharmacokinetics of the resulting particles have been lacking. Also, although the uptake of particles by the reticulo-endothelial system (RES) has been the subject of many studies using inhibitors such as polyinosinic acid[19], and clodronate[20, 21], systemic analyses of RES uptake and changes with these inhibitors have been limited.
Previous scintillation studies of liposome kinetics have involved radionuclides attached to the liposome surface using chelators covalently linked to lipid soluble anchors [22], or encapsulated the imaging probe inside the liposome hydrophilic cavity [23, 24]. Here, in order to facilitate high-resolution pharmacodynamic measurements, we employ 18F for PET trafficking of peptide-targeted liposomes (Figure 1), using an 18F-labeled lipid ([18F]fluorodipalmitin, [18F]FDP) inserted into the lipid membrane [25]. Within a liposome bilayer, the [18F]FDP probe stably associates with the vehicle in vitro and during in vivo circulation [25]. When free lipid is injected or the vehicle is recognized and metabolized by the RES, the 18F-containing metabolite rapidly clears from the liver, facilitating real-time evaluation of the dynamics [25]. Other advantages of the [18F]FDP label include the absence of any change in surface properties due to the effects of chelators, the small size of the 18F group attached to the lipid head group, the simplicity of the labeling, and the generality of the approach.
Figure 1.
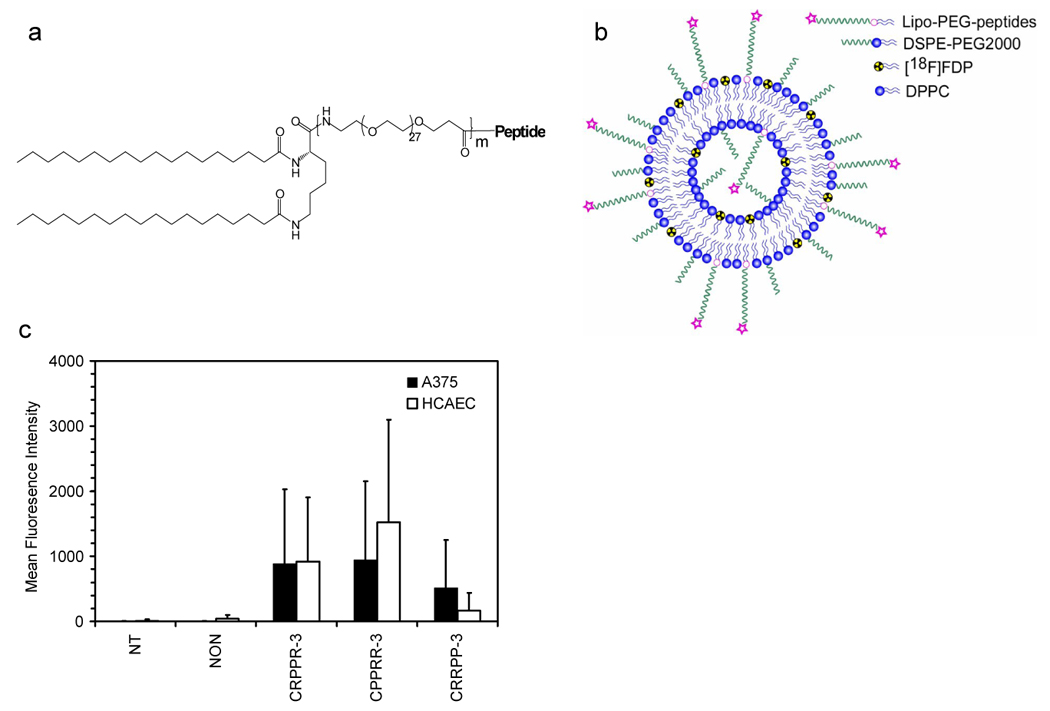
(a) Chemical structures of lipo-PEG-peptides (LPPs) and (b) schematic of radiolabeled targeted liposome. Molecular weights of the PEG spacer are 1200, 2400, 3600 for m =1, 2, 3, respectively. Abbreviations for the LPPs are as follows. CRPPR-3: peptide = CRPPR, m = 3; CRPPR-2: peptide = CRPPR, m = 2; CRPPR-1: peptide = CRPPR, m = 1; RGD-3: peptide = c(RGDY(OMe)KE), m = 3; CPPRR-3: peptide = CPPRR, m = 3; CRRPP-3: peptide = CRRPP, m = 3; CRRRR-3: peptide = CRRRR, m = 3; NON indicates no LPP (but 12% DSPE-PEG2000); NT indicates in vitro incubation without liposomes. LPPs are incorporated within a liposome prior to injection with a formulation of LPP : DSPE-PEG2000 : DPPC = 6:6:88 (mol/mol), except in figure 4d. (c) Fluorescence intensity, as measured by flow cytometry, for a melanoma cell line, A375, and an endothelial cell line, Human Coronary Artery Endothelial Cells (HCAEC) incubated with liposomes containing CRPPR-3 lipo-PEG-peptides. Fluorescent, viable cells were quantified after incubation and washing.
2 Materials and Methods
2.1 Targeted-liposome preparation
2.1.1 Peptide synthesis
Protected 9-fluorenylmethyloxycarbonyl (Fmoc) amino acids and coupling agents (O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) and O-benzotriazolyl-N',N',N',N'-tetramethyluronium hexafluorophosphate (HBTU)) were purchased from GL Biochem (Shanghai) Ltd. Fmoc-PAL-PEG-PS resin (0.16–0.21 mmol/g) was from Applied Biosystems (Foster City, CA). Solvents and other agents were all of analytical purity and from Sigma-Aldrich (Milwankee, WI) and VWR (Brisbane, CA). The following amino acid side chain protections were used: t-But (Asp), Pbf (Arg), Trt (Cys), Mmt(Lys) and OAll(Glu) (Applied Biosystems). Five peptides were synthesized in this paper: CRPPR, CPPRR, CRRPP, CRRRR and c(RGDY(OMe)KE). For c(RGDY(OMe)KE), after the linear peptide Fmoc-R(Pbf)GD(But)Y(OMe)K(Mmt)E(OAll) was synthesized, OAll was removed with catalyst Pd, Fmoc was removed with 20% piperidine in dimethylformamide, and head-to-tail cyclization was performed. Mmt was removed by 1% trifluoroacetic acid (TFA) in dicloromethane (DCM) to produce a free amino group on lysine for further coupling. After peptide synthesis, part of the peptidyl resin was cleaved from the resin using 94% TFA, 1.0% triisopropylsilane (TIPS), 2.5% ethanedithiol (EDT), and 2.5% water followed by precipitation with diethylether (for c(RGDY(OMe)KE), TFA:TIPS:Water = 95:2.5:2.5 was used). The products were purified using reversed-phase high performance liquid chromatography (HPLC). Matrix Assisted Laser Desorption Ionisation time-of-flight (MALDI-TOF) mass spectrometry (MS) confirmed the mass of the free peptide measured with the ABI-4700 TOF-TOF (Applied Biosystems) using matrix Sinapic acid with 3 layer sample preparation method [26]. Reversed-phase HPLC was performed using a Phenomenex Jupiter 4μ Proteo 90A (250×4.6 mm, analytical), and a Phenomenex Jupiter 10μ Proteo 90A (250×21.2 mm, preparative) with a gradient from 10–90% B in 30min (solvent A: 0.05% TFA, solvent B: 0.05% TFA/acetonitrile 10:90 (v/v)) and a flow rate of 1.5 /15 ml/min for analytical/preparative column.
2.1.2 Lipo-PEG-Peptide (LPP) Synthesis
Fmoc-NH-(PEG)27-COOH and Fmoc-Lys(Fmoc)-OH were purchased from Novabiochem (Darmstadt, Germany) and Stearic Acid was purchased from Sigma-Aldrich. Polyethylene glycol (PEG) was coupled onto peptidyl resin with HBTU as the coupling agent, which was repeated until the expected PEG length was reached. After finishing the pegylation step, Fmoc-Lys(Fmoc)-OH and stearic acid were coupled in sequence. LPPs (Figure 1a) were cleaved from resin and purified with HPLC and molecular weights were confirmed with MALDI-TOF MS. Yields were 20% and 12% for linear and cyclic peptides. For cysteine containing LPP, dimerization was carried out as described in [27]. LPP were dissolved in 0.01 M ammonium bicarbonate at a concentration of 1–2mM and the solution was left open to the air and stirred. The reaction was monitored with HPLC until all of the monomer reacted, which normally requires less than 4 hours. Our initial in vitro experiments showed that dimerization could enhance targeting (data not shown), therefore we used the dimerized LPP in this work. Molar fractions were calculated based on the peptides prior to dimerization.
2.1.3 Preparation of [18F]FDP and Fluorescently-labeled Liposomes
1,2-dipalmitoyl-snglycero-3-phosphocholine (DPPC), 1,2 distearoyl-sn-glycero-3-phosphoethanolamine-N-[Methoxy(Polyethylene glycol)-2000] (DSPE-PEG2000), and a mini-extruder were purchased from Avanti Polar Lipids Inc. (Alabaster, AL). An appropriate amount of DPPC, DSPE-PEG2000, and LPP in chloroform was mixed and chloroform was removed by lyophilization.
[18F]FDP Liposomes: The lipid mixture was resuspended with 20 mM PBS buffer (pH 7.4) and added to the fresh prepared [18F]FDP. The solution was sonicated at 60°C for 1 min, and extruded through polycarbonate membranes at 60°C (21 passes through 100-nm-diameter pore membrane) and then the extruded liposomes were purified with Sephadex G-50 columns (GE Healthcare, NJ) to obtain the radiolabeled targeted liposomes (Figure 1b). After the radioactivity decayed, the liposomal size distribution was measured with the Nanotrac (Microtrac Inc., FL), and the zeta potential was characterized by the Zeta Potential/Particle Sizer Nicomp™ 380ZLS (Particle Sizing Systems, CA). The phospholipid concentration was tested with the Phospholipids C kit (Wako Chemicals USA, Inc., VA) and the LPP concentration was measured by HPLC.
Fluorescent Liposomes: liposome synthesis was as above except that the fluorescent dye Alexa 555 (Invitrogen Corporation, Carlsbad, CA) was dissolved into the buffer at a concentration of 0.3 mM, and the labeled buffer was used to resuspend the dried lipid mixture. After sonication and extrusion, the liposomes were purified with a G-75 column (GE Healthcare, NJ).
2.1.4 In vitro incubation and internalization
Binding and internalization of NON-, CRPPR-, CPPRR- and CRRPP- targeted liposomes were studied with a malignant melanoma cell line, A375 (American Type Culture Collection, Manassas, VA), and an endothelial cell line, Human Coronary Artery Endothelial Cells (HCAEC, Lonza, NJ). Cells were seeded at ~4.8 × 104 cells per dish on 60nmm petri dishes and were grown to approximately 90% confluency. To each dish, calcein (Sigma-Aldrich)-loaded liposomes were added with final concentrations of lipids and calcein in media of ~ 0.5–0.9 mg/ml and 0.75–1.25 mM, respectively. Cells were incubated at 37°C in 5% CO2 for 16 hours; including a wash after 2 hours to remove lipsomes. Then, cells were collected via trypsinization with 0.05% trypsin-EDTA (Invitrogen), and incubated 10 min with 10ul 7-AAD viability staining solution (BioLegend, San Diego, CA). Flow cytometry was performed using a FACScan flow cytometer and CELLQuest software (Becton Dickinson, Franklin Lakes, NJ) for calcein and 7-AAD. For each sample, 10,000 gated events were collected in low-speed mode at a collection rate of approximately 100 counts per second. Mean fluorescence intensities were recorded for all samples and viable cells containing calcein quantified.
2.2 Animal studies
All animal studies were conducted under a protocol approved by the University of California, Davis Animal Use and Care Committee (Davis, CA). A total of 160 animals (male FVB mice, 8–12 weeks, 25–30g, Charles River, MA) were examined over the course of this study, with four animals imaged with each day’s formulation. In order to detect a difference between groups (as compared with the heart targeting of CRPPR-3 without an inhibitor), 4, 8, or 12 animals were studied to detect an expected difference of 30, 20 or 17%, respectively, based on preliminary data (not shown), using a power of 0.8 and alpha error level of 0.05. All mice were housed four animals per cage in a 12 hour light cycle environment and allowed access to water and a standard mouse diet. For all procedures, induction of anesthesia was achieved at 3.0–3.5% isoflurane (Halocarbon Laboratory, River Edge, NJ) and maintained at 2.0–2.5%. Respiratory rate and temperature were monitored throughout each procedure to ensure proper levels of anesthesia and comfort. Body temperature was maintained by placing the animals on a heating pad during all procedures and scans. The mice were catheterized to ensure proper injection of lipid formulations, after which bolus injections of [18F]FDP-labeled liposomes (0.05 mg lipids, 2 mg lipids per kg of body weight, mg/kg) were administered as PET scans were initiated, using a manually-controlled injection that was timed for uniform administration over 15 seconds.
2.2.1 Positron emission tomography (PET) scans and Time–activity curves (TAC)
PET, a nuclear medicine medical imaging technique producing a three-dimensional image of radiotracer concentration over time, was employed to study the pharmacodynamics of injected liposomes. PET scans were conducted with microPET Focus (Siemens Medical Solutions, Knoxville ,TN) over 90 minutes and maximum a posteriori (MAP) files were created with ASIPro software (Siemens Medical Solutions, Knoxville, TN) and used to obtain quantitative activity levels in each organ of interest as a function of time. TACs were obtained with region-of-interest (ROI) analysis using ASIPro software and expressed as percentage of injected dose per cubic centimeter (%ID/cc).
2.2.2 Well counts
After the PET scans, the mice were euthanized by cervical dislocation and organs of interest were harvested and radioactivity measured using a 1470 Automatic Gamma Counter (Perkin Elmer Life Sciences, MA). Validation of the time activity curve with well counts is described in the Supplementary methods.
2.2.3 Comparison between well counts and TAC
A regression analysis was performed to validate the estimates of radioactivity obtained using microPET imaging with those obtained using well counts. For the primary organs of interest, the slope of the linear regression (%ID/cc vs %ID/g) was 1.07 for blood measured within the heart on microPET (R2=0.92) and 0.8 for the heart tissue (R2=0.96).
2.2.4 Pharmacokinetics in blood pool
Estimates of radioactivity within the blood pool over time were fit with a biphasic clearance curve,
| (1) |
where Ct is the blood isotope concentration at time t, A and B are pre-exponential constants, and α and β are first-order hybrid time constants describing the biphasic nature of the concentration-time profile.
2.2.5 Autoradiography
In a limited set of studies (four mice in total), after the gamma count was obtained, the heart tissue was imaged with autoradiography. The heart was affixed to a 30 mm specimen disc (Leica, Bannockburn, IL) using Tissue-Tek Optimum Cutting Temperature (OCT) Compound (Sakura, Torrance, CA). The sample was frozen using a dry ice/isopropanol bath. Once the OCT compound was solidified, the specimen disc was placed in a Leica CM 1850 cryotome chamber (Bannockburn, IL) for 15 minutes to uniformly equilibrate to −22°C. The tissue cutting thickness was set to 60 um, and slices were mounted onto microscope slides. Slides were then placed in 10% natural buffered formalin for approximately one minute and then briefly (10 seconds) dipped in 100% EtOH for drying. After drying, the slides were placed in an Exposure Cassette with a Phosphor screen (Amersham Biosciences, Piscataway, NJ). The screen was exposed to the slide overnight and the resulting images were processed by an Amersham Biosciences Storm 860.
2.2.6 Confocal microscopy
Fluorescently- labeled NON-, and CRPPR- targeted liposomes were injected into mice (n=4), which were euthanized 15 minutes later. Cardiac tissue was harvested and sliced to ~1 mm in thickness. Confocal microscopy (LSM-510, Zeiss, Thornwood, NY) images were recorded with excitation at 555 nm and emission at 565 nm. For the region of interest, 16 x/y images were acquired as z-stacks each separated by 15 µm and the projection images were obtained.
2.2.7 Inhibitor preparation and administration
Blank liposomes: blank liposomes (DPPC:DSPE-PEG2000 = 98:2 (mol/mol), with diameter 100 nm) with 300 µg lipids (12mg/kg) in 100 µl saline were injected 15 minutes before the radiolabeled targeted liposomes. Polyinosinic acid (PI): PI (P4154), purchased from Sigma-Aldrich, was dissolved in 0.9% sterile injection saline at 2 mg/ml. A small amount of sodium hydroxide solution was added to dissolve PI powder and the pH of the PI solution was adjusted to 7.4 with HCl solution. 5 µl of PI solution (~10 µg of PI, 0.4mg/kg) was diluted with 100 µl saline and injected 1 minute before the injection of the radiolabeled targeted liposomes. Control animals received an equal volume of saline solution. Clodronate liposomes: Clodronate liposomes were prepared as in [21]. 100 µl of clodronate liposomes (~0.05 mg, 2mg/kg) were injected 24 hours in advance of the administration of radiolabeled targeted liposomes. Control animals received an equal volume of saline solution. Free CRPPR Peptide: 700µg CRPPR peptides (25mg/kg) were injected 1 minutes before the administration of radiolabeled targeted liposomes. Control animals received an equal volume of injection saline solution.
2.3 Statistical Analysis
Data were recorded as a (mean ± standard deviation) for continuous data. Significant differences were assessed using a one-tailed Student’s t test, with α of 0.05, and by linear regression analysis. All statistical analyses were performed by using software (Excel 11.0, Microsoft, Seattle, WA; GraphPrism 4, Graphpad Inc., San Diego, CA). A p value less than 0.05 indicated a statistically significant difference.
3 Results
3.1 Liposome diameter, zeta potential, Lipo-PEG-Peptide (LPP) incorporation, and in vitro binding
The LPP (structure shown in Figure 1a), radiolabeled lipid ([18F]FDP), phospholipid (DPPC), and pegylated phospholipid (DSPE-PEG2000) were combined to produce a particle (Figure 1b, with formulations and notation detailed in Figure 1 caption), with a polymer brush layer of 2000 molecular weight (MW) and a ligand that was either exposed (m=3 in Figure 1a) or buried (m=1). Initial in vitro studies indicated that the formulation of CRPPR-3:DSPE-PEG2000:DPPC = 6%:6%:88% (mol/mol) produced effective targeting, therefore this formulation was employed in in vitro studies and as the baseline formulation in in vivo studies. In vivo results with the CRPPR-3:DSPE-PEG2000:DPPC = 6%:6%:88% were compared with matched total PEG concentration (12%), LPP concentration (6%), DSPE-PEG2000 concentration (6%), or LPP:DSPE-PEG2000 ratio (1:1), while other parameters were varied (Table 1).
Table 1.
Liposome diameter, zeta potential, and lipoPEGpeptide incorporation as a function of formulation*
| Liposome Formulation (mol %) | Diameter (nm) | Zeta Potential (mv) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| LPP type | LPP | DSPE-PEG 2000 | DPPC | Mean | SD | Mean | SD | Mean | SD |
| CRPPR-3 | 6 | 6 | 88 | 110 | 38 | 31 | 9 | 135 | 30 |
| CRPPR-3 | 10 | 2 | 88 | 192 | 89 | 33 | 18 | 74 | 9 |
| CRPPR-3 | 3 | 3 | 94 | 72 | 27 | 3 | 2 | 74 | 8 |
| CRPPR-3 | 6 | 6 | 92 | 80 | 36 | −24 | 2 | 83 | 1 |
| NON | 0 | 12 | 88 | 71 | 26 | −48 | 10 | N/A | N/A |
| RGD-3 | 6 | 6 | 88 | 84 | 32 | −31 | 2 | 72 | 13 |
| CRPPR-2 | 6 | 6 | 88 | 93 | 35 | 26 | 4 | 79 | 18 |
| CRPPR-1 | 6 | 6 | 88 | 82 | 26 | 34 | 2 | 137 | 29 |
| CPPRR-3 | 6 | 6 | 88 | 138 | 11 | 42 | 3 | 120 | 7 |
| CRRPP-3 | 6 | 6 | 88 | 148 | 10 | 41 | 9 | 116 | 7 |
| CRRRR-3 | 6 | 6 | 88 | 169 | 13 | 39 | 8 | 83 | 2 |
For abbreviations: please refer to the caption of Figure 1.
Using flow cytometry (Figure 1c), both endothelial and melanoma cells incubated in culture with calcein-containing particles targeted by CRPPR-3 or CPPRR-3 showed a significantly higher fluorescence intensity than cells incubated with particles without the lipopeptide (p<0.05). Endothelial cells incubated with particles containing CRRPP-3 demonstrated a lower fluorescence intensity than those incubated with particles containing CRPPR-3 or CPPRR-3.
3.2 PET images
Ninety-minute accumulative PET images (Figure 2a–i) acquired with the [18F]FDP and CRPPR-3 incorporated into the liposomal vectors (Figure 2a, d, g) demonstrate the high level of the radiotracer within the heart, a lower density within the liver, and a low concentration within the spleen and bladder. Images acquired with the shorter PEG length LPP (CRPPR-1) loaded onto an identical vehicle demonstrate that radioactivity has accumulated within the liver and bladder at 90 minutes and a low level of activity is present in the heart (Figure 2b, e, h). Clearance of the radiotracer occurs through the bladder, after metabolism in the liver separates the fatty acid chains (each with a molecular weight of 256) from the head group containing the isotope (with a molecular weight of 94). By comparison, images acquired with the identical vehicle, without a peptide attached to the liposome, demonstrate that the radioisotope is primarily circulating within the blood volume throughout the ninety-minute scan, visualized in the heart chamber and carotid vessels (Figure 2c, f, i).
Figure 2.
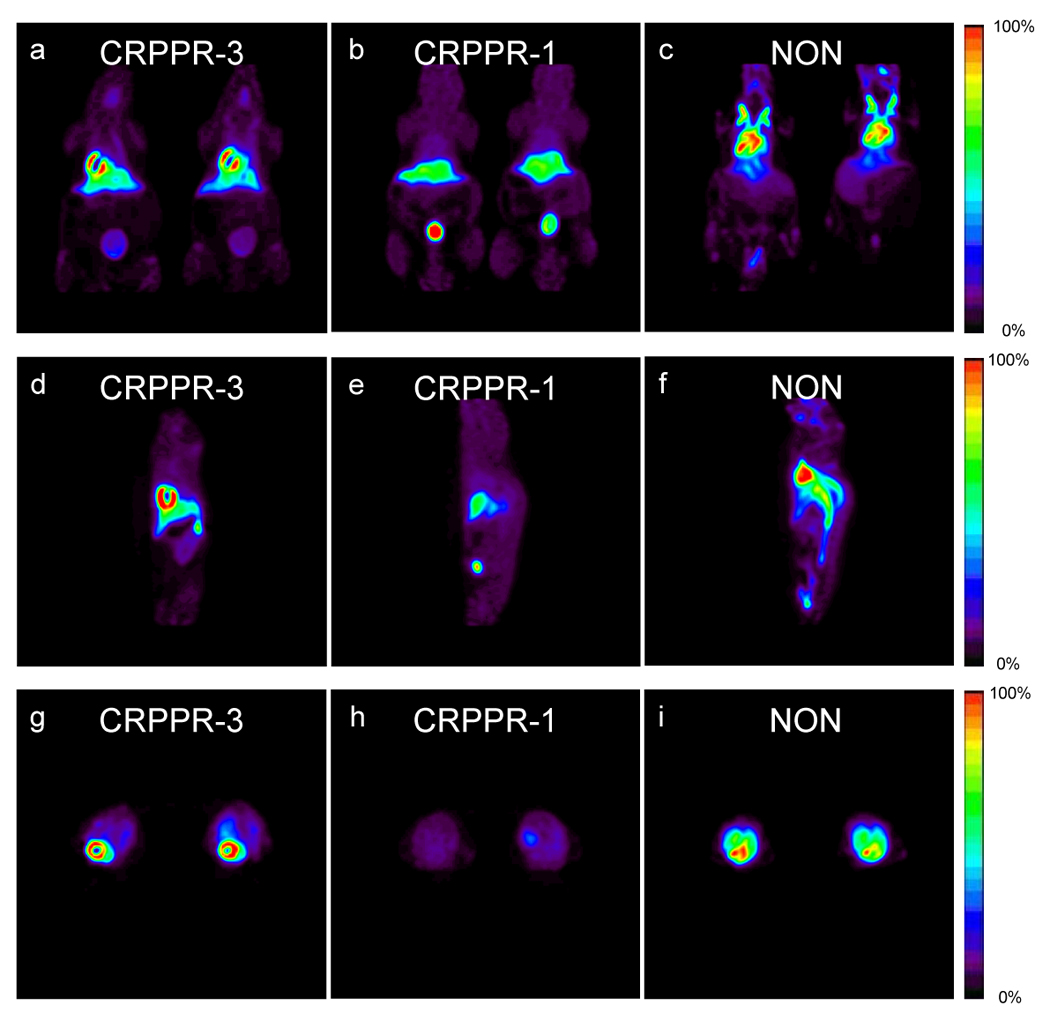
(a–i) 90-minute accumulative PET images acquired after injection of radiolabeled liposomes from coronal (a–c), sagittal (d–f) and transverse views (g–i) with LPPs CRPPR-3 (a,d,g), CRPPR-1 (b,e,h), and NON (c,f,i).
Autoradiography confirmed that the radiotracer was present throughout the atria and both ventricles (Figure 3 a–f), although the highest counts were observed within the thick ventricular walls. Confocal microscopy confirmed that CRPPR-targeted liposomes bind to blood vessel walls within the heart (Figure 3 g–j).
Figure 3.
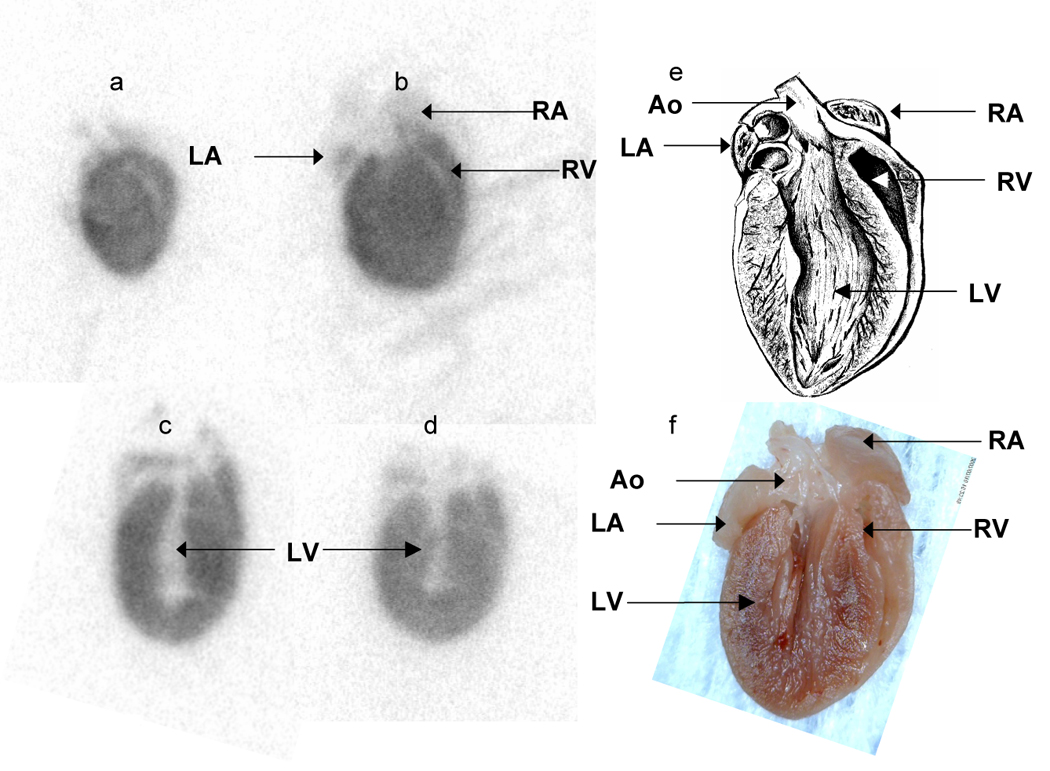

High resolution autoradiography and optical imaging of the heart after injection of targeted and control particles. (a–d) Autoradiography images acquired from 60 µm tissue slices 90 minutes after injection of CRPPR-3 liposomes. (e) Anatomic drawing of a mouse heart with the same orientation as the processed tissue slices (picture by William Moroski). (f) Digital photograph of a mouse heart fixed in 10% formalin. A for atrium, V for ventricle, L for left, R for right and Ao for aorta. (g–j) Confocal microscopy images of heart tissue after intravenous injection of CRPPR-3- (g, i), and NON- (h, j) targeted liposomes with low (g, h) and high (i, j) magnification.
3.3 Effect of peptide and surface architecture on biodistribution at 90 minutes
Well counts obtained from harvested tissues at the ninety-minute time point quantify the differences in biodistribution produced by the peptide (Figure 4a and b), the length of the PEG spacer between the fatty acid and CRPPR peptide portions of the LPP (Figure 4c), and the molar fraction of the CRPPR-3 incorporated within the vehicle (Figure 4d). When injected on a vehicle containing CRPPR-3, the concentration of the tracer was significantly higher within the heart than other organs (p<0.001), with a target to skeletal muscle ratio as high as 100 in individual animals (averaging 32) and a mean heart concentration of 44 ± 9% injected dose per gram of tissue (ID/g). For CRPPR-3, activity within the liver and spleen was significantly lower than the heart at 22 ± 9 and 12 ± 4% ID/g (p<0.001). Other arginine-rich peptidyl liposomes bound to the heart (Figure 4b) at levels of 39 ± 13, 26 ± 13, 17 ± 5% ID/g for CPPRR-3, CRRRR-3, and CRRPP-3, respectively, with target to skeletal muscle ratios of 32, 23 and 19. Injection of vehicles with the cyclized RGD peptide and an otherwise identical liposome surface architecture did not produce radioactivity above the baseline (no–peptide) case. The cyclized RGD and no-peptide controls also resulted in significantly lower activity levels within the liver and urine at 90 minutes as compared with arginine-rich peptidyl vehicles (p<0.05).
Figure 4.
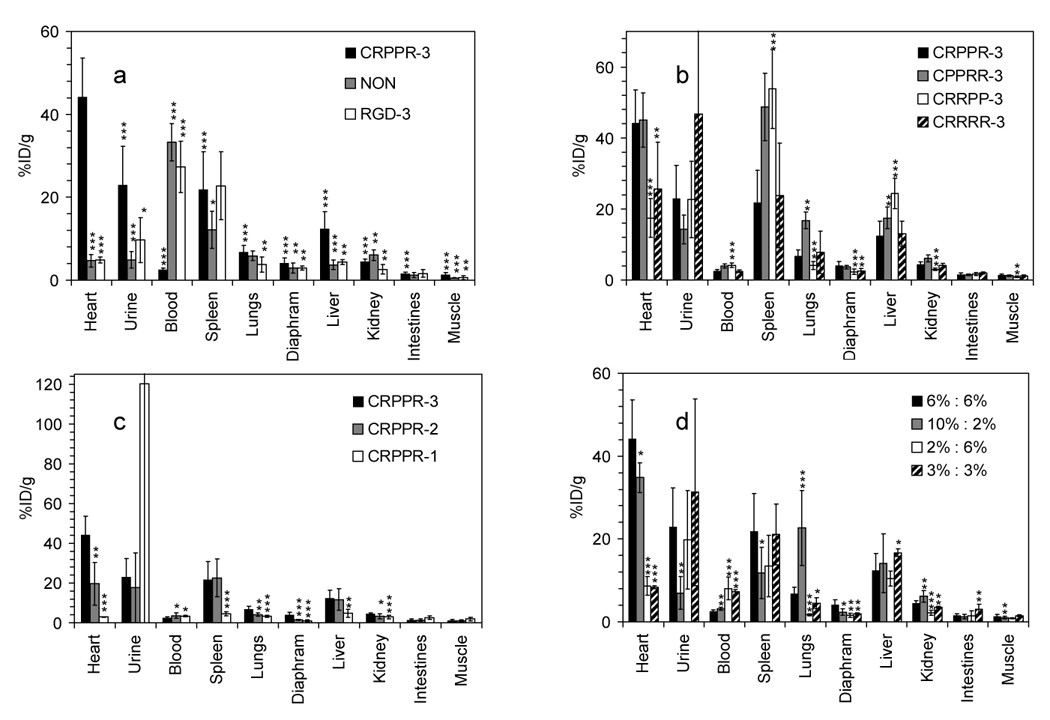
Well counts (%ID/g) obtained 90 minutes after injection. (a), (b) and (c), with different LPP; (d), with varied ratios of CRPPR-3:DSPE-PEG2000. Significance of the accumulation of particles targeted with CRPPR-3:DSPE-PEG2000 6%:6% tested against other peptides and surface architectures is shown by ***, p<0.001; ** , p<0.01; *, p<0.05. In 4a with CRPPR-3, accumulation in each organ is tested against accumulation in the heart. Significance of accumulation is otherwise tested against CRPPR-3 in the same organ.
When the PEG spacer length supporting the peptide was decreased from 3600 to 1200 MW within a surrounding brush layer of DSPE-PEG2000, such that the peptide was shielded by the brush layer, binding of the isotope-containing particle decreased ~10 fold (Figure 4c). For particles targeted with CRPPR-1, the isotope concentration within the urine at 90 minutes was greatly increased (p<0.01), demonstrating the rapid clearance of the tracer. For particles targeted with CRPPR-2, isotope accumulation within the heart was in all cases less than particles containing CRPPR-3 (p<0.05), and greater than particles containing CRPPR-1 (p<0.001).
Radioactivity detected within the heart increased with increasing CRPPR-3 content from 2 to 6%, with the molar percent of DSPE-PEG2000 held constant at 6% (p<0.001, Figure 4d). Neither LPP incorporation or resulting radioactivity increased further as the CRPPR-3 content was increased to 10% (with 2% DSPE-PEG2000), although we note that the 10% CRPPR-3 particles were difficult to extrude and had a higher mean diameter of 192 ± 89nm (Table 1). For particles with 2% CRPPR-3, increasing the molar percentage of DSPE-PEG2000 from 6 to 10% increased the percentage circulating within the blood (p<0.001) at 90 minutes but further decreased the activity within the heart (p<0.01) (data not shown).
3.4 Real-time pharmacokinetics
Dynamic PET analysis provides the opportunity to evaluate the rate of accumulation of the isotope at the target site and to detect accumulation in unexpected targets in real time (Supplemental Video 1 and 2). Accumulation of radiolabeled-particles containing CRPPR-3 was very rapid (tens of seconds) within the heart (Figure 5a–b). Particles containing CRPPR-1 cleared rapidly from the heart, with activity significantly below CRPPR-3 from 40 seconds after the start of the injection (Figure 5b) (p<0.01). Since the heart region of interest (ROI) evaluated with microPET also includes circulating blood within the heart, the heart TAC resulting from the injection of particles containing CRPPR-1 (which did not bind but had similar blood pharmacokinetics to CRPPR-3) was subtracted from the CRPPR-3 TAC. The resulting plot (Figure 5c) indicates that the bound activity within the heart increases rapidly over an interval less than 100 seconds, and continues to increase at a slower rate over the duration of the scan. When fit to a single exponential, a time constant of ~30 seconds for accumulation of activity for particles containing CRPPR-3 was estimated based on Figure 5c.
Figure 5.
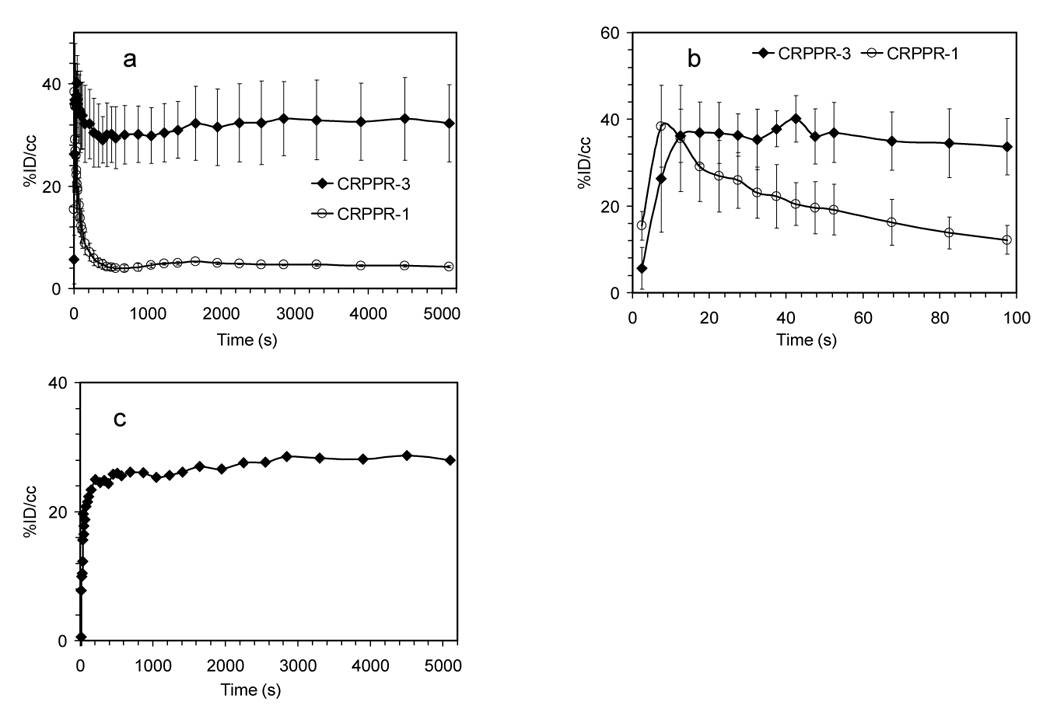
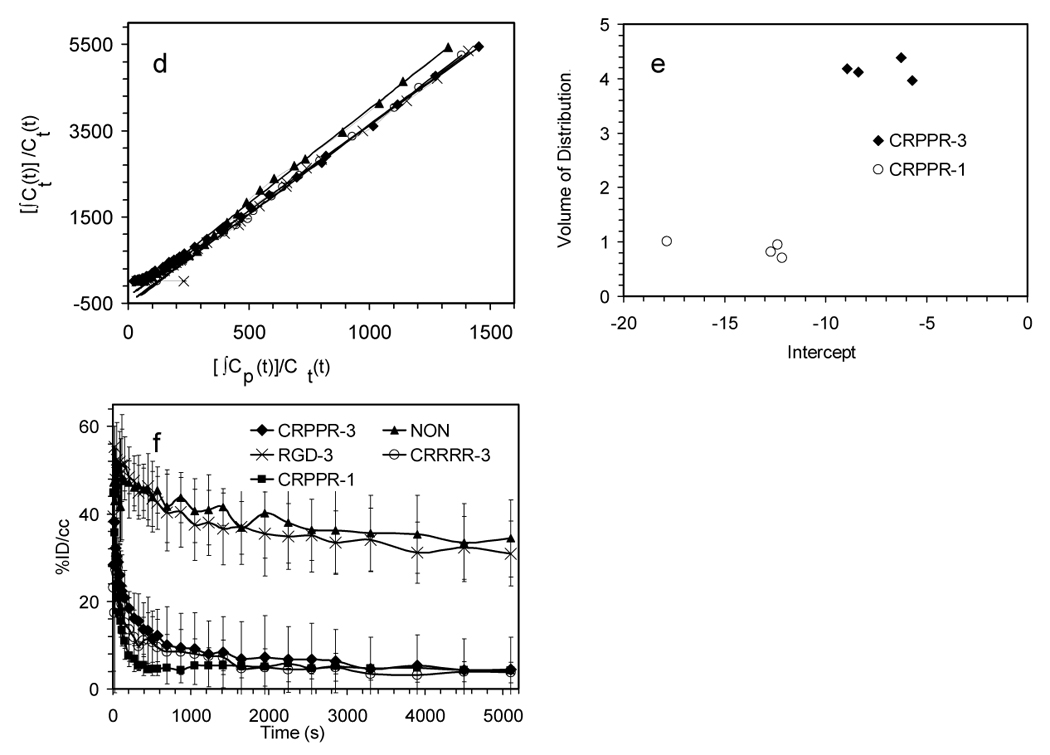
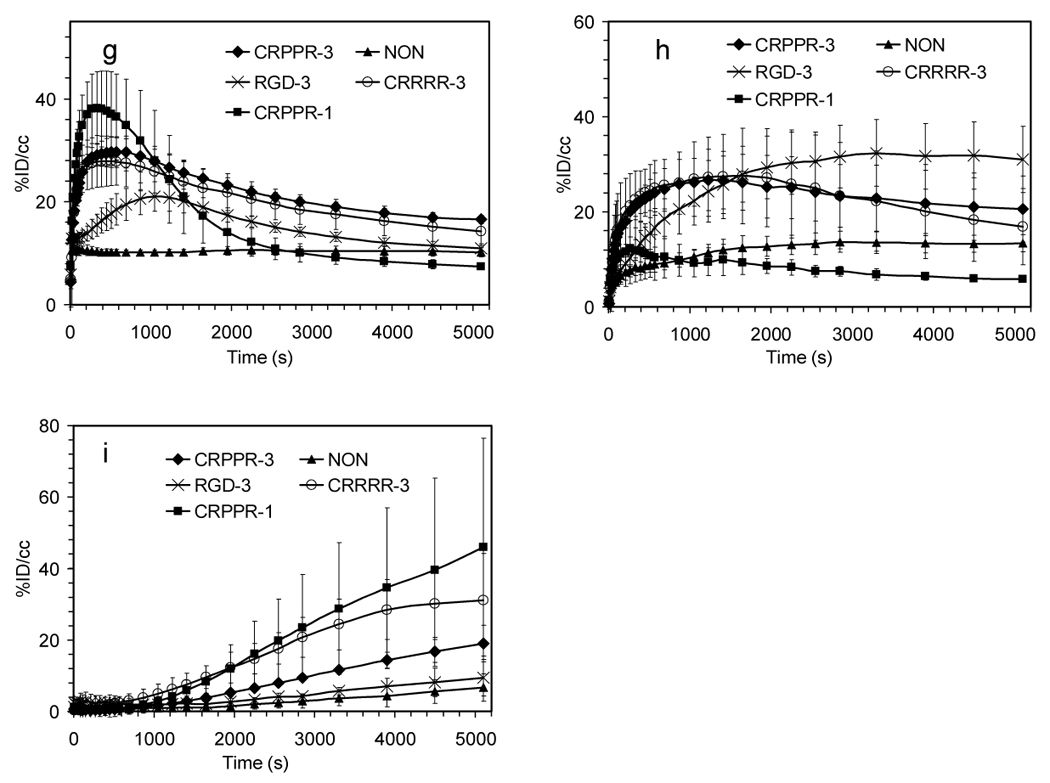
Time activity curves (TACs) and Logan Plot from dynamic PET analysis of various LPP liposomes. (a)–(b) TACs for heart muscle, (c) difference between CRPPR-3 and CRPPR-1 liposomes in TAC from heart muscle. Subtraction of non-binding CRPPR-1 particles removes the effect of the blood pool, showing accumulation of particles within heart muscle. (d) results of Logan analysis for 4 injections of CRPPR-3 liposomes, plotting the time integral of activity at target, Ct(t) against the integral of activity in blood, Cp(t), each normalized by Ct(t). (e) summary of slope and intercept for plots as shown in (d). The higher volume of distribution of CRPPR-3 particles indicates higher avidity. (f – i) TACs for regions of interest. (f) blood within the heart chamber, (g) liver, (h) spleen, and (i) bladder.
The volume of distribution of different tracers in the myocardium was calculated using a Logan plot [28]. The volume of distribution of particles containing CRPPR-3 (for example Figure 5d) is significantly larger than CRPPR-1 (Figure 5e) (p<0.001), indicating higher binding avidity. Note that the estimated values are conservative, particularly for CRPPR-3, as the effects of possible metabolites in blood and the contribution of the blood activity in the myocardial region were not accounted for. Following correction for circulating metabolites, the true volume of distribution of particles containing CRPPR-3 can be significantly greater than four.
Gross differences between PET images were visible in the blood clearance rate of particles depending on the attached peptide (Figure 5f). Estimates of radioactivity within the blood pool over time were fit with a biphasic clearance curve (1), with the constants as shown in Table 2. Without an LPP and with 12% DSPE-PEG2000 (NON in Table 2), the particle was long circulating, with an α value of 104 seconds. Without an LPP and with 6% DSPE-PEG2000, the particle is similarly long circulating (data not shown). Particles coated with RGD-3 were long circulating as indicated by α and β values of 103 and 105 seconds, respectively. The presence of CRPPR, CPPRR, CRRPP, or CRRRR on the liposome substantially reduced the circulation time (even with the peptide shielded by a longer brush layer) with α values of ~102 seconds.
Table 2.
Circulation time constants for [18F] in the blood pool and peak concentrations and accumulation time constants for organs as assessed from PET TACs
| LPP type | Blood Clearance | Heart Muscle | Liver | Spleen | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| A (%ID/cc) | α (sec) | B (%ID/cc) | β (sec) | Peak time (sec) | Peak value (%ID/cc) | Peak time (sec) | Peak value (%ID/cc) | Peak time (sec) | Peak value (%ID/cc) | |
| CRPPR-3 | 24 | 194 | 11 | 4724 | 23 | 39 | 570 | 30 | 1230 | 27 |
| NON | 46 | 9653 | 0.45 | −1835 | N/A | N/A | N/A | 11 | N/A | 10 |
| RGD-3 | 17 | 996 | 34 | 57537 | N/A | N/A | 1050 | 21 | 3300 | 32 |
| CPPRR-3 | 21 | 160 | 16 | 5977 | 23 | 44 | 570 | 28 | 2850 | 30 |
| CRRPP-3 | 25 | 110 | 13 | 8410 | 23 | 33 | 510 | 33 | 2850 | 38 |
| CRRRR-3 | 17 | 216 | 9 | 4202 | 53 | 38 | 510 | 28 | 1650 | 28 |
| CRPPR-1 | 44 | 66 | 5.3 | 32383 | 8 | 30 | 330 | 38 | 270 | 12 |
3.5 RES recognition and clearance
For particles containing the arginine-rich linear peptide, accumulation in the liver is very rapid (570 seconds or less in all cases) (Figure 5g, h, i and Table 2). Alternatively, particles containing RGD-3 reach a greater peak concentration within the spleen as compared with the liver (p<0.05), with the peaks observed at 3300 and 1050 seconds for spleen and liver, respectively.
With CRPPR-1-targeted particles, clearance of the radioisotope from the liver (and accumulation in the bladder) was more rapid than observed with the longer PEG spacer (CRPPR-3). Particles targeted with CRRRR-3 also accumulated more rapidly in the bladder than CRPPR-3 (Figure 5i).
For particles containing RGD-3, the concentration within liver, spleen and bladder (Figure 5g–i) indicate that a fraction was metabolized through the liver (concentration peaking at 1050 seconds) and cleared through the urine. At the 90 minute time point, accumulation of activity within the liver was not significantly different than the no-peptide control (p=0.17).
3.6 RES inhibitors
In order to increase the accumulation at the target site, several methods for the reduction of liver and spleen uptake were evaluated, including the pre-administration of polyinosinic acid, blank liposomes, clodronate liposomes, or free CRPPR peptide. Pre-administration of 12 milligram per kilogram of body weight (mg/kg) of blank liposomes did not significantly change the biodistribution of the targeted liposomes (p=0.10, data not shown).
When clodronate liposomes were administered 24 hours before particles containing CRPPR-3, the circulation time of the particles and binding of the particles to the heart increased at the early time points (before 500sec), each as compared with matched controls not receiving clodronate (p<0.001, Figure 6a–b). However, radioactivity in the heart decreased by 10% over the 90-minute scan (not observed in the absence of clodronate), and activity simultaneously increased within the spleen (Figure 6a, d).
Figure 6.
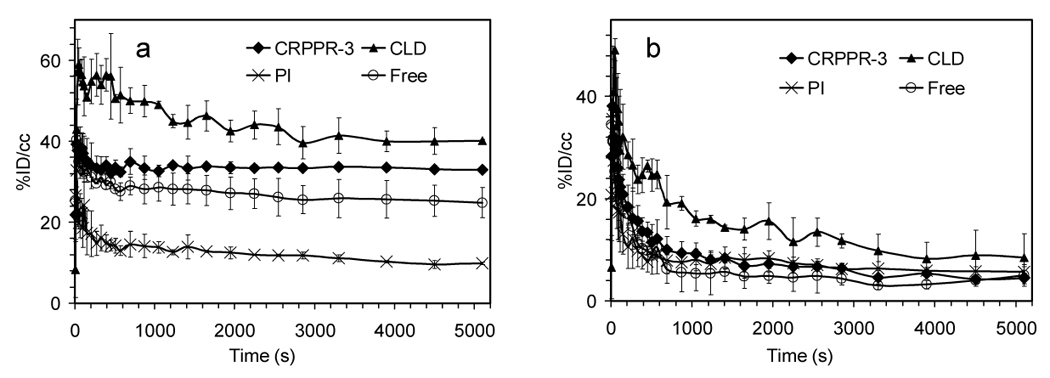
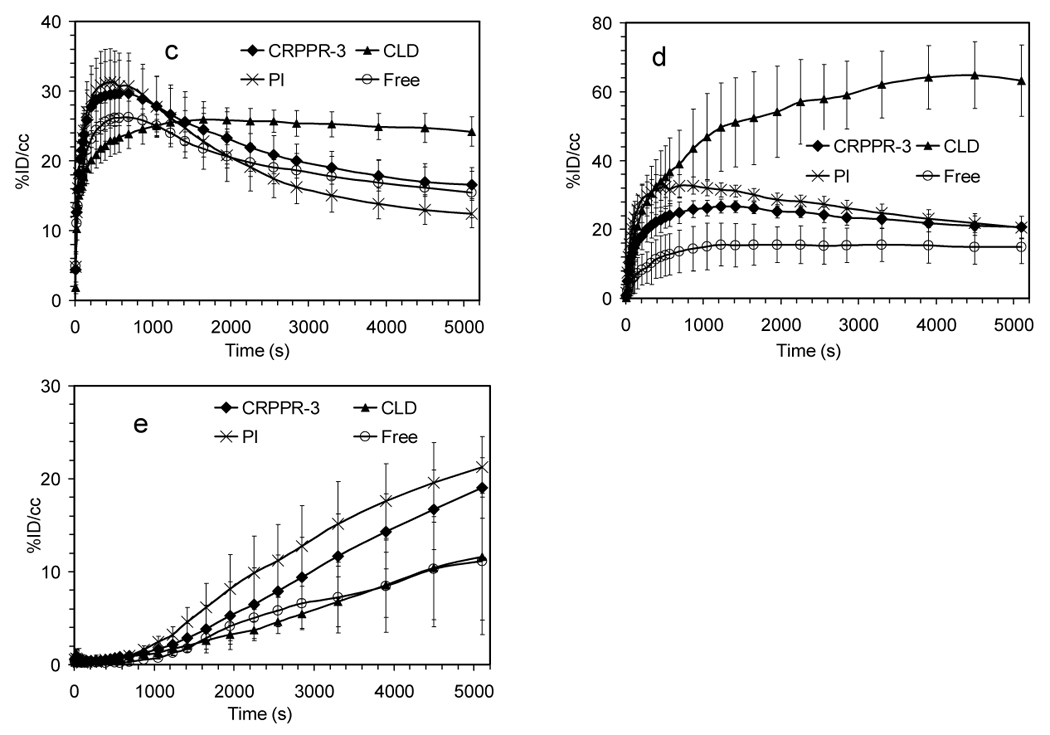
TACs from dynamic PET analysis for (a) heart muscle (b) blood pool (c) liver (d) spleen (e) bladder after injection of CRPPR-3 liposomes and various inhibitors. Abbreviations: CRPPR-3: no inhibitors; CLD: clodronate liposomes injected 24 hours in advance; PI: poly(inosinic acid) injected 1 minute in advance; Free: free CRPPR peptide injected 1 minute in advance of CRPPR-3 liposome injection. Well counts at 90 minutes after injection: the ratios of radioactivity in the heart with and without inhibitors are 1.30, 0.59 and 0.81 for CLD, PI and Free, p = 0.10, < 0.01, and < 0.05, respectively.
Early liver uptake (before 1000 seconds) was significantly lower than observed without clodronate (p<0.05) (Figure 6c). Liver activity does not decrease over the scan (ending higher than without clodronate) (Figure 6c), indicating that the probe is not metabolized and cleared, and the accumulation of activity within the urine at the 90-minute time point was significantly decreased (p<0.05) (data not shown).
Pre-administration of polyinosinic acid (0.4mg/kg) decreased accumulation within the heart by 41% at the 90 minute time point (p<0.05), and this decrease was significant from the six-minute time point forward (p<0.01, Figure 6a). Pre-administration of the free CRPPR peptide (25mg/kg) decreased the accumulation within the heart (p<0.05, Figure 6a) by ~19% at the 90 minute time point (p<0.05), showing the specificity of the targeting.
4 Discussion
Particles targeted with short linear peptides (CRPPR and CPPRR) rapidly and efficiently bound to blood vessel walls in the heart at a significantly higher level than control particles (targeted with CRRPP, CRRRR, c(RGDY(OMe)KE) or without a peptide), showing the potential to carry a substantial payload to heart vessels. Accumulation of CRPPR-targeted particles within the target region increased rapidly over the first 100 seconds after injection (averaging 44% ID/g), reaching a target-to-muscle ratio of 32. Attachment of the CRPPR peptide to a lipid particle decreased the organ specificity of targeting as compared with the phage targeting of CRPPR described in [4], where the target-to-muscle ratio was greater than 300 fold.
Comparing both in vitro endothelial cell binding and the in vivo target-to-muscle ratio assessed by PET (~32 fold), targeting of particles using CRPPR and CPPRR was similar. Alternatively, for CRRPP-targeted particles with an identical charge but without the final arginine amino acid, in vitro binding was reduced, the target to muscle ratio decreased to ~20 fold, and RES uptake was increased (as compared with CRPPR). Finally, CRRRR-targeted particles with a greater positive charge and larger number of arginine amino acids also accumulated in the heart at a lower concentration than CRPPR but at a greater rate than the no-peptide control. Preadministration of the free CRPPR peptide did significantly decrease in vivo binding of the CRPPR-targeted particles, typically indicating specificity. However, given that each of these short, arginine-rich peptides bound to the heart vasculature in vivo at levels above the controls (with the highest binding for peptides with a final arginine), the net positive charge and the guanidine moiety of the arginine amino acid may also contribute to the accumulation of these particles in the heart vasculature.
Dynamic imaging has great promise for effective optimization of nanoparticle drug delivery systems; and this is important due to the vast parameter space of materials, vehicle diameter, charge, surface architecture, ligands, molecular targets and release mechanisms for the dissociation of the vehicle and drug. While imaging has played a role, quantitative measurement of the pharmacokinetics with dynamic imaging has thus far been limited. In our study, dynamic PET facilitated the evaluation of the circulation, targeting, and metabolism of the lipid particle.
The literature on the pharmacokinetics of peptide-targeted particles includes contradictory reports as to whether the presence of a peptide on the surface substantially reduces the circulation lifetime of the particle [29]. Here, particles with a charged linear peptide (CRPPR, CPPRR, CRRRR or CRRPP) on the surface were cleared very rapidly. Alternatively, particles with a neutrally-charged cyclized RGD peptide circulate far longer than the imaging interval, although binding of particles coated with this peptide to circulating platelets could also enhance the circulation time of this RGD peptide.
A comparison of the volume distribution of particles demonstrated that the particles accumulate at the target site at a rate that is proportional to their availability within the blood. Uptake within the heart appears to be limited by the rapid uptake within the liver, as the ratio of target on to off rate remains constant over time. Depletion of macrophages prior to injection of the particles increased circulation lifetime and targeted accumulation at the early time points. Preadministration of polyinosinic acid (a scavenger receptor competitor) significantly decreased accumulation within the heart but also produced lesser changes in circulation and metabolism of the particles.
The use of a radiolabeled lipid also facilitated an evaluation of targeting dynamics with differing particle surface architecture. The accumulation in the heart of targeted liposomes with the CRPPR peptide supported on a PEG-spacer of 3600 molecular weight (MW) was ~10 times higher than particles with a PEG-spacer of 1200 MW (the surrounding PEG brush of the liposome was 2000 MW). The presence of a brush layer, extending beyond the targeting ligand, blocked adhesion of the particles to the target, but did not block uptake and rapid metabolism by the liver. The presence of a high peptide concentration (6 mol% or approximately ~6000 peptide groups per liposome) was required to maximize local uptake of the particle. Increased targeting with a dense peptide coating is consistent with previous studies in which antibodies were targeted to the endothelium of the lung [8].
Dynamic PET analysis of [18F]FDP was also used to measure the clearance of this radiolabeled lipid from the liver, as excretion of the 18F label requires cleavage of the fatty acids. With the exception of studies involving CRRRR, in all data reported here and in [25], activity in the bladder increased only after a delay of ~1000 seconds. With particles containing CRRRR, activity in the bladder increased immediately after injection of the particles, with this difference hypothesized to result from immediate cellular internalization of these particles or rapid extracellular enzymatic breakdown of the radiolabeled lipid. The presence of large numbers of arginine amino acids has otherwise been reported to facilitate internalization[10]. Also, a shorter PEG spacer (CRPPR-1) facilitated rapid metabolism of the radioactive lipid.
5 Conclusion
Liposomes with a 6 molar percent coating of CRPPR or CPPRR lipoPEGpeptide, a PEG spacer of 3600 MW, and a PEG brush of 2000 MW bound to heart vasculature within 100 seconds. The rapid and specific targeting of liposomes to the heart using a surface coating of peptide suggests the potential for use in drug and gene delivery applications and provides a model system to engineer targeted particles.
Supplementary Material
Supplemental Video 1. Sequence of dynamic PET images recorded after the injection of liposomes containing the CRPPR-3.
Supplemental Video 2. Sequence of dynamic PET images recorded after the injection of control liposomes (no peptide).
Supplemental Video 3. 3D Confocal Microscopy Images recorded after the injection of liposomes containing CRPPR-3.
Supplemental Video 4. 3D Confocal Microscopy Images recorded after the injection of control liposomes (no peptide).
Acknowledgments
This work was supported by NIH CA R01 103828 and NIH CA R24 110804. The assistance of Jennifer Fung and Chris Griesemer in imaging studies and the development of autoradiography methods, Sven Hausner and Julia Choi in the development of methods for lipoPEGpeptide synthesis, Jinxiu Liao and Guobao Wang in the reconstruction and analysis of the dynamic PET images, and Roger Adamson in the confocal microscopy are gratefully acknowledged.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ding BS, Dziubla T, Shuvaev VV, Muro S, Muzykantov VR. Advanced drug delivery systems that target the vascular endothelium. Molecular Interventions. 2006;6(2):98–112. doi: 10.1124/mi.6.2.7. [DOI] [PubMed] [Google Scholar]
- 2.Hajitou A, Pasqualini R, Arap W. Vascular targeting: Recent advances and therapeutic perspectives. Trends in Cardiovascular Medicine. 2006;16(3):80–88. doi: 10.1016/j.tcm.2006.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruoslahti E. Vascular zip codes in angiogenesis and metastasis. Biochem Soc Trans. 2004;32:397–402. doi: 10.1042/BST0320397. [DOI] [PubMed] [Google Scholar]
- 4.Zhang LL, Hoffman JA, Ruoslahti E. Molecular profiling of heart endothelial cells. Circulation. 2005;112(11):1601–1611. doi: 10.1161/CIRCULATIONAHA.104.529537. [DOI] [PubMed] [Google Scholar]
- 5.Brissette R, Prendergast JKA, Goldstein NI. Identification of cancer targets and therapeutics using phage display. Curr Opin Drug Discov Dev. 2006;9(3):363–369. [PubMed] [Google Scholar]
- 6.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nature Reviews Drug Discovery. 2005;4(2):145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 7.Wilson A, Zhou W, Champion HC, Alber S, Tang ZL, Kennel S, et al. Targeted delivery of oligodeoxynucleotides to mouse lung endothelial cells in vitro and in vivo. Molecular Therapy. 2005;12(3):510–518. doi: 10.1016/j.ymthe.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Maruyama K, Kennel SJ, Huang L. Lipid-composition is important for highly efficient target binding and retention of immunoliposomes. Proc Natl Acad Sci U S A. 1990;87(15):5744–5748. doi: 10.1073/pnas.87.15.5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wiewrodt R, Thomas AP, Cipelletti L, Christofidou-Solomidou M, Weitz DA, Feinstein SI, et al. Size-dependent intracellular immunotargeting of therapeutic cargoes into endothelial cells. Blood. 2002;99(3):912–922. doi: 10.1182/blood.v99.3.912. [DOI] [PubMed] [Google Scholar]
- 10.Patel LN, Zaro JL, Shen WC. Cell penetrating peptides: intracellular pathways and pharmaceutical perspectives. Pharmaceutical Research. 2007;24(11):1977–1992. doi: 10.1007/s11095-007-9303-7. [DOI] [PubMed] [Google Scholar]
- 11.Sen Gupta A, Huang G, Lestini BJ, Sagnella S, Kottke-Marchant K, Marchant RE. RGD-modified liposomes targeted to activated platelets as a potential vascular drug delivery system. Thrombosis and Haemostasis. 2005;93(1):106–114. doi: 10.1160/TH04-06-0340. [DOI] [PubMed] [Google Scholar]
- 12.Gerlag DM, Borges E, Tak PP, Ellerby HM, Bredesen DE, Pasqualini R, et al. Suppression of murine collagen-induced arthritis by targeted apoptosis of synovial neovasculature. Arthritis Research. 2001;3(6):357–361. doi: 10.1186/ar327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sutcliffe-Goulden JL. The synthesis of novel 18F-labelled peptides for PET [Ph.D] London: King's College London; 2002. [Google Scholar]
- 14.Raffaghello L, Pagnan G, Pastorino F, Cosimo E, Brignole C, Marimpietri D, et al. Immunoliposomal fenretinide: a novel antitumoral drug for human neuroblastoma. Cancer Lett. 2003;197(1–2):151–155. doi: 10.1016/s0304-3835(03)00097-1. [DOI] [PubMed] [Google Scholar]
- 15.Lukyanov AN, Elbayoumi TA, Chakilam AR, Torchilin VP. Tumor-targeted liposomes: doxorubicin-loaded long-circulating liposomes modified with anticancer antibody. Journal of Controlled Release. 2004;100(1):135–144. doi: 10.1016/j.jconrel.2004.08.007. [DOI] [PubMed] [Google Scholar]
- 16.Shadidi M, Sioud M. Selective targeting of cancer cells using synthetic peptides. Drug Resistance Updates. 2003;6(6):363–371. doi: 10.1016/j.drup.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 17.Schiffelers RM, Koning GA, ten Hagen TLM, Fens M, Schraa AJ, Janssen A, et al. Anti-tumor efficacy of tumor vasculature-targeted liposomal doxorubicin. Journal of Controlled Release. 2003;91(1–2):115–122. doi: 10.1016/s0168-3659(03)00240-2. [DOI] [PubMed] [Google Scholar]
- 18.Lestini BJ, Sagnella SM, Xu Z, Shive MS, Richter NJ, Jayaseharan J, et al. Surface modification of liposomes for selective cell targeting in cardiovascular drug delivery. Journal of Controlled Release. 2002;78(1–3):235–247. doi: 10.1016/s0168-3659(01)00505-3. [DOI] [PubMed] [Google Scholar]
- 19.Kamps J, Morselt HWM, Swart PJ, Meijer DKF, Scherphof GL. Massive targeting of liposomes, surface-modified with anionized albumins, to hepatic endothelial cells. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(21):11681–11685. doi: 10.1073/pnas.94.21.11681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Simberg D, Duza T, Park JH, Essler M, Pilch J, Zhang LL, et al. Biomimetic amplification of nanoparticle homing to tumors. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(3):932–936. doi: 10.1073/pnas.0610298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vanrooijen N, Sanders A. Liposome-mediated depletion of macrophages - mechanism of action, preparation of liposomes and applications. Journal of Immunological Methods. 1994;174(1–2):83–93. doi: 10.1016/0022-1759(94)90012-4. [DOI] [PubMed] [Google Scholar]
- 22.Tilcock C, Ahkong QF, Fisher D. Tc-99m-Labeling of Lipid Vesicles Containing the Lipophilic Chelator Pe-Dtta - Effect of Tin-to-Chelate Ratio, Chelate Content and Surface Polymer on Labeling Efficiency and Biodistribution Behavior. Nuclear Medicine and Biology. 1994;21(1):89–96. doi: 10.1016/0969-8051(94)90134-1. [DOI] [PubMed] [Google Scholar]
- 23.Bao AD, Goins B, Klipper R, Negrete G, Phillips WT. Direct Tc-99m labeling of pegylated liposomal doxorubicin (Doxil) for pharmacokinetic and non-invasive imaging studies. Journal of Pharmacology and Experimental Therapeutics. 2004;308(2):419–425. doi: 10.1124/jpet.103.059535. [DOI] [PubMed] [Google Scholar]
- 24.Bao A, Goins B, Klipper R, Negrete G, Phillips WT. Re-186-liposome labeling using Re-186-SNS/S complexes: In vitro stability, imaging, and biodistribution in rats. J Nucl Med. 2003;44(12):1992–1999. [PubMed] [Google Scholar]
- 25.Marik J, Tartis MS, Zhang H, Fung JY, Kheirolomoom A, Sutcliffe JL, et al. Long-circulating liposomes radiolabeled with [F-18]fluorodipalmitin ([F-18]FDP) Nuclear Medicine and Biology. 2007;34(2):165–171. doi: 10.1016/j.nucmedbio.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Keller BO, Li L. Three-layer matrix/sample preparation method for MALDI MS analysis of low nanomolar protein samples. Journal of the American Society for Mass Spectrometry. 2006;17(6):780–785. doi: 10.1016/j.jasms.2006.02.012. [DOI] [PubMed] [Google Scholar]
- 27.Chan WC, White PD. Fmoc solid phase peptide synthesis : a practical approach. New York: Oxford University Press; 2000. [Google Scholar]
- 28.Logan J. Graphical analysis of PET data applied to reversible and irreversible tracers. Nuclear Medicine and Biology. 2000;27(7):661–670. doi: 10.1016/s0969-8051(00)00137-2. [DOI] [PubMed] [Google Scholar]
- 29.Koning GA, Schiffelers RM, Wauben MHM, Kok RJ, Mastrobattista E, Molema G, et al. Targeting of angiogenic endothelial cells at sites of inflammation by dexamethasone phosphate-containing RGD peptide liposomes inhibits experimental arthritis. Arthritis and Rheumatism. 2006;54(4):1198–1208. doi: 10.1002/art.21719. [DOI] [PubMed] [Google Scholar]
- 30.Moghimi SM, Hamad I, Andresen TL, Jorgensen K, Szebeni J. Methylation of the phosphate oxygen moiety of phospholipid-methoxy(polyethylene glycol)conjugate prevents PEGylated liposome-mediated complement activation and anaphylatoxin production. Faseb Journal. 2006;20(14):2591–2593. doi: 10.1096/fj.06-6186fje. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Video 1. Sequence of dynamic PET images recorded after the injection of liposomes containing the CRPPR-3.
Supplemental Video 2. Sequence of dynamic PET images recorded after the injection of control liposomes (no peptide).
Supplemental Video 3. 3D Confocal Microscopy Images recorded after the injection of liposomes containing CRPPR-3.
Supplemental Video 4. 3D Confocal Microscopy Images recorded after the injection of control liposomes (no peptide).