


Abstract
The selective degradation of mutated mitochondrial DNA (mtDNA) molecules is a potential strategy to re-populate cells with wild-type (wt) mtDNA molecules and thereby alleviate the defective mitochondrial function that underlies mtDNA diseases. Zinc finger nucleases (ZFNs), which are nucleases conjugated to a zinc-finger peptide (ZFP) engineered to bind a specific DNA sequence, could be useful for the selective degradation of particular mtDNA sequences. Typically, pairs of complementary ZFNs are used that heterodimerize on the target DNA sequence; however, conventional ZFNs were ineffective in our system. To overcome this, we created single-chain ZFNs by conjugating two FokI nuclease domains, connected by a flexible linker, to a ZFP with an N-terminal mitochondrial targeting sequence. Here we show that these ZFNs are efficiently transported into mitochondria in cells and bind mtDNA in a sequence-specific manner discriminating between two 12-bp long sequences that differ by a single base pair. Due to their selective binding they cleave dsDNA at predicted sites adjacent to the mutation. When expressed in heteroplasmic cells containing a mixture of mutated and wt mtDNA these ZFNs selectively degrade mutated mtDNA, thereby increasing the proportion of wt mtDNA molecules in the cell. Therefore, mitochondria-targeted single-chain ZFNs are a promising candidate approach for the treatment of mtDNA diseases.
INTRODUCTION
One of the main functions of mitochondria is to provide the cell with energy in the form of ATP; however, mitochondria also play a role in other important cellular processes such as Fe-S cluster biosynthesis, calcium homoeostasis, apoptosis or tumorigenesis. Mammalian mitochondria have their own small, circular, 16.5-kb genome that encodes two rRNAs, 22 tRNAs and 13 subunits of the oxidative phosphorylation machinery. Correct expression of mitochondrial DNA (mtDNA) is essential for mitochondrial function; therefore, mutation to mtDNA can have severe consequences for the cell by disrupting oxidative phosphorylation. Currently, more than 100 point mutations and large-scale rearrangements in the human mtDNA are known to be associated with a wide spectrum of clinical manifestations, ranging from progressive muscle weakness to fatal infantile diseases (1,2).
There are no effective treatments for diseases caused by mtDNA mutation, in part due to the difficulty of expressing exogenous nucleic acids within mitochondria; consequently, alternative approaches to the treatment of these diseases have been sought (3,4). One approach is to use the fact that each cell harbours hundreds of mtDNA molecules (1), so that deleterious mtDNA mutations commonly co-exist in the same cell with wild-type (wt) mtDNA, a phenomenon called heteroplasmy. In heteroplasmic cells, the phenotype of a pathogenic mtDNA mutation is determined by the ratio of mutant and wt genomes, and pathology is only observed above a threshold proportion of mutated mtDNAs (5). As cells have a tendency to maintain a fixed number of mtDNA molecules (6), one treatment option is the selective elimination of mutated mtDNA, thus allowing the cell to re-populate itself with wt mtDNA, thereby correcting the defective phenotype (7). This has been achieved by targeting the restriction endonuclease (RE) SmaI or XmaI to mitochondria in cells in culture, where it selectively cleaved a unique site created in mtDNA by the m.8993T>G mutation underlying the mtDNA diseases neurogenic muscle weakness, ataxia and retinitis pigmentosa (NARP) and maternally inherited Leigh's syndrome (MILS) (8,9). Similarly, in a heteroplasmic murine cell line containing two types of mtDNA molecules, one with two PstI recognition sites and another with none, mitochondrial expression of the PstI restriction enzyme significantly depleted the haplotype with two PstI sites (10). This strategy can also be applied in vivo, as was shown in a heteroplasmic mouse model containing the BALB and NZB mtDNA haplotypes, where only the BALB haplotype has a recognition site for cleavage by the ApaLI RE (11). Mice transfected with mitochondrially targeted ApaLI showed a rapid shift in heteroplasmy toward the NZB variant in muscle and brain (11). Furthermore, the same mouse system was used to verify that mitochondrially expressed REs could shift heteroplasmy when a mutation created a new restriction site in addition to the ones normally present in the wt mtDNA (12).
These studies show that targeting mutated mtDNA molecules for degradation has potential as a therapeutic strategy. However, the use of natural REs limits its application to those sequences created by a pathological mutation that do not occur in wt mtDNA, and for which an RE exists. One way around this constraint is to develop sequence-specific nucleases that can be designed to cleave any target sequence. Zinc finger technology allows the engineering of zinc-finger proteins (ZFPs) that can bind any predetermined DNA sequence (13). Fusing a particular ZFP to a nuclease domain creates a zinc-finger nuclease (ZFN) that can cleave DNA adjacent to the specific ZFP-binding site, thus providing virtually universal sequence specificity for DNA cleavage (14,15). The nuclease domain generally used in ZFNs is the cleavage domain of the type IIs restriction enzyme FokI, which requires dimerization to cleave double-stranded DNA (14). To achieve dimerization, pairs of ZFNs are used that bind to adjacent sites on complementary DNA strands spanning the target sequence, thus allowing the FokI catalytic domains to cleave the DNA (16). Such customized ZFN technology has been used with human cells in culture to drive site-specific gene correction and gene addition via homologous recombination stimulated by double-strand breaks to nuclear DNA (17,18).
Therefore, targeting ZFNs to mitochondria is an appealing prospect to selectively deplete mutated mtDNA sequences from heteroplasmic cells. In earlier work we developed an efficient method to deliver engineered ZFPs to mitochondria and showed that zinc finger technology can be used to target and alter mtDNA in a sequence-specific manner (19). The selective binding of mitochondria-specific ZFPs to mtDNA was exemplified by targeting the m.8993T>G mutation, which causes two mitochondrial diseases, NARP and MILS. In the present article, we report targeting the same mtDNA m.8993T>G mutation using a ZFN. In applying ZFN technology to mitochondria, we initially used conventional pairs of heterodimeric ZFNs; however, we were unable to find suitable pairs of ZFNs for our specific mtDNA target (see Supplementary Data). Therefore, we extended the ZFN approach by developing a novel, single-chain ZFN. This arrangement proved effective in selectively cleaving mutated, but not wt, mtDNA in cells. Here, we report on the development and application of mitochondria-targeted, single-chain ZFNs.
MATERIALS AND METHODS
Engineering ZFPs for targeting human mtDNA
A number of potential target sites for binding ZFPs have been identified in mtDNA. These include sites in the non-coding region of the wt mtDNA as well as sequences containing point mutations involved in the genetic disorders (e.g. NARP m.8993T>G). The potential ZFPs that bind to these targets have been assembled from the archives of pre-selected modules by Sangamo BioSciences Inc., Richmond, CA, USA (20) and are based on Zif268 randomized libraries described by Isalan et al. (21). DNA-binding domains were engineered either as 1×4 zinc finger, where individual zinc fingers are connected via canonical TGEKP linkers (22) (e.g. NARPd) or as a 2 × two-finger design (23) (e.g. NARPa or NARPb). The ZFPs were then tested for their ability to selectively bind their target sequences. The protein sequence of the ZFP targeting the m.8993T>G mutation is given in Supplementary Figure 1. The ZFP targeting the human mitochondrial non-coding region is clone 18 as shown in Figure 1 and Supplementary Figure 1 of our earlier publication (19).
Figure 1.

Interactions of ZFNs with wt and mutated mtDNA. The sequence surrounding the m.8993T>G mutation in mtDNA was used to exemplify targeting of pathogenic mitochondrial mutations by ZFNs. The changed base at position 8993 is indicated in yellow. DNA cleavage is marked by the green scissors symbol. (A) Schematic diagram of ZFN heterodimer in the standard configuration bound to the mutated mtDNA target (upper). Each of the monomeric ZFN consists of the FokI nuclease domain linked to a ZFP. One of the ZFNs (red) was designed to bind to the mutated mtDNA site, whereas its companion ZFN binds a native sequence on the opposite DNA strand (blue). In the case of wt mtDNA (lower) mutation-specific ZFN does not bind the target therefore precluding a formation of a heterodimer and DNA cleavage. (B) Schematic diagram of the partner ZFN (blue) bound to the wt sequence and producing DNA cleavage by dimerization with another copy of the same construct non-specifically bound to mtDNA when the mutation-specific ZFN (red) is not bound. This outcome is undesirable when a mutation is targeted in heteroplasmic population of mtDNA molecules.(C) Schematic diagram of ‘single-chain ZFN’ consisting of two FokI nuclease domains tethered together by a long protein linker and fused to a ZFP. The ZFP is designed to bind exclusively to the mutated mtDNA site; therefore, only mtDNA molecules harbouring this mutation are cleaved (upper) while the wt copies are spared (lower).
Construction of ZFN expression vectors
The DNA encoding appropriate engineered zinc finger peptides was modified by PCR to introduce the HA epitope tag (YPYDVPDYA) to the C-terminus of the ZFP and flanked with unique XhoI (5′) and EcoRI (3′) restriction sites. These were joined with PCR-amplified DNA fragments flanked with unique XbaI (5′) and XhoI (3′) restriction sites, which encoded the 51-aa mitochondrial targeting sequence (MTS) from human ATP synthase F1β subunit (denoted F). The nuclear export signal (NES) was added by PCR-amplification of DNA fragments encoding F-ZFP and epitope tag using 5′ F-specific primer containing XbaI restriction site and a 3′ epitope tag-specific primer containing additional NES coding sequence and a BamHI site added at the extreme 3′ end. The NES used here was the one from the NS2 protein of Minute Virus of Mice (VDEMTKKFGTLTIHDTEK) (24). The resulting F-ZFP-NES XbaI–BamHI fragments were cloned into XbaI and BamHI sites of pcDNA3.1(–) (Invitrogen, Paisley, Renfrewshire, UK). The F-ZFP-NES constructs were used for in vitro binding tests to verify the specificity of the ZFPs as described (19).
In order to construct the F-ZFP-Fok constructs (ZFN monomers) the FokI catalytic domain (amino acids 388–583) was fused at the C-terminus of F-ZFP-NES via a 17-aa-long flexible linker having the following sequence [SGGGG]3SS in a two-part PCR reaction using four primers. The amplified DNA fragments flanked with unique XbaI (5′) and BamHI (3′) restriction sites were cloned into pcDNA3.1(–), pTRACER/CMV/BGH (Invitrogen) or pIRESpuro/EF1α (Clontech-Takara Bio Europe, Saint-Germain-en-Laye, France).
In order to create the single-chain quasi-dimeric ZFNs the second FokI domain was fused at the C-terminus of the F-ZFP-Fok constructs via the following flexible linkers:
18 aa: SSGGGGSGGGGSGGGGSG
22 aa: SSGGGGSGGGGSGGGGSGGGGS
27 aa: SSGGGGSGGGGSGGGGSGGGGSGGGGS
32 aa: SSGGGGSGGGGSGGGGSGGGGSGGGGSGGGGS
35 aa: SSGGGGSGGGGSGGGGSGSSGGGGSGGGGSGGGGS
40 aa: SSGGGGSGGGGSGGGGSGGSGGGSSGGGGSGGGGSGGGGS
The second domain of FokI was introduced in a two-part PCR reaction using four primers and the entire construct encoding F-ZFP-Fok-linker-Fok was cloned between XbaI (5′) and BamHI (3′) restriction sites into pcDNA3.1(–), pTRACER/CMV/BGH (Invitrogen) or pIRESpuro/EF1α (Clontech).
In vitro digestion assay
The DNA probes for the NARPd ZFNs were constructed by cloning a 200-bp fragment of wt mtDNA, a mtDNA fragment containing the m.8993T>G mutation or a fragment containing the m.8993T>C mutation into the pCR4 vector (Invitrogen), the resulting plasmids were named pCR4-NARP-T, pCR4-NARP-G and pCR4-NARP-C, respectively. The DNA probes for the NCR ZFN were constructed by cloning a 562-bp fragment containing the human mitochondrial non-coding region (positions 163–725 of the human mitochondrial genome) into the pCR4 vector (Invitrogen), the resulting plasmid was named pCR4-NCR. The above DNA plasmids were linearized with NcoI and radioactively end-labelled using Klenow polymerase. Probes for the experiments showed in Figure 3 were constructed by long range PCR (the positions of the primers are indicated in Figure 3) and end-labelled using T7 Kinase (New England Biolabs, Hitchin, Herts, United Kingdom).
Figure 3.

The specificity of single-chain ZFNs designed to target the mitochondrial point mutation. (A) In vitro assay verifying the specificity of the F-NARPd-Fok-L35-Fok (line 3) and F-NARPd-Fok-L40-Fok (line 4) constructs to a single site in human mtDNA. The assay was performed similarly to the experiment shown in Figure 2. The DNA probes used here though were generated in a long-range PCR reaction so that the two resulting products represented the entire mtDNA molecule harbouring the m.8993T>G substitution (grey curved bars with the mtDNA coordinates given on each end). The PCR products were radioactively labelled using T4 Kinase. (B) Mapping the main cleavage sites of F-NARPd-Fok-L35-Fok and F-NARPd-Fok-L40-Fok constructs by primer extension. Unlabelled DNA probes (as described above) were subjected to the in vitro digestion assay with F-NARPd-Fok-L35-Fok (line 3) or F-NARPd-Fok-L40-Fok (line 4). Next the digested DNA served as a template for the extension reaction of the 5' labelled primer that anneals 100-bp upstream from the NARPd-binding site. DNA strand breaks introduced by F-NARPd-Fok-L35-Fok or F-NARPd-Fok-L40-Fok map to several sites 2–7-bp upstream from the NARPd-binding site.
The ZFP constructs were synthesized in the in vitro transcription/translation reaction assay based on a reticulocyte lysate (Promega, Southampton, Hampshire UK) using 10 μl of lysate + 1 μl of each 300 ng/ul DNA template). Proteins as indicated on the figures (1 μl each or 1 μl of protein and 1 μl reticulocyte lysate) were incubated (either individually or in pairs) with the 1-nM DNA targets in 10 μl of the reaction buffer (20 mM Tris–acetate, 50 mM potassium acetate, 10 mM magnesium acetate, 1 mM DTT, 100 μM zinc chloride, 0.05 μg/ml BSA and 5% glycerol, pH 7.9) for 1 h at 37°C. Next, 5 μl of the reactions were loaded on a 1% agarose gel, resolved and, after drying, subjected to autoradiography. Specific digestion at the m.8993T>G mutation site resulted in the formation of 2.7- and 1.5-kb DNA fragments.
Mapping the cleavage site by primer extension
In order to map the double-strand breaks introduced by ZFNs a primer extension reaction was performed. Unlabelled DNA probes were subjected to the in vitro digestion assay (see above) and served as a template for the extension reaction. The reactions were purified using QIAquick PCR Purification Kit (Qiagen, Crawley, Sussex, UK) and eluted in 30 μl of water. For the primer extension 20 μl PCR reaction mix containing 5 μl of the purified template and 20 pmoles of 5′ T7 kinase (New England Biolabs) labelled primer were cycled 20–30 times. The reactions were then precipitated and resolved in 4–5% urea sequencing gels.
Maintenance, transfection and selection of mammalian cell lines
Cell lines 143B (TK-) and 143B (TK-) NARP cybrids, which contained the various percentage of the m.8993T>G mutation in their mtDNA (kindly donated by Eric Schon of Columbia University, New York) were all grown in high-glucose DMEM containing 2 mM L-glutamine (Gibco) supplemented with 20% of FCS (Invitrogen). Transfections of 143B (TK-) and cybrid cells were performed using Cell Line Nucleofector (Amaxa Biosystems, Cologne, Germany), buffer kit V (Amaxa Biosystems), plasmid DNA (1 μg) purified by Qiafilter MidiPrep Kit (Qiagen) and applying programme I-13. For immunodetection studies zinc finger constructs were transiently expressed for 24–36 h. For FACS sorting experiments as presented in Figure 5 and 6, cells were transfected with appropriate pTRACER vector 48 h prior to a cell sorting. The FACS sorting using GFP as a marker was performed as described (25). For long-term selection experiments, cells as presented in Supplementary Figure 5 cybrid cells were transfected with appropriate pIRESpuro/EF1α vector and 24 h after transfection puromycin (2 µg/ml) was added and the selective medium was replaced every 3–4 days. The cell clones were picked up using plastic cloning cylinders (Sigma, Gillingham, Dorset, UK).
Figure 5.

Mitochondrial heteroplasmy in cells expressing mutation-specific ZFNs. The m.8993T>G mutation-specific or control ZFNs were co-expressed with eGFP from a single vector in heteroplasmic cybrid cells containing ∼85% of mtDNA molecules with the NARP m.8993T>G mutation (dashed line). Two days post-transfection, cells were FACS sorted using eGFP as a marker and the degree of heteroplasmy was assessed in the sorted cells. Mock indicates cells transfected with vector alone. Symbols: *P < 0.05; **P < 0.01 in two-tailed t-test, unequal variance. (A) Schematic illustration of the experimental approaches taken. (B) Differences in the proportion of the wt and mutant mtDNA in cells transfected with F-NARPd-Fok or F-NARPd-Fok-L35-Fok specific for the m.8993T>G. The result is an average of three independent experiments. The difference between mock and F-NARPd-Fok-L35-Fok transfected cells is highly significant (P = 0.015), while the difference between mock and the F-NARPd-Fok monomer is not significant (P = 0.2).(C) Total (sum of mutant and wt) mtDNA copy number in the cells transfected with the m.8993T>G mutation-specific ZFNs expressed in arbitrary units (a.u.). The results are normalized to mock-transfected cells. (D) Differences in the proportion of the wt and mutant mtDNA in cells transfected with F-NCR-Fok or F-NCR-Fok-L35-Fok targeting a site in the mitochondrial non-coding region. The result is an average of three independent experiments. No statistically significant differences between the constructs have been found. (E) Total mtDNA copy number in the cells transfected with the mitochondrial NCR-specific ZFNs expressed in arbitrary units (a.u.). The results are normalized to mock-transfected cells.
Figure 6.

Stable shift in mitochondrial heteroplasmy in cells expressing mutation-specific ZFNs. The m.8993T>G mutation-specific ZFNs, F-NARPd-Fok or F-NARPd-Fok-L35-Fok, were co-expressed with eGFP from a single vector in heteroplasmic cybrid cells containing ∼85% of mtDNA molecules with the NARP m.8993T>G mutation (dashed line). Two days after the transfection cells were FACS sorted using eGFP as a marker, then the sorted cells were grown on non-selective medium for a further 28 days. The percentage of mutant mtDNA was assessed again at 2 and 30 days. Mock indicates cells transfected with vector alone. Symbols: *P < 0.05; **P < 0.01 in two-tailed t-test, unequal variance. (A) Schematic illustration of the experimental approaches taken. (B) Differences in the proportion of the wt and mutant mtDNA in bulk population of transfected cells at 2 and 30 days. The difference between mock and F-NARPd-Fok-L35-Fok transfected cells after 30 days is highly significant (P = 0.007), whereas the difference between mock and the F-NARPd-Fok monomer is not significant (P = 0.54). (C) Total mtDNA copy number in the transfected cells measured at 2 and 30 days expressed in arbitrary units (a.u.). The results are normalized to mock transfected cells. (D) Differences in the proportion of the wt and mutant mtDNA in individual clones, randomly picked at 30 days post-transfection. The red horizontal bar indicates an average value for each construct. The difference between mock and F-NARPd-Fok-L35-Fok transfected cells after 30 days is highly significant (P = 0.003), the difference between mock and the F-NARPd-Fok monomer is of much lower significance (P = 0.4).
The Flp-In TREx HEK293 cell-line (Invitrogen), used to generate stable inducible expression of the ZFN constructs, was grown in DMEM, 10% FCS (Invitrogen) supplemented with 100 µg/ml Zeocin (Invivogen) and 15 µg/ml Blasticidin (Invivogen). Twenty-four hours prior to transfection cells were split to 15-cm plates and grown to 80–90% confluence. Flp-In TREx HEK293 cells were transfected according to the manufacturer's instruction using Cell Line Nucleofector (Amaxa Biosystems), buffer kit V (Amaxa Biosystems) applying programme A-23. The pcDNA5/FRT/TO plasmid DNA encoding appriopriate ZFNs was purified by Qiafilter MidiPrep Kit (Qiagen). Twenty four hours after transfection the selective antibiotics hygromycin (100 µg/ml) and blasticidin (15 µg/ml) were added and the selective medium was replaced every 3–4 days. The expression was induced with the indicated amount of doxycycline (Sigma) and medium was changed every 2 days.
Immunodetection of ZFNs
Transiently expressed ZFNs were analysed by either immunofluorescence or immunoblotting. Adherent cells intended for immunofluorescence were grown on coverslips and if required, stained with Mitotracker CMX Red (Molecular Probes, Paisley, Renfrewshire UK) added to the culture medium for 30 min in order to visualize mitochondria. Cells were then washed in PBS and fixed with 4% formaldehyde/PBS directly on cover slips. Following permeabilization with 1% Triton X-100 and washing in PBS, the ZFNs were visualized using rat monoclonal antibody 3F10 against the HA epitope tag (Roche, Burgess Hill, Sussex, UK). This was followed by incubation with a secondary antibody, anti-rat IgG conjugated to FITC (Vector, Peterborough, Cambridgeshire). The immunofluorescence was then viewed using a Zeiss LSM 510 META confocal microscope.
For immunoblot analysis equal amounts of proteins corresponding to total cell lysates, protein fractions (see below) or reticulocyte lysates were subjected to SDS–PAGE, semi-dry transferred to nitrocellulose membranes and incubated with specific antibodies. The blots were further incubated with HRP-conjugated secondary antibodies and visualized using ECL (Amersham, Little Chalfont, Buckinghamshire, UK).
Cell fractionation
Mitochondria from the wt or cybrid cells were isolated as described by Minczuk et al. (26). The mitochondrial fractions were then incubated in 1 × IB buffer (40 mM Tris–HCl, pH 7.4, 25 mM NaCl and 5 mM MgCl2) supplemented with proteinase K (BDH) at the concentrations indicated in Figure 4A and B. The sub-cellular fractions normalized for protein contents were analysed with anti-HA mAb in order to detect ZFP protein constructs. Blotting using antibodies against marker proteins [anti-TFAM serum, anti-Cox2 mAb (Molecular Probes), anti Tom22 mAb (Abcam) or anti-GAPDH mAb (Abcam)] was also performed in order to verify the fractions.
Figure 4.
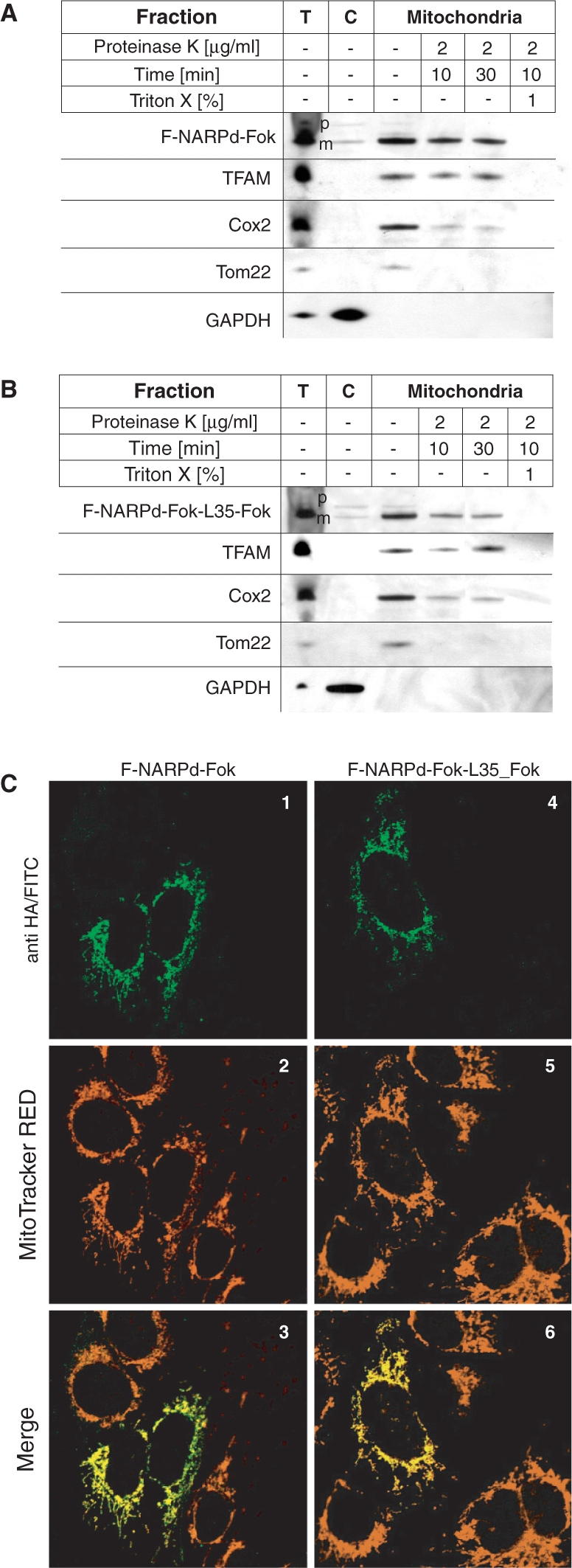
ZFNs are localized in mitochondria in cells. (A) and (B) The F-NARPd-Fok and F-NARPd-Fok-L35-Fok ZNFs localize inside mitochondria. The 143B cells harbouring wt mtDNA were transiently transfected with monomeric F-NARPd-Fok (A) or single-chain ZFN—F-NARPd-Fok-L35-Fok (B), fractionated 48 h post-transfection and the protein fractions were analysed by western blotting using anti-HA mAb. The localization of the ZFN precursors (‘p’) and their mature (‘m’) form in total cell lysate (‘T’), cytosolic (‘C’) and a mitochondrial fraction treated with proteinase K under various conditions as indicated, was compared with the localization of marker proteins. The precursors of mitochondrial ZFNs, found in the mitochondrial fractions, were located outside the mitochondria, since they were accessible to protease digestion. In contrast, the mature form of ZFNs was protected and became accessible to proteolysis only after the mitochondria were lysed with Triton X-100. The following endogenous proteins were used as fractionation markers: (i) TFAM: the transcription factor that is localized in the mitochondrial matrix; (ii) Cox2; a subunit of the cytochrome oxidase complex localized in the mitochondrial inner membrane; (iii) Tom22: a subunit of mitochondrial translocase of outer membrane; and (iv) GAPDH: a protein localized in cytoplasm. (C) The F-NARPd-Fok and F-NARPd-Fok-L35-Fok ZFNs co-localize with mitochondria. The intra-cellular localization of ZFNs was additionally analysed by immunofluorescence in transiently transfected 143B cells. ZFNs were detected with antibodies against the HA epitope-tag followed by secondary antibodies conjugated to FITC (1 and 4; green) Mitochondria were stained with MitoTracker CMX Red (2 and 5; red). Both ZFNs exhibited mitochondrial-staining pattern that in represented by yellow staining on digitally overlaid pictures (3 and 6).
Measurement of mitochondrial heteroplasmy and mtDNA copy number
The ratio between the wt and mutant mtDNA was measured by RFLP. Total DNA from cells was isolated using DNasy Blood and Tissue Kit (Qiagen). A DNA fragment corresponding to nucleotide positions 8339–9334 of the human mitochondrial genome was PCR amplified and radioactive dCTP (GE Healthcare, Bucks, Buckinghamshire) was added for the last cycle. The final PCR product was digested with SmaI recognizing a site containing the m.8993T>G NARP/MILS mutation, and electrophoresed in 2% agarose gels. The radioactive signal was quantified using a phosphorimager system (GE Healthcare).
The relative copy number of mtDNA was determined by real-time PCR of the DNA encoding subunit 2 of cytochrome oxidase (Cox2) using the nuclear, single-copy gene for amyloid precursor protein (APP) as a reference. Total cellular DNA was isolated using DNasy Blood and Tissue Kit (Qiagen). Copy numbers of Cox2 and APP were determined by Taqman PCR on an ABI Prism 7000 (Applied Biosystems, Foster City, CA, USA) using the standard curve method.
RESULTS
In vitro characterization of a single-chain zinc finger nuclease
With the aim of eliminating mutant mtDNA from heteroplasmic populations of mtDNA molecules, we first designed a ZFN that should specifically bind and cleave a mutated mtDNA sequence containing the m.8993T>G mutation. In the standard ZFN configuration, pairs of ZFNs bind to adjacent DNA sequences within the target sequence, enabling their FokI nuclease domains to dimerize and cleave DNA selectively (16,18) (Figure 1A). To cleave mtDNA containing the m.8993T>G mutation we created two monomer ZFNs, one specific for the m.8993T>G mutation and a companion ZFN that binds upstream of nucleotide position 8993 to the complementary mtDNA strand (Figure 1A). This combination should ensure the dimerization of the ZFN FokI nuclease domains necessary for introducing DNA double-strand breaks adjacent to the m.8993T>G mutation site, but not in wt mtDNA (Figure 1A). However, under circumstances of non-optimal expression the dimerization of the companion ZFNs may occur, resulting in cleavage of wt mtDNA if the specificity of the companion ZFN is insufficient to prevent homodimer formation at the target (Figure 1B). Despite extensive investigation of more than 30 ZFN pairs (Supplementary Data), we failed to find a suitably active and specific ZFN pair that would cut our specific target sequence. Therefore, we developed an alternative approach by designing a single-chain ZFN, in which a ZFP is linked to two FokI catalytic domains present on a single polypeptide chain (Figure 1C). Such an arrangement should enable intra-molecular association of the two FokI domains allowing more efficient DNA cleavage than with a monomeric ZFN. By choosing a ZFP selective for the mtDNA containing the m.8993T>G mutation it should be possible to selectively cleave mtDNA containing this mutation, but leave wt mtDNA unmodified (Figure 1C).
The initial single-chain ZFNs are designated as F-ZFP-Fok-L(n)-Fok, where L(n) denotes an n-residue linker between FokI domains (Figure 2A). The ZFP chosen, NARPd, (Supplementary Figure 1, Top), has been shown to bind selectively to the m.8993T>G sequence (data not shown). This ZFP was linked to the mitochondria targeting sequence from human ATP synthase F1β subunit (denoted F) at its N-terminus and a HA tag and a nuclear export sequence (NES) at its C-terminus [previously we have shown that a NES greatly assists in the delivery of ZFPs to mitochondria in cells (19)]. The C-terminus was fused to a FokI domain via a flexible linker and in turn this FokI domain was linked to a second FokI domain by flexible linkers of various lengths (Figure 2A).
Figure 2.

Designing a single-chain ZFN to target a mitochondrial point mutation. (A) Schematic structure of mitochondrially targeted single-chain nucleases. The mitochondrial targeting sequence of F1β subunit of mitochondrial ATP synthase (MTS F) was fused to the N-terminus of the ZFP. The HA epitope tag and the NES facilitating mitochondrial targeting were added to the C-terminus of the ZFP. Two FokI domains were fused on the C-terminus of the ZFP and linked together via flexible linkers of various lengths denoted L(n), where n is the length of the linker in amino acids. (B) Optimization of the single-chain ZFN based on the NARPd construct (see Figure 1C). Variants of F-NARPd-Fok-L(n)-Fok were subjected to the in vitro assay as described in Materials and methods section using the specific target DNA in order to determine the optimal length of the linker between the two tethered FokI domains. The plot shows the results of the assay performed three times. (C) In vitro cleavage assay testing the specificity of the F-NARPd-Fok-L(n)-Fok constructs. The assay was performed as described in Materials and methods section. The following probes have been used: pCR4-NARP-G—containing the m.8993T>G substitution, pCR4-NARP-T —wt (i.e ‘T’) at the 8993 position and pCR4-NARP-C— containing the m.8993T>C substitution. The f.p denotes free probe. Specific digestion at the m.8993T>G mutation site results in the formation of 2.7- and 1.5-kb DNA fragments. The percentage of cleavage of the pCR4-NARP-G probe is given in the top panel. (D) Western blot illustrating that the constructs used in the test above are produced with the same efficiency in the in vitro transcription/translation system. ZFNs were detected with anti-HA antibody.
To cleave DNA efficiently the linker between the two FokI domains must allow them to form an intra-molecular quasi-dimer. To optimize linker length we tested the effects of six different linker lengths on in vitro cleavage of a DNA target, pCR4-NARP-G, containing the m.8993T>G substitution (Figure 2B and C). Linkers of 35 or more amino acids gave almost complete cleavage (Figure 2C, lines 7 and 8). In contrast, cleavage of the same DNA substrate with the F-NARPd-Fok monomer was much less efficient, and even increasing the incubation time from 1 to 4 h resulted in only ∼20% cleavage (data not shown). DNA cleavage by F-ZFP-Fok-L(n)-Fok was specific for the m.8993T>G substitution (pCR4-NARP-G) as DNA substrates harbouring T or C instead of G in this position (pCR4-NARP-T or pCR4-NARP-C, respectively) were not cleaved by any of the F-NARPd-Fok-L(n)-Fok constructs (Figure 2C).
To obtain additional insight into the specificity of the F-ZFP-Fok-L(n)-Fok nucleases, the two most active constructs, with 35 and 40 amino acid linkers, were tested in vitro with two large DNA molecules (9176 and 8776 bp), that covered the entire human mtDNA sequence and both of which contained the m.8993T>G mutation (Figure 3A). Incubation of these DNA molecules with F-NARPd-Fok-L35-Fok (Figure 3A, line 3) or F-NARPd-Fok-L40-Fok (Figure 3A, line 4) resulted in digestion products of ∼350 or ∼330 bp that corresponds to DNA cleavage at the m.8993T>G mutation sites. Cleavage generates a small product (∼350 or ∼330 bp), and also a large product (∼8820 or ∼8450 bp) slightly smaller than the starting material that is not separated from the starting material by this gel system. Thus, F-NARPd-Fok-L35-Fok and F-NARPd-Fok-L40-Fok only cleave DNA at the site of the m.8993T>G mutation, leaving the rest of the wt mtDNA genome unaltered. Primer extension experiments performed on the DNA digested by these nucleases showed that the DNA double-strand breaks occur 2–7 bp upstream from the ZFP-binding site (Figure 3B, lines 3 and 4), consistent with the binding position of the nuclease relative to the ZFP-binding site. Therefore, we have successfully constructed a single-chain ZFN that selectively cleaves mutated, but not wt, mtDNA.
Localization of single-chain ZFNs to mitochondria within cells
The MTS and NES on F-ZFP-Fok-L(n)-Fok should ensure their effective localization to mitochondria in cells (19). To see if this was the case, we studied the distribution of F-NARPd-Fok-L35-Fok within 143B cells and compared it to its monomeric counterpart F-NARPd-Fok. To see if these proteins were localized within mitochondria, we transiently expressed them and then fractionated the cells into different compartments (Figure 4A and B). Both ZFNs were present in the mitochondrial fraction and the MTS was cleaved off from the precursor protein (p) leaving the mature (m) protein, consistent with uptake through the conventional mitochondrial import pathway. To determine where within mitochondria the proteins were located, the isolated mitochondria were incubated with proteinase K. The mature forms of F-NARPd-Fok (Figure 4A) and F-NARPd-Fok-L35-Fok (Figure 4B) were protected from proteolysis, to the same extent as the mitochondrial matrix protein TFAM (27), while a protein associated with the mitochondrial outer membrane (Tom22) (28) and one associated with the inner membrane (Cox2) were progressively degraded over time by proteinase K (Figure 4A and B). To further confirm the mitochondrial localization of F-NARPd-Fok-L35-Fok and F-NARPd-Fok, we carried out immunofluorescence experiments on the cells. The location of the proteins within cells was visualized using an antibody against the HA tag, and co-staining with the mitochonodrial probe Mitotracker-RED showed that these proteins were present mainly in mitochondria (Figure 4C). Together, these data indicate that both proteins F-NARPd-Fok-L35-Fok and F-NARPd-Fok are present in the mitochondrial matrix within cells.
Mitochondria-targeted specific ZFNs cleave wt mtDNA in cells
We have shown that F-NARPd-Fok-L(n)-Fok constructs are taken up by mitochondria in cells and can selectively cleave their target mtDNA sequence in vitro. However, the mtDNA environment in vivo is quite different from that in vitro and therefore it is important to confirm that F-ZFP-Fok-L(n)-Fok constructs can cleave mtDNA within mitochondria inside cells. To assess this, we used a F-ZFP-Fok-L(n)-Fok with a ZFP against a sequence in the wt mtDNA non-coding region (NCR) that enabled us to test the efficacy of the F-ZFP-Fok-L(n)-Fok at cleaving mtDNA in cells containing only wt mtDNA. We constructed a F-NCR-Fok-L35-Fok designed to cleave a sequence in the control region of wt mtDNA, as well as its monomeric form, F-NCR-Fok, that should bind to the same sequence. The specificity of cleavage of the mtDNA target sequence was tested in the in vitro assay as above and ZFN F-NCR-Fok-L35-Fok almost completely cleaved the specific DNA target, while its monomeric counterpart, F-NCR-Fok, cleaved the same target with ∼15% efficiency (Supplementary Figure 3). Cellular localization of F-NCR-Fok and F-NCR-Fok-L35-Fok was mitochondrial as shown by imunocytochemistry and subcellular fractionation (data not shown).
In order to see if F-NCR-Fok-L35-Fok cleaved wt mtDNA in cells, tetracycline-inducible HEK293 TREx cell lines expressing F-NCR-Fok and F-NCR-Fok-L35-Fok were constructed. Induction of the single-chain ZFN F-NCR-Fok-L35-Fok resulted in a rapid and almost complete loss of mtDNA after 2 days, consistent with the rapid cleavage of wt mtDNA (Supplementary Figure 4). Expression of the F-NCR-Fok monomer also significantly decreased mtDNA copy number, but to a lesser extent than the single-chain ZFN, consistent with its lower nuclease activity in vitro (Supplementary Figure 4). Therefore, when expressed in cells mitochondria-targeted single-chain ZFN is capable of binding and cleaving mtDNA.
Selective depletion of mutated mtDNA molecules in heteroplasmic cells by a mitochondrially targeted ZFN
As shown in vitro, F-NARPd-Fok-L35-Fok can selectively degrade mutated mtDNA, while leaving wt mtDNA uncut, and it is readily taken up by mitochondria in cells. Furthermore, a related F-ZFP-Fok-L35-Fok designed to bind to wt mtDNA was active within mitochondria in cells, as indicated by the depletion of mtDNA. Therefore, within heteroplasmic cells F-NARPd-Fok-L35-Fok should selectively degrade mutated but not wt mtDNA and thereby decrease the proportion of mutated mtDNA molecules in a cell.
As observed for F-NCR-Fok, a mitochondrially targeted monomeric ZFN was also capable of depleting mtDNA in cells even when present without a binding partner. Thus, ZFN monomers designed to bind mutated DNA sites, could to some extent eliminate mutant mtDNA. Therefore, in subsequent experiments we compared monomeric F-ZFN-Fok with the single-chain ZFN with a quasi-dimeric FokI domain, to determine which configuration is most effective.
To assess efficiency of cleavage, we used heteroplasmic cybrid cells containing 90% of mtDNA molecules with the m.8993T>G mutation (the term cybrid refers to a clonal cell line resulting from the fusion of heteroplasmic cells bearing mutated mtDNA derived from patients with mtDNA-less cells). Plasmids with a puromycin-selectable marker expressing F-NARPd-Fok-L35-Fok or the F-NARPd-Fok were used to transfect cells, which were grown for 10 days using puromycin to ensure retention of the transgene. Clones derived from individual transfected cells were then isolated and the percentage of the wt mtDNA was assessed for each clone using PCR/RFLP. Those clones transfected with F-NARPd-Fok-L35-Fok had significantly higher levels of wt mtDNA compared to the mock transfected controls or to those transfected with the F-NARPd-Fok monomer (Supplementary Figure 5). Some clones transfected with the F-NARPd-Fok-L35-Fok ZFP-single-chain nuclease had 40% wt mtDNA, compared to 5–20% in the controls. Transfection with the F-NARPd-Fok monomer did not increase wt mtDNA content to a statistically significant extent. However, by 14 days post-transfection none of the clones initially transfected with F-NARPd-Fok or F-NARPd-Fok-L35-Fok expressed the protein at detectable levels and the number of clones that survived selection was much lower for the ZFN constructs than for the control. This suggests a possible growth disadvantage associated with a prolonged mitochondrial expression of these ZFNs under conditions that led to loss of the corresponding plasmid DNA over time. Even so, the increased proportion of wt mtDNA in the transfected cells was promising, and suggested that expression of the transgene initially led to the selective depletion of mutated mtDNA that was retained, even after the transgene was no longer expressed in those cells.
As experiments with F-NCR-Fok-L35-Fok showed rapid depletion of mtDNA with a maximum effect evident after 2 days, prolonged expression of mitochondrial ZFNs should not be necessary to deplete mtDNA. Therefore, we next assessed the changes in heteroplasmy that occurred over 2 days post-transfection in heteroplasmic cybrid cells containing 85% of the m.8993T>G mutation. For these experiments, the F-NARPd-Fok or F-NARPd-Fok-L35-Fok constructs were co-expressed with GFP encoded by the same vector. This enabled us to enrich the population of F-NARPd-Fok or F-NARPd-Fok-L35-Fok expressing cells by FACS sorting using GFP as a marker. The levels of wt mtDNA in the cells enriched for F-NARPd-Fok or F-NARPd-Fok-L35-Fok 2 days after transfection were then measured by PCR/RFLP. The F-NARPd-Fok-L35-Fok transfected cells, but not those transfected with F-NARPd-Fok, showed significantly higher levels of wt mtDNA as compared to the mock-transfected controls in three independent experiments (Figure 5B). The total mtDNA copy number in the F-NARPd-Fok or F-NARPd-Fok-L35-Fok transfected and FACS sorted cells was also lowered compared to controls by ∼40% and ∼60%, respectively (Figure 5C). The drop in the mtDNA level for F-NARPd-Fok-L35-Fok was consistent with the selective removal of mutated mtDNA. Initially the cells contained 15% wt and 85% mutated mtDNA; if the 40% decrease in mtDNA copy number was due exclusively to loss of mutated mtDNA the cell would then contain 25% wt mtDNA, close to what was found. In contrast, the decrease in mtDNA copy number caused by F-NARPd-Fok was greater than expected from the selective removal of the mutant mtDNA. If loss of 60% of the wt mtDNA was due exclusively to loss of mutated mtDNA then we would expect 37.5% wt mtDNA, instead we found 20%, perhaps implying an non-specific interaction of the mononmeric ZFN with the wt mtDNA in these experiments. The number of GFP positive cells was much lower for F-NARPd-Fok or F-NARPd-Fok-L35-Fok transfected cells compared with the mock-transfected controls, implying a general cytotoxic effect of ZFN expression at the dose levels tested (Supplementary Table 1). To test whether the shift in heteroplasmy observed above was simply caused by this non-specific effect of ZFN expression in mitochondria, we transiently expressed the F-NCR-Fok-L35-Fok or F-NCR-Fok constructs in heteroplasmic cybrid cells containing 85% of the m.8993T>G mutation. As these constructs are specific for the mtDNA non-coding region, they should affect mutated and the wt mtDNA molecules to the same extent. As before, cells expressing F-NCR-Fok or F-NCR-Fok-L35-Fok were enriched by FACS sorting using GFP as a marker, 2 days after transfection. In this case, the ratio between the mutant and the wt mtDNA was unaltered by expression of the ZFNs (Figure 5D). As expected, the mtDNA copy number was significantly lowered compared to mock-transfected controls (Figure 5E) and with respect to the NARPd constructs (Figure 5C). This is consistent with elimination of both the wt and mutated mtDNA by the F-NCR-Fok or F-NCR-Fok-L35-Fok and indicates that the increased proportion of wt mtDNA upon expression of F-NARPd-Fok-L35-Fok was a specific effect due to the selective elimination of the mutated mtDNA.
We next investigated whether the changes in heteroplasmy caused by the F-NARPd-Fok-L35-Fok were stable and whether the removal of the mutant mtDNA by ZFNs observed in the first 48 h can be maintained over an extended period. To do this, heteroplasmic cybrids containing 85% of the m.8993T>G mutation were transiently transfected with F-NARPd-Fok or F-NARPd-Fok-L35-Fok as described above and isolated by FACS sorting 2 days post-transfection. These cells were then grown in a high-glucose medium without any selection pressure to retain the plasmids coding for ZFNs and under these conditions the plasmids were lost within 1–3 days (3–5 days post-transfection). The level of wt mtDNA and the mtDNA copy number were then measured 2 and 30 days post-transfection. The elimination of mutated mtDNA detected 2 days after transfection with the F-NARPd-Fok-L35-Fok construct was maintained at almost the same level after 30 days (Figure 6B). Most interestingly, the mtDNA copy number in the transfected cells that had been lowered 2 days post-transfection was restored to 80–90% of control levels at 30 days (Figure 6C), while retaining the higher proportion of wt mtDNA.
The level of wt mtDNA in cybrids was additionally assessed in clones derived from single cells isolated by FACS sorting and then grown for 30 days. The average level of wt mtDNA in ten randomly selected clones (Figure 6D) was similar to that in the bulk population (Figure 6B). Three out of the 10 clones transfected with the F-NARPd-Fok-L35-Fok construct had >40% wt mtDNA.
DISCUSSION
The difficulties of developing therapies for mitochondrial diseases have led to the development of mitochondrial genome-targeted strategies to selectively deplete mutated mtDNA molecules from cells and thereby enable the wt mtDNA molecules to re-populate the cell (3,4). The selective degradation of mutated mtDNA molecules has been achieved by targeting restriction enzymes to mitochondria, but these are limited to the small number of sequences that are present in mutated but not in wt mtDNA molecules and for which an RE is available. To overcome this limitation we developed mitochondria-targeted ZFNs in order to specifically target a mutated mtDNA for degradation. Initial attempts were made using the conventional configuration of heterodimeric ZFNs (Figure 1A), where one monomer binds a mutated mtDNA sequence while its partner binds an adjacent wt sequence enabling dimerization between two FokI domains to cleave dsDNA between the ZFPs-binding sites. However, this approach has a number of potential limitations to its applicability to targeting mutated mtDNA molecules. First, one of the monomers must bind very selectively to a sequence differing from the wt sequence by a single base pair out of 9–12 bp. Second, a heterodimeric ZFN designed to target a mtDNA mutation could lead to the monomer that binds a wt sequence forming an active nuclease that cleaves a wt mtDNA molecule if the specificity of this ZFN was insufficient to prevent homodimer formation at the target (Figure 1B). Consistent with this, we frequently observed cleavage of wt mitochondrial targets by ZFN monomers in vitro (Supplementary Figure 2), and a rapid mtDNA depletion was observed when mitochondrially targeted monomeric ZFN binding to the mtDNA NCR was expressed in normal (Supplementary Figure 5B) and mutant cells (Figure 5E) under our experimental conditions.
To address these concerns, we constructed and characterized a single-chain ZFN, in which a zinc finger peptide is linked to two FokI catalytic domains that are present on a single polypeptide. Unlike a heterodimeric ZFN that usually recognizes 24 bp (12 bp by each monomer), our single-chain ZFN only recognizes 12 bp. However, this lower specificity is sufficient for targeting sites in human mtDNA as the genome is only 16.5 kb making it unlikely that identical 12-bp binding sites exists more then once in mtDNA (statistically a 12-bp-long sequence would be present once per 1.68 × 107 bp). Our novel construct of a ZFP conjugated to two FokI catalytic domains allows for correct dimerisation of the catalytic domains and formation of an active nuclease that is far more efficient in vitro than its counterpart with only one FokI domain. Furthermore, within mitochondria in heteroplasmic cells (Supplementary Figure 5 and Figure 5), a mitochondrially targeted single-chain ZFN was also far more efficient in cleaving the mutated sequence than the nuclease containing a single FokI domain. However, there was some cleavage by the monomer, presumably due to cross-dimerization between DNA-bound and free F-NARPd-Fok. The ability of a single-chain quasi-dimeric ZFN to generate ds DNA breaks opens up the possibility of designing specific ZFNs without a need for generating pairs of ZFNs. This significantly simplifies design and could be particularly useful for targeting unique sequences in relatively short genomes, such as in organelles, bacteria or viruses. Furthermore, our novel design allows for generation of nucleases with novel specificities that could be used in standard in vitro laboratory tests such as RFLP.
Previous studies indicated that cellular expression of non-optimized ZFNs can be associated with significant cytotoxicity due to cleavage at off-target sites (29,30). Consistent with this possibility, the expression of the ZFN constructs (both monomeric and single-chain dimers) used in this study was associated with cytotoxic effects. Although in order to minimize general cytotoxicity linked to generation of nuclear DNA breaks, we equipped mitochondrially targeted ZFNs with the nuclear export signal (NES), it may be that the NES used is not fully effective and it may be necessary to minimize cytotoxicity by using a tightly regulated expression system or by optimizing the NES. Other ways of lowering the cytotoxicty of ZFNs could be the optimization of the ZFP protein sequence (18). However, toxicity associated with prolonged over-expression is not a major barrier to this ZFN technology, because it causes a permanent change at the DNA level. This was shown by the fast shift in the heteroplasmy observed 2 days after the transfection of a mutation-specific ZFN was maintained at the same level for a further 28 days (Figure 6). Therefore, long-term exposure to a ZFN should not be necessary to bring about a phenotypic change, in contrast to engineered ZFP transcription factors. This may be particularly relevant for the elimination of mutated mtDNA from heteroplasmic cells, which require only a certain threshold proportion of wt mtDNA to be reached in order to restore the phenotype.
In summary, here we have demonstrated that selective, single-chain quasi-dimeric ZFN can be generated. When single-chain ZFNs specific for a pathogenic point mutation in mtDNA were targeted to mitochondria in heteroplasmic cells harbouring mutated mtDNA there was selective elimination of the mutated mtDNA and the proportion of wt mtDNA in these cells increased. Therefore, ZFN technology may prove useful in the therapeutic manipulation of heteroplasmy in the treatment of mtDNA diseases.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
Funding was provided by the Medical Research Council, UK. M.M. has been supported by a Federation of European Biochemical Societies Long-Term Fellowship. We are indebted to E. Schon of Columbia University for providing us with the m.8993T>G heteroplasmic cell lines. We thank E. Rebar of Sangamo BioSciences, Inc. for his suggestions and comments on the article. The authors also would like to thank D. Kang of Kyushu University for antibodies against TFAM. Funding to pay the Open Access publication charges for this article was provided by Medical Research Council, UK.
Conflict of interest statement. None declared.
REFERENCES
- 1.Schapira AH. Mitochondrial disease. Lancet. 2006;368:70–82. doi: 10.1016/S0140-6736(06)68970-8. [DOI] [PubMed] [Google Scholar]
- 2.Schon EA. Mitochondrial genetics and disease. Trends Biochem. Sci. 2000;25:555–560. doi: 10.1016/s0968-0004(00)01688-1. [DOI] [PubMed] [Google Scholar]
- 3.DiMauro S, Hirano M, Schon EA. Approaches to the treatment of mitochondrial diseases. Muscle Nerve. 2006;34:265–283. doi: 10.1002/mus.20598. [DOI] [PubMed] [Google Scholar]
- 4.Taylor RW, Wardell TM, Lightowlers RN, Turnbull DM. Molecular basis for treatment of mitochondrial myopathies. Neurol. Sci. 2000;21:S909–S912. doi: 10.1007/s100720070002. [DOI] [PubMed] [Google Scholar]
- 5.Dimauro S, Davidzon G. Mitochondrial DNA and disease. Ann. Med. 2005;37:222–232. doi: 10.1080/07853890510007368. [DOI] [PubMed] [Google Scholar]
- 6.Tang Y, Manfredi G, Hirano M, Schon EA. Maintenance of human rearranged mitochondrial DNAs in long-term cultured transmitochondrial cell lines. Mol. Biol. Cell. 2000;11:2349–2358. doi: 10.1091/mbc.11.7.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Taylor RW, Chinnery PF, Turnbull DM, Lightowlers RN. Selective inhibition of mutant human mitochondrial DNA replication in vitro by peptide nucleic acids. Nat. Genet. 1997;15:212–215. doi: 10.1038/ng0297-212. [DOI] [PubMed] [Google Scholar]
- 8.Tanaka M, Borgeld HJ, Zhang J, Muramatsu S, Gong JS, Yoneda M, Maruyama W, Naoi M, Ibi T, Sahashi K, et al. Gene therapy for mitochondrial disease by delivering restriction endonuclease SmaI into mitochondria. J. Biomed. Sci. 2002;9:534–541. doi: 10.1159/000064726. [DOI] [PubMed] [Google Scholar]
- 9.Alexeyev MF, Venediktova N, Pastukh V, Shokolenko I, Bonilla G, Wilson GL. Selective elimination of mutant mitochondrial genomes as therapeutic strategy for the treatment of NARP and MILS syndromes. Gene Ther. 2008;15:516–523. doi: 10.1038/gt.2008.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Srivastava S, Moraes CT. Manipulating mitochondrial DNA heteroplasmy by a mitochondrially targeted restriction endonuclease. Hum. Mol. Genet. 2001;10:3093–3099. doi: 10.1093/hmg/10.26.3093. [DOI] [PubMed] [Google Scholar]
- 11.Bayona-Bafaluy MP, Blits B, Battersby BJ, Shoubridge EA, Moraes CT. Rapid directional shift of mitochondrial DNA heteroplasmy in animal tissues by a mitochondrially targeted restriction endonuclease. Proc. Natl Acad. Sci. USA. 2005;102:14392–4397. doi: 10.1073/pnas.0502896102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bacman SR, Williams SL, Hernandez D, Moraes CT. Modulating mtDNA heteroplasmy by mitochondria-targeted restriction endonucleases in a ‘differential multiple cleavage-site’ model. Gene Ther. 2007;14:1309–1318. doi: 10.1038/sj.gt.3302981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Papworth M, Kolasinska P, Minczuk M. Designer zinc-finger proteins and their applications. Gene. 2006;366:27–38. doi: 10.1016/j.gene.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 14.Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc. Natl Acad. Sci. USA. 1996;93:1156–1160. doi: 10.1073/pnas.93.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu J, Kandavelou K, Chandrasegaran S. Custom-designed zinc finger nucleases: what is next? Cell Mol. Life Sci. 2007;64:2933–2944. doi: 10.1007/s00018-007-7206-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smith J, Bibikova M, Whitby G, Reddy AR, Chandrasegaran S, Carroll D. Requirements for double-strand cleavage by chimeric restriction enzymes with zinc finger DNA-recognition domains. Nucleic Acids Res. 2000;28:3361–3369. doi: 10.1093/nar/28.17.3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moehle EA, Rock JM, Lee YL, Jouvenot Y, DeKelver RC, Gregory PD, Urnov FD, Holmes MC. Targeted gene addition into a specified location in the human genome using designed zinc finger nucleases. Proc. Natl Acad. Sci. USA. 2007;104:3055–3060. doi: 10.1073/pnas.0611478104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Urnov FD, Miller JC, Lee YL, Beausejour CM, Rock JM, Augustus S, Jamieson AC, Porteus MH, Gregory PD, Holmes MC. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 19.Minczuk M, Papworth MA, Kolasinska P, Murphy MP, Klug A. Sequence-specific modification of mitochondrial DNA using a chimeric zinc finger methylase. Proc. Natl Acad. Sci. USA. 2006;103:19689–19694. doi: 10.1073/pnas.0609502103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jamieson AC, Miller JC, Pabo CO. Drug discovery with engineered zinc-finger proteins. Nat. Rev. Drug Discov. 2003;2:361–368. doi: 10.1038/nrd1087. [DOI] [PubMed] [Google Scholar]
- 21.Isalan M, Klug A, Choo Y. A rapid, generally applicable method to engineer zinc fingers illustrated by targeting the HIV-1 promoter. Nat. Biotechnol. 2001;19:656–660. doi: 10.1038/90264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Laity JH, Dyson HJ, Wright PE. DNA-induced alpha-helix capping in conserved linker sequences is a determinant of binding affinity in Cys(2)-His(2) zinc fingers. J. Mol. Biol. 2000;295:719–727. doi: 10.1006/jmbi.1999.3406. [DOI] [PubMed] [Google Scholar]
- 23.Moore M, Klug A, Choo Y. Improved DNA binding specificity from polyzinc finger peptides by using strings of two-finger units. Proc. Natl Acad. Sci. USA. 2001;98:1437–1441. doi: 10.1073/pnas.98.4.1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eichwald V, Daeffler L, Klein M, Rommelaere J, Salome N. The NS2 proteins of parvovirus minute virus of mice are required for efficient nuclear egress of progeny virions in mouse cells. J. Virol. 2002;76:10307–10319. doi: 10.1128/JVI.76.20.10307-10319.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Papworth M, Moore M, Isalan M, Minczuk M, Choo Y, Klug A. Inhibition of herpes simplex virus 1 gene expression by designer zinc-finger transcription factors. Proc. Natl Acad. Sci. USA. 2003;100:1621–1626. doi: 10.1073/pnas.252773399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Minczuk M, Piwowarski J, Papworth MA, Awiszus K, Schalinski S, Dziembowski A, Dmochowska A, Bartnik E, Tokatlidis K, Stepien PP, et al. Localisation of the human hSuv3p helicase in the mitochondrial matrix and its preferential unwinding of dsDNA. Nucleic Acids Res. 2002;30:5074–5086. doi: 10.1093/nar/gkf647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Garrido N, Griparic L, Jokitalo E, Wartiovaara J, van der Bliek AM, Spelbrink JN. Composition and dynamics of human mitochondrial nucleoids. Mol. Biol. Cell. 2003;14:1583–1596. doi: 10.1091/mbc.E02-07-0399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saeki K, Suzuki H, Tsuneoka M, Maeda M, Iwamoto R, Hasuwa H, Shida S, Takahashi T, Sakaguchi M, Endo T, et al. Identification of mammalian TOM22 as a subunit of the preprotein translocase of the mitochondrial outer membrane. J. Biol. Chem. 2000;275:31996–32002. doi: 10.1074/jbc.M004794200. [DOI] [PubMed] [Google Scholar]
- 29.Cornu TI, Thibodeau-Beganny S, Guhl E, Alwin S, Eichtinger M, Joung J, Cathomen T. DNA-binding specificity is a major determinant of the activity and toxicity of zinc-finger nucleases. Mol. Ther. 2008;16:352–358. doi: 10.1038/sj.mt.6300357. [DOI] [PubMed] [Google Scholar]
- 30.Miller JC, Holmes MC, Wang J, Guschin DY, Lee YL, Rupniewski I, Beausejour CM, Waite AJ, Wang NS, Kim KA, et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat. Biotechnol. 2007;25:778–785. doi: 10.1038/nbt1319. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.