


Abstract
The structures of DNA–protein complexes have illuminated the diversity of DNA–protein binding mechanisms shown by different protein families. This lack of generality could pose a great challenge for predicting DNA–protein interactions. To address this issue, we have developed a knowledge-based method, DNA-binding Domain Hunter (DBD-Hunter), for identifying DNA-binding proteins and associated binding sites. The method combines structural comparison and the evaluation of a statistical potential, which we derive to describe interactions between DNA base pairs and protein residues. We demonstrate that DBD-Hunter is an accurate method for predicting DNA-binding function of proteins, and that DNA-binding protein residues can be reliably inferred from the corresponding templates if identified. In benchmark tests on ∼4000 proteins, our method achieved an accuracy of 98% and a precision of 84%, which significantly outperforms three previous methods. We further validate the method on DNA-binding protein structures determined in DNA-free (apo) state. We show that the accuracy of our method is only slightly affected on apo-structures compared to the performance on holo-structures cocrystallized with DNA. Finally, we apply the method to ∼1700 structural genomics targets and predict that 37 targets with previously unknown function are likely to be DNA-binding proteins. DBD-Hunter is freely available at http://cssb.biology.gatech.edu/skolnick/webservice/DBD-Hunter/.
INTRODUCTION
With the progress of structural genomics projects, an increasing number of protein structures have become available (1). As of early February 2008, a total of 160 792 proteins have been registered as targets by structural genomics centers worldwide, with the structures of 5396 targets already deposited in the PDB (http://targetdb.pdb.org/). Since many targets are representatives of previously uncharacterized protein families, the function of a large number of these proteins is unknown. Identifying their function is an important challenge. In recent years, many computational methods have been developed to assist in functional annotation (2–4). Compared to experimental studies, computational methods have the advantage of high efficiency and low cost. Most are based on the idea of functional inference through homology. While sequence comparison methods (5,6) are very powerful (7–9), they may offer limited help with the task of assigning functions for structural genomics targets because many have low-sequence similarity to previously characterized proteins. Structure-based methods may provide additional clues to a protein's function because structure is better conserved than sequence (10). However, since a common fold may be shared by proteins with very different functions, it remains a challenge to infer protein function on the basis of structure alone (11).
An area where protein structure could potentially be useful is in the identification of DNA-binding proteins. Such proteins play an essential role in a cell and are involved in transcription, replication, packaging, repair and rearrangement. It has been estimated that 2–3% of prokaryotic proteins and 6–7% of eukaryotic proteins bind DNA (12). To understand the basic rules, many efforts have investigated the patterns of DNA–protein interactions observed in the structures of DNA–protein complexes (13–15). In contrast to the strict base pairing rule observed in double-stranded DNA (dsDNA), it is now clear that there is no simple code for protein–DNA recognition. Instead, numerous mechanisms are exhibited by different protein families (12).
Given the structure of a protein whose function is unknown, one wishes to answer the following questions: (i) Does this protein have a DNA-binding function? (ii) If so, where are its DNA-binding sites? (iii) What specific DNA sequences, if any, does the protein recognize? To address the first problem, several knowledge-based approaches have been developed (16–20). Shanahan et al. (18) used structural comparison to detect three types of well-defined DNA-binding structural motifs, helix-turn-helix (HTH), helix-hairpin-helix (HhH) and helix-loop-helix (HLH), which appear in ∼30% of 54 structural families based on an earlier classification of DNA–protein complex structures (12). Sophisticated machine-learning methods have also been attempted (16,17,19,20). These utilize techniques such as neural networks (16,19), logistic regression (20) and support vector machines (17). Features used by these methods include the composition of amino acids, the charge/dipole moment of the protein molecule and the presence of positively charged surface patches. On average, these studies reported sensitivities ranging from 70% to 90% and specificities ranging from 65% to 95%. To address the second question, similar machine-learning methods have been developed to predict individual DNA-binding residues on proteins (21–23), with an average accuracy ranging from 65% to 80%. To address the third problem, statistical models have been introduced to characterize the specificity of DNA sequences for a given DNA-binding protein (13,24–26), with a few successful examples reported.
Since DNA-binding proteins likely comprise only a small fraction of structural genomics targets, for practical applications, it is necessary to develop a method with high precision. Otherwise, the number of false positives could easily outnumber the true positives, rending such an approach is impractical for automatic function assignment. All machine-learning methods mentioned above, however, reported relatively low specificities for non-DNA-binding proteins (16,17,19,20). In addition, these rates were obtained on small sets of less than 250 structures. It is not clear whether similar specificity would be obtained on a much larger data set of thousands of structures, a scenario relevant to proteomic-scale applications. To address these issues, we describe a new method, DNA-binding Domain Hunter (DBD-Hunter), for the prediction of DNA-binding proteins and associated DNA-binding sites. The method uses both structural comparisons and a DNA–protein statistical potential to assess whether or not a given protein binds DNA. We demonstrate that DBD-Hunter achieves an extremely high specificity of 99.5% and a precision of 84% in benchmark tests, with a sensitivity of 47% obtained on unbound and a sensitivity of 58% on DNA bound protein structures. By way of illustration as to its applicability, we apply our method to 1697 structural genomics targets and predict that 37 previously unknown targets are DNA-binding proteins.
METHODS
Availability
All datasets listed below, the statistical potential parameters and a web-server implementation of DBD-Hunter are available at http://cssb.biology.gatech.edu/skolnick/files/.
Data sets
DB179
A set of 179 DNA–protein complex structures (DB179) were selected by the following procedure: The July 2007 release of the PDB was queried to retrieve all X-ray structures of protein–DNA complexes with resolution better than 3.0 Å. The resulting 1045 complex structures were further partitioned into chains and analyzed. We only consider protein–DNA complexes that satisfy the following three conditions: (i) the protein has a minimum of 40 amino acids; (ii) the DNA molecule is dsDNA with at least six base pairs and (iii) the protein has at least five protein residues within 4.5 Å of the DNA molecule. The analysis led to 1676 DNA-binding protein chains. An all-against-all global sequence alignment was performed on these protein chains using the ALIGN0 program (27) from the FASTA2 package. Pairwise sequence identity is defined as the ratio of the number of identical residues over the length of the shorter sequence. We used the number of DNA–protein contacts, structure resolution and the available literature to select only one representative among protein chains with larger than 35% sequence identity, leading to a nonredundant set such that any two protein chains have <35% sequence identity. SCOP annotations (28) and visual inspection were used to identify the DNA-binding domain (DBD) for each protein chain. The resulted 179 DNA-binding protein domains and associated DNA chains are listed in Supplementary Table 1.
NB3797
A control set of 3797 non-DNA-binding protein chains (NB3797) was created from a nonredundant set of 7037 protein structures, which comprise the PROSPECTOR threading template library built from the May 2006 PDB release by clustering all PDB protein structures using a 35% global sequence identity cutoff and choosing one representative from each cluster (29). From these 7037 chains, we select the 5636 proteins with SCOP annotations. We further discarded any chain if its PDB record contains a DNA molecule or its SCOP annotation contains the keyword ‘hypothetical’. Protein chains were manually inspected to determine whether its SCOP superfamily, family and domain annotations contain the keyword ‘DNA’. Those whose function is associated with DNA-binding were removed. All ribosomal proteins were also excluded. For each of the resulting 3911 protein chains, the keyword ‘DNA’ was searched in the title, abstract and keyword sections of its primary citation. Positive hits were inspected by reading the literature to exclude DNA-binding proteins. The final 3797 protein chains compose NB3797.
APO104/HOLO104
A total of 104 pairs of DNA-binding protein structures determined both in the absence and presence of DNA were constructed from the PDB May 2007 release. The PDB was queried to retrieve two sets of proteins: the holo structure set consists of 759 proteins cocrystallized with a dsDNA molecule; the apo set comprises 35 899 crystal/NMR structures determined without any DNA. An all-against-all sequence alignment between the two sets was performed following the same procedure described above. The alignment procedure resulted in 679 holo–apo pairs, which have sequence identity >35% between each pair. The 679 holo chains were further culled by excluding redundant sequences with an identity cutoff of 35%. One representative was selected among proteins with pairwise sequence identity >35%, using literature and structure resolution information for guidance. The final results are 104 holo-structures and their corresponding apo-structures, denoted as HOLO104 and APO104, respectively. Most of these holo–apo pairs have high-sequence identity, 100% for 62 pairs and >95% for 91 pairs. The remaining 13 pairs, which have sequence identities ranging from 45% to 95% are composed of apo–holo homologs from the same SCOP family.
SG1697
A set of structural genomics targets was selected from the Jan 2008 PDB release. The PDB was queried with the classification keyword ‘structural genomics’, resulting in 1886 PDB entries. These were split into protein chains, which were further clustered at a 90% sequence identity cutoff with the program CD-HIT (30). From each cluster, we randomly select one representative. These 1697 representatives compose the set SG1697.
Statistical pair potential
To derive a statistical pair potential for describing DNA–protein interactions, we consider a contact between a protein residue and a functional group of DNA defined as that when at least one heavy atom from the protein residue is within 4.5 Å of at least one heavy atom from the corresponding DNA functional group. Four types of functional groups were considered for DNA nucleotides (Figure 1). Pyrimidines C and T have the phosphate (PP), the sugar (SU) and the pyrimidine (PY) groups. In addition, purines A and G have a fourth group, the imidazole (IM) group. Note that all DNA groups are residue specific. To differentiate them, we add their nucleotide names as prefixes, e.g. A.PP represents the phosphate group of an adenine.
Figure 1.

Scheme of DNA nucleotide functional groups considered for protein–DNA contact analysis. The phosphate, sugar, pyrimidine and imidazole groups are colored in green, orange, red and blue, respectively. Note that the functional groups are base specific, e.g. the phosphate group of adenine is different from the phosphate group of thymine.
A knowledge-based statistical pair potential was developed from an analysis in the DNA–protein complex set DB179 (Supplementary Figure 1). The derivation of the potential was based on the assumption that the frequencies of observed pair interaction states follow a Boltzmann distribution (31,32). The pair interaction energy between protein residue type and a reduced DNA atom is defined as:
| 1 |
where 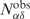 is the number of observed contacts for the αδ pair and
is the number of observed contacts for the αδ pair and 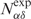 is the number of expected contacts assuming no preferential interaction. The reference state, , is defined by the product of the total number of observed contacts
is the number of expected contacts assuming no preferential interaction. The reference state, , is defined by the product of the total number of observed contacts 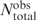 and the mole fractions of α and δ, namely
and the mole fractions of α and δ, namely
| 2 |
The statistical potential energy E (also named the interfacial energy) of a DNA–protein complex structure is defined as the sum of pair interactions for all protein–DNA contacts.
The Z-score of a native complex structure is defined as:
| 3 |
where Eave and σ are the average and SD of the statistical potential energies of all random structures, and Enat is the statistical potential energy of the native complex. In specificity tests, random structures were obtained by replacing interfacial DNA or protein residues with random nucleotides or amino acids. We assume that a contact is formed in a random structure at the same location as observed in the native structure. Since the imidazole group only appears in purines, we ignore any contact involving a purine imidazole group if the purine is replaced by a pyrimidine.
DNA-binding interaction prediction protocol
Using structural alignment and the statistical potential, we developed a new method, DBD-Hunter, to predict DNA–protein interactions, given a target structure. First, the target structure is scanned against the template library DB179 for similar protein structures with the structural alignment program TM-align (9). Only Cα backbone coordinates are used for the structural alignment and for root mean squared deviation (RMSD) calculations. A TM-score >0.4 indicates significant structural alignment (9). To reduce the number of false positives, we employed the higher TM-score threshold of 0.55 for template selection (see below). For templates with a TM-score better than the threshold, the statistical potential energy between the target protein and the template DNA is calculated by evaluating contacts within the structurally aligned regions. The contact evaluation follows the similar procedure adopted in structure threading by replacing original template protein residues with corresponding aligned template residues (29). The templates are then ranked according to their interfacial energies. If the lowest interfacial energy is below a (to be determined) energy threshold, the target is predicted to be a DNA-binding protein. If no template is found in either the structural alignment or satisfying the energy criterion, the target is classified as a non-DNA-binding protein. For proteins predicted as a DNA-binding protein, we further infer DNA-binding protein residues from their templates. A DNA-binding protein residue is defined as a residue with at least one heavy atom within 4.5 Å of a DNA functional group.
Optimization of parameters for DNA-binding protein prediction
The DNA-binding protein prediction method requires two threshold parameters: the TM-score threshold and the statistical potential energy threshold. Here, we present two strategies for optimizing these two parameters by maximizing the Matthews correlation coefficient (MCC) (33) of predictions on DB179 and NB3797. In the first strategy, we simply search for the best parameter pair that gives the highest MCC. Supplementary Figure 2 shows the contour representation of MCC in threshold space. The best MCC of 0.64 is given by a TM-score threshold of 0.62 and an energy threshold of −4.8, corresponding to a sensitivity of 0.49 and a specificity of 0.997. The high (>0.60) MCC region is located within the TM-score threshold range from 0.53 to 0.67 and the energy threshold range from −10 to −2.5. As the TM-score threshold increases, the optimal energy threshold corresponding to the highest MCC at a given TM-score threshold increases as well. This can be easily understood: since structures with higher similarity are more likely to share a similar function, the energy criterion can be softened as the level of structural similarity increases and vice versa.
The observation leads to the second optimization strategy: rather than using a constant energy threshold, the optimal energy threshold can be dependent on the value of the TM-score. Specifically, we divided the TM-score range from 0.4 to 1.0 into nine regions (Table 1). Starting from template hits within the top region, we select the optimal energy threshold that gives the highest MCC of predictions on DB179 and NB3797. Positive targets under the optimal energy criteria were removed from both sets, and we re-run predictions on the reduced target sets for the next TM-score region of template hits. The process was repeated for all nine TM-score regions and generated an optimal energy threshold for each region. However, for TM-scores below 0.55, the number of false positives greatly exceeds the number of true positives at the maximum MCCs, which are invariably low (<0.15). Therefore, the minimum TM-score threshold is set at 0.55 to reduce false positives. The list of optimized parameters is provided in Table 1. Using these parameters, a sensitivity of 0.58 and a specificity of 0.995 were achieved on the training set DB179 and NB3797, with a corresponding MCC of 0.69. The optimal parameters were used in validation tests on APO104/HOLO104 and the application on SG1697.
Table 1.
Optimization of TM-score and energy threshold parameters for DBD-Hunter on DB179 and NB3797
| TM-score threshold range | Energy threshold | TP | FN | FP | TN | Precision | Max. MCC |
|---|---|---|---|---|---|---|---|
| 0.74–1.00 | 1.1 | 52 | 8 | 6 | 63 | 0.90 | 0.78 |
| 0.62–0.74 | −4.8 | 43 | 23 | 10 | 368 | 0.81 | 0.69 |
| 0.58–0.62 | −9.5 | 4 | 23 | 2 | 568 | 0.67 | 0.30 |
| 0.55–0.58 | −12.3 | 4 | 28 | 1 | 968 | 0.80 | 0.31 |
| 0.52–0.55 | −2.8 | 16 | 34 | 188 | 1496 | 0.08 | 0.11 |
| 0.49–0.52 | −3.0 | 17 | 31 | 321 | 2029 | 0.05 | 0.09 |
| 0.46–0.49 | −14.1 | 4 | 34 | 20 | 2676 | 0.17 | 0.12 |
| 0.43–0.46 | −2.3 | 22 | 16 | 981 | 2070 | 0.02 | 0.06 |
| 0.40–0.43 | −13.7 | 2 | 14 | 36 | 2189 | 0.05 | 0.07 |
The optimal energy threshold that gives the highest MCC of predictions is listed for each TM-score range. When TM-scores are below 0.55, the numbers of false positives greatly exceed the number of true positives at the maximum MCCs. Therefore, we set the minimum TM-score threshold at 0.55. The optimized threshold values adopted in this study were represented in bold.
Assessment of DNA-binding protein prediction methods
We compared our prediction method with three different approaches: TM-align (9), PSI-BLAST (5) and the method proposed by Szilagyi and Skolnick (denoted as the SS method) (20). The protein structures of DB179 were used as the template library for TM-align. When applying TM-align, a target is classified as a DNA-binding protein if it hits a template with a TM-score higher than a specified threshold. Otherwise, the target is classified as non-DNA-binding. When applying PSI-BLAST (version 2.2.17), up to four rounds of scanning on the NCBI-NR nonredundant protein sequence library (the July 2007 release) were performed to derive a position-specific sequence profile for each target sequence. An inclusion E-value threshold of 0.001 and the default values for other arguments were employed. Using this profile, a last PSI-BLAST run was performed on the DB179 sequence library. If a target hits a template with an E-value higher than the specified threshold, the target is classified as a DNA-binding protein; otherwise, it is classified as a non-DNA-binding protein. The default parameters were employed for the SS method. During the benchmark tests on DB179, APO104 and HOLO104, homologs with global sequence identity >35% were excluded from the template library.
The prediction outcome can be classified and counted for each method. The numbers of true positives, false positives, true negatives and false negatives are designated as TP, FP, TN and FN, respectively. Performance measures are defined as the following:
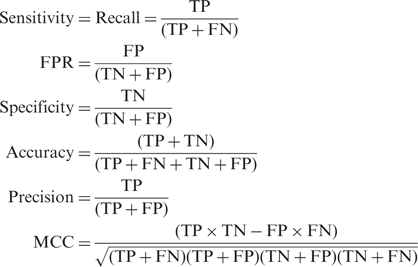 |
4 |
where FPR denotes false positive rate.
RESULTS
We first develop the statistical potential and examine its specificity to both protein sequences and DNA sequences. The statistical potential energy and structural similarity, two features used by our DNA–protein prediction method, were analyzed on DB179/NB3797. The performance of our method was assessed by leave-one-out cross-validation and compared to the three other methods described above. Conformational changes occurred during the apo-to-holo transition were subsequently studied for DNA-binding proteins. The performance of our method was tested on both apo- and holo-structure sets APO/HOLO104. Finally, a total of 1679 structural genomics targets were scanned for DNA-binding proteins as a real world test of the methodology.
Statistical pair potential
The pair potential parameters have been derived for 20 amino acids and 14 nucleotide functional groups using the Boltzmann principle (Supplementary Figure 1). A total of 12 771 DNA–protein contacts observed in the nonredundant DNA–protein complex structure set DB179 have been considered. The positively charged amino acids Arg and Lys are the most preferred contact partners by DNA nucleotides. The result is expected due to the negative charge carried by DNA. The polar amino acids Asn, His, Tyr, Gln, Thr and Ser are attracted to the DNA backbone phosphate and sugar groups, but are less preferred by base groups. The hydrophobic residue Leu and positively charged residue Glu have the most energetically unfavorable interactions with DNA nucleotides. In general, DNA base groups have more specific interactions with amino acids than backbone groups. Imidazole groups, for example, are favored by only two to three amino acids. One case of such favorable pairs is the guanine imidazole group and Arg, which is expected because hydrogen bonding between them has been frequently observed. By comparison, most polar and positively charged amino acids are attracted to the phosphate and sugar groups.
A basic requirement for any good statistical potential is the capability to characterize favorable energetic interactions, given a DNA–protein complex structure. To examine whether our potential meets this requirement, as shown in Figure 2, we performed both a self-consistent test and a jackknife test on DB179. In the self-consistent test, the interfacial energy E for a complex structure is evaluated with the parameter set determined from all 179 complexes. The result shows that 97% of these complexes have a favorable interfacial energy (E < 0). In the jackknife test, also termed leave-one-out cross-validation, the target structure is excluded from the statistical potential derivation, and the interfacial energy for the target structure is then calculated with the corresponding parameter set. As shown in Figure 2, energies calculated from both tests are closely correlated with a correlation coefficient >0.99. The average potential energy from the jackknife test is −24.2, which is 2.1 kT units higher than the average of −26.3 from the self-consistent test. The fraction of complexes with favorable interfacial energy in the jackknife test is 94%, which is 3% lower than the self-consistent test.
Figure 2.

Distribution of the statistical potential energy for 179 DNA–protein complexes in the self-consistent test. The insert shows the potential energy calculated in a Jackknife test versus the energy calculated in the self-consistent test.
Specificity of the statistical potential
The requirement of favorable energy interaction of the native DNA–protein complex is a necessary, but not sufficient condition for characterizing specific recognitions between proteins and DNA. We further assess the specificity of our potential parameter set by generating random DNA or protein sequences separately for each complex. The DNA/protein specificity is measured by the Z-score (Equation 3) of the native potential energy compared with energies calculated for random DNA/protein sequences. All energies were calculated with the individual jackknife parameter set corresponding to each target complex.
In the DNA specificity test, up to one million DNA sequences were randomly generated for the interfacial DNA base pairs of each complex. Equal probabilities of 0.25 were assigned for the four types of nucleotides. For structures with less than 10 DNA base pairs, we conducted an exhaustive investigation of all possible combinations. As shown in Figure 3A, 109 proteins with specific DNA recognition, e.g. transcription factors and restriction endonucleases, have an average Z-score of −1.2, which is one unit lower than the average Z-score of −0.2 for 70 proteins recognizing nonspecific DNA. The result demonstrates a modest Z-score difference between specific DNA recognition and nonspecific DNA recognition on average.
Figure 3.

(A) Distribution of native structure Z-scores among randomly generated DNA sequences. (B) Distribution of native structure Z-scores for randomly generated protein sequences.
In the protein specificity test, one million random protein sequences were generated for DNA-binding protein residues of each complex. To avoid overrepresentation of rare amino acids, we assign the frequency of an amino acid type observed in DB179 as the probability of the corresponding amino acid in random sequences. As shown in Figure 3B, the mean and SD of the Z-score for native complex structures is −3.2 and 1.1. Only seven, or 3.9%, of the complex structures have a Z-score higher than −1.0. The result suggests that the statistical potential is reasonably specific to DNA-binding proteins. We can, therefore, utilize the statistical potential as a feature to discriminate DNA-binding proteins from non-DNA-binding proteins.
Characteristic features of DNA-binding proteins
For the purpose of discriminating DNA-binding from non-DNA-binding proteins, we consider two features: structure similarity and statistical potential energy. In our approach, a target structure is scanned against the template library DB179 for similar structures with TM-align. Using the TM-score as the structural similarity metric, we identify templates that have a score higher than a given TM-score threshold. The statistical potential energy is then calculated for the target using the structural alignment to a qualified template. The two features were examined on DB179 and the non-DNA-binding set NB3797. In the test of DB179, the target structure was excluded from the template library for both the template scanning and the statistical potential derivation.
The distributions of the top TM-score-ranked template for DB179 and NB3797 are shown in Figure 4A. About 93% of DNA-binding proteins and 70% of non-DNA-binding proteins hit at least one template with a TM-score higher than a significant value of 0.50. As one raises the value of the TM-score threshold, the fraction of non-DNA-binding protein structures with a qualified template decreases more rapidly than that of DNA-binding protein structures. At a TM-score threshold of 0.62, only 10% of non-DNA-binding proteins have at least one template hit, whereas 68% of DNA-binding proteins satisfy this criterion. However, since the size of NB3797 is much larger than that of DB179, the absolute number of positives from NB3797 is over three times the number of positives from DB179.
Figure 4.

Discriminatory feature analysis for DNA-binding and non-DNA-binding proteins. (A) Distribution of the highest TM-score-ranked template on DB179/NB3797. The numbers of template hit are given above the histogram bars. (B) Cumulative faction of the top energy-ranked template versus the statistical potential energy. Only templates higher than the TM-score threshold of 0.62 were considered.
To further reduce false positives, we use the statistical potential energy to re-rank the templates preselected from the structural alignment procedure. Figure 4B shows the distribution of the top energy ranked template for DB179 and NB3797 at a TM-score threshold of 0.62. About 1.3% (69) of non-DNA-binding proteins and 57% (102) of DNA-binding proteins have a template with a favorable energy value (E < 0). At an energy threshold of −4.8, the fraction of DNA-binding proteins satisfying the energy criterion drops slightly to 49%, while only 0.003% (12) of non-DNA-binding protein's top templates satisfy the same criteria. Use of the statistical potential dramatically reduces the number of false positive hits.
We have not employed the Z-score of the target sequence/structure relative to the randomized target sequence aligned to the selected template, as we find that the performance is essentially the same as when the energy cutoff is used. Since about 25% of DNA-binding residues are missed on average by the structural alignment and the DNA sequence is that of the template, the average Z-score of −2.1 for the top Z-score ranked template is not surprisingly larger (less negative) than that for the native protein–DNA complex whose average is −3.2. Given the rather small range of Z-scores, this is the origin of the comparable performance as to whether an energy cutoff or Z-score criterion is used.
Assessment of DNA-binding protein prediction methods
By combining structural comparison with a statistical potential, we developed DBD-Hunter for DNA-binding protein prediction (see Methods section for details). To assess the performance of our method, we compared it with three other methods: the SS method, PSI-BLAST and TM-align. Figure 5 shows the receiver operator characteristic (ROC) curves and precision-recall (PR) curves for benchmark tests on DB179 and NB3797. The ROC and the PR curves of our method were obtained by varying the energy threshold while fixing the TM-score threshold at 0.62. For the other three methods, the variable used to obtain the ROC and PR curves are: the threshold defined in ref. (20) for SS, the E-value for PSI-BLAST and the TM-score for TM-align. For comparison, the results of DBD-Hunter using TM-score-dependent optimized energy threshold, denoted as DBD-Hunteropt, are also provided; the corresponding sensitivity of 58%, specificity of 99.5%, precision of 84% and MCC of 0.69 are the best in our benchmark tests (Table 2).
Figure 5.

Performance comparison of methods for DNA-binding protein prediction. All tests were performed on DB179 and NB3797. The result obtained by DBD-Hunter using optimized threshold parameters is indicated by a star symbol. (A) ROC (sensitivity versus FPR) curves. (B) PR (precision versus sensitivity) curves.
Table 2.
Comparison of the maximum MCC by four DNA-binding protein prediction methods on DB179/NB3797
| Method | Max. MCC | Sensitivity | FPR | Precision |
|---|---|---|---|---|
| DBD-Hunteropt | 0.69 | 0.58 | 0.005 | 0.84 |
| DBD-Hunter | 0.64 | 0.49 | 0.003 | 0.87 |
| TM-align | 0.47 | 0.52 | 0.028 | 0.47 |
| PSI-BLAST | 0.56 | 0.44 | 0.007 | 0.76 |
| SS | 0.31 | 0.40 | 0.044 | 0.93 |
Clearly, our method outperforms all other three methods within the low FPR regime (<10%), which is relevant for practical applications. The maximum MCCs of the four methods are listed in Table 2. DBD-Hunter achieves the highest maximum MCC of 0.69, compared with 0.47 for TM-align, 0.56 for PSI-BLAST and 0.31 for SS. As shown in the ROC plot (Figure 5A), the sensitivity of our method jumps to 49% at a very low FPR of 0.3%, then gradually increases to 60% at a FPR of 1.6% and finally reaches a plateau of 68% at a FPR of 6.6%. The 68% sensitivity limit is due to the TM-score threshold imposed. If one only considers structural similarity, inferior performance was obtained. For example, TM-align gives a sensitivity of 50% with a FPR of 2.8%, which is more than nine times the FPR of our method at the same sensitivity. PSI-BLAST is generally less sensitive than the structure-based methods. At a permissive FPR of 4%, PSI-BLAST recognizes about half of the targets. The performance of the SS method is the worst among these methods. We note that its FPR is much higher on NB3797 than previous reported FPRs on small control sets (20). The threshold used to obtain an FPR of 2% on smaller sets yield an FPR of 5.7% on NB3797. One advantage of our method is that it delivers a high precision at a reasonable sensitivity. As shown in the PR plot (Figure 5B), the precision of DBD-Hunter stays at a high level above 88% for a sensitivity up to 50%. By comparison, none of the other three methods can achieve this level of precision at a sensitivity better than 30%. The high precision is important for application to targets on a proteomic scale.
Prediction of DNA-binding residues on proteins
Since DBD-Hunter identifies a template for each target, it is attempting to infer DNA-binding sites from the template, whose DNA-binding site is known. The most straightforward way is to assign DNA-binding function to target residues aligned with DNA-binding residues of the template. This approach was conducted on 103 proteins predicted as DNA-binding proteins by DBD-Hunter using the TM-score-dependent optimal thresholds. These proteins include 42 enzymes, 48 transcription factors and 13 other types of DNA-binding proteins. For each target structure, we make a binary prediction (DNA binding or non-DNA binding) on each residue aligned with the top energy-ranked template. Performance measures, sensitivity, specificity, accuracy and precision were calculated for each target structure. The box plot of the results is shown in Figure 6. On average, a sensitivity of 72%, a specificity of 93%, an accuracy of 90% and a precision of 71% were obtained. For 81% of the target structures, we achieved a precision >60%. The results imply that the closely related target-template pairs were correctly identified in most cases.
Figure 6.

Performance on the prediction of DNA-binding residues. A total of 103 DNA-binding proteins predicted by DBD-Hunter were examined. The lower, middle and upper quartiles of each box are 25th, 50th and 75th percentile, respectively. Whiskers extend to a distance of up to 1.5 times the interquartile range. Outliers and averages are represented by circles and squares, respectively.
Examples of DNA-binding protein prediction
Six examples of successful predictions by our method are illustrated in Figure 7A–F. In these examples, the sequence identity between a target and its template ranges from 9% to 17%. The lack of sequence similarity makes it difficult for the sequence-based PSI-BLAST method to identify these templates. In fact, none of them was hit by PSI-BLAST for the corresponding targets.
Figure 7.

Examples of DNA-binding protein predictions on DB179. (A–F) Structural alignment of the target structure and the template in cartoon representations. In each panel, the left snapshot shows the overall alignment, together with the cocrystallized DNA molecules. The color codes for protein and DNA representations are red and purple for the target, and green and cyan for the template, respectively. The right snapshot highlights DNA-binding residues of both the target and the template in the same color code as the left snapshot. Non-DNA-binding residues of the target were dimmed in gray. For a clear view of the binding interface, the two snapshots were taken from different orientations. In parentheses, each structure was labeled in the format of xxxxX, where xxxx is the four-digit PDB code and X is the chain identifier of the protein. If the PDB record contains no chain identifier, X is replaced with an underscore. Sequence identity (SID), TM-score (TMS), RMSD and the statistical potential energy E are provided at the bottom of each panel. Graphic images were made with the program VMD (62).
In the first example (Figure 7A), the target, the bipartite DBD of Tc3 transposase Tc3A (34), consists of two sub-domains that belong to the homeodomain-like superfamily defined in SCOP. The target hits three templates above a TM-score threshold of 0.62. As expected, they all share a homeodomain-like structure with a classic HTH DNA-binding motif. Each template yielded an interfacial energy strong enough for making a positive prediction, despite the fact that only one sub-domain of the target was aligned with the template. The best energy-ranked template, telomeric protein TRF1 DBD (35), has the lowest TM-score of 0.64 among these three templates, but it generated the most correct predictions of DNA-binding residues (15 versus 10 for both of the other two cases) and only one false positive.
The second example involves the target, the DBD of catabolite gene activator (36), and the template, the DBD of replication terminator protein (Figure 7B) (37). They share a similar structure, the winged helix DBD, which is a common DNA-binding motif. In fact, the target hits 10 templates with a TM-score higher than 0.62. The top energy-ranked template has the lowest TM-score among these 10 templates, but it made the highest number of correct predictions for DNA-binding residues (12 of 14).
In the third example, we examine the target, acute myeloid leukemia 1 protein RUNT domain (38), and the template, the DBD of p53 tumor suppressor (Figure 7C) (39). They closely resemble each other with a β-sandwich fold. Although the DNA-binding sites are located in a largely disordered region composed of two loops and two β-strands, the target was successfully predicted through the template. We note that the same template was hit by 15 non-DNA-binding proteins above the TM-score threshold of 0.62. Fourteen of these negative cases are correctly classified as non-DNA-binding by the energy criterion, because they exhibit repulsive energies. The only exception, actinoxantin (PDB code 1acx_), belongs to an antitumour antibiotic chrompoprotein family, whose members recruit chromophores that cleave DNA substrates (40). Although it is not clear whether actinoxantin itself binds to DNA, our prediction suggests that actinoxantin binds DNA and that this leads to subsequent DNA cleavage by the chromophore.
Two restriction endonucleases, HinP1I (41) and MspI (42), are presented in the fourth example. Both consist of two domains, aligned with an RMSD of 3.3 Å, the largest among these examples. The two enzymes extensively interact with DNA; there are 47 DNA-binding residues in the target HinP1I and 36 in the template MspI. Our method successfully identified 22 DNA-binding residues and produced nine false positives.
In the fifth example, we investigate the target, transcriptional repressor CopG (43), and the template, the DBD of methionine repressor MetJ (44). A DNA-binding motif, the so-called ribbon-helix-helix motif, is selected. The interfacial energy of −7.8 is relatively weak, mainly due to the small number of DNA-binding residues involved. All seven DNA-binding residues of the target are correctly predicted.
In the last example, the target, the DBD of Epstein-Barr nuclear antigen 1 (45), hits the template, the DBD of human papillomavirus-18 E2 (46). The two virus proteins share a low-sequence identity of 10%, yet have high-structural similarity with a TM-score of 0.75. Their structure is a ferredoxin-like fold, which was found in many non-DNA-binding proteins. In fact, 41 non-DB proteins from NB3797 hit the same template. All, but one, of these false hits were eliminated on the basis of the interfacial energy.
Conformational changes between apo- and holo-forms
For any structure-based method for DNA-binding protein prediction, it is necessary to examine its performance on structures determined in the absence of DNA. The reason is that the conformational changes occurring on DNA binding may affect the accuracy of the method. To address this issue, we have collected 104 pairs of apo- and holo-form DNA-binding proteins (APO104/HOLO104) and analyzed their conformational changes. Two RMSD metrics were calculated: RMSDglobal measures the overall conformational changes by superposing the two forms in the sequence aligned regions; and RMSDTM measures the conformational changes in the structural aligned regions identified by TM-align. As shown in Supplementary Figure 3, the majority (70%) of pairs have a RMSDglobal <3 Å, but a few (14%) have an RMSDglobal >5 Å. The latter are mainly due to flexible termini or relative domain movement of multiple-domain proteins (see examples below). If we consider structural alignment instead, the corresponding RMSDTM is <5 Å for all pairs and is within 3 Å for 89% of pairs. The corresponding coverage of the structural alignment is usually high, better than 90% of the shorter chain for 87% of the pairs (Supplementary Figure 3 insert). The results reveal that apo-to-holo conformational changes are mostly localized with a RMSD <3 Å for more than 70% of DNA-binding proteins.
Prediction of DNA binding using apo-structures
We further benchmarked our method on APO104/HOLO104 using the optimized threshold parameters determined on DB179/NB3797. For a given target, any template with sequence identity >35% was excluded from the template library and the statistical potential derivation in our tests. As shown in Figure 8A, about the same number of APO104 and HOLO104 members hit at least one template above the TM-score threshold of 0.52, 94 for HOLO104 versus 95 for APO104. However, the distributions of the best structural templates are somewhat different. The holo target set hit more closely resembled templates than the apo set did. The latter has 28% less templates above the TM-score cutoff of 0.68 than the former. In particular, nine holo queries have one template with a TM-score better than 0.88, but no apo-structure has a template with such a high level structural similarity. Despite the relatively lower structural similarity to their templates, APO104 yielded only slightly fewer number of correct predictions as HOLO104 by DBD-Hunter. The number of positive predictions is 57 for the holo set and 49 for the apo set. These numbers correspond to a sensitivity of 55% for HOLO104 and 47% for APO104, compared with the value of 58% observed for DB179. DNA-binding residues were further inferred from the top-ranked template for predicted DNA-binding proteins from the apo/holo sets (Figure 8B), except for target 2frhA that has a controversial DNA-binding site (see examples below). On average, the predictions yield sensitivities of 68%/66%, specificities of 93%/93%, accuracies of 89%/87% and precisions of 67%/66% for HOLO104/APO104.
Figure 8.

Prediction of DNA-binding interactions for APO104 and HOLO104. (A) Distribution of the top TM-score ranked templates. Using the statistical potential energy threshold parameters optimized on the benchmark set DB179, DBD-Hunter predicted 48 and 57 targets of DNA-binding proteins for APO104 and HOLO104, respectively. For each predicted DNA-binding protein, DNA-binding residues were predicted. The performance measures of these predictions were shown in (B). The box plots are drawn as in Figure 6.
Our method was compared with PSI-BLAST on APO104. For a fair comparison, an E-value of 1E-8.5 was chosen for PSI-BLAST such that it provided a similar precision rate (82%) to DBD-Hunter (precision rate 84%) on DB179/NB3797. Only 31 apo-structures were identified correctly as DNA-binding proteins by PSI-BLAST. DBD-Hunter, therefore, is about 60% more sensitive than PSI-BLAST on APO104. A much more permissive E-value of 0.001 generates 45 hits for PSI-BLAST, which is still 10% less than the correct predictions made by our method.
Examples of DNA-binding protein prediction on apo-structures
Four positive predictions on APO104 are illustrated in Figure 9A–D. In these examples, the target apo-forms and their holo-forms exhibit large RMSDglobe values ranging from 3 to 19 Å. Despite these significant conformational changes upon DNA binding, the apo-structures were successfully predicted as DNA-binding proteins.
Figure 9.

Examples of DNA-binding protein prediction on APO104. (A–D) In each panel, the left snapshot shows the structural alignment of the apo-structure and its corresponding holo-structure, and the right snapshot shows the alignment of the target apo-structure versus its template. The apo-, holo- and template structures are colored in red, blue and green, respectively. In (B), all proteins are composed of two monomers. The monomer studied is shown in solid color, while the other monomer is dimmed. PDB codes are given in parentheses.
The first example is the bacteriophage λ integrase protein, a tyrosine recombinase possessing two DBDs, the catalytic domain and the central domain (Figure 9A). The latter domain is missing in the apo-structure (47), but is present in the holo-structure (48). In the apo-to-holo transition, dramatic movement occurred at the C-terminal region (residues 331–356) of the catalytic domain, which brought a crucial catalytic residue Tyr342 in contact with a scissile phosphate of DNA from a distance of 20 Å away. The movement made the major contribution to the large RMSDglobal of 10 Å, because the remainder (residues 170–330) of the catalytic domain is virtually unchanged with an RMSDTM of 0.4 Å. It is the static core region of the target that allows a hit to a template, the N-terminal domain of Flp recombinase (49). The target and the template have a high TM-score of 0.71, in spite of a low-sequence identity of 12%. Major DNA-binding sites, including the catalytic triad of Arg212-His308-Arg311, were correctly identified as DNA-binding residues. Based on the strong interfacial energy of −24, a positive prediction was made for the target apo-structure.
The second example is the Max protein, a transcription factor from the basic/HLH/zipper (bHLH-Zip) family of DNA-binding proteins (Figure 9B). Members of this family form a stable dimeric structure when complexed with DNA, but they are notoriously difficult to stabilize under DNA-free conditions. The NMR structures of the apo-form were determined after cross-linking two monomers at the C-termini and introducing two stabilizing point mutations (50). As shown in Figure 9B, the apo-structure closely resembles the holo-form (51), except for the first 14 N-terminal amino acids of the basic region, which are unfolded in the apo-structure but form a helix in the presence of DNA. Nevertheless, half of the 14 DNA-binding residues are aligned with DNA-binding residues of the template from the sterol regulatory element binding protein (52), and the apo-structure is correctly classified as a DNA-binding protein.
The third example is the p65 subunit (also known as RelA) of nuclear factor-κB (Figure 9C). The p65 subunit consists of two β-sandwich domains connected by a flexible 10 amino acid linker. In the DNA-bound form of p65, the N-terminal domain provides most of the DNA-binding residues, while the C-terminal domain interacts with the p50 subunit (not shown) to form a heterodimer complex (53). The DNA-binding activity of p65 can be inhibited by IκBα, a protein recognizing p65 that induces a domain rotation of p65 (54). As shown in Figure 9C, the N-terminal domains from the apo- and the holo-structures overlap, whereas the C-terminal domain undergoes about a 40° rotation around the interdomain linker. Similar conformational changes have also been observed in the alignment of the target to the template NFAT1, a nuclear factor of activated T cell (55). Despite such a dramatic in-block movement of the C-terminal domain, p65 was correctly classified as a DNA-binding protein because most DNA-binding residues located in its N-terminal domain were correctly identified through the template.
The last example, the protein SarA, is the most intriguing (Figure 9D). The apo-structure (56) of the single-domain transcription factor adopts a dramatically different topology from its holo-structure (57). The RMSDglobal is 19 Å between these two structures. A notable difference is a winged HTH motif present in the apo-structure but missing in the holo-form, which instead has a unique DNA-binding motif. However, it has been suggested that the apo-structure represents the native form of SarA and that the unique DNA-binding mode observed in the holo-structure is due to crystallization artifacts (56). In our test, the winged HTH motif of the apo-structure was predicted to be DNA-binding by three templates (1qbjB, 1sfuA and 1cpgA). In particular, Arg90 is predicted to be a DNA-binding residue. This is consistent with the mutagenesis study (56), which shows that the residue is critical to the DNA-binding function of the SarA. Overall, our prediction provides evidence for the hypothesis that the apo-form structure is functionally relevant.
Application to structural genomics targets
Finally, we have applied our method to 1697 protein structures of unknown function determined by the structural genomics initiative. The optimized threshold parameters corresponding to a precision of 84% were employed for this application. A total of 37 targets predicted to be DNA-binding proteins are listed in Table 3. Fourteen of these targets were previously hypothesized to have a function associated with DNA binding, such as transcription factor activity. Three targets (1nogA, 1t6sA and 1vbkA) have a putative function not related to DNA binding. These three predictions are probably false positives. The putative function of the remaining 20 targets was not assigned. By comparison, PSI-BLAST predicted 29 targets as DNA-binding proteins using an E-value of 1E-8.5, which corresponds to a similar precision of 82% in benchmark tests. Among PSI-BLAST predictions, eight targets have a putative DNA-binding function and two targets have a putative function not related to DNA binding. One (1i60A) of the latter two targets has the fold of endonuclease IV, a DNA repair enzyme, but it has been proposed to have a function other than that of endonuclease IV due to an altered Zn-binding site (58). DBD-Hunter identified an endonuclease IV template for 1i60A with a high TM-score of 0.76 and predicts that the target is non-DNA-binding based on a repulsive statistical potential energy. Since all but four DNA-binding targets predicted by DBD-Hunter have sequence identity <25% to their templates, it is difficult for PSI-BLAST to identify these targets due to low-sequence similarity. In fact, only nine positive predictions are common to both methods. Among targets predicted by PSI-BLAST but missed by DBD-Hunter, only one target has a putative function related to DNA binding. In principle, one can combine these two methods to improve sensitivity.
Table 3.
A list of structural genomics targets predicted as DNA-binding proteins from SG1697
| Target | Template | TM-score | RMSD | SID | Energy | Putative function |
|---|---|---|---|---|---|---|
| 1j27A | 2bdpA | 0.63 | 2.40 | 0.035 | −5.4 | UK |
| 1nogA | 1sknP | 0.58 | 3.28 | 0.043 | −15.9 | NB |
| 1s7oA | 1gdtA | 0.67 | 1.46 | 0.116 | −7.1 | DB |
| 1sfxA | 1h0mD | 0.59 | 2.76 | 0.155 | −22.7 | DB |
| 1t6sA | 1u8rJ | 0.65 | 2.25 | 0.14 | −7.3 | NB |
| 1tuaA | 1jj4A | 0.55 | 2.37 | 0.123 | −14.8 | UK |
| 1vbkA | 1jj4A | 0.63 | 2.21 | 0.188 | −5.0 | NB |
| 1wi9A | 1qbjB | 0.68 | 2.31 | 0.133 | −12.2 | UK |
| 1wj5A | 1qbjB | 0.70 | 2.16 | 0.177 | −16.0 | UK |
| 1x58A | 1w0tA | 0.87 | 1.22 | 0.275 | 3.8 | DB |
| 1xg7A | 2bzfA | 0.59 | 3.46 | 0.096 | −13.4 | UK |
| 1z7uA | 1h0mD | 0.61 | 2.51 | 0.138 | −15.6 | DB |
| 1zelA | 1qbjB | 0.61 | 2.65 | 0.183 | −12.1 | UK |
| 2da4A | 1pufA | 0.64 | 1.72 | 0.211 | −29.1 | UK |
| 2dceA | 1qbjB | 0.59 | 2.14 | 0.179 | −10.2 | UK |
| 2e1oA | 1jkqC | 0.55 | 2.91 | 0.143 | −21.4 | UK |
| 2eshA | 1cgpA | 0.62 | 1.56 | 0.137 | −19.4 | UK |
| 2esnA | 1u8rJ | 0.62 | 2.21 | 0.121 | −12.2 | DB |
| 2ethA | 1u8rJ | 0.71 | 1.84 | 0.186 | −17.3 | DB |
| 2f2eA | 1sfuA | 0.65 | 1.89 | 0.143 | −11.7 | DB |
| 2fiuA | 1jj4A | 0.66 | 2.50 | 0.057 | −4.9 | UK |
| 2fmlA | 1qbjB | 0.57 | 2.44 | 0.075 | −12.7 | UK |
| 2fnaA | 1qbjB | 0.66 | 1.83 | 0.204 | −5.0 | UK |
| 2fyxA | 2a6oB | 0.79 | 1.67 | 0.289 | −20.9 | DB |
| 2hytA | 1jt0A | 0.75 | 1.61 | 0.212 | −14.1 | DB |
| 2g7uA | 1cgpA | 0.69 | 1.72 | 0.20 | −15.2 | DB |
| 2iaiA | 1jt0A | 0.64 | 3.36 | 0.25 | −5.4 | DB |
| 2jn6A | 1gdtA | 0.70 | 2.16 | 0.14 | −10.7 | UK |
| 2nr3A | 1sfuA | 0.70 | 2.34 | 0.103 | −10.3 | UK |
| 2nx4A | 1jt0A | 0.76 | 2.07 | 0.143 | −2.6 | DB |
| 2od5A | 1gdtA | 0.58 | 2.36 | 0.122 | −9.7 | DB |
| 2p8tA | 1h0mD | 0.57 | 2.27 | 0.123 | −16.8 | UK |
| 2pg4A | 1z9cF | 0.72 | 2.05 | 0.181 | −8.5 | UK |
| 2qc0A | 1sfuA | 0.65 | 2.18 | 0.111 | −9.6 | UK |
| 2qvoA | 1sfuA | 0.72 | 1.94 | 0.043 | −10.5 | UK |
| 3b73A | 1cgpA | 0.65 | 1.54 | 0.204 | −23.7 | UK |
| 3bddA | 1cgpA | 0.65 | 1.48 | 0.259 | −17.4 | DB |
RMSD and sequence identity were calculated for the structurally aligned region between the target and the template with TM-align. Targets are labeled according to their putative function annotated in their PDB records: DB (DNA-binding), NB (function not related to DNA-binding) and UK (unknown).
DISCUSSION
The main goal of the current study is to develop a knowledge-based method for predicting DNA-binding proteins and associated DNA-binding residues from structural genomics targets. For this purpose, the method had to satisfy three conditions: First, it must be capable of predicting DNA-binding proteins that have low or no sequence similarity (<35% identity) to their templates. If a target has high-sequence similarity (>40%) to any template, typically it can be detected using a sequence-based method such as PSI-BLAST, which is computationally more efficient than structure-based approaches. Second, the method must have a very low FPR because only a small fraction of proteins bind DNA. Assuming that a method with a 10% FPR and 90% sensitivity is applied to a target set, 10% of which are DNA-binding proteins, these numbers translate into a precision rate of about 50%. That is, half of the predictions are false positives, which is generally unacceptable for systematic application on thousands of genomics targets. Third, the method has to be validated on structures in the DNA-free form, since all target structures with unknown DNA-binding function are solved without DNA present. And the concern that DNA-binding proteins undergo conformational changes during the apo-to-holo transition has to be addressed. In the current study, we have demonstrated that DBD-Hunter satisfies all three conditions. In benchmark tests, it consistently outperforms the three other knowledge-based methods: the sequence-based method PSI-BLAST, the structural-based method TM-align and the SS method, which uses both sequence and structural information. Furthermore, we applied DBD-Hunter to 1697 structural genomics targets and predicted that 37 proteins bind DNA.
The current method employs two features, structural similarity and the statistical potential energy, for the purpose of discriminating DNA-binding proteins from non-DNA-binding proteins. Since protein structures with similar function are more likely conserved than their sequences (10), this allows us to identify a target that has low-sequence similarity but high-structural similarity to a homologous template. In tests on DB179 and APO104, the structural alignment procedure identifies 60% more DNA-binding proteins than PSI-BLAST does. Six pairs of target/template examples from DB179 are given in Figure 7. Invariably, they have low-sequence identify (<17%) but high-structural similarity (TM-score ≥0.62). In addition, the vast majority of negatives were filtered out during the structural comparison procedure. In the test on NB3797, 65% negatives were eliminated by structure alone.
To achieve high accuracy, however, structural similarity to known DNA-binding proteins is not enough. We note that the one-third of non-DNA-binding proteins from NB3797 have a significant structural alignment to DNA-binding proteins with a TM-score higher than 0.55. To further reduce false positives, an interfacial potential has been introduced. The potentials are specific to DNA-binding proteins with an average Z-score of −3.2 in the randomized sequence test. By requiring that the target structure not only be structurally similar to a known DNA-binding protein but that it also has a favorable interfacial potential, we reduced the number of false positives from 1327 to 19 in the test on NB3797, corresponding to an extremely low FPR of 0.5%. By comparison, FPRs ranging from 5% to 20% were reported in previous studies (16,17,19,20). Due to the reasons mentioned above, these high FPRs limit the potential application of these methods to structural genomics targets.
In previous machine-learning studies (16,17,19,20), the sizes of the non-DNA-binding protein sets used for training were small, typically ranging from 100 to 250 structures. This raises the concerns that the discriminatory features may be over-trained and that the FPR may be under-estimated as a result. For example, we tested the SS method (20) on a much larger control dataset NB3797. Indeed, we found that the FPR on NB3797 is much higher than that on smaller size data sets. The previously reported FPR of 2% increases to a FPR of 5.7% on the larger set. Since similar features such as the composition of amino acids and/or the charge/dipole distribution have also been used in the other studies (16,17,19), the FPRs reported in these studies should be viewed with caution.
A major concern with the use of structure-based methods is whether discriminatory features derived from holo-structures are transferable to apo-form structures. Two previous studies have examined performance on small sets of apo-structures, 52 targets in ref. (20) and 11 targets in ref. (19), and reported similar performance on both holo and apo sets. Here, we constructed much larger apo/holo sets composed of 104 targets for validation. We found that the sensitivities of our method on these two sets are very close, being just 8% lower on the apo set. The small difference can be understood from structural comparison analysis. 89% of apo–holo pairs have an RMSD of <3 Å in their structurally aligned region (typically >90% coverage), which is consistent with the suggestion that the conformational changes of DBDs are mostly localized (59). Notable conformational changes can be categorized into two major types: (i) refolding at the DNA-binding interface, e.g. the basic region of the leucine-zipper-like protein Max (Figure 9B), and (ii) domain reorientation of multiple-domain proteins, e.g. p65 of NF-κB (Figure 9C). The conformational changes observed at the binding interface may cause difficulty for approaches using strict DNA-binding motif comparisons. The HTH motif searching method, for example, requires an RMSD of <1.6 Å between the target and the template (60). Our method is less restrictive because structural comparison is performed for the entire DBD, the core region of which may have relatively small conformational changes. This is reflected by the similar performance on APO/HOLO104 sets, a few examples of which are provided in Figure 9. In one rare case, the transcription factor SarA adopts different folds in the apo- and holo-forms (56,57). Surprisingly, a winged HTH DNA-binding motif was observed in the apo-structure but not in the holo-structure. Our method correctly identified the DNA-binding region of the apo-structure, including an Arginine crucial to the DNA-binding function of SarA. The holo-structure, however, did not yield a positive prediction. Our results support the view that the apo-form of SarA more likely represents the native conformation (56).
One advantage of a template-based approach is that one can infer functionally relevant details from the template. For example, specific DNA-binding sites can be transferred from the template. In this respect, DBD-Hunter achieves an average sensitivity of 66% and an average accuracy of 87% on predicted DNA-binding proteins in their DNA-free conformation forms (Figure 8B). By comparison, machine-learning algorithms designed specifically for DNA-binding site prediction provide an average accuracy ranging from 60% to 82% (21–23).
Worldwide structural genomics centers have deposited thousands of protein structures in the PDB. It is of great importance to characterize the functions of these targets. With respect to DNA-binding protein prediction, only one method has been applied to structural genomics targets so far, despite numerous methods proposed. In their report, Jones et al. predicted one DNA-binding protein from 30 targets using a structural motif-based approach (60), which is limited to DBDs with a HTH motif. In the current study, we have applied our method to 1697 structural genomics targets and identified 37 potential DNA-binding proteins. To our knowledge, this is the first time a structural-based method for DNA-binding protein prediction has been systematically applied on a genome scale. These predictions provide valuable clues for assessing protein function experimentally, and it is of great interest to conduct experimental validations of these predictions in the future.
Like all knowledge-based approaches, our method is limited by the completeness of the template library. It cannot predict DNA-binding proteins with novel structures or binding modes that are not included in the template library, which is the main disadvantage of the current approach.
Future work entails the extension of the methodology to the case when the structure of the protein is not yet solved but has to be predicted. Here, an unanswered question is how good the predicted structure has to be to provide for the accurate prediction of DNA binding. Another issue is to attempt to model the conformation change on DNA binding. While this is not crucial for the majority of known DNA-binding proteins, it is important for a significant minority of cases. One promising approach is the extension of TASSER, a protein structure prediction algorithm (61), to include DNA binding. Thus, while DBD-Hunter is a promising approach, additional extensions and improvements are required to increase its range of applicability.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
We thank Dr Shashi Pandit for stimulating discussions. This work was supported in part by NIH grant GM-37408. Funding to pay the Open Access publication charges for this article was provided by NIH.
Conflict of interest statement. None declared.
REFERENCES
- 1.Burley SK. An overview of structural genomics. Nat. Struct. Biol. 2000;7:932–934. doi: 10.1038/80697. [DOI] [PubMed] [Google Scholar]
- 2.Lee D, Redfern O, Orengo C. Predicting protein function from sequence and structure. Nat. Rev. Mol. Cell Biol. 2007;8:995–1005. doi: 10.1038/nrm2281. [DOI] [PubMed] [Google Scholar]
- 3.Watson JD, Laskowski RA, Thornton JM. Predicting protein function from sequence and structural data. Curr. Opin. Struct. Biol. 2005;15:275–284. doi: 10.1016/j.sbi.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 4.Whisstock JC, Lesk AM. Prediction of protein function from protein sequence and structure. Q. Rev. Biophys. 2003;36:307–340. doi: 10.1017/s0033583503003901. [DOI] [PubMed] [Google Scholar]
- 5.Altschul SF, Madden TL, Schaffer AA, Zhang JH, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krogh A, Brown M, Mian IS, Sjolander K, Haussler D. Hidden markov models in computational biology – applications to protein modeling. J. Mol. Biol. 1994;235:1501–1531. doi: 10.1006/jmbi.1994.1104. [DOI] [PubMed] [Google Scholar]
- 7.Shindyalov IN, Bourne PE. Protein structure alignment by incremental combinatorial extension (CE) of the optimal path. Protein Eng. 1998;11:739–747. doi: 10.1093/protein/11.9.739. [DOI] [PubMed] [Google Scholar]
- 8.Holm L, Sander C. Protein structure comparison by alignment of distance matrices. J. Mol. Biol. 1993;233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 9.Zhang Y, Skolnick J. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302–2309. doi: 10.1093/nar/gki524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chothia C, Lesk AM. The relation between the divergence of sequence and structure in proteins. EMBO J. 1986;5:823–826. doi: 10.1002/j.1460-2075.1986.tb04288.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Skolnick J, Fetrow JS. From genes to protein structure and function: novel applications of computational approaches in the genomic era. Trends Biotechnol. 2000;18:34–39. doi: 10.1016/s0167-7799(99)01398-0. [DOI] [PubMed] [Google Scholar]
- 12.Luscombe NM, Austin SE, Berman HM, Thornton JM. An overview of the structures of protein-DNA complexes. Genome Biol. 2000;1 doi: 10.1186/gb-2000-1-1-reviews001. REVIEWS001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kono H, Sarai A. Structure-based prediction of DNA target sites by regulatory proteins. Proteins Struct. Funct. Genet. 1999;35:114–131. [PubMed] [Google Scholar]
- 14.Luscombe NM, Laskowski RA, Thornton JM. Amino acid-base interactions: a three-dimensional analysis of protein-DNA interactions at an atomic level. Nucleic Acids Res. 2001;29:2860–2874. doi: 10.1093/nar/29.13.2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mandel-Gutfreund Y, Margalit H. Quantitative parameters for amino acid-base interaction: implications for prediction of protein-DNA binding sites. Nucleic Acids Res. 1998;26:2306–2312. doi: 10.1093/nar/26.10.2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ahmad S, Sarai A. Moment-based prediction of DNA-binding proteins. J. Mol. Biol. 2004;341:65–71. doi: 10.1016/j.jmb.2004.05.058. [DOI] [PubMed] [Google Scholar]
- 17.Bhardwaj N, Langlois RE, Zhao GJ, Lu H. Kernel-based machine learning protocol for predicting DNA-binding proteins. Nucleic Acids Res. 2005;33:6486–6493. doi: 10.1093/nar/gki949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shanahan HP, Garcia MA, Jones S, Thornton JM. Identifying DNA-binding proteins using structural motifs and the electrostatic potential. Nucleic Acids Res. 2004;32:4732–4741. doi: 10.1093/nar/gkh803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stawiski EW, Gregoret LM, Mandel-Gutfreund Y. Annotating nucleic acid-binding function based on protein structure. J. Mol. Biol. 2003;326:1065–1079. doi: 10.1016/s0022-2836(03)00031-7. [DOI] [PubMed] [Google Scholar]
- 20.Szilagyi A, Skolnick J. Efficient prediction of nucleic acid binding function from low-resolution protein structures. J. Mol. Biol. 2006;358:922–933. doi: 10.1016/j.jmb.2006.02.053. [DOI] [PubMed] [Google Scholar]
- 21.Ahmad S, Gromiha MM, Sarai A. Analysis and prediction of DNA-binding proteins and their binding residues based on composition, sequence and structural information. Bioinformatics. 2004;20:477–486. doi: 10.1093/bioinformatics/btg432. [DOI] [PubMed] [Google Scholar]
- 22.Bhardwaj N, Lu H. Residue-level prediction of DNA-binding sites and its application on DNA-binding protein predictions. FEBS Lett. 2007;581:1058–1066. doi: 10.1016/j.febslet.2007.01.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kuznetsov IB, Gou ZK, Li R, Hwang SW. Using evolutionary and structural information to predict DNA-binding sites on DNA-binding proteins. Proteins Struct. Funct. Bioinform. 2006;64:19–27. doi: 10.1002/prot.20977. [DOI] [PubMed] [Google Scholar]
- 24.Donald JE, Chen WW, Shakhnovich EI. Energetics of protein-DNA interactions. Nucleic Acids Res. 2007;35:1039–1047. doi: 10.1093/nar/gkl1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu ZJ, Mao FL, Guo JT, Yan B, Wang P, Qu YX, Xu Y. Quantitative evaluation of protein-DNA interactions using an optimized knowledge-based potential. Nucleic Acids Res. 2005;33:546–558. doi: 10.1093/nar/gki204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robertson TA, Varani G. An all-atom, distance-dependent scoring function for the prediction of protein-DNA interactions from structure. Proteins Struct. Funct. Bioinform. 2007;66:359–374. doi: 10.1002/prot.21162. [DOI] [PubMed] [Google Scholar]
- 27.Myers EW, Miller W. Optimal alignments in linear space. Comput. Appl. Biosci. 1988;4:11–17. doi: 10.1093/bioinformatics/4.1.11. [DOI] [PubMed] [Google Scholar]
- 28.Hubbard TJP, Ailey B, Brenner SE, Murzin AG, Chothia C. SCOP, structural classification of proteins database: applications to evaluation of the effectiveness of sequence alignment methods and statistics of protein structural data. Acta Crystallogr. D Biol. Crystallogr. 1998;54:1147–1154. doi: 10.1107/s0907444998009172. [DOI] [PubMed] [Google Scholar]
- 29.Skolnick J, Kihara D, Zhang Y. Development and large scale benchmark testing of the PROSPECTOR_3 threading algorithm. Proteins Struct. Funct. Bioinform. 2004;56:502–518. doi: 10.1002/prot.20106. [DOI] [PubMed] [Google Scholar]
- 30.Li WZ, Godzik A. CD-HIT: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics. 2006;22:1658–1659. doi: 10.1093/bioinformatics/btl158. [DOI] [PubMed] [Google Scholar]
- 31.Sippl MJ. Knowledge-based potentials for proteins. Curr. Opin. Struct. Biol. 1995;5:229–235. doi: 10.1016/0959-440x(95)80081-6. [DOI] [PubMed] [Google Scholar]
- 32.Lu H, Lu L, Skolnick J. Development of unified statistical potentials describing protein-protein interactions. Biophys. J. 2003;84:1895–1901. doi: 10.1016/S0006-3495(03)74997-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Matthews BW. Comparison of predicted and observed secondary structure of T4 phage lysozyme. Biochim. Biophys. Acta. 1975;405:442–451. doi: 10.1016/0005-2795(75)90109-9. [DOI] [PubMed] [Google Scholar]
- 34.Watkins S, van Pouderoyen G, Sixma TK. Structural analysis of the bipartite DNA-binding domain of Tc3 transposase bound to transposon DNA. Nucleic Acids Res. 2004;32:4306–4312. doi: 10.1093/nar/gkh770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Court R, Chapman L, Fairall L, Rhodes D. How the human telomeric proteins TRF1 and TRF2 recognize telomeric DNA: a view from high-resolution crystal structures. EMBO Rep. 2005;6:39–45. doi: 10.1038/sj.embor.7400314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schultz SC, Shields GC, Steitz TA. Crystal structure of a CAP-DNA Complex – the DNA is bent by 90 degrees. Science. 1991;253:1001–1007. doi: 10.1126/science.1653449. [DOI] [PubMed] [Google Scholar]
- 37.Wilce JA, Vivian JP, Hastings AF, Otting G, Folmer RHA, Duggin IG, Wake RG, Wilce MCJ. Structure of the RTP-DNA complex and the mechanism of polar replication fork arrest. Nat. Struct. Biol. 2001;8:206–210. doi: 10.1038/84934. [DOI] [PubMed] [Google Scholar]
- 38.Tahirov TH, Inoue-Bungo T, Morii H, Fujikawa A, Sasaki M, Kimura K, Shiina M, Sato K, Kumasaka T, Yamamoto M, et al. Structural analyses of DNA recognition by the AML1/Runx-1 Runt domain and its allosteric control by CBF beta. Cell. 2001;104:755–767. doi: 10.1016/s0092-8674(01)00271-9. [DOI] [PubMed] [Google Scholar]
- 39.Cho YJ, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a P53 tumor suppressor DNA complex – understanding tumorigenic mutations. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 40.Tanaka T, Fukuda-Ishisaka S, Hirama M, Otani T. Solution structures of C-1027 apoprotein and its complex with the aromatized chromophore. J. Mol. Biol. 2001;309:267–283. doi: 10.1006/jmbi.2001.4621. [DOI] [PubMed] [Google Scholar]
- 41.Horton JR, Zhang X, Maunus R, Yang Z, Wilson GG, Roberts RJ, Cheng XD. DNA nicking by HinP1I endonuclease: bending, base flipping and minor groove expansion. Nucleic Acids Res. 2006;34:939–948. doi: 10.1093/nar/gkj484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu QS, Roberts RJ, Guo HC. Two crystal forms of the restriction enzyme MspI-DNA complex show the same novel structure. Protein Sci. 2005;14:2590–2600. doi: 10.1110/ps.051565105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Costa M, Sola M, del Solar G, Eritja R, Hernandez-Arriaga AM, Espinosa M, Gomis-Ruth FX, Coll M. Plasmid transcriptional repressor CopG oligomerises to render helical superstructures unbound and in complexes with oligonucleotides. J. Mol. Biol. 2001;310:403–417. doi: 10.1006/jmbi.2001.4760. [DOI] [PubMed] [Google Scholar]
- 44.Garvie CW, Phillips SEV. Direct and indirect readout in mutant Met repressor-operator complexes. Structure. 2000;8:905–914. doi: 10.1016/s0969-2126(00)00182-9. [DOI] [PubMed] [Google Scholar]
- 45.Bochkarev A, Bochkareva E, Frappier L, Edwards AM. The 2.2 angstrom structure of a permanganate-sensitive DNA site bound by the Epstein-Barr virus origin binding protein, EBNA1. J. Mol. Biol. 1998;284:1273–1278. doi: 10.1006/jmbi.1998.2247. [DOI] [PubMed] [Google Scholar]
- 46.Kim SS, Tam JK, Wang AF, Hegde RS. The structural basis of DNA target discrimination by papillomavirus E2 proteins. J. Biol. Chem. 2000;275:31245–31254. doi: 10.1074/jbc.M004541200. [DOI] [PubMed] [Google Scholar]
- 47.Kwon HJ, Tirumalai R, Landy A, Ellenberger T. Flexibility in DNA recombination: structure of the lambda integrase catalytic core. Science. 1997;276:126–131. doi: 10.1126/science.276.5309.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aihara H, Kwon HJ, Nunes-Duby SE, Landy A, Ellenberger T. A conformational switch controls the DNA cleavage activity of lambda integrase. Mol. Cell. 2003;12:793. doi: 10.1016/s1097-2765(03)00268-5. [DOI] [PubMed] [Google Scholar]
- 49.Conway AB, Chen Y, Rice PA. Structural plasticity of the Flp-Holliday junction complex. J. Mol. Biol. 2003;326:425–434. doi: 10.1016/s0022-2836(02)01370-0. [DOI] [PubMed] [Google Scholar]
- 50.Sauve S, Tremblay L, Lavigne P. The NMR solution structure of a mutant of the max b/HLH/LZ free of DNA: insights into the specific and reversible DNA binding mechanism of dimeric transcription factors. J. Mol. Biol. 2004;342:813–832. doi: 10.1016/j.jmb.2004.07.058. [DOI] [PubMed] [Google Scholar]
- 51.Nair SK, Burley SK. X-ray structures of Myc-Max and Mad-Max recognizing DNA: molecular bases of regulation by proto-oncogenic transcription factors. Cell. 2003;112:193–205. doi: 10.1016/s0092-8674(02)01284-9. [DOI] [PubMed] [Google Scholar]
- 52.Parraga A, Bellsolell L, Ferre-D’Amare AR, Burley SK. Co-crystal structure of sterol regulatory element binding protein 1a at 2.3 angstrom resolution. Structure. 1998;6:661–672. doi: 10.1016/s0969-2126(98)00067-7. [DOI] [PubMed] [Google Scholar]
- 53.Chen FE, Huang DB, Chen YQ, Ghosh G. Crystal structure of p50/p65 heterodimer of transcription factor NF-kappa B bound to DNA. Nature. 1998;391:410–413. doi: 10.1038/34956. [DOI] [PubMed] [Google Scholar]
- 54.Huxford T, Huang DB, Malek S, Ghosh G. The crystal structure of the I kappa B alpha/NF-kappa B complex reveals mechanisms of NF-kappa B inactivation. Cell. 1998;95:759–770. doi: 10.1016/s0092-8674(00)81699-2. [DOI] [PubMed] [Google Scholar]
- 55.Giffin MJ, Stroud JC, Bates DL, von Koenig KD, Hardin J, Chen L. Structure of NFAT1 bound as a dimer to the HIV-1 LTR kappa B element. Nat. Struct. Biol. 2003;10:800–806. doi: 10.1038/nsb981. [DOI] [PubMed] [Google Scholar]
- 56.Liu YF, Manna AC, Pan CH, Kriksunov IA, Thiel DJ, Cheung AL, Zhang GY. Structural and function analyses of the global regulatory protein SarA from Staphylococcus aureus. Proc. Natl Acad. Sci. USA. 2006;103:2392–2397. doi: 10.1073/pnas.0510439103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schumacher MA, Hurlburt BK, Brennan RG. Crystal structures of SarA, a pleiotropic regulator of virulence genes in S-aureus. Nature. 2001;409:215–219. doi: 10.1038/35051623. [DOI] [PubMed] [Google Scholar]
- 58.Zhang RG, Dementieva I, Duke N, Collart F, Quaite-Randall E, Alkire R, Dieckman L, Maltsev N, Korolev O, Joachimiak A. Crystal structure of Bacillus subtilis IolI shows endonuclase IV fold with altered Zn binding. Proteins Struct. Funct. Genet. 2002;48:423–426. doi: 10.1002/prot.10159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wright PE, Dyson HJ. Intrinsically unstructured proteins: re-assessing the protein structure-function paradigm. J. Mol. Biol. 1999;293:321–331. doi: 10.1006/jmbi.1999.3110. [DOI] [PubMed] [Google Scholar]
- 60.Jones S, Barker JA, Nobeli I, Thornton JM. Using structural motif templates to identify proteins with DNA binding function. Nucleic Acids Res. 2003;31:2811–2823. doi: 10.1093/nar/gkg386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Y, Skolnick J. Automated structure prediction of weakly homologous proteins on a genomic scale. Proc. Natl Acad. Sci. USA. 2004;101:7594–7599. doi: 10.1073/pnas.0305695101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.