


Abstract
Nucleic acid-based biochemical assays are crucial to modern biology. Key applications, such as detection of bacterial, viral and fungal pathogens, require detailed knowledge of assay sensitivity and specificity to obtain reliable results. Improved methods to predict assay performance are needed for exploiting the exponentially growing amount of DNA sequence data and for reducing the experimental effort required to develop robust detection assays. Toward this goal, we present an algorithm for the calculation of sequence similarity based on DNA thermodynamics. In our approach, search queries consist of one to three oligonucleotide sequences representing either a hybridization probe, a pair of Padlock probes or a pair of PCR primers with an optional TaqMan™ probe (i.e. in silico or ‘virtual’ PCR). Matches are reported if the query and target satisfy both the thermodynamics of the assay (binding at a specified hybridization temperature and/or change in free energy) and the relevant biological constraints (assay sequences binding to the correct target duplex strands in the required orientations). The sensitivity and specificity of our method is evaluated by comparing predicted to known sequence tagged sites in the human genome. Free energy is shown to be a more sensitive and specific match criterion than hybridization temperature.
INTRODUCTION
Successful nucleic acid-based detection assays must be able to uniquely identify small amounts of potentially diverse pathogen DNA or RNA in samples that contain large amounts of nucleic acid sequence from background species. For example, PCR assays to detect the presence of HIV in human blood samples must be able to amplify imperfectly conserved regions of the HIV genome without amplifying any part of the human genome.
In silico assay screening can identify unreliable detection assays and reduce assay development costs by querying sequence databases with assay oligonucleotides (oligos) to predict both potential false positive matches and the absence of expected matches (i.e. false negatives). While there are a number of existing software programs for assay-dependent database searching, they are limited in the choice of biochemical assay format, target database size (a critical consideration for sampling in the presence of a complex background) and sensitivity of the search algorithm. Many existing tools can search a database with a pair of PCR primers, including e-PCR (1), me-PCR (2), PRIMEX (3), simPCR (4), BiSearch (5), SPCR (6) and iPCRess (7). However, none of these programs support additional assay formats (e.g. TaqMan™ PCR, an assay commonly used for pathogen detection). Additionally, web-based and interactive tools, including PUNS (8), the website insilico.ehu.es (9) and Amplify (10), are typically restricted to searching a single genome. While searching a single genome is useful for validating or annotating expected true positive matches, generation of robust detection assays requires testing a target database that includes all available near neighbor and background sequences. Given the ability to screen a large number of targets, tools must also be able to accurately predict possible assay matches in the target database. To identify matches, most of the existing tools for in silico PCR rely on a heuristic definition of sequence similarity based on the number of mismatches (non-Watson and Crick base pairs) and the number of gaps (insertions and deletions in the primer-template duplexes). As we will demonstrate subsequently, using a thermodynamic similarity measure yields improved sensitivity and specificity. Finally, many programs fail to consider all possible configurations of assay oligos that can result in a positive detection. For example, in addition to the PCR amplicon produced by the forward and reverse primers, palindromic template sequences can generate unintended amplicons when a single oligo serves as both forward and reverse primer. Our approach tests for these unintended matches.
MATERIALS AND METHODS
A common feature of many nucleic acid-based biochemical assays is that detection requires an initial temperature-dependent hybridization of assay oligos to complementary target sequences (which may then be followed by enzymatic base extension or ligation reactions). The first step in predicting potential matches between assay oligos and target sequences is to identify all potential binding sites in the target sequences that are consistent with user-defined thresholds in hybridization melting temperature (TM) and/or free energy change (ΔG = ΔH − TΔS). The melting temperature is calculated using
| 1 |
where ΔH and ΔS are computed using the nearest neighbor free energy parameters of SantaLucia (11), R is the universal gas constant and CT is the total molar concentration of strands.
Binding site locations are identified in two steps. First, initial match candidates are identified by an exhaustive enumeration of exact short word matches between query and target sequences (3–8 bases, as specified by the user). Queries that contain degenerate bases (e.g. R = A or G) are expanded into multiple queries containing only real bases (i.e. A, T, G and C). Second, a thermodynamic alignment that minimizes the computed free energy change (ΔG = ΔH − TΔS) between the unbound and bound states is used to predict the binding of the assay oligo to the target DNA. The free-energy change is a function of a pair-wise sequence alignment and is parameterized in terms of the stacking contributions of pairs of nearest neighbor bases (12): each term represents the free-energy contribution of four bases, two pairs of adjacent bases facing each other on the opposite strands of a DNA duplex. Due to this nearest neighbor dependence, the exact dynamic programming calculation of the thermodynamically most probable alignment (that minimizes ΔG) for a sequence of length N requires O(N3) operations (13). While O(N3) thermodynamic alignment algorithms have been implemented for DNA (14,15), the need to screen large numbers of assay oligos against large sequence databases makes the O(N3) cost very expensive, even for short (20–25 base) sequences. A computationally feasible O(N2) algorithm for computing the alignment that minimizes the free energy at either a specified annealing temperature or at the predicted TM has been proposed (16,17). This approach uses an O(N2) dynamic programming algorithm to align two sequences at a specified alignment temperature. The calculation of the alignment that minimizes the free energy at the predicted TM uses the Dinkelbach algorithm to iteratively adjust the alignment temperature until it equals the TM of the aligned duplex. However, calculation of alignments using O(N2) dynamic programming requires simplification of the duplex free-energy function and the introduction of free-energy parameters not specified in the formal duplex free-energy function. In particular, the exact free-energy dependencies on base-dependent duplex initiation terms, gaps and consecutive mismatches are not included in the simplified free-energy function. These omissions and modifications decrease the accuracy of the computed duplex free energy.
To avoid both the high cost of the exact O(N3) alignment and to reduce the approximation error due to a simplified free-energy function, we developed a novel, O(N2) thermodynamic alignment algorithm that incorporates and improves on the algorithm of Leber et al. (17). Our approach treats the simplified free-energy function of Leber et al. as a surrogate (18) free-energy function whose only purpose is to generate an alignment that approximately minimizes the full duplex free-energy function. Our algorithm divides the calculation of a thermodynamic alignment into two stages. In the first stage, dynamic programming is used to compute the duplex alignment that minimizes a surrogate free-energy function at the specified alignment temperature. In the second stage, the alignment generated in the first stage is used to evaluate the full duplex free energy function described in ref. (13). The full duplex free energy function provides all thermodynamic parameters of interest: ΔH, ΔS and TM. Both stage one and two can be incorporated into the Dinkelbach algorithm (17) and iterated to produce alignments that approximately minimize the full free energy of the duplex at its predicted melting temperature.
To insure that the surrogate free-energy function produces alignments that approximately minimize the full duplex free-energy function, we determined a new, optimized set of dynamic programming parameters. The parameters in the surrogate function that are not explicitly specified by the full duplex free-energy function were determined by simulated annealing-based minimizations of the average full duplex free-energy function (evaluated at the alignments that exactly optimize the surrogate function) for a test set of 104 randomly generated sequence pairs and a range of hybridization temperatures (35–65°C). The random sequence pairs were approximately 25 bases long, had 50% G + C composition and contained insertions, deletions and mismatches. We assumed that all parameters depend linearly on temperature and used linear regressions to compute the slope and intercept for each surrogate parameter, as shown in Table 1.
Table 1.
Optimized surrogate function parameters for computing thermodynamic alignments
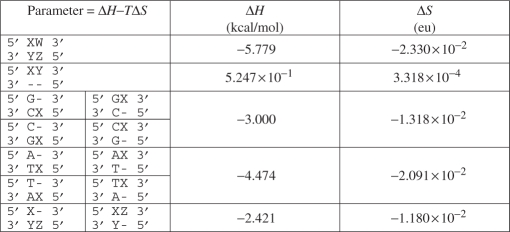 |
The bases W, X, Y and Z represent any of the four bases (A, T, G or C), with the constraint that neither X and Y nor W and Z form a Watson and Crick base pair. The ‘–’ symbol represents a gap. Each parameter is a linear function of temperature: ΔH − TΔS. While the ΔS and ΔH values have units of entropy and enthalpy, they are for alignment purposes only.
To assess the accuracy of our thermodynamic alignment algorithm, alignments that minimized the surrogate free-energy function were computed for randomly generated test sets of 104 sequence pairs (containing insertions, deletions and mutations) with 25%, 50% and 75% G + C composition and compared to the exact O(N3) alignments produced by the DINAmelt server (14). The temperature-dependent error in ΔG is shown in Figure 1. Both the single stage method (of Leber et al.) and the two stage method systematically overestimate ΔG compared to the values produced by the DINAmelt server. With the exception of the region above 76°C for the 75% G + C test set, our two stage thermodynamic alignment algorithm (i.e. the full duplex ΔG evaluated at the alignment produced using optimized surrogate parameters) produces more accurate estimates of duplex free energy than the fixed temperature alignment, dynamic programming algorithm of Leber et al. The observed improvement in ΔG is significant, considering that the free-energy contribution of a A-T pair at 37°C ranges from −0.58 to −1.45 kcal/mol [depending on the identity of the neighboring pair (13)]. The accuracy of our two stage ΔG approximation improves as duplex G + C content increases. However, this is most likely an artifact that arises from whether A-T pairs at the ends of a duplex are treated as Watson and Crick base pairs or dangling single-stranded DNA. Our implementation considers them to be Watson and Crick base pairs, but the DINAmelt server treats them as dangling single stranded DNA [which is the lower energy configuration (19)].
Figure 1.

The temperature-dependent thermodynamic alignment approximation error evaluated using randomly generated test sets with (A) 25% G + C content, (B) 50% G + C content and (C) 75% G + C content. The solid black lines show the average error in the free energy computed using only the non-optimized dynamic programming parameters of Leber et al. The dotted red lines show the average error in the two-stage free energy computed by first producing an alignment using the dynamic programming parameters of Leber et al. and then evaluating the free energy using the full duplex free-energy function of SantaLucia. Finally, the solid red lines show the average error in the two-stage free energy computed by first producing an alignment using optimized dynamic programming parameters and then evaluating the free energy using the full duplex free-energy function of SantaLucia. For all curves, the approximation error, ΔΔG, is defined as the approximate ΔG minus the exact ΔG computed by the DINAmelt server (14). The standard deviations about each point in all graphs are less than or equal to 1.5 × 10−2 kcal/mol. Each randomly generated test set of 104, 25-base sequence pairs (containing insertions, deletions and mutations) was pre-screened to insure that no sequence pairs in the test set were also in the training set used to optimize the dynamic programming parameters.
To measure the relative speedup of our O(N2) two-stage thermodynamic algorithm compared to the exact O(N3) alignments produced by the DINAmelt server, we computed the average ratio of wall clock execution times for both algorithms using randomly generated test sets with a range of duplex sizes relevant for nucleic acid-based assays. As shown in Figure 2, the size of the observed speedup varies from a factor of 1 (no speedup) for a 15 base duplex (the size of a small PCR primer) to a factor of 8.5 for a 35 base duplex (the size of a TaqMan™ probe). As one might expect, the two-stage thermodynamic alignment algorithm has an increased computational cost compared to the single stage (fixed temperature, dynamic programming) algorithm of Leber et al. For all duplex lengths shown, the increased computational cost of the second stage (evaluating the full duplex free energy function using the alignment produced by the first stage) makes the two-stage algorithm about 20% slower than the single-stage method. The average wall clock time required to compute a single two-stage thermodynamic alignment using a 2.4 GHz Xeon CPU ranges from 1 × 10−4 s to 1.8 × 10−4 s for a 15 base and 35 base duplex, respectively. As we will demonstrate subsequently, searches using our O(N2) two-stage algorithm and thousands of PCR primers against the human genome are feasible on small to mid-sized computer clusters.
Figure 2.

Comparing the time to compute single stage (fixed temperature, dynamic programming only) and two-stage thermodynamic alignments to the time to compute O(N3) exact alignments. Speedup is computed as the average of the wall clock time required to compute an exact O(N3) alignment divided by the wall clock time required to compute either a one or two-stage thermodynamic alignment. Wall clock time was computed using the Unix time command to measure the run time required to align 105 random duplexes (containing insertions, deletions and mutations) of the specified approximate length (insertions and deletions can change the duplex length). The exact alignments were performed using the hybrid-min program that is included with the DINAMelt server (UNAfold) package (14). Error bars were computed by averaging the evaluation times of 10 different randomly generated sets of duplexes for each duplex length.
The algorithms presented in this article have been implemented in ‘ThermonucleotideBLAST’, a cross-platform command line tool written in C++ and freely distributed under the BSD open source license from http://public.lanl.gov/jgans/tntblast. The code has been tested on Linux, Windows and OS X, and has been parallelized using the MPI API for distributed memory machines and the OpenMP API for shared memory machines. Additional documentation for building and running the program is provided at the website listed above.
RESULTS AND DISCUSSION
In silico PCR is a common example of assay-specific searching for which a number of tools already exist. We compared the specificity and sensitivity of the e-PCR program and our approach (implemented in the ThermonucleotideBLAST program) for the task of identifying sequence-tagged sites (STSs), which are unique sites in the human genome defined by a pair of PCR primers and an amplicon length. Using a methodology based on Rotmistrovsky et al. (1), we constructed an STS test set that contained 2185 STS primer pairs. Primer pairs were included in the test set if they occurred once and only once in both the GeneBridge 4 (20) and Stanford G3 (21) radiation hybrid panels, and were reported to match a single unique site on a single human chromosome. When evaluating specificity and sensitivity, the first predicted match of an STS to the correct chromosome was counted as a true positive, while any subsequent matches to the correct chromosome were counted as false positives (1). Each match of an STS to an incorrect chromosome was counted as a false positive. An STS that was not correctly assigned to the proper chromosome was counted as a false negative.
While significantly slower than e-PCR, ThermonucleotideBLAST can search thousands of PCR primers against the human genome. Using a 200 node cluster of 1.2 GHz Pentium III Mobile CPUs and reading the sequence data from an NFS file server, ThermonucleotideBLAST required 2 h to search the human genome using the 2185 STS primer pairs. Over 99% of this search time was spent computing two-stage thermodynamic alignments. The e-PCR performed the same search in a significantly shorter time: 25 min when run on a single 1.2 GHz cluster node with 2 GB of RAM (with two gaps and two mismatches allowed and a word size of 12 bases).
Figure 3 has five panels that show the same receiver operator curve for e-PCR plotted with the different receiver operator curves for ThermonucleotideBLAST that were supplemented with an additional match criterion of 0–4 bases of exact match between the 3′ end of the PCR primer and the target sequence. The additional use of an exact match criterion (i.e. at the 3′ end of the PCR primer) is a heuristic rule to account for the DNA polymerase's reduced efficiency extending primers that contain 3′ terminal mismatches. A similar strategy is employed by e-PCR, which searches for primer-target matches using a hash table-based search of the last W bases at the 3′ end of the primer (1). The primary match criteria used by ThermonucleotideBLAST were (i) a ΔG match criteria ranging from −20 to −9 kcal/mol (solid black line), (ii) a TM (computed at 37°C) match criteria ranging from 35°C to 60°C and (iii) a TM (computed using the Dinkelbach algorithm) match criteria ranging from 35°C to 60°C.
Figure 3.

Comparing e-PCR and ThermonucleotideBLAST in STS search specificity and sensitivity. Comparisons were made by searching a set of 2185 STS primer pairs against the complete human genome (build 35, version 1). Specificity is defined as TP/(TP + FP) and sensitivity is defined as TP/(TP + FN), where TP is the number of true positives, FP is the number of false positives and FN is the number of false negatives. The e-PCR was run using W = 12 (word size) and M = 200 (allowed deviation from the expected amplicon size) and discontiguous words (DW), as opposed to contiguous words (CW), were activated with F = 3, as described in ref. (1). In order of increasing sensitivity, the plotted e-PCR points were computed using the following parameters: (CW, g = 0, n = 0), (CW, g = 0, n = 1), (CW, g = 1, n = 1), (DW, g = 0, n = 1), (DW, g = 1, n = 1), (DW, g = 2, n = 1), (DW, g = 1, n = 2) and (DW, g = 2, n = 2), where g is the number of allowed gaps and n is the number of allowed mismatches within the 12 base word. ThermonucleotideBLAST was run using single-primer-PCR = False (to disable searching for PCR amplicons produced by a single amplicon), W = 7 (requiring an exact match of seven bases to initiate a thermodynamic alignment), s = 0.05 (salt concentration in M), t = 9 × 10−7 (strand concentration in M), l = 1000 (maximum allowed amplicon size) and dangle5 = False and dangle3 = False (to disable the use of dangling end bases at both ends of a thermodynamic alignment). Each of the five graphs shows the effects of requiring an increasing the number of exact matches at the 3′ end of each PCR primer—however, the same e-PCR curve is reproduced in each graph. To compare the specificity and sensitivity of different match criteria, ThermonucleotideBLAST searches were performed using ΔG (solid black curve), TM at 37°C (solid red curve) and TM computed using the Dinkelbach algorithm (solid blue curve).
As shown in Figure 3, the ΔG match criterion consistently outperforms e-PCR in search sensitivity. Furthermore, it matches e-PCR in specificity when supplemented with the additional, PCR-specific heuristic requiring an exact match of at least three bases at the 3′ end of each primer. Finally, the ΔG match criterion is both more sensitive and specific than either of the melting temperature-based criteria.
ACKNOWLEDGEMENTS
The authors would like to thank N. Pawley for helpful discussions. This research was supported in part by the Department of Homeland Security Science and Technology Directorate, award HSHQDC-07-X-00 055 and the Los Alamos National Laboratory Directed Research and Development Program (LDRD 20070010DR). Funding to pay the Open Access publication charges for this article was provided by Los Alamos National Laboratory Directed Research and Development Program (LDRD 20070010DR).
Conflict of interest statement. None declared.
REFERENCES
- 1.Rotmistrovsky K, Jang W, Schuler G. A web server for performing electronic PCR. Nucleic Acids Res. 2004;32:W108–W112. doi: 10.1093/nar/gkh450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Murphy K, Raj T, Winters R, White P. me-PCR: a refined ultrafast algorithm for identifying sequence-defined genomic elements. Bioinformatics. 2004;20:588–590. doi: 10.1093/bioinformatics/btg466. [DOI] [PubMed] [Google Scholar]
- 3.Lexa M, Valle G. PRIMEX: rapid identification of oligonucleotide matches in whole genomes. Bioinformatics. 2003;19:2486–2488. doi: 10.1093/bioinformatics/btg350. [DOI] [PubMed] [Google Scholar]
- 4.Rubin E, Levy A. A mathematical model and a computerized simulation of PCR using complex templates. Nucleic Acids Res. 1996;24:3538–3545. doi: 10.1093/nar/24.18.3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tusnady G, Simon I, Varadi A, Aranyi T. BiSearch: primer-design and search tool for PCR on bisulfite-treated genomes. Nucleic Acids Res. 2005;33:e9. doi: 10.1093/nar/gni012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cao Y, Wang L, Xu K, Kou C, Zhang Y, Wei G, He J, Wang Y, Zhao L. Information theory-based algorithm for in silico prediction of PCR products with whole genomic sequences as templates. BMC Bioinform. 2005;6:190. doi: 10.1186/1471-2105-6-190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Slater G. iPCRess; 2007. Available online at http://www.ebi.ac.uk/~guy/exonerate/ [Google Scholar]
- 8.Boutros P, Okey A. PUNS: transcriptomic- and genomic-in silico PCR for enhanced primer design. Bioinformatics. 2004;20:2399–2400. doi: 10.1093/bioinformatics/bth257. [DOI] [PubMed] [Google Scholar]
- 9.Bikandi J, Millan R, Rementeria A, Garaizar J. In silico analysis of complete bacterial genomes: PCR, AFLP-PCR and endonuclease restriction. Bioinformatics. 2004;20:798–799. doi: 10.1093/bioinformatics/btg491. [DOI] [PubMed] [Google Scholar]
- 10.Engels W. Contributing software to the internet: the amplify program. Trends Biochem. Sci. 1993;18:448–450. doi: 10.1016/0968-0004(93)90148-g. [DOI] [PubMed] [Google Scholar]
- 11.SantaLucia J. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl Acad. Sci. USA. 1998;95:1460–1465. doi: 10.1073/pnas.95.4.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crothers D, Zimm B. Theory of the melting transition of synthetic polynucleotides: evaluation of the stacking free energy. J. Mol. Biol. 1964;9:1–9. doi: 10.1016/s0022-2836(64)80086-3. [DOI] [PubMed] [Google Scholar]
- 13.SantaLucia J, Hicks D. The thermodynamics of DNA structural motifs. Annu. Rev. Biophys. Biomol. Struct. 2004;33:415–440. doi: 10.1146/annurev.biophys.32.110601.141800. [DOI] [PubMed] [Google Scholar]
- 14.Markham N, Zuker M. DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Res. 2005;33:W577–W581. doi: 10.1093/nar/gki591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zuker M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003;31:3406–3415. doi: 10.1093/nar/gkg595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaderali L, Schliep A. Selecting signature oligonucleotides to identify organisms using DNA arrays. Bioinformatics. 2002;18:1340–1349. doi: 10.1093/bioinformatics/18.10.1340. [DOI] [PubMed] [Google Scholar]
- 17.Leber M, Kaderali L, Schonhuth A, Schrader R. A fractional programming approach to efficient DNA melting temperature calculation. Bioinformatics. 2005;21:2375–2382. doi: 10.1093/bioinformatics/bti379. [DOI] [PubMed] [Google Scholar]
- 18.Queipo N, Haftka R, Shyy W, Goel T, Vaidyanathan R, Tucker P. Surrogate-based analysis and optimization. Progress in Aerospace Sciences. 2005;41:1–28. [Google Scholar]
- 19.Bommarito S, Peyret N, SantaLucia J. Thermodynamic parameters of DNA sequences with dangling ends. Nucleic Acids Res. 2000;28:1929–1934. doi: 10.1093/nar/28.9.1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gyapay G, Schmitt K, Fizames C, Jones H, Vega-Czarny N, Spillett D, Muselet D, Prud'homme J, Dib C, Auffray C, et al. A radiation hybrid map of the human genome. Hum. Mol. Genet. 1996;5:339–346. doi: 10.1093/hmg/5.3.339. [DOI] [PubMed] [Google Scholar]
- 21.Stewart E, McKusick K, Aggarwal A, Bajorek E, Brady S, Chu A, Fang N, Hadley D, Harris M, Hussain S, et al. An STS-based radiation hybrid map of the human genome. Genome Res. 1997;7:422–433. doi: 10.1101/gr.7.5.422. [DOI] [PubMed] [Google Scholar]