

Abstract
Parenteral immunization of transgenic mouse models of Alzheimer disease (AD) with synthetic amyloid β-peptide (Aβ) prevented or reduced Aβ deposits and attenuated their memory and learning deficits. A clinical trial of immunization with synthetic Aβ, however, was halted due to brain inflammation, presumably induced by a toxic Aβ, T-cell- and/or Fc-mediated immune response. Another issue relating to such immunizations is that some AD patients may not be able to raise an adequate immune response to Aβ vaccination due to immunological tolerance or age-associated decline. Because peripheral administration of antibodies against Aβ also induced clearance of amyloid plaques in the model mice, injection of humanized Aβ antibodies has been proposed as a possible therapy for AD. By screening a human single-chain antibody (scFv) library for Aβ immunoreactivity, we have isolated a scFv that specifically reacts with oligomeric Aβ as well as amyloid plaques in the brain. The scFv inhibited Aβ amyloid fibril formation and Aβ-mediated cytotoxicity in vitro. We have tested the efficacy of the human scFv in a mouse model of AD (Tg2576 mice). Relative to control mice, injections of the scFv into the brain of Tg2576 mice reduced Aβ deposits. Because scFvs lack the Fc portion of the immunoglobulin molecule, human scFvs against Aβ may be useful to treat AD patients without eliciting brain inflammation.
Keywords: Alzheimer disease, Amyloid, Single-chain antibody, Animal model, Immune therapy, Spatial memory
Alzheimer disease (AD) is pathologically characterized by amyloid plaques in the brain. To date, no satisfactory treatment is available for AD. The main constituent of amyloid plaques is amyloid β-peptide (Aβ). Increasing evidence supports the notion that Aβ and its precursors play pathogenetic roles in AD. Overexpression of mutant forms of APP in transgenic mice led to AD-like pathologies, including amyloid plaques. Immunization of these AD mouse models with synthetic Aβ by repeated injection prevented or reduced Aβ deposits [1] and attenuated their memory and learning deficits [2,3]. These groundbreaking results led to clinical trials of immunizing AD patients with synthetic Aβ. These clinical trials, however, had to be halted due to brain inflammation, presumably induced by T-cell-mediated and/or Fc-mediated immune responses [4–6]. In other experiments, peripheral administration of antibodies against Aβ induced clearance of preexisting amyloid plaques in an AD mouse model, despite modest serum levels of the antibodies [7], indicating that an active T-cell-mediated immune response was unnecessary. Moreover, topical injection of the F(ab′)2 fragment of antibody against Aβ led to clearance of amyloid deposits in an AD mouse model [8], indicating that non-Fc-mediated mechanisms were also involved in the clearance. Based upon these experimental and clinical observations, intravenous injection of humanized Aβ antibodies lacking Fc has been proposed as a potential therapy for AD.
While human or humanized antibodies are extremely useful for diagnostic and therapeutic purposes, they are difficult to obtain from hybridoma or immunizing animals. Such antibodies are readily selected from human scFv phage libraries [9]. scFv consists of a single polypeptide chain, comprising an antibody heavy chain variable domain (VH) associated by a flexible polypeptide linker to a light chain variable domain (VL). In order to evaluate the therapeutic potential of human scFv for AD, we have isolated an scFv by screening a human single-chain antibody (scFv) library for Aβ1–42 immunoreactivity and characterized scFv for immunoreactivity against amyloid plaques, as well as against oligomeric and fibrillar Aβ. Here, we assessed the efficacy of the scFv in ameliorating Aβ aggregates and cytotoxicity in vitro and in vivo (Tg2576 mice, a transgenic animal model of AD).
Materials and methods
Isolation and purification of human scFvs against Aβ
A synthetic peptide containing amino acid residues 1–42 of Aβ was purchased from U.S. Peptide (Rancho Cucamonga, CA). scFvs that specifically react with the synthetic peptide were obtained through screening the ETH-2 human antibody phage library (Eidgenössische Technische Hochschule, Zurich, Swiss). The synthetic peptide was coated onto immunotubes at a concentration of 20 μg/ml in PBS overnight at 4 °C. Unbound antigen was washed away and the tubes were blocked by incubation with 2% dried milk in PBS for 2 h at room temperature. A library of approximately 1012 phages, encompassing 100–1000 copies of 109 distinct clones, was incubated with the immobilized antigen. After 2 h at room temperature, unbound phages were washed away and the remaining phages eluted by exposure to 100 mM triethylamine. Logarithmically growing Escherichia coli (suppressor strain TG1) were infected with a portion of the eluted phages and the titers of eluted phages were determined by serial dilution. The remaining phages were grown in bacterial culture overnight, packaged and expressed through co-infection with helper phage, and precipitated from the bacterial supernatant. The precipitated phages were then used for subsequent rounds of antigen panning. Phages were isolated from single ampicillin-resistant colonies of infected TG1 cells using helper phage, and binding specificity for antigen was determined by enzyme-linked immunosorbent assay (ELISA). Single ampicillin-resistant colonies were used to inoculate 200 μl of culture broth in microtiter plates, and the expression of soluble scFv fragments was induced by addition of 1 mM isopropyl-β-D-thiogalactopyranoside to the cultures. Bacteria were pelleted, and the supernatants containing monoclonal phage populations were screened for binding to antigen by ELISA. Binding specificity was determined by comparing signals obtained from plates coated with the relevant antigen versus those obtained with the negative control antigen. Phages with high ELISA titers were isolated and used to infect the E. coli HB2151 (non-suppressor). Soluble scFv fragments induced by addition of 1 mM isopropyl- β-D-thiogalactopyranoside to the cultures were used to further screen scFv phage clones for binding to the synthetic Aβ1–42 peptide by ELISA using anti-FLAG M2 monoclonal antibody (scFv contains Flag sequences as a marker) (Sigma, St. Louis, MO) as the detecting reagent. An scFv clone that demonstrated the highest ELISA titer was for Aβ immunoreactivity were selected for immunohistochemical and Western blot analyses.
For other experiments, purified scFv was used. Infected HB2151 bacteria were grown in 1 L of 2× TY medium supplemented with 0.1% glucose and 100 μg/ml ampicillin, and induced overnight with 1 mM isopropyl- β-D-thiogalactopyranoside at 30 °C. Bacteria were removed via centrifugation and the supernatant filtered to remove the remaining pellet. Soluble scFv was purified by passing the filtered supernatant over a protein A agarose column. scFv was eluted using low pH buffer, neutralized, and dialyzed against PBS for storage and use. Protein concentration was determined spectrophotometrically assuming A280 of 1.0 = 0.7 mg/ml.
Immunoreactivity of scFv to amyloid deposits in the brain and muscle
To assess immunoreactivity of scFv with amyloid deposits in the brain and muscle, serial sections of formalin fixed, paraffin embedded brain and muscle from a 15-month-old Tg2576 [10] and a 22-month-old Tg13592 (an animal model of inclusion-body myositis) [11] mouse, respectively, were subjected to the immunoperoxidase method. Endogenous peroxidase was eliminated by treatment with 3% H2O2 for 30 min after deparaffinization of the sections. After washing with distilled water, the sections were treated with 88% formic acid and rinsed with water and 0.1M Tris-buffered saline (TBS) (pH 7.4). The sections were blocked with 15% horse serum in TBS for 60 min at room temperature and incubated with scFv in 0.1M TBS containing 5% horse serum for 16 h at 4 °C. The sections were rinsed in 0.1 MTBS containing 1% serum and incubated with an anti-Flag M2 antibody (secondary antibody) for 60 min at room temperature. After washing with 0.1M TBS containing 1% serum, the sections were incubated with biotinylated anti-mouse IgG antibody (tertiary antibody) for 60 min at room temperature. After washing, the sections were incubated with Vectastain ABC reagent (Vector, Burlingame, CA) for 60 min at room temperature. Peroxidase activity was detected by treatment with 3,3′-diaminobenzidine. The sections were counterstained with hematoxylin. As a positive control, amyloid deposits in the brain and muscle were stained with 6E10 (1 μg IgG/ml; a mouse monoclonal antibody raised against amino acid residues 1–16 of Aβ, Signet, Dedham, Massachusetts) using the avidin–biotin immunoperoxidase method (Vectastain ABC kit) [12]. For comparison, a serial brain section was also stained with thioflavine-S for detection of neuritic plaques [13].
Immunoreactivity of scFv to oligomeric and fibrillar Aβ
Western blot analysis was performed to evaluate the immunoreactivity of scFv to oligomeric and fibrillar Aβ. Synthetic Aβ1–42 was purchased from US peptide. Oligomeric Aβ was prepared as described by Dahlgren et al. [16]. The peptide was dissolved in 1 mM hexafluoroisopropanol (Sigma) and then removed under vacuum in a Speed Vac (Savant, Holbrook, NY). The residual peptide was re-suspended in dry dimethyl sulfoxide (Sigma) to a concentration of 5 mM. By adding phenol red free Ham’s F-12 medium (Mediatech, Herndon, VA) to the re-suspended peptide, the concentration was made to 100 μM and the peptide was kept at 4 °C for 24 h. The samples were diluted in NuPage sample buffer (Invitrogen, Carlsbad, CA) and separated by 16.5% Tris–Tricine SDS–PAGE.
Monomeric and fibrillar Aβ was prepared from a 12-month-old Tg2576 mouse by extracting brain amyloid plaques in formic acid. The brain was weighed and homogenized in 10× weight of 10% SDS. The homogenized samples were centrifuged at 100,000g for 1 h and the pellets were washed with 0.1M TBS (pH 7.4). The pellets were re-suspended in 88% formic acid using Dounce homogenizers and then centrifuged at 100,000g for 20 min. The supernatant was dried using a vacuum concentrator (SpeedVac, Savant). The dried samples were re-suspended in SDS buffer (10% SDS, 25% glycerol, 300 mM Tris, pH 6.8, and 100 mM Tricine). The samples were boiled for 5 min before loading onto a 16.5% Tris/Tricine gel. The sample on each lane was derived from 30 mg wet weight of the brain.
After electrotransfer to polyvinylidine difluoride (PVDF) membranes (Immobilon-P, Millipore, Bedford, MA), monomeric, oligomeric, and fibrillar Aβ were stained with scFv and anti-Flag M2 antibody using the avidin–biotin immunoperoxidase method (Vectastain ABC kit) followed by the enhanced chemiluminescence method (Amersham, Arlington Heights, IL) according to the manufacturers’ protocols. For comparison, the mono- and oligomeric Aβ was similarly visualized by 6E10 (1 μg IgG/ ml) and the avidin–biotin immunoperoxidase method.
Detection of amyloid fibrils by thioflavine T fluorescence assay
Inhibition of Aβ fibril formation by scFv59 was studied by thioflavine T fluorescence and electron microscopy. Aβ1–42 stock solution (5 mM) in dimethyl sulfoxide was diluted to the final concentration of 15 μM by adding PBS (pH 7.4) and, then, incubated at 37 °C for 22 days with or without an equimolar concentration of scFv59. Aβ fibrils were stained by adding 3 μl of the protein samples (n = 4) to 200 μl of 5 μM thioflavine T solution (50 mM phosphate buffer, pH 6.5). Sample was analyzed under a fluorescence microscope (Olympus BX61 microscope and Olympus Fluoview system). Other aliquots of the protein samples were dropped on 300 mesh formar grids, dried, and treated with 1% phosphotungstic acid for 1 min. The samples were scanned by a transmission electron microscope (JEOL, JEM100C).
Cytotoxicity assay
Human embryonic kidney 293 (HEK293) cells were cultured in Dulbecco’s minimum essential medium(DMEM) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin in a humidified atmosphere of 5% CO2/95% air, at 37 °C. HEK293 cells were plated into a 96-well plate with 1.25 × 103 cells per 100 μl of medium per well 24 h before addition of oligomeric Aβ. Oligomeric Aβ1–42 was prepared as described above. Oligomeric Aβ1–42 (1.4 μM) alone or oligomeric Aβ1–42 (1.4 μM) with an equimolar concentration of scFv59 (n = 3) was preincubated for 9 days at 4 °C in the serum-free medium. The peptide samples were then added to cells at the final Aβ1–42 concentration of 700 nM and incubated for 72 h. Cell viability was determined using Cell Titer 96 Non-Radioactive Cell Proliferation Assay (Promega) according to the manufacturer’s protocol. The absorbance was measured using a Model 680XR plate reader (Bio-Rad). Averages from three replicate wells were used for each sample and control, and each experiment was repeated three times. Cell viability was calculated by dividing the absorbance of wells containing samples (corrected for background) by the absorbance of wells containing medium alone (corrected for background).
Stereotaxic injection of scFv to the hippocampus and cortex of Tg2576 mice
Six Tg2576 mice at the age of 16 months were subjected to scFv injection and, as controls, 6 age- and sex-matched Tg2576 mice were injected with PBS. All animal procedures used for this study were prospectively reviewed and approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham. Mice were anesthetized by pentobarbital and placed on a stereotaxic instrument with a motorized stereotaxic injector (Stoelting, Wood Dale, IL). A hole on the skull was made by a drill 0.2 cm posterior to the bregma and 0.15 cm to the midline for the cortico hippocampal injection. Two bilateral injections (2 μl each, 1 μg/μl scFv) were performed at the depth of 0.17 (hippocampus) and 0.07 (cortex) cm at a rate of 1 μl/min. After allowing the needle to remain in place for 5 min, the needle was slowly raised at a rate of 0.1 cm/min. A total of 8 μl (8 μg scFv)/mouse was injected.
Amyloid load by immunohistochemistry and thioflavine-S fluorescence
The immunohistochemical method has been described previously in Li et al. [14]. The brains were subjected to immunohistochemical quantification of amyloid load. The mice were deeply anesthetized by intraperitoneal injection of sodium pentobarbital and the brains were quickly removed. The brains were fixed in 10% formaldehyde: 90% alcohol and embedded in paraffin. For the hippocampus and cerebral cortex (coronal sections at the injection site spreading 1.2 mm on a rostro-caudal axis), five sections, each separated by consecutive 120 μm intervals, were analyzed. Three micron tissue sections were subjected to the avidin–biotin immunoperoxidase method using 6E10 antibody and Vectastain ABC kit. Ten micron brain sections were subjected to thioflavine-S staining for quantification of amyloid plaques. The amyloid burden in the cortex and the hippocampus of the mouse brain was quantified by histomorphometry consisting of a Leica DMR research microscope equipped for fluorescence, polarizer/analyzer, and brightfield microscopy, a SPOT RT Slider digital camera (Diagnostic Instruments, Sterling Heights, Michigan), and the Image Pro Plus v4 image analysis software (Media Cybernetics, Silver Spring, Maryland) capable of color segmentation and automation via programmable macros. The entire hippocampus and cerebral cortex within 3 mm from the longitudinal fissure in each slide was scanned. Thirty-two to forty fields (1 mm2 each, using a 10× objective and a 1× eyepiece lens) from five coronal brain sections from each mouse at the injection site were analyzed with each staining method. Amyloid burden was expressed as a percentage of total area covered by Aβ immunoreactivity or thioflavine-S fluorescence.
Statistical analysis
Data were expressed as means ± standard error. Comparison of treatment groups was performed by ANOVA using the SigmaStat software (SPSS Science, Chicago, IL). A value of P < 0.05 was considered statistically significant.
Results
Characterizing scFv by immunohistochemistry
After screening approximately 1 × 109 clones of a certain human scFv phage library, a scFv clone (scFv59) with the highest ELISA against Aβ1–42 was chosen to test its immunoreactivity against amyloid deposits in tissues. Serial brain sections from a 15-month-old Tg2576 mouse were immunostained with scFv59 using anti-Flag M2 mouse monoclonal antibody (secondary antibody), biotinylated anti-mouse IgG antibody (tertiary antibody), and the avidin–biotin immunoperoxidase method. As a positive control, 6E10 (a mouse monoclonal antibody against Aβ1–16) and the avidin–biotin immunoperoxidase method were used. For comparison, one of the serial sections was also stained with thioflavine-S for detection of amyloid fibrils. The results are shown in Figs. 1A–D. scFv59 reacted to amyloid plaques (Fig. 1C). The amyloid plaque staining by scFv59 was comparable to that by 6E10 antibody (Fig. 1B). When the same fraction was pre-absorbed with synthetic Aβ1–42, no immunoreactivity in the brain was observed, demonstrating specificity of the scFv clones to Aβ deposits (Fig. 1D). scFv59 was also immunoreactive with Aβ deposits in skeletal muscle from a Tg13592 mouse, an animal model of inclusion-body myositis [11] (Fig. 1F).
Fig. 1.
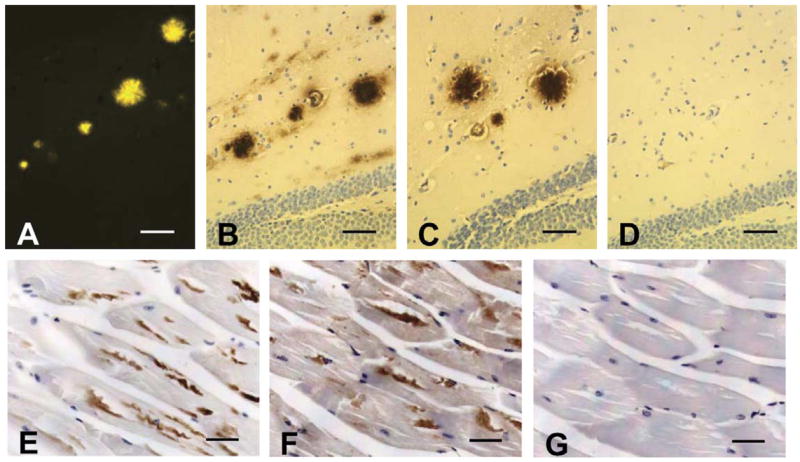
Characterization of scFv by immunohistochemistry. Amyloid deposits detected by immunohistochemistry in the hippocampus (B–D) and in muscle (E–G): serial brain sections from a 15-month-old Tg2576 mouse were stained with (A) thioflavine-S (observed under fluorescence), (B) 6E10, (C) scFv59, and (D) scFv 59 pre-absorbed with synthetic Aβ1–42. Serial skeletal muscle sections from a 22-month-old Tg13592 mouse were stained with (E) 6E10, (F) scFv59, and (G) normal serum. Scale bars 50 μm (A–D) and 40 μm (E–G).
Characterizing scFv by Western blot analysis
Immunoreactivity to monomeric, oligomeric, and fibrillar Aβ was determined by Western blot analysis. Monomeric and oligomeric Aβ species were prepared by the method described by Dahlgren et al. [16] and separated by 16.5% Tris–Tricine SDS–PAGE. When 6E10 antibody was used, Aβ monomer and tetramer were visualized as major species in addition to minor species of dimer, trimer, and pentamer (Fig. 2A). On the other hand, scFv59 reacted strongly with tetrameric Aβ but barely with monomeric Aβ (Fig. 2A). Aβ aggregates/fibrils as well as monomeric Aβ extracted in formic acid from the brain of a 12-month-old Tg2576 mouse were also analyzed by Western blotting. Only Aβ fibrils at the stacking gel were visualized by scFv59, while both monomeric and fibrillar Aβ were visualized by 6E10 antibody (Fig. 2B). Thus, scFv59 is specifically immunoreactive with oligomeric and fibrillar/aggregated Aβ but little to no with monomeric Aβ.
Fig. 2.
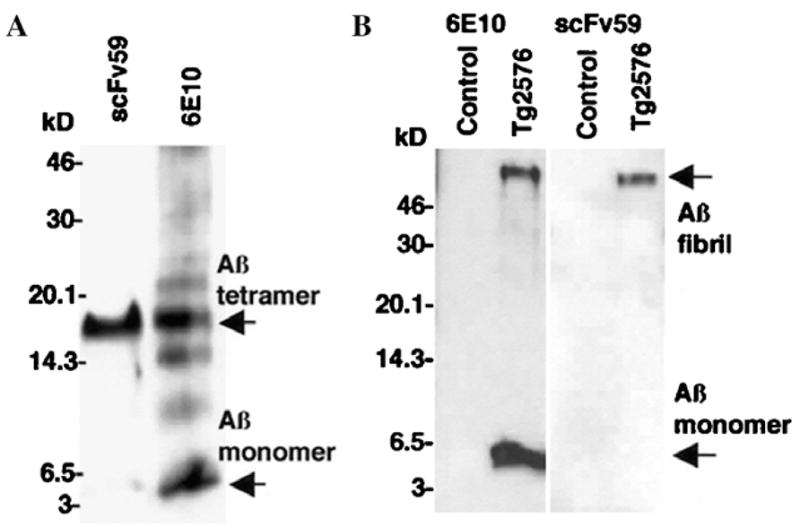
Characterization of scFv by Western blot analysis. (A) Oligomeric Aβ species were prepared from synthetic Aβ1–42 and separated 16.5% Tris–Tricine SDS–PAGE. After electroblotting to a PVDF membrane, oligomeric and monomeric Aβ species were detected using 6E10 antibody or scFv59. Both Aβ oligomers and monomer were visualized by 6E10. scFv59 reacted strongly with tetrameric Aβ but barely with monomeric Aβ. (B) The SDS–insoluble/formic acid-soluble fraction was prepared from the brain of a 12-month-old Tg2576 mouse. After separating the proteins by 16.5% Tris–tricine SDS–PAGE, Western blot analysis was carried out with 6E10 or scFv59. scFv59 detected fibrillar Aβ at the interface between the stacking and separating gel (the top of the gel) but barely monomeric Aβ, while 6E10 detected both. Each lane corresponds to 30 mg (wet weight) of the brain.
Inhibition of Aβ fibril formation by scFv
Aβ1–42 was incubated in PBS for 22 days with or without scFv59 and inhibition of Aβ fibril formation by scFv59 was studied by thioflavine T fluorescence assay and electron microscopy. Under fluorescence microscopy, numerous fluorescent Aβ aggregates with large sizes (up to ~70 μm in diameter) were observed in all the samples (n = 4) without scFv (Fig. 3A) while Aβ aggregates were much smaller (less than 20 μm) in all the samples (n = 4) with scFv59 (Fig. 3B). By electron microscopy, numerous fibrils ~13 nm in diameter were observed in the samples without scFv59 (Fig. 3C). Such fibrils were rarely found in the samples with scFv59 but appeared to be amorphous (Fig. 3D). Thus, scFv59 inhibited Aβ fibril formation.
Fig. 3.
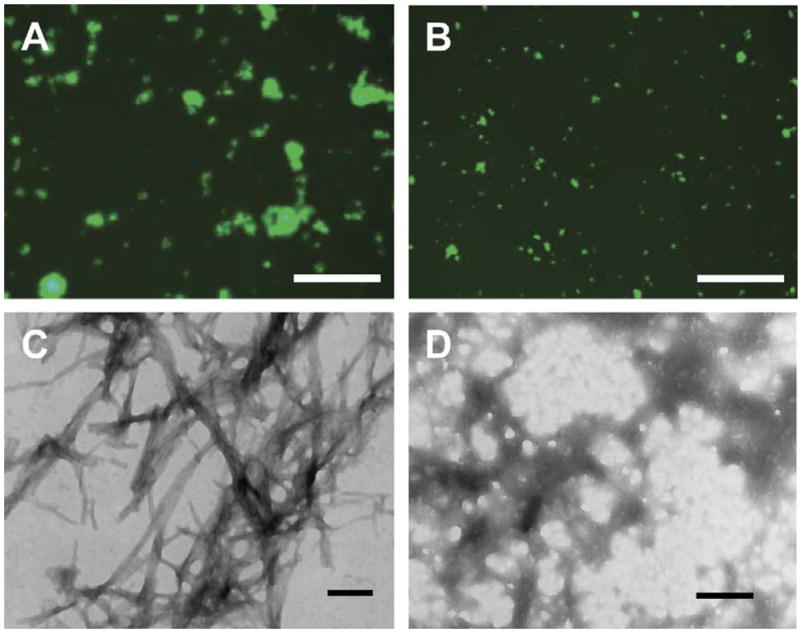
scFv59 inhibits Aβ amyloid fibril formation. Aβ1–42 (15 μM) was incubated with and without scFv59 (15 μM) for 22 days. Aliquots of the protein samples were stained by thioflavine T solution for fluorescence microscopy (A, B) and other aliquots were subjected to transmission electron microscopy (C, D). Numerous Aβ aggregates with large sizes (up to ~70 μm in diameter) are observed in the sample without scFv59 (A) under a fluorescence microscope and are composed of ~13 nm fibrils under an electron microscope (C). Aβ aggregates in the sample with scFv59 are much smaller (less than 20 μm) (B) and show amorphous structures (D). Scale bars 125 μm (A, B) and 142 nm (C, D).
Amelioration of Aβ cytotoxicity by scFv
To determine if scFv59 inhibits cytotoxicity of Aβ oligomers, HEK293 cells were incubated with Aβ oligomers alone or Aβ oligomers plus scFv59. Cell viability was determined by MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium] assay 72 h after incubation. The cells incubated with Aβ oligomers alone showed 45.2 ± 0.78% of cell survival compared to cells without Aβ oligomers. The mean of cell survival in the cultures (n = 3) treated with Aβ oligomers and scFv59 was 58.8 ± 5.45% (P = 0.069). Thus, scFv59 tends to inhibit cytotoxicity of Aβ oligomers in HEK293 cell culture.
Amyloid load by histochemical and immunohistochemical analyses
To evaluate the efficacy of scFv59 in clearing Aβ deposits in the hippocampus and neocortex at the injection site spreading 1.2 mm on a rostro-caudal axis and within 3 mm from the midline, diffuse and fibrillar Aβ deposits were detected by 6E10 antibody, while fibrillar Aβ deposits (neuritic plaques) were stained by thioflavine-S. Representative results are shown in Fig. 4. The β-amyloid load (average percentage of area showing Aβ immunoreactivity or thioflavine-S fluorescence) in the hippocampus and cortex at the injection sites was quantified by morphometric analysis. The Aβ loads detected by 6E10 immunoreactivity were on average 0.991 ± 0.136% and 2.23 ± 0.364% for scFv59 treated Tg2576 and control mice, respectively (P < 0.01) (Fig. 5A). The Aβ loads showing thioflavine-S fluorescence were 0.132 ± 0.0236% and 0.412 ± 0.120% for scFv59 treated Tg2576 and control mice, respectively (P < 0.05) (Fig. 5B). Thus, Tg2576 mice subjected to scFv59 injection had fewer diffuse and fibrillar Aβ deposits than Tg2576 mice injected with PBS.
Fig. 4.
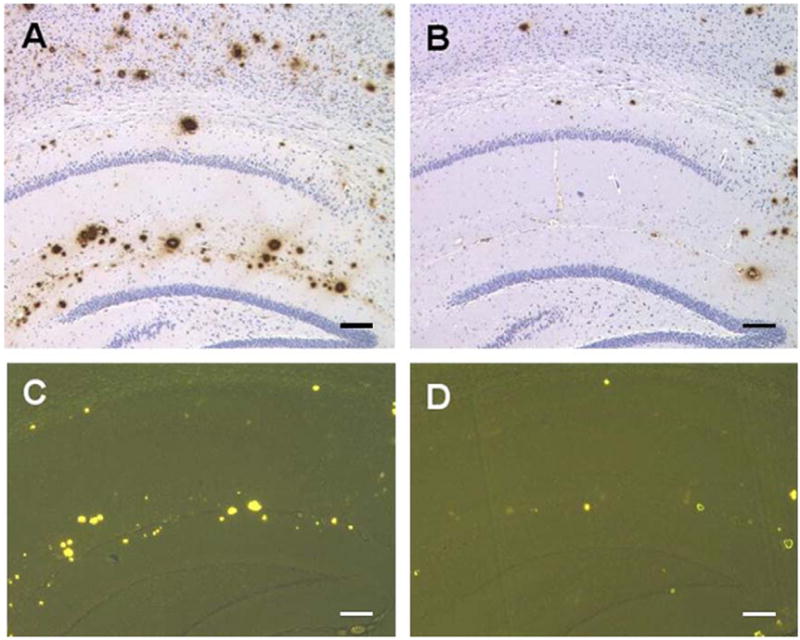
scFv59 injection reduces Aβ deposits in the brain of Tg2576 mice. scFv59- or PBS-injected Tg2576 mice were sacrificed and the brains were subjected to immunohistochemical or histochemical analyses to evaluate the efficacy of scFv59 in clearing Aβ deposits in the hippocampus and in cerebral cortex. Diffuse and fibrillar Aβ deposits were detected with 6E10 antibody (A, B) while fibrillar Aβ deposits (neuritic plaques) were stained with thioflavine-S (C, D). Representative photographs are shown: (A, C) are PBS-injected; (B, D) are scFv59-injected brain. Scale bars 150 μm (A, B) and 140 μm (C, D).
Fig. 5.
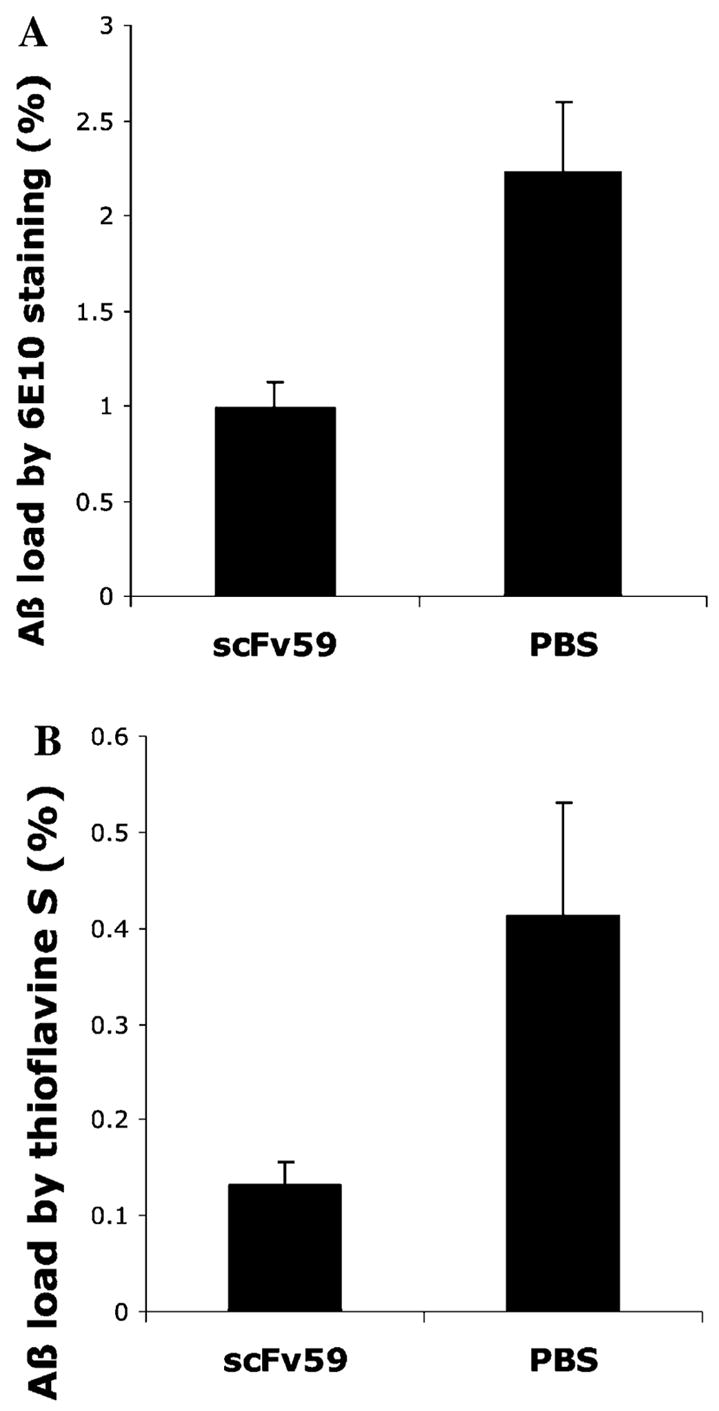
scFv 59 is effective in clearing both diffuse and fibrillar Aβ deposits in Tg2576 mice. The β-amyloid load in the hippocampus and cortex at the injection sites was quantified by morphometric analysis (n = 6 for each treatment). Average percentages of area showing Aβ immunoreactivity (A) or thioflavine-S fluorescence (B) are shown. Aβ immunoreactive area represents diffuse and fibrillar Aβ deposits (P < 0.01) while thioflavine-S fluorescent area represents fibrillar Aβ deposits (P < 0.05). The results are means ± SEM.
Discussion
Transgenic mouse models of AD have previously been used in attempts to develop novel therapeutic means. Early success in treating such AD models by Aβ immunization immediately led to human clinical trials. These clinical trials resulted in brain inflammation in a small fraction of patients, presumably caused by a T-cell and/or Fc-mediated immune response [17–19]. A possible solution to this problem is the use of a passive immunization, that is, injections of human or humanized antibody into patients with AD. To explore this therapeutic possibility, several groups isolated scFvs by screening scFv libraries for Aβ immunoreactivity and demonstrated that scFvs reactive to Aβ can inhibit/reverse Aβ aggregation and ameliorate Aβ-mediated cytotoxicity in vitro [20–22]. Until this report, however, no scFv had been tested for efficacy in treating AD or animal models of AD. We have successfully isolated an scFv antibody that specifically reacts against Aβ deposits as well as against oligomers of Aβ. The scFv inhibited Aβ fibril formation in vitro. When the scFv against Aβ was injected into the hippocampus and the cortex of an AD mouse model, brain Aβ deposits were reduced. Thus, the scFv treatment was effective in clearing Aβ deposits in the brain of an AD mouse model.
In a human clinical trial of synthetic Aβ (AN1792) immunization, Hock et al. [23] reported that antibodies in the AD patients did not cross-react with monomeric as well as oligomeric Aβ, but strongly reacted with amyloid deposits in the brain. In a later study, Lee et al. [24] found that anti-sera from AD patients vaccinated with AN1792 reacted to both Aβ monomer and Aβ plaques, but were much less sensitive to Aβ oligomers. The latter investigators speculated that this discrepancy might be caused by a very low sensitivity of Western blot compared with immunohistochemical staining. The human scFv59 described here showed little to no reactivity against monomeric Aβ, but demonstrated a strong reactivity against oligomeric as well as fibrillar/aggregated Aβ by Western blot analysis (Fig. 2). On the other hand, almost all previous antibodies raised against Aβ1–42 in mice have reacted with monomeric Aβ [25,26]. Antibodies that react with oligomeric and/or fibrillar Aβ but not with monomeric Aβ could have more efficacy in therapeutic use than antibodies that react with monomeric Aβ, because monomeric Aβ may have important physiological functions. Moreover, soluble Aβ oligomers are more toxic than Aβ fibrils [27–29]. In accordance with this view, 3-month-old APP23 mice expressing APP751 with the Swedish double mutation (K670N, M671L) displayed learning and memory deficits during acquisition and the probe trial of the Morris water maze before the onset of amyloid deposits [30]. Likewise, APP/IND mice with the Indiana V717F mutation showed altered synaptic morphology and electro-physiological changes without Aβ deposits [31]. Thus, soluble oligomeric Aβ is thought to be a better target of AD immunotherapy than monomeric and fibrillar Aβ [32,33]. Our preliminary data indicate that intracranial injection of scFv59 is effective in improving learning and memory deficits in Tg2576 mice.
Aβ makes deposits in the brain of the AD mouse model; however, the precise mechanisms through which this happens are not clearly known. A number of studies have demonstrated that cultured microglial cells are capable of internalizingAβ1–42 aggregates [34,35]. Consistent with this view, research reports in the early stage suggested Fc receptor- mediated phagocytosis by microglia as the mechanism of Aβ clearance [36,37]. Later, however, several groups suggested that it is another mechanism, disaggregation, and removal of Aβ deposits by direct interaction of antibodies with Aβ. Solomon et al. [38] demonstrated in vitro that Aβ antibodies physically interacted with Aβ deposits resulting in disaggregation of the deposits in vitro. Bacskai et al. [39] demonstrated that F(ab′)2 fragments prepared from an mouse anti-Aβ antibody reduced amyloid deposits as effectively as the intact antibody when applied topically to the cortex of transgenic mice. Wilcock et al. [40] reported that mouse anti-Aβ antibodies reducedAβ deposition by mechanisms, both independent of and associated with microglial activation. Our results also support non-Fc-mediated mechanisms in clearing Aβ deposits in vivo.
Another postulated mechanism is sequestration of Aβ by antibodies in blood and CNS, resulting in removal of Aβ from the brain (peripheral antibodies acting as a sink for Aβ). DeMattos et al. [41] demonstrated that peripheral administration of mouse monoclonal antibody to an AD mouse model reduced Aβ deposits in the brain, without direct interaction of the antibody with the Aβ deposits in the brain, and postulated that sequestration of Aβ by antibodies in blood and CNS might result in removal of Aβ. We are currently testing whether other therapeutic modalities by which anti-Aβ scFv is peripherally delivered in the circulation are effective in treating an AD mouse model.
Acknowledgments
We thank Dr. Karen Hsiao Ashe for providing Tg2576 mice, Dr. Maria Belousova and Timothy Gunn for technical assistance, and Karen Minter for manuscript preparation. This research was supported in part by grants from the National Institutes of Health (NS43947) and the Alzheimer’s Association (ZEN-03-5834).
Footnotes
Publisher's Disclaimer: This PDF receipt will only be used as the basis for generating PubMed Central (PMC) documents. PMC documents will be made available for review after conversion (approx. 2–3 weeks time). Any corrections that need to be made will be done at that time. No materials will be released to PMC without the approval of an author. Only the PMC documents will appear on PubMed Central -- this PDF Receipt will not appear on PubMed Central.
References
- 1.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 2.Janus C, Pearson J, McLaurin J, Mathews PM, Jiang Y, Schmidt SD, Chishti MA, Horne P, Heslin D, French J, Mount HT, Nixon RA, Mercken M, Bergeron C, Fraser PE, George-Hyslop P, Westaway D. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- 3.Morgan D, Diamond DM, Gottschall PE, Ugen KE, Dickey C, Hardy J, Du K, Jantzen P, DiCarlo G, Wilcock D, Connor K, Hatcher J, Hope C, Gordon M, Arendash GW. A beta peptide vaccination prevents memory loss in an animal model of Alzheimer’s disease. Nature. 2000;408:982–985. doi: 10.1038/35050116. [DOI] [PubMed] [Google Scholar]
- 4.Check E. Nerve inflammation halts trial for Alzheimer’s drug. Nature. 2002;415:462. doi: 10.1038/415462a. [DOI] [PubMed] [Google Scholar]
- 5.Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 6.Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- 7.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 8.Bacskai BJ, Kajdasz ST, McLellan ME, Games D, Seubert P, Schenk D, Hyman BT. Non-Fc-mediated mechanisms are involved in clearance of amyloid-beta in vivo by immunotherapy. J Neurosci. 2002;22:7873–7878. doi: 10.1523/JNEUROSCI.22-18-07873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Viti F, Nilsson F, Demartis S, Huber A, Neri D. Design and use of phage display libraries for the selection of antibodies and enzymes. Methods Enzymol. 2000;326:480–505. doi: 10.1016/s0076-6879(00)26071-0. [DOI] [PubMed] [Google Scholar]
- 10.Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 11.Fukuchi K, Pham D, Hart M, Li L, Lindsey JR. Amyloid-beta protein deposition in skeletal muscle of transgenic mice: possible model of inclusion body myopathy. Am J Pathol. 1998;153:1687–1693. doi: 10.1016/s0002-9440(10)65682-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fukuchi K, Li L, Hart M, Lindsey JR. Accumulation of amyloid-beta protein in exocrine glands of transgenic mice overexpressing a carboxyl terminal portion of amyloid protein precursor. Int J Exp Pathol. 2000;81:231–239. doi: 10.1046/j.1365-2613.2000.00156.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yamamoto T, Hirano A. A comparative study of modified Bielschowsky, Bodian and thioflavine S stains on Alzheimer’s neurofibrillary tangles. Neuropathol Appl Neurobiol. 1986;12:3–9. doi: 10.1111/j.1365-2990.1986.tb00677.x. [DOI] [PubMed] [Google Scholar]
- 14.Li L, Cao D, Garber DW, Kim H, Fukuchi K. Association of aortic atherosclerosis with cerebral beta-amyloidosis and learning deficits in a mouse model of Alzheimer’s disease. Am J Pathol. 2003;163:2155–2164. doi: 10.1016/s0002-9440(10)63572-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dahlgren KN, Manelli AM, Stine WB, Jr, Baker LK, Kraffit GA, LaDu MJ. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- 17.Check E. Nerve inflammation halts trial for Alzheimer’s drug. Nature. 2002;415:462. doi: 10.1038/415462a. [DOI] [PubMed] [Google Scholar]
- 18.Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- 19.Orgogozo JM, Gilman S, Dartigues JF, Laurent B, Puel M, Kirby LC, Jouanny P, Dubois B, Eisner L, Flitman S, Michel BF, Boada M, Frank A, Hock C. Subacute meningoencephalitis in a subset of patients with AD after Abeta42 immunization. Neurology. 2003;61:46–54. doi: 10.1212/01.wnl.0000073623.84147.a8. [DOI] [PubMed] [Google Scholar]
- 20.Lee M, Bard F, Johnson-Wood K, Lee C, Hu K, Griffith SG, Black RS, Schenk D, Seubert P. Abeta42 immunization in Alzheimer’s disease generates Abeta N-terminal antibodies. Ann Neurol. 2005;58:430–435. doi: 10.1002/ana.20592. [DOI] [PubMed] [Google Scholar]
- 21.Liu R, Yuan B, Emadi S, Zameer A, Schulz P, McAllister C, Lyubchenko Y, Goud G, Sierks MR. Single chain variable fragments against beta-amyloid (Abeta) can inhibit Abeta aggregation and prevent abeta-induced neurotoxicity. Biochemistry. 2004;43:6959–6967. doi: 10.1021/bi049933o. [DOI] [PubMed] [Google Scholar]
- 22.Paganetti P, Calanca V, Galli C, Stefani M, Molinari M. beta-site specific intrabodies to decrease and prevent generation of Alzheimer’s Abeta peptide. J Cell Biol. 2005;168:863–868. doi: 10.1083/jcb.200410047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hock C, Konietzko U, Papassotiropoulos A, Wollmer A, Streffer J, von Rotz RC, Davey G, Moritz E, Nitsch RM. Generation of antibodies specific for beta-amyloid by vaccination of patients with Alzheimer disease. Nat Med. 2002;8:1270–1275. doi: 10.1038/nm783. [DOI] [PubMed] [Google Scholar]
- 24.Lee M, Bard F, Johnson-Wood K, Lee C, Hu K, Griffith SG, Black RS, Schenk D, Seubert P. Abeta42 immunization in Alzheimer’s disease generates Abeta N-terminal antibodies. Ann Neurol. 2005;58:430–435. doi: 10.1002/ana.20592. [DOI] [PubMed] [Google Scholar]
- 25.Town T, Tan J, Sansone N, Obregon D, Klein T, Mullan M. Characterization of murine immunoglobulin G antibodies against human amyloid-beta1–42. Neurosci Lett. 2001;307:101–104. doi: 10.1016/s0304-3940(01)01951-6. [DOI] [PubMed] [Google Scholar]
- 26.Spooner ET, Desai RV, Mori C, Leverone JF, Lemere CA. The generation and characterization of potentially therapeutic Abeta antibodies in mice: differences according to strain and immunization protocol. Vaccine. 2002;21:290–297. doi: 10.1016/s0264-410x(02)00464-4. [DOI] [PubMed] [Google Scholar]
- 27.Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Kraffit GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1–42 are potent central nervous system neurotoxins. Proc Natl Acad Sci USA. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dahlgren KN, Manelli AM, Stine WB, Jr, Baker LK, Krafft GA, LaDu MJ. Oligomeric and fibrillar species of amyloid-beta peptides differentially affect neuronal viability. J Biol Chem. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- 29.Wang SS, Becerra-Arteaga A, Good TA. Development of a novel diffusion-based method to estimate the size of the aggregated Abeta species responsible for neurotoxicity. Biotechnol Bioeng. 2002;80:50–59. doi: 10.1002/bit.10347. [DOI] [PubMed] [Google Scholar]
- 30.Van Dam D, D’Hooge R, Staufenbiel M, Van GC, Van MF, De Deyn PP. Age-dependent cognitive decline in the APP23 model precedes amyloid deposition. Eur J Neurosci. 2003;17:388–396. doi: 10.1046/j.1460-9568.2003.02444.x. [DOI] [PubMed] [Google Scholar]
- 31.Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer’s disease mouse models. Proc Natl Acad Sci USA. 1999;96:3228–3233. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klein WL. Abeta toxicity in Alzheimer’s disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem Int. 2002;41:345–352. doi: 10.1016/s0197-0186(02)00050-5. [DOI] [PubMed] [Google Scholar]
- 33.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 34.Paresce DM, Chung H, Maxfield FR. Slow degradation of aggregates of the Alzheimer’s disease amyloid beta-protein by microglial cells. J Biol Chem. 1997;272:29390–29397. doi: 10.1074/jbc.272.46.29390. [DOI] [PubMed] [Google Scholar]
- 35.Webster SD, Yang AJ, Margol L, Garzon-Rodriguez W, Glabe CG, Tenner AJ. Complement component C1q modulates the phagocytosis of Abeta by microglia. Exp Neurol. 2000;161:127–138. doi: 10.1006/exnr.1999.7260. [DOI] [PubMed] [Google Scholar]
- 36.Schenk D, Barbour R, Dunn W, Gordon G, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Liao Z, Lieberburg I, Motter R, Mutter L, Soriano F, Shopp G, Vasquez N, Vandevert C, Walker S, Wogulis M, Yednock T, Games D, Seubert P. Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature. 1999;400:173–177. doi: 10.1038/22124. [DOI] [PubMed] [Google Scholar]
- 37.Bard F, Cannon C, Barbour R, Burke RL, Games D, Grajeda H, Guido T, Hu K, Huang J, Johnson-Wood K, Khan K, Kholodenko D, Lee M, Lieberburg I, Motter R, Nguyen M, Soriano F, Vasquez N, Weiss K, Welch B, Seubert P, Schenk D, Yednock T. Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med. 2000;6:916–919. doi: 10.1038/78682. [DOI] [PubMed] [Google Scholar]
- 38.Solomon B, Koppel R, Frankel D, Hanan-Aharon E. Disaggregation of Alzheimer beta-amyloid by site-directed mAb. Proc Natl Acad Sci USA. 1997;94:4109–4112. doi: 10.1073/pnas.94.8.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bacskai BJ, Kajdasz ST, McLellan ME, Games D, Seubert P, Schenk D, Hyman BT. Non-Fc-mediated mechanisms are involved in clearance of amyloid-beta in vivo by immunotherapy. J Neurosci. 2002;22:7873–7878. doi: 10.1523/JNEUROSCI.22-18-07873.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wilcock DM, DiCarlo G, Henderson D, Jackson J, Clarke K, Ugen KE, Gordon MN, Morgan D. Intracranially administered anti-Abeta antibodies reduce beta-amyloid deposition by mechanisms both independent of and associated with microglial activation. J Neurosci. 2003;23:3745–3751. doi: 10.1523/JNEUROSCI.23-09-03745.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.DeMattos RB, Bales KR, Cummins DJ, Dodart JC, Paul SM, Holtzman DM. Peripheral anti-Abeta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA. 2001;98:8850–8855. doi: 10.1073/pnas.151261398. [DOI] [PMC free article] [PubMed] [Google Scholar]