

Abstract
Insulin activates glucose transport by promoting translocation of the insulin-sensitive fat/muscle-specific glucose transporter GLUT4 from an intracellular storage compartment to the cell surface. Here we report that an optimal insulin effect on glucose uptake in 3T3-L1 adipocytes is dependent upon expression of both PIKfyve, the sole enzyme for PtdIns 3,5-P2 biosynthesis, and the PIKfyve activator, ArPIKfyve. Small-interfering RNAs that selectively ablated PIKfyve or ArPIKfyve in this cell type, depleted the PtdIns 3,5-P2 pool and reduced insulin-activated glucose uptake to a comparable degree. Combined loss of PIKfyve and ArPIKfyve caused further PtdIns 3,5-P2 ablation that correlated with greater attenuation in insulin responsiveness. Loss of PIKfyve/ArPIKfyve reduced insulin-stimulated Akt phosphorylation and the cell-surface accumulation of GLUT4 or IRAP, but not GLUT1-containing vesicles without affecting overall expression of these proteins. ArPIKfyve and PIKfyve were found to physically associate in 3T3-L1 adipocytes and this was insulin independent. In vitro labeling of membranes isolated from basal or insulin-stimulated 3T3-L1 adipocytes documented substantial insulin-dependent increases of PtdIns 3,5-P2 production on intracellular membranes. Together, the data demonstrate for the first time a physical association between functionally related PIKfyve and ArPIKfyve in 3T3-L1 adipocytes and indicate that the novel ArPIKfyve-PIKfyve-PtdIns 3,5-P2 pathway is physiologically linked to insulin-activated GLUT4 translocation and glucose transport.
Keywords: PtdIns 3,5-P2; ArPIKfyve-PIKfyve complexes; glucose transport; GLUT4 translocation; GLUT1; IRAP; protein knockdown; Akt; siRNAs
Insulin effect in stimulating glucose uptake in fat and muscle is mediated by the facilitative tissue-specific glucose transporter GLUT4 [reviewed in Ref. 1]. In basal states, GLUT4 is localized in the cell interior and cycles slowly between the plasma membrane and one or more intracellular compartments. Insulin stimulates GLUT4 translocation to the cell surface by >10-fold, thereby increasing the rate of glucose transport. Studies over the last two decades revealed a complex effect of insulin on both the signaling and the trafficking itinerary of GLUT4 vesicles through the endosomal/endomembrane system. Despite intensive investigation that culminated in the identification of key protein or phospholipid intermediates [reviewed in Refs. 2–5], the precise molecular mechanism of insulin-regulated GLUT4 membrane dynamics that leads to stimulation of glucose transport is still incompletely understood.
PIKfyve (PhosphoInositide Kinase for five position containing a fyve finger) [6] is an evolutionarily conserved low-abundance enzyme that produces phosphatidylinositol (PtdIns) 5-P and PtdIns 3,5-P2 in mammalian cells. Both phospholipids are low abundance and are now recognized as signaling molecules in their own right [6–9]. PIKfyve is a large protein harboring a FYVE domain membrane-localization sequence at the N-terminus and a kinase domain at the C-terminus [reviewed in Ref. 10]. In addition, it displays a DEP domain, whose function is unknown, and a Chp60/TCP1 domain that assures protein-protein interactions, some of which are already identified [11]. PIKfyve is a product of a single-copy gene with at least two splice variants whose cDNAs were initially isolated from a mouse adipocyte library through a screen for transcripts that, like GLUT4, are expressed in a fat/muscle-specific/enriched manner [12,13]. Subsequent studies in mouse 3T3-L1 cells revealed considerable up-regulation of PIKfyve protein levels upon transition from a fibroblastic to adipocytic phenotype, implying a key role in 3T3-L1 adipocyte specific functions [14]. Consistent with this notion are our findings for an unprecedented PIKfyve-mediated PtdIns 3,5-P2 rise in hyperosmotically stressed 3T3-L1 adipocytes, likely associated with osmo-protective response mechanisms in these fat-laden cells [15]. However, evolutionarily conserved PIKfyve is present to various degrees in all mammalian cells tested thus far, where it likely operates in diverse signaling pathways and membrane trafficking events. More advanced are studies in the context of insulin action, where PIKfyve has been characterized as a positive stimulatory intermediate in several acute and long-term insulin-regulated cellular responses. [14]. Furthermore, the ability of insulin to induce GLUT4 translocation to the 3T3-L1 adipocyte cell surface was found to be markedly inhibited by expression of dominant-negative lipid kinase-deficient PIKfyve mutants [14]. Because these mutants harbor substitutions within the catalytic domain to abrogate the PtdIns 5-P/PtdIns 3,5-P2-producing activity [16], the data are consistent with PIKfyve lipid products functioning as a positive regulator in insulin-mobilized GLUT4 translocation. Recently, serine at position 318 of mouse PIKfyveL has been determined as an Akt phosphorylation site, with phosphorylation up-regulating PtdIns 3,5-P2 production [17]. Unexpectedly, in this report the authors found enhanced insulin responsiveness of GLUT4 translocation to the adipocyte cell surface in the presence of the Akt-phosphorylation-deficient PIKfyveS318A mutant whose enzyme activity is presumably compromised [17]. Clearly, whereas PIKfyve-dependent PtdIns 3,5-P2 production undoubtedly plays a role in insulin responsiveness of GLUT4 translocation, whether it functions as a positive or negative regulator is a topic of debate.
Recent studies indicate that the PtdIns 3,5-P2 pool in mammalian cells, including 3T3-L1 adipocytes, is controlled not only by the PIKfyve enzyme but also by ArPIKfyve (Associated regulator of PIKfyve [15,18]. The physiological roles of the newly emerging ArPIKfyve-PIKfyve-PtdIns 3,5-P2 pathway in mammalian cells are largely uncharacterized. Like PIKfyve, ArPIKfyve is an evolutionarily conserved protein whose single copy gene is found in all eukaryotes with a sequenced genome. Studies with the endogenous proteins, conducted thus far only in PC12 cells, are consistent with the conclusion that ArPIKfyve physically interacts with PIKfyve to up-regulate its activity [18]. This association mechanism is likely evolutionarily variant because the ortholog proteins that regulate the steady-state or activated PtdIns 3,5-P2 levels in S. cerevisiae, i.e., Vac14 (ArPIKfyve) and Fab1 (PIKfyve), do not assemble in a complex [19,20]. The present study was designed to examine the role of ArPIKfyve and PIKfyve in insulin-responsiveness of glucose transport and the mode of interaction between the two proteins in 3T3-L1 adipocytes. We demonstrate here that the ArPIKfyve-PIKfyve-PtdIns 3,5-P2 pathway is among the mechanisms linking insulin signaling to its effect on GLUT4 vesicle dynamics and glucose transport.
MATERIALS AND METHODS
Cell cultures, antibodies, and constructs
Mouse 3T3-L1 fibroblasts were differentiated into adipocytes following a standard differentiation protocol detailed elsewhere [13,14]. 3T3-L1 adipocytes were used on day 5 of the differentiation program in the siRNA transfection experiments and between day 8–10 in the other studies. Polyclonal anti-PIKfyve (R7069; East-Acres, MA) and anti-ArPIKfyve antisera (WS047; Covance, Denver, PA), characterized previously [13,18], were used for immunoprecipitation. For immunoblotting, anti-ArPIKfyve antiserum was affinity purified on His- or glutathione-S-transferase-tagged recombinant ArPIKfyve as detailed previously [18]. Anti-phosphoThr308, anti-phosphoSer473-Akt or anti-Akt antibodies were from Cell Signaling (Beverly, MA). Polyclonal antibodies against GLUT4 (R1288), GLUT1 (R480), hemagglutinin (HA)-epitope (R4289), and insulin-responsive aminopeptidase (IRAP) were gifts by Drs. Mike Czech and Paul Pilch, respectively. HA-GLUT4-eGFP and GLUT1-HA constructs were gifts by Drs. Jeff Pessin and Tony Carruthers, respectively.
siRNAs and 3T3-L1 adipocyte transfection
Smart pool™ small-interfering RNA (siRNA) duplexes directed to mouse sequences of ArPIKfyve (cat# M-040510-01-0005) or cyclophilin B (D-001136-01-20), designed and synthesized by Dharmacon (Lafayette, CO), were characterized previously [15]. Smart pool™ siRNA duplexes directed to mouse PIKfyve was a new design (cat# M-040127-01-0005) and achieved higher depletion levels vs. the already reported siRNA pool (cat# M-040127-00-05) [15]. Differentiated 3T3-L1 adipocytes were transfected by electroporation with siRNA duplexes (0.2–0.4 nmol/5 × 106 cells) with a Bio-Rad gene pulser II system on day 5 of the differentiation program under conditions specified previously [15]. After electroporation, cells were mixed with fresh medium, seeded onto 24-well (for glucose transport), 6-well (for IRAP biotinylation, GLUT4/GLUT1 translocation or immunoblotting) or 60-mm plates (fractionation and [γ-32P]ATP labeling) and used 55–72 h post-transfection in the indicated assays (see below).
Glucose transporter translocation and fluorescence microscopy
To analyze GLUT4 or GLUT1 translocation, 3T3-L1 adipocytes were co-electroporated with HA-GLUT4-eGFP plasmid or GLUT1-HA (45 μg) and siRNAs on day 5 of the differentiation program. Cells were seeded on glass coverslips placed into a 6-well plate. Fifty five h post-transfection, cells were serum-starved for 3 h in low-glucose DMEM, incubated in the presence or absence of insulin (100 nM; 30 min at 37°C), washed with PBS, fixed in formaldehyde (4% in PBS) and then stained with anti-HA antibodies without permeabilization (for GFP-GLUT4-HA) or following permeabilization with 0.5% Triton X-100 in PBS/1% FBS (for GLUT1-HA). Following incubation with Cy3-conjugated goat anti-rabbit IgG (Kirkegard and Perry Laboratories), cells were washed and mounted on slides as detailed previously [8,16]. Transporter surface abundance was quantified in two independent manners. First, randomly selected GFP-positive (for GLUT4) or HA-positive (for GLUT1) adipocytes (at least 100 cells/condition) observed by a Nikon Eclipse TE 200 inverted fluorescence microscope with a 40x objective and a standard green-fluorescence filter, were evaluated for the presence of a complete HA-plasma membrane rim on the red channel. Cells with an incomplete rim were considered negative. The percentage of positive cells was calculated. In the second approach applied for GLUT4, we quantified were observed by inverted confocal microscope (model 1X81, Olympus) with a cooled charge-coupled device 12 bit camera (Hamamatsu). GFP-positive adipocytes were selected at random for analysis (10–20 cells/condition). Images were collected using a 40x objective. The exposure times for each fluorescence channel were always set below the saturation threshold of the camera. The area of the selected cell was individually analyzed for an average fluorescence intensity per pixel for Cy3 (cell-surface HA-GLUT4-eGFP) or GFP signals (total HA-GLUT4-eGFP) by IPLab Software (Scanalytics, Inc., Fairfax, VA). After subtracting the background fluorescence of a non-transfected neighboring cell, quantified in a similar way, the Cy3/GFP ratio was calculated. Slides from separate experiments were analyzed simultaneously.
Glucose transport
Glucose transport was determined by measuring 2-deoxyglucose (2DG) uptake as previously described [14]. Briefly, following several washes in PBS, 3T3-L1 adipocytes (24-well dish) were starved for 3 h in low-glucose DMEM supplemented with 0.5% FBS. Cells were then washed and incubated for 30 min in KRH buffer (NaCl, 120 mM; KCl, 6 mM; Mg2SO4, 1.2 mM; CaCl2.2H20, 1 mM; Na2HPO4, 0.6 mM; NaH2PO4, 0.4 mM and HEPES, pH 7.4, 30 mM), containing 0.5% FBS and 2 mM sodium pyruvate, in the presence or absence of insulin (100 nM). 2-[1,2-3H]deoxy-D-glucose (NEN Du-Pont; Boston, MA) was then added to a final concentration of 100 μM (0.5 μCi per well) for 5 min at 37° C. The reaction was stopped by addition of 2DG (20 mM). After extensive washing in KRH, cells were lysed in 1% Triton X-100. Aliquots of the solubilized cells were analyzed for radioactivity and protein concentration. Nonspecific glucose uptake was measured in the presence of 10 μM cytochalasin B and was subtracted for each determination. All values were normalized for protein content.
3T3-L1 adipocyte subcellular fractionation
3T3-L1 adipocytes, with or without siRNA transfection, were serum starved for 4 h in DMEM. Cells were then stimulated or not, with insulin (100 nM) for an additional 10 min and subjected to subcellular fractionation as described previously [21]. Briefly, cells were washed twice with PBS, once in the homogenization buffer [“HES++ buffer”, 20 mM Hepes/NaOH, pH 7.5, 1 mM EDTA, 255 mM sucrose, containing 1x protease (1 mM phenylmethylsulphonylfluoride, 5 μg/ml leupeptin, 5 μg/ml aprotinin, 1 μg/ml pepstatin and 1 mM benzamidine) and 1x phosphatase inhibitor cocktails (50 mM NaF, 10 mM Na pyrophosphate, 25 mM Na β-glycerophosphate and 2 mM Na metavanadate)] at 25°C, and then scraped at 4°C in HES++ buffer. Cells were homogenized by a motor-driven homogenizer with 10 strokes at 225 rpm and 5 strokes at 800 rpm. Subcellular fractionation was performed by consecutive centrifugations with an SS-34 rotor (Sorvall Instrument Division) and a Beckman TLA 100.3 rotor (Beckman Instruments Inc.) following established protocols (21) with a slight modification thereby the separation of the high density from low-density microsomal fractions was omitted. The resultant fraction is therefore a combined microsomal fraction, called herein intracellular membranes (IM). The plasma membrane (PM) fraction was obtained by the conventional techniques (21). Pellets were resuspended in HES++ buffer to a protein concentration of ~2 mg/ml. Membrane fractions were used in a labeling with [γ-32P]ATP.
In some experiments, after insulin treatment, 3T3-L1 adipocytes were fractionated into total membranes and cytosol. In this case, following cell homogenization at 4°C in HES++ buffer, total membranes were obtained by sequential centrifugations: 5 min at 800 × g and 30 min at 200,000 × g at 4°C. Total membrane fractions were solubilized in RIPA++ buffer (50 mM Tris/HCl buffer, pH 8.0, containing 150 mM NaCl, 1% Nonidet P-40, 0.5% Na deoxycholate and the inhibitor cocktails). Fractions were clarified by centrifugation and protein concentration was measured. Equal protein amounts were subjected to immunoprecipitation analyses without freezing.
In vitro labeling of membrane lipids and 32P-PI detection
Equal amounts of membrane protein (100 μg) were subjected to labeling with [γ– 32P]ATP (100 μCi) for 30 min at 30° C in a reaction mix consisting of 50 mM Tris/HCl buffer, pH 7.4, 2.5 mM MnCl2, 2.5 mM MgCl2 and 15 μM ATP. Lipids were then extracted, deacylated and the glycerophosphorylinositols (GroPIns) were subjected to high pressure liquid chromatography (HPLC) analysis on a Whatman 235-mm × 4.60-mm Partisphere SAX column as described elsewhere [6,15,18]. Co-injected internal HPLC standards, [3H]GroPIns 4-P, [3H]GroPIns 4,5-P2 and [3H]GroPIns 3-P, were deacylated from [3H]PtdIns 4-P, [3H]PtdIns 4,5-P2 and [3H]PtdIns 3-P, respectively. [32P]GroPIns 3,5-P2 and [32P]GroPIns 3,4-P2 external standards were deacylated from [32P]PtdIns 3,5-P2 and [32P]PtdIns 3,4-P2 enzymatically synthesized with PIKfyve and PI 3-K as described previously [6,15,18]. The radioactivity incorporated was analyzed with an on-line flow scintillation analyzer (Packard, Radiomatic 525TR). FLO-ONE radiochromatography software (Packard) was used for data evaluation and documentation. Individual peak radioactivity was quantified by area integration and is presented as a percentage of the summed radioactivity from the [32P]-labeled GroPIns 3-P, GroPIns 4-P, GroPIns 3,5-P2, GroPIns 3,4-P2 and GroPIns 4,5-P2 peaks (“total radioactivity”).
IRAP surface biotinylation
The presence of IRAP on the cell surface was determined by an IRAP biotinylation assay that uses the cell-impermeant biotin analog sulfo-NHS-LC-LC-biotin (Pierce) as detailed elsewhere [22–24]. Briefly, 3 days post-transfection with the indicated siRNAs, 3T3-L1 adipocytes seeded on a 6-well dish were serum starved for 3 h and then treated with insulin (100 nM; 20 min) or left untreated. Biotinylation was performed at 4°C in KRPH, containing 0.75 mM sulfo-NHS-LC-LC-biotin for 30 min under constant shaking. Cells were then washed with 20 mM glycine in KRH buffer to quench the reaction and solubilized in 20 mM Tris/HCl buffer, pH 7.4, containing 150 mM NaCl, 5 mM EDTA, 1% Triton X-100 and 1 × protease inhibitor cocktail. Lysates were clarified by centrifugation and subjected to immunoprecipitation with anti-IRAP antibodies for 16 h at 4°C, with Protein A-Sepharose added during the last 3 h of incubation. Washed immunoprecipitates, resolved by SDS-PAGE and transferred to a PVDF membrane were probed with streptavidin-horseradish peroxidase (HRP; Pierce, Rockford, IL).
Immunoprecipitation and immunoblotting
Unless otherwise stated, RIPA lysates supplemented with 1 × protease inhibitor cocktail derived from insulin-treated (100 nM; 0–60 min) or untreated 3T3-L1 adipocytes were first clarified by centrifugation (14, 000 × g, 15 min, 4°C) and then subjected to immunoprecipitation with anti-PIKfyve or anti-ArPIKfyve antiserum (16 h, 4°C) with Protein A-Sepharose added during the last 1.5 h of incubation. Detergent-solubilized cytosol or membrane fractions obtained as described above were clarified and subjected to immunoprecipitation under similar conditions, determined to consistently remove grater than 90% of PIKfyve or ArPIKfyve from the supernatant. Immunoprecipitates were washed in RIPA buffer, supplemented with protease inhibitors, then separated by SDS-PAGE and, after electrotransfer onto nitrocellulose membranes, analyzed by immunoblotting with the indicated antibodies as described previously [6,7]. To control on-target and off-target protein ablation under the siRNA-targeted gene silencing, RIPA lysates derived from 3T3-L1 adipocytes 72 h following electroporation were analyzed for PIKfyve, ArPIKfyve, GLUT1, GLUT4, IRAP and Akt levels by immunoblotting with corresponding antibodies.
PIKfyve lipid and protein kinase activities
PIKfyve lipid and protein kinase activities were measured in parallel using PIKfyve immunoprecipitates derived from 3T3-L1 adipocytes. For the lipid kinase assay, washed PIKfyve immunoprecipitates were subjected to 15 min incubation at 30°C in an assay buffer (25 mM Hepes, pH 7.4, 120 mM NaCl, 2.5 mM MgCl2 and 2.5 mM MnCl2, 5 mM β–glycerophosphate and 1 mM DTT) supplemented with 50 μM ATP, [γ-32P]ATP (12.5 μCi) and 100 μM PtdIns (from soybean, Avanti Polar Lipids Inc). Lipids were extracted and analyzed by TLC using the acidic solvent system (2M acetic acid/propanol, 1:2 v/v) as previously described (6,18). PIKfyve protein kinase was measured by the PIKfyve autophosphorylation activity in a reaction mix composed of [γ-32P]ATP (5 μCi), 25 μM ATP, 2.5 mM MnCl2, 12 mM MgCl2 in 50 mM Hepes, pH 7.4, and carried out for 30 min at 25°C as described elsewhere [16].
Others
Protein concentration was determined by bicinchoninic acid protein assay kit (Pierce). Protein levels were quantified from the intensity of the bands with a laser scanner (Microteck) and UN-SCAN-IT software (Silk Scientific). Several films of different exposure times were quantified to assure the signals were within the linear range. Data are expressed as means ± SEM. Statistical analysis was performed by Student’s t test, with p<0.05 considered as significant.
RESULTS
Ablation of ArPIKfyve or PIKfyve reduces PtdIns 3,5-P2 and dampens insulin responsiveness in cultured adipocytes
siRNA pools directed against mouse PIKfyve or ArPIKfyve used in this study were tested for their ability to knock down expression of the endogenous proteins following electroporation in 3T3-L1 adipocytes. Both PIKfyve and ArPIKfyve protein levels were found to be selectively attenuated by ~75% as revealed by immunoblotting conducted 72 h post-transfection (Fig. 1A). It should be emphasized that the siRNA pools used in this study were highly efficient as evidenced by the low siRNA doses (0.2–0.4 nmol/5 × 106 cells) yielding marked ablation of either ArPIKfyve or PIKfyve without affecting other proteins. This could be appreciated by the identical intensities among the cross-reacting protein bands seen in Fig. 1A. Consistent with our previous observations, the siRNA-mediated depletion of PIKfyve and ArPIKfyve reduced steady-state PtdIns 3,5-P2 levels in 32P-labeled 3T3-L1 adipocytes by 28 and 14%, respectively [15, and data not shown]. Simultaneous siRNA targeting of ArPIKfyve and PIKfyve silenced both proteins to an extent equal to that of the individual protein ablation (Fig. 1A) but produced a greater reduction (40 ± 5%) in accumulated 32P-PtdIns 3,5-P2. These data are consistent with the idea that PtdIns 3,5-P2 levels in 3T3-L1 adipocytes are dependent on both PIKfyve and ArPIKfyve presence.
Fig. 1. siRNA-based ablation of PIKfyve and/or ArPIKfyve inhibits insulin-induced stimulation of glucose transport in 3T3-L1 adipocytes.
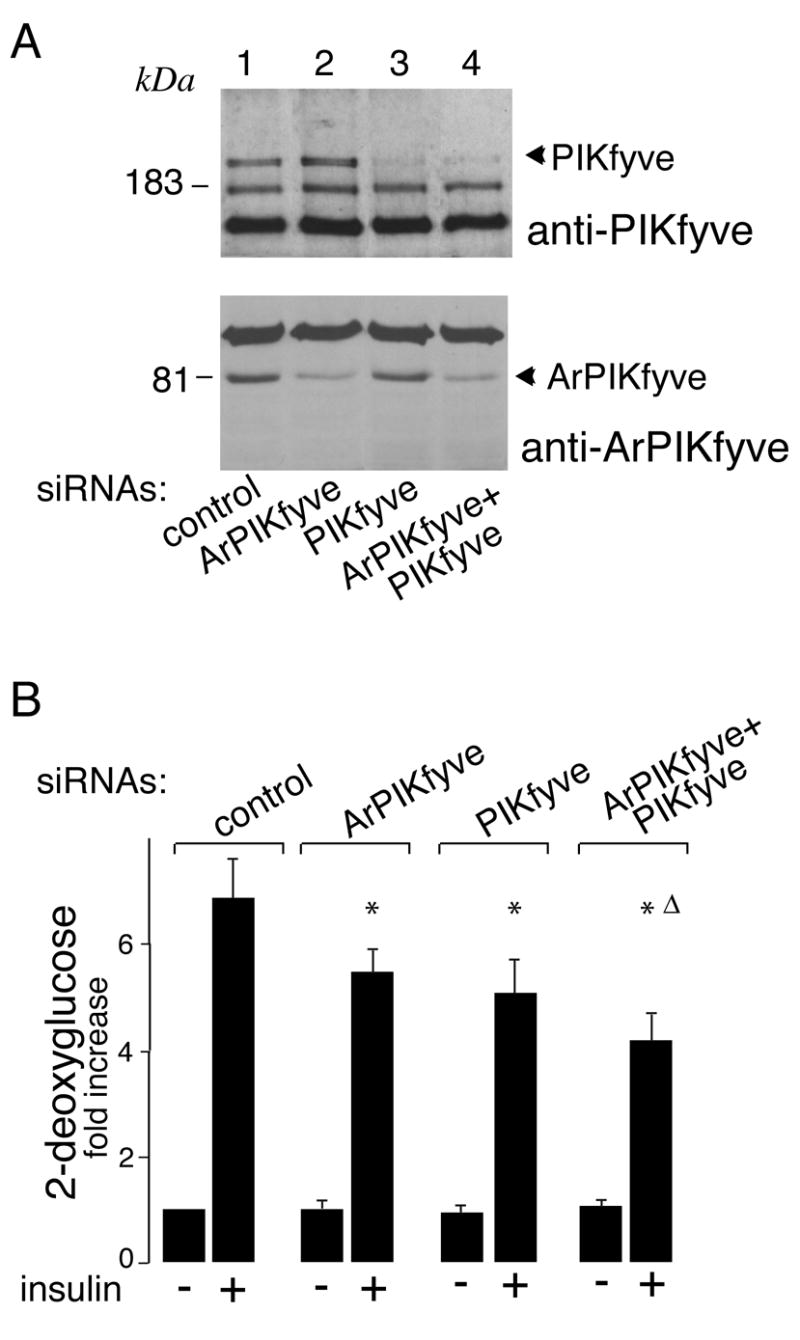
A. 3T3-L1 adipocytes were transfected by electroporation with the indicated siRNA duplexes targeting specific regions of mouse PIKfyve (0.2 nmol/5 × 106 cells), ArPIKfyve (0.4 nmol/5 × 106 cells), the combination of the two, or cyclophilin B (control, 0.6 nmol/5 × 106 cells). Cells were seeded and 72 h post-transfection, lysed as detailed in Materials and Methods. Equal amounts (100 μg protein) were resolved by SDS-PAGE and analyzed by immunoblotting with the indicated antibodies. Shown is a chemiluminescence detection of representative immunoblots out of five independent experiments with similar results. B. 3T3-L1 adipocytes were electroporated with the indicated siRNA pools as in A, and then seeded on 24-well plates. Control siRNA pools (at concentrations identical to the examined siRNAs) were to mouse cyclophilin B or to sequences in ArPIKfyve found ineffective in silencing ArPIKfyve protein expression levels. After 72 h, serum-starved cells were stimulated with or without insulin (100 nM; 30 min). 2DG uptake was measured as detailed in Materials and Methods. Shown is a quantitation from 3 independent experiments in triplicates and expressed relative to the basal control; *, different vs. insulin-stimulated control, P<0.01; Δ, different vs. insulin-stimulated ArPIKfyve knockdown, P<0.05.
To examine whether the depleted PtdIns 3,5-P2 pool has any impact on insulin-induced glucose transport activation, we measured 2-deoxyglucose uptake in 3T3-L1 adipocytes under conditions of ablated PIKfyve, ArPIKfyve, or both. As illustrated in Figure 1B, we found that the depletion of ArPIKfyve or PIKfyve significantly dampened the insulin-induced stimulation of glucose transport (20–25%) without affecting significantly the basal uptake. When both proteins were simultaneously ablated, the inhibition in insulin responsiveness was greater (~40%; Fig. 1B). Together, these data indicate that both ArPIKfyve and PIKfyve, likely through maintaining optimal PtdIns 3-P-to-PtdIns 3,5-P2 conversion, positively regulate insulin-induced glucose transport activation.
Normal glucose transporter protein expression but reduced insulin-dependent GLUT4 translocation in ArPIKfyve/PIKfyve-depleted adipocytes
Whereas differentiated 3T3-L1 adipocytes express both GLUT1 and GLUT4 glucose transporters, the increased capacity for sugar uptake in response to insulin appears to be catalyzed mainly by the GLUT4 carrier [1,25,26]. Therefore, we next examined whether the observed inhibition in insulin responsiveness of glucose transport was associated with reduced expression levels of GLUT4 protein as a result of a potential toxicity, off-target effect of ArPIKfyve/PIKfyve siRNAs and/or enhanced degradation. Immunoblotting with anti-GLUT4 antibodies demonstrated unaltered expression levels of GLUT4 protein under depletion of ArPIKfyve and PIKfyve either individually (data not shown) or in combination (Fig. 2). Concordantly, overall protein expression of IRAP, a constituent of GLUT4-containing vesicles [27,28], remained unaltered under the siRNA-mediated ArPIKfyve/PIKfyve silencing (Fig. 2). Likewise, protein expression of GLUT1, the principal transporter responsible for basal glucose uptake, was unaltered under the siRNA-directed ArPIKfyve/PIKfyve-targeted depletion (Fig. 2).
Fig. 2. Protein expression levels of GLUT4 and IRAP are unaltered under siRNA-mediated ablation of ArPIKfyve/PIKfyve.
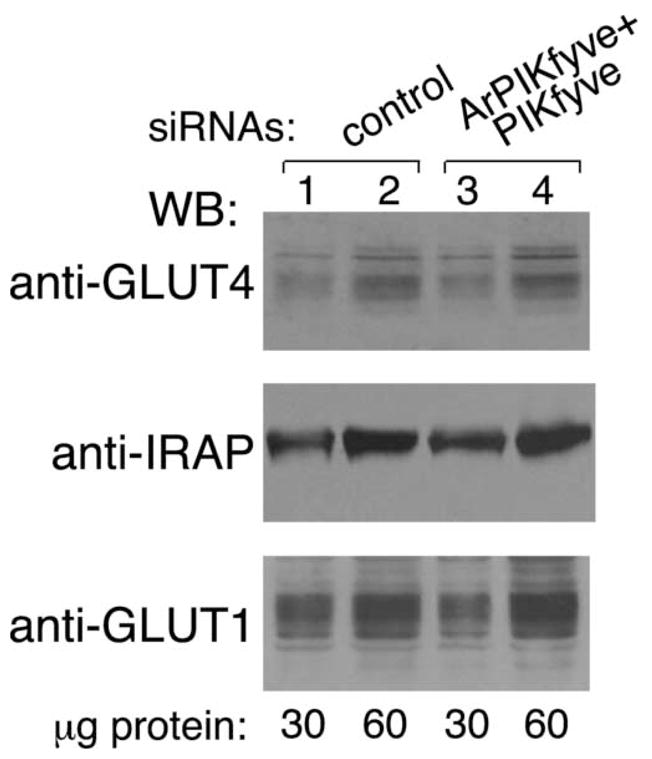
3T3-L1 adipocytes were transfected by electroporation with cyclophilin B (control) or mouse ArPIKfyve+PIKfyve siRNAs at concentrations indicated in Fig. 1A. Cells were seeded on 6-well plates and 72 h post-transfection, lysed as described in MATERIALS AND METHODS. Indicated protein amounts were resolved by SDS-PAGE. Duplicate 10% gels were used for GLUT1 or GLUT4 and a 6% gel, for IRAP. After transfer, the nitrocellulose membranes were analyzed by immunoblotting with the indicated antibodies. Shown is a chemiluminescence detection of representative immunoblots out of two independent experiments with similar results.
The finding for unaltered overall GLUT4 protein expression levels suggests that reduced insulin responsiveness of glucose transport under ablation of PIKfyve/ArPIKfyve might be due to altered insulin-regulated trafficking of GLUT4 to the 3T3-L1 adipocyte cell surface. We addressed this possibility by two independent approaches. In the first biochemical approach, we examined the extent of cell-surface biotinylation of endogenous IRAP as a marker of the GLUT4 vesicle cell surface translocation. Reportedly IRAP and GLUT4 exhibit almost identical trafficking characteristics but, unlike GLUT4, cell surface IRAP is susceptible to biotinylation owing to numerous Lys positioned at the predicted extracellular domain [22–24,27]. Quantitation of cell surface IRAP biotinylation by IRAP immunoprecipitation and subsequent blotting with streptavidin-HRP demonstrated ~30% loss in surface IRAP following insulin stimulation of cells depleted of ArPIKfyve/PIKfyve vs. controls (Fig. 3A and B). In the second fluorescence microscopy approach, we tested the effect of ArPIKfyve/PIKfyve protein ablation on insulin responsiveness of GLUT4 translocation in 3T3-L1 adipocytes cotransfected with doubly labeled HA-GLUT4-eGFP (Fig. 3C). This widely used construct harbors an eGFP fused to the C-terminus and an HA-epitope at the first exofacial loop of GLUT4, allowing quantitation of the transporter cell-surface translocation/insertion in non-permeabilized cells [29]. The ratio of cell-surface HA (Cy3) fluorescence over the total GFP signal in transfected cells revealed that the loss of ArPIKfyve/PIKfyve resulted in a ~35% decrease in insulin-stimulated gain of cell-surface HA-GLUT4-eGFP (Fig. 3D). Similar reduction in insulin responsiveness (~30%) associated with the ArPIKfyve/PIKfyve loss was quantified by counting the number of HA-GLUT4-eGFP-expressing adipocytes that displayed a full HA-plasma- membrane rim in response to insulin (Fig. 3E).
Fig. 3. Reduced insulin-dependent cell surface accumulation of GLUT4 and IRAP, but not that of GLUT1, under loss of ArPIKfyve/PIKfyve.
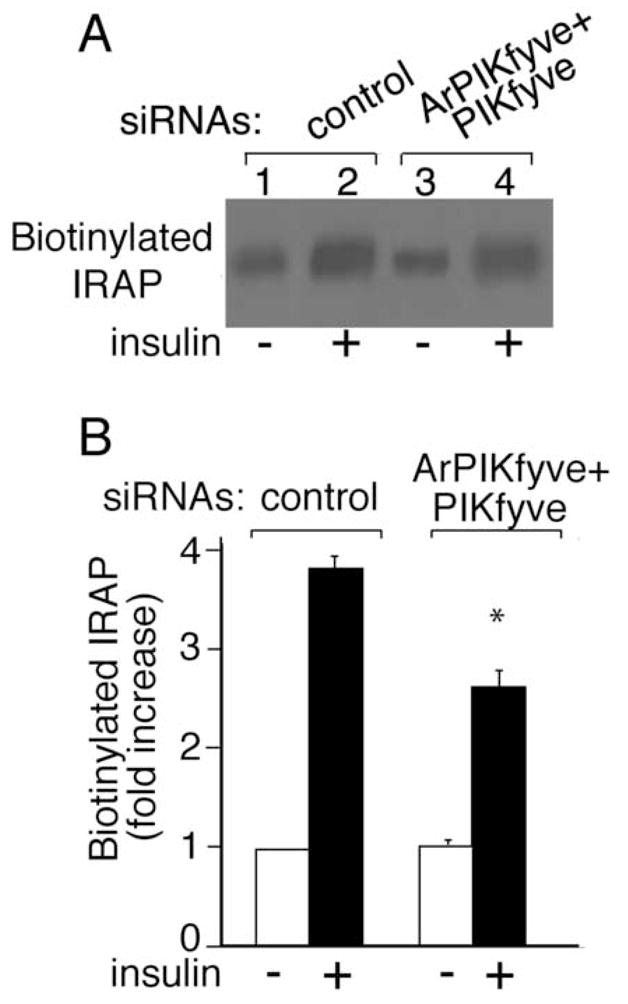
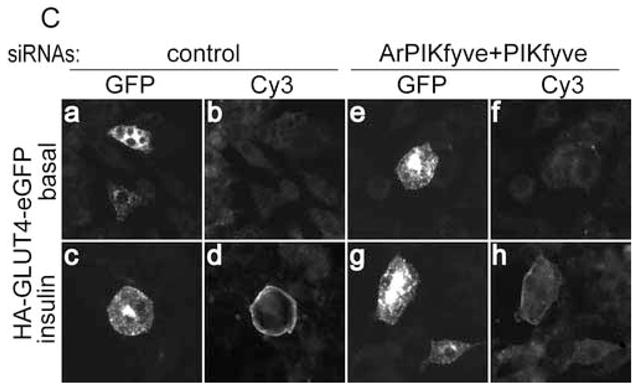
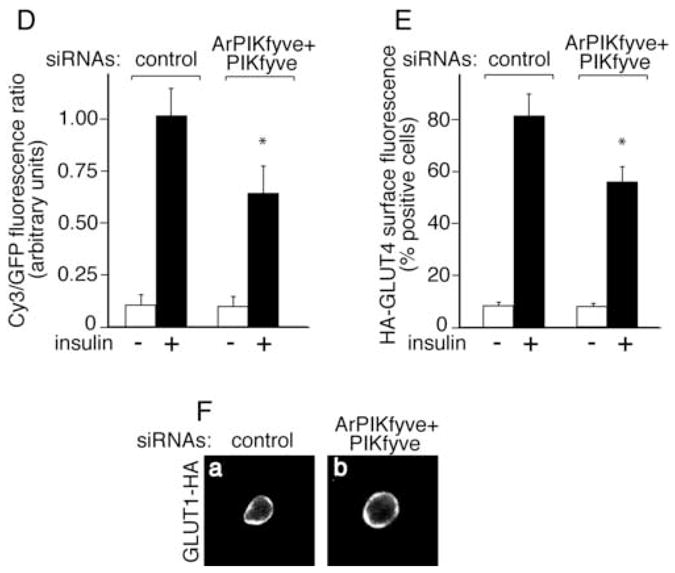
A and B, 3T3-L1 adipocytes were transfected by electroporation with the siRNA duplexes targeting specific regions in cyclophilin B (control) or ArPIKfyve+PIKfyve at concentrations indicated in Fig. 1A and then seeded on 6-well plates. After 72 h, serum-starved cells were stimulated with insulin (100 nM; 20 min; 37°C) or left untreated, and then biotinylated for 30 min at 4°C as detailed in Materials and Methods. Cleared cell lysates were immunoprecipitated with anti-IRAP antibodies. Washed immunoprecipitates were resolved by SDS-PAGE and the proteins were transferred to a PVDF membrane. Biotinylated IRAP was detected by streptavidin-HRP. Shown is a chemiluminescence detection of a representative blot (A) and the quantitation of the band intensity, normalized to non-stimulated control, mean ± SEM, n=3; *, different vs. insulin-stimulated control, P<0.01 (B). C-E, 3T3-L1 adipocytes were coelectroporated with the above indicated siRNAs and HA-GLUT4-eGFP cDNA. Cells were then seeded on coverslips that were placed into a 6-well plate. After 55 h, serum-starved cells were stimulated with insulin for 30 min at 37°C or left untreated, then washed, fixed and processed for immunofluorescence microscopy analysis with anti-HA polyclonal and Cy3-conjugated anti-rabbit IgG as described in Materials and Methods. C. Typical images in three independent experiments depicting the GFP fluorescence and exofacial HA (Cy3) staining in basal (panels a,b,e and f) or insulin-stimulated 3T3-L1 adipocytes (panels c,d,g and h) by confocal microscopy (model 1X81, Olympus) under control (panels a,b, c and d) or ArPIKfyve/PIKfyve protein silencing (panels e, f, g and h). D. Quantitation of the ratio of cell-surface HA (Cy3)-signal to total GFP fluorescence in the HA-GLUT4-eGFP-expressing cells from three independent experiments, in which 10–20 cells/condition/experiment were analyzed by quantitative fluorescence microscopy as described in Materials and Methods (mean ± SEM; * different vs. insulin-stimulated control, P<0.01). E. Quantitation of the cell-surface HA (Cy3) fluorescence based on counting cells displaying a complete plasma membrane rim and presented as a percentage of the counted HA-GLUT4-eGFP expressing cells (~100 cells/condition) in three independent experiments (mean ± SEM *, different vs. insulin-stimulated control, P<0.01). F, 3T3-L1 adipocytes were coelectroporated with the above indicated siRNAs and GLUT1-HA cDNA. Cells were seeded on coverslips placed into a 6-well plate. After 55 h, serum-starved cells were stimulated with insulin for 30 min at 37°C, then washed, fixed, permeabilized and processed for immunofluorescence microscopy with anti-HA polyclonal and Cy3-conjugated anti-rabbit IgG as described in Materials and Methods. Shown are typical confocal microscope (model 1X81, Olympus) images in three independent experiments indicating no significant changes in GLUT1-HA surface abundance under ArPIKfyve/PIKfyve loss.
Since GLUT1 could also contribute to insulin responsiveness of glucose transport, and, in certain conditions, to a substantial extent (26,30), we examined GLUT1 trafficking by immunofluorescence microscopy in transfected 3T3-L1 adipocytes. Importantly, there was no difference in surface GLUT1-HA between cells transfected with ArPIKfyve/PIKfyve and control siRNAs at both basal or insulin-stimulated conditions as based on quantifying the number of cells displaying a complete HA-plasma membrane rim (Fig. 3F and not shown). This result indicates that ArPIKfyve/PIKfyve loss did not significantly affect insulin-regulated GLUT1 trafficking. Collectively, these data are consistent with the conclusion that PIKfyve-catalyzed and ArPIKfyve-regulated PtdIns 3,5-P2 synthesis is among the mechanisms of insulin-activated GLUT4 mobilization to the adipocyte cell surface.
Insulin-dependent increases of PtdIns 3,5-P2 production on isolated membranes
If PtdIns 3,5-P2 positively affects insulin action on glucose influx and GLUT4 translocation, one would expect an insulin-dependent elevation of this lipid. HPLC inositol-head-group analysis in 3T3-L1 adipocytes metabolically labeled with [32P]orthophosphate, however, failed to detect any significant changes in 32P-PtdIns 3,5-P2 accumulation in response to insulin stimulation over a period of 1 to 30 min [15]. We believe that this lack of effect is associated with technical limitations, whereby expected increases could not be registered by the HPLC-measurable total 32P-PtdIns 3,5-P2 accumulation upon [32P]orthophosphate cell labeling. Therefore, in this study we reinvestigated the insulin effect on membrane PtdIns 3,5-P2 by using a 32P-labeling assay conducted in vitro. We compared the ability of membrane fractions freshly isolated from insulin-stimulated or non-stimulated 3T3-L1 adipocytes to synthesize 32P-PtdIns 3,5-P2 in the presence of exogenous [γ-32P]ATP. As illustrated in Fig. 4A and B, the HPLC inositol-head-group separation of extracted lipids documented a substantial increase (64 ± 12%) of PtdIns 3,5-P2 production by intracellular membranes in response to insulin in four separate experiments. A slight insulin-dependent increase in 32P-PtdIns 3,5-P2 levels was also observed with the plasma membrane fraction (Fig. 4B). Consistent with insulin-dependent PI 3-kinase activation, we also documented slight increases in 32P-PtdIns 3-P on both plasma and intracellular membrane fractions (Fig. 4B). It should be noted that PtdIns 4-P, incorporating the vast majority of the radiolabel under the conditions of the phosphorylation assay, remained unchanged in response to insulin.
Fig. 4. Insulin action raises PtdIns 3,5-P2 synthesis on inner membranes isolated from 3T3-L1 adipocytes.
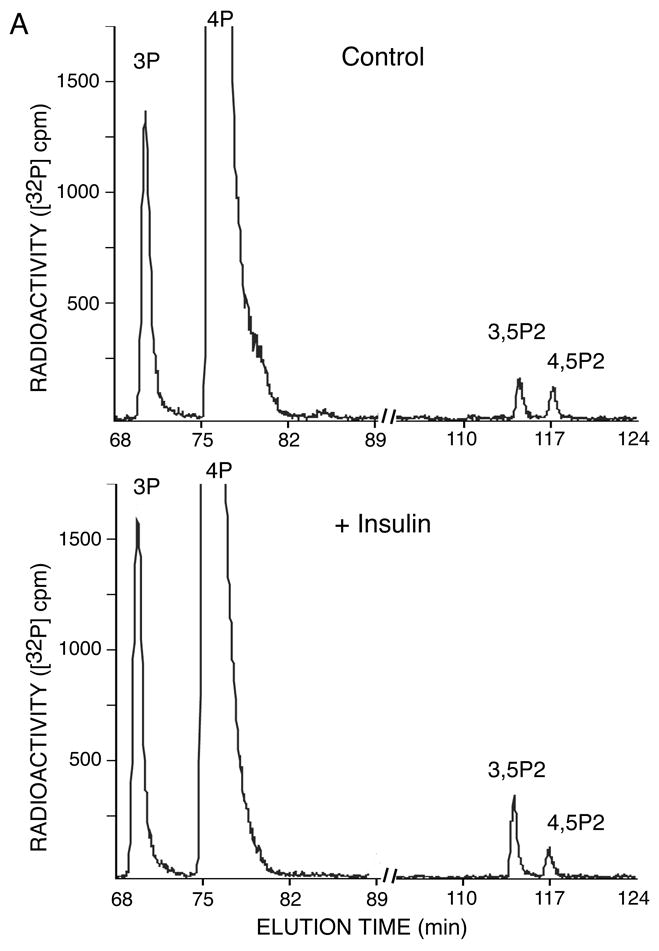
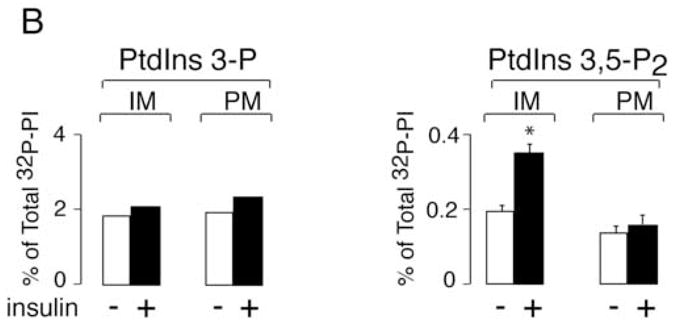
Cells were serum-starved and then stimulated with insulin (10 min, 37°C) or left untreated. Intracellular microsomal (IM) or plasma membrane fractions (PM) were isolated and labeled (100 μg) with exogenous [γ-32P]ATP. [32P]-labeled lipids were extracted, deacylated and co-injected with [3H]-labeled internal or [32P]-labeled external HPLC standards (indicated) as described in Materials and Methods. Radioactivity was monitored automatically with an online flow scintillation analyzer. Shown are HPLC elution profiles of a representative labeling experiment for IM fraction out of four experiments with similar results (A) and the quantitation with respect to PtdIns 3,5-P2 and PtdIns 3-P changes found on IM and PM (B). In the quantitation, the 32P-PtdIns 3,5-P2 radioactive peaks were calculated as a percentage of total PI radioactivity as detailed in Materials and Methods. Insulin-dependent increase of PtdIns 3,5-P2 production on IM was 64 ± 12% (mean ± SEM) after normalization to basal PtdIns 3,5-P2 in four independent experiments. *, different vs. control, P<0.01
Because PIKfyve is the only enzyme known thus far to produce PtdIns 3,5-P2 via biosynthesis, it is expected that both the basal and insulin-dependent increase in PtdIns 3,5-P2 are dependent on PIKfyve and ArPIKfyve. Consistent with this prediction, our [γ-32P]ATP-labeling experiments with membranes isolated from 3T3-L1 adipocytes in which ArPIKfyve/PIKfyve were knocked down did not detect basal PtdIns 3,5-P2 nor was there a significant increment in response to insulin (Suppl. Fig. 1). This indicates that ArPIKfyve/PIKfyve are responsible for both basal 32P-PtdIns 3,5-P2 synthesis and that increased by insulin.
Physical interaction of PIKfyve with ArPIKfyve in 3T3-L1 adipocytes and the effect of insulin
Whereas both the PIKfyve enzyme and the ArPIKfyve regulator appear to control the intracellular PtdIns 3,5-P2 pool [15,18], how this is coordinated in time and space in the context of 3T3-L1 adipocytes is unknown. One plausible mechanism is a physical association between the two proteins. Such a link between the endogenous PIKfyve and ArPIKfyve proteins has been recently established in PC12 cells but not in S. cerevisiae, where only a functional, but not physical, interaction was documented with the yeast counterparts [19,20]. Therefore, we examined the mode of interaction between the endogenous PIKfyve and ArPIKfyve in 3T3-L1 adipocytes by coimmunoprecipitation analysis. We found that PIKfyve readily and specifically coimmunoprecipitated with ArPIKfyve and vice versa, ArPIKfyve coimmunoprecipitated with PIKfyve in either Triton X-100 (0.8%, not shown) or RIPA lysates (1% Nonidet P-40 and 0.5% Na deoxycholate) derived from quiescent 3T3-L1 adipocytes (Fig. 5A). These data imply a relatively stable ArPIKfyve-PIKfyve association in 3T3-L1 adipocytes, since it could sustain the above detergents at the indicated concentrations. Under these conditions (Fig. 5A), we have estimated that >25% of each protein is engaged in this association. This number, however, might be underestimated due to the presence of detergents during the coimmunoprecipitation. Consistent with this, addition of only 0.05% SDS to the RIPA lysates abrogated completely the ArPIKfyve coimmunoprecipitation without affecting the levels of immunoprecipitated PIKfyve (Fig. 5B), indicating that ionic detergents are detrimental to the PIKfyve-ArPIKfyve complex integrity. Importantly, under these conditions, the PIKfyve lipid kinase activity, unlike the PIKfyve protein kinase activity [16], assayed in parallel in the PIKfyve immmunoprecipitates was largely abrogated (Fig. 5C and D). Together, these data are consistent with ArPIKfyve physically associating with PIKfyve and functioning as an activator of PIKfyve lipid kinase activity in 3T3-L1 adipocytes.
Fig. 5. In the PIKfyve-ArPIKfyve physical complex, ArPIKfyve upregulates PIKfyve lipid kinase activity in 3T3-L1 adipocytes.
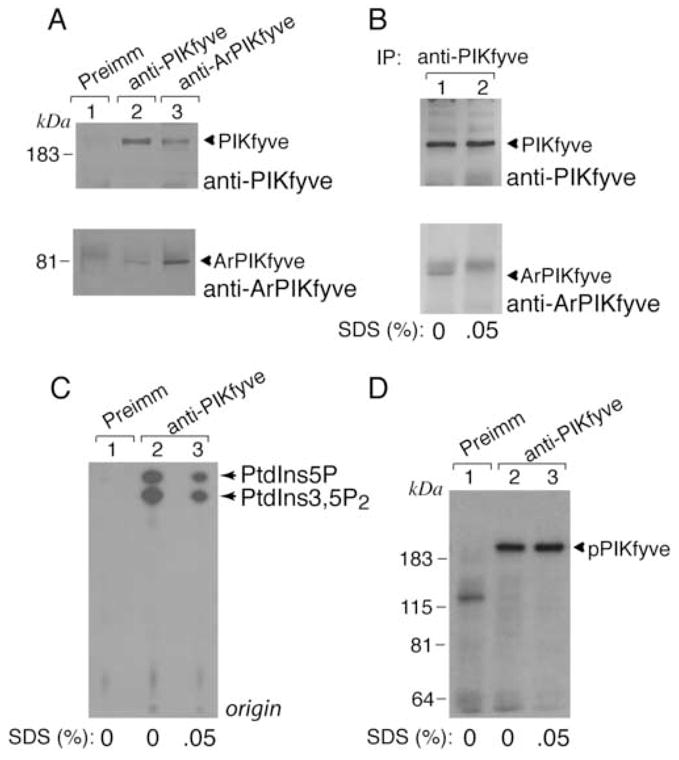
A-D, Cells were lysed in RIPA buffer supplemented with or without 0.05% SDS and immunoprecipitated with the preimmune (Preimm) serum from the ArPIKfyve antibody production, anti-PIKfyve, and anti-ArPIKfyve antisera as indicated. A and B. Immunoprecipitates were washed in RIPA buffer, resolved by SDS-PAGE (6% acrylamide) and electro-transferred onto a nitrocellulose membrane. The membrane was cut horizontally at the 115 kDa protein marker and immunoblotted with anti-PIKfyve (upper panels) and affinity purified anti-ArPIKfyve antibodies (lower panels). Shown are chemiluminescence detections of immunoblots from representative experiments out of three independent experiments with similar results. The amounts of coimmunoprecipitated PIKfyve and ArPIKfyve were determined relative to the corresponding total immunoprecipitated amounts, considering 90% removal of both PIKfyve and ArPIKfyve under these conditions. C and D. Washed immunoprecipitates were analyzed in lipid kinase (C) or protein kinase assays of autophosphorylated PIKfyve (D) as described under Materials and Methods. Shown are autoradiograms of a plate with TLC-separated radiolabeled lipids (C) and of the autokinase reaction resolved by SDS-PAGE and transferred onto membranes (D) from representative experiments out of three independent experiments with similar results. Depicted PtdIns 5-P and PtdIns 3,5-P2 lipid products (C) were confirmed by HPLC analysis.
The observed high correlation between the extent of depleted intracellular PtdIns 3,5-P2 pool and the dampened insulin-regulated glucose transport activation in 3T3-L1 adipocytes, together with increased PtdIns 3,5-P2 production in the microsomal fraction isolated from insulin-stimulated 3T3-L1 adipocytes demonstrated above, suggest that the ArPIKfyve-PIKfyve association might be regulated by activation of the insulin-signaling pathway. We therefore examined the effect of insulin on the complex formation. As illustrated in Fig. 6A the amount of ArPIKfyve coprecipitated with anti-PIKfyve antibodies from 3T3-L1 adipocyte RIPA lysates remained largely unaltered over a 0–60 min time course of insulin challenge.
Fig. 6. Unaltered PIKfyve-ArPIKfyve assembly in response to insulin in 3T3-L1 adipocytes.
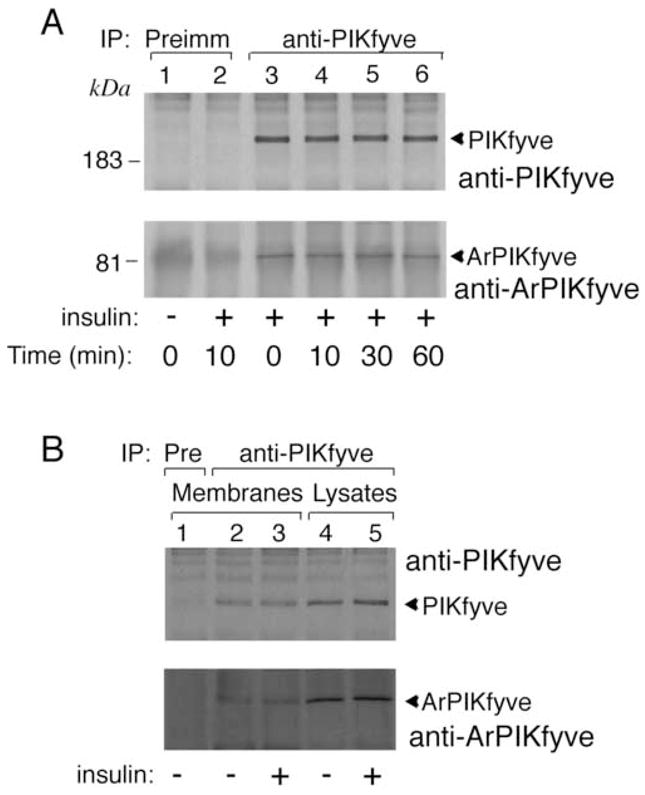
Serum-starved cells were treated with or without insulin (100 nM) at 37° C for the indicated time intervals (A) or for 10 min (B). Cells were then lysed in RIPA buffer either directly (A and B, lanes 4 and 5) or following fractionation to isolate a nuclei-free total membrane fraction (B, lanes 1–3) as detailed in Materials and Methods. Immunoprecipitation was then performed with the preimmune (Pre) serum or anti-PIKfyve antiserum as indicated. Immunoprecipitates were washed in RIPA buffer, resolved by SDS-PAGE (6% acrylamide) and electro-transferred onto nitrocellulose membrane. The membrane was cut horizontally at the 115 kDa protein marker and immunoblotted with anti-PIKfyve (upper halves) and anti-ArPIKfyve antibodies (lower halves) as indicated. Shown are chemiluminescence detections of immunoblots from representative experiments out of 3 to 7 independent experiments with similar results.
The phosphoinositide products synthesized by PIKfyve enzymatic activity are membrane bound. Therefore, we reasoned that insulin action might selectively increase the relative content of the ArPIKfyve activator in the PIKfyve membrane complexes, which would account for the observed insulin-dependent increases in membrane PtdIns 3,5-P2. To test this possibility we next examined whether insulin treatment alters the relative amounts of ArPIKfyve associated with PIKfyve on 3T3-L1 adipocyte membranes. Similarly to our data with the RIPA lysates, the relative amounts of ArPIKfyve coprecipitated with anti-PIKfyve antibodies from total membrane fractions remained unchanged in response to acute insulin (Fig. 6B). Similar analyses with IM and PM, yielded the same results (data not shown). Therefore, whereas acute insulin action is associated with elevated PIKfyve-catalyzed PtdIns 3,5-P2 membrane production, the regulation is not at the level of formation of ArPIKfyve-PIKfyve membrane complexes as determined by coimmunoprecipitation analysis under the specified conditions.
Akt activation in ArPIKfyve/PIKfyve-depleted adipocytes
An essential step in the insulin-signaling circuit leading to GLUT4 translocation and glucose transport is the phosphorylation and activation of Akt [2–5,31] Therefore we examined Akt phosphorylation in response to insulin in the ArPIKfyve/PIKfyve-depleted adipocytes. As illustrated in Fig. 7, insulin-regulated activation of Akt was not significantly altered by ArPIKfyve or PIKfyve depletion as revealed by immunoblotting with anti-phosphoSer473-Akt. Unaltered Akt activation was also documented with anti-phosphoThr308-Akt antibodies (data not shown). Combined PIKfyve/ArPIKfyve depletion, however, resulted in a small but significant decrease in insulin-induced Akt phosphorylation (19±2%; Fig. 7). This indicates that Akt activation is positively affected by PIKfyve/ArPIKfyve and the concomitant increase in PtdIns 3,5-P2 production by insulin.
Fig. 7. Reduced Akt phosphorylation by insulin under combined siRNA-based ablation of PIKfyve and ArPIKfyve in 3T3-L1 adipocytes.
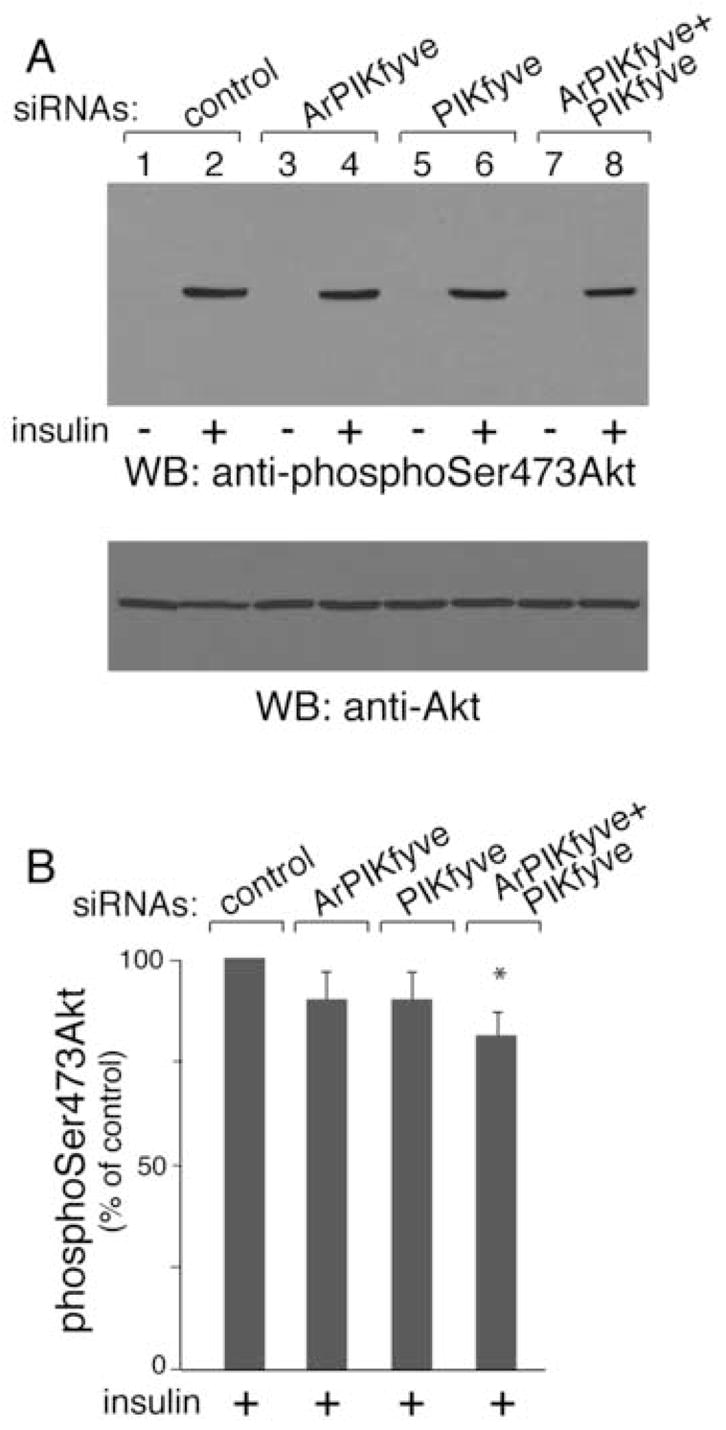
Cells were electroporated with the siRNA pools indicated in Fig. 1A and then seeded on 6-well plates. After 72 h, serum-starved cells were stimulated with insulin (100 nM; 15 min) or left untreated and then were lysed. Equal amounts of protein (100 μg) were subjected to SDS-PAGE and immunoblotted with anti-phosphoSer473-Akt and anti-Akt antibodies as indicated (A). Shown are chemiluminescence detections of representative blots (A) and the quantitation of the band intensity of the insulin-stimulated condition normalized for total Akt, and presented as a percentage of the insulin-stimulated control, mean ± SEM, n=3; *, different vs. insulin-stimulated control, P<0.025 (B).
DISCUSSION
Although PIKfyve, the sole enzyme producing PtdIns 3,5-P2 and PtdIns 5-P via biosynthesis, has been identified in the context of insulin-sensitive fat and muscle tissues [12,13], its role in the fundamental metabolic effect of insulin to stimulate glucose transport in those tissues remains unknown. Moreover, in the light of recent data that increased surface levels of GLUT4 do not always result in accelerated glucose transport [3], the observed positive effect of PIKfyve activity on insulin-regulated GLUT4 translocation in cultured adipocytes [14] can not be extrapolated to the hormonal effect on hexose uptake. Furthermore, recent studies have characterized a novel regulator of intracellular PtdIns 3,5-P2 production, ArPIKfyve [18], but how the functional interaction between ArPIKfyve and PIKfyve is achieved in 3T3-L1 adipocytes and whether ArPIKfyve contributes to insulin responsiveness of glucose transport is also unidentified. The present study was designed to assess the role of the newly emerging ArPIKfyve-PIKfyve-PtdIns 3,5-P2 pathway in insulin-stimulated glucose transport and the mode of interaction between the two proteins in 3T3-L1 adipocytes. To this end, we used siRNA-based selective depletion of endogenous PIKfyve and ArPIKfyve in 3T3-L1 adipocytes. Our studies unequivocally demonstrated that ablation of PIKfyve and/or ArPIKfyve was associated with reduced insulin responsiveness manifested by loss of cell-surface HA-GLUT4-eGFP or IRAP and a drop in glucose transport (Figs. 1 and 3). The inhibitory effect highly correlated with the extent of the PtdIns 3,5-P2 depletion, allowing the conclusion that the ArPIKfyve-PIKfyve-PtdIns 3,5-P2 pathway is a key positive regulator in insulin responsiveness of GLUT4 translocation and glucose transport in cultured adipocytes. Using coimmunoprecipitation analysis we further demonstrated that the functional connection between the endogenous PIKfyve and ArPIKfyve in 3T3-L1 adipocytes was achieved by formation of relatively stable physical complexes containing the two proteins (Fig. 5).
One important observation in our study was the apparent increase of PtdIns 3,5-P2 produced by membrane fractions isolated from insulin-treated 3T3-L1 adipocytes (Fig. 4). Together with the observed drop in insulin responsiveness under depleted PtdIns 3,5-P2 concomitant with siRNA-mediated ArPIKfyve/PIKfyve ablation, these data support the notion that insulin action on GLUT4 is associated with up-regulation of intracellular PtdIns 3,5-P2 levels. This conclusion is consistent with our previous observations based on expression of the kinase-deficient PIKfyveK1831E mutant in 3T3-L1 adipocytes [14] and indicates that arrested GLUT4 surface exposure in response to insulin is not due to an adverse dominant-negative effect of PIKfyveK1831E, as speculated by others [17]. Based on our recent observations for a key role of the PIKfyve-catalyzed PtdIns 3-P-to-PtdIns 3,5-P2 conversion in proper membrane processing at early/post-early endosomes [32], it is conceivable that PtdIns 3,5-P2 production affects GLUT4 trafficking through endosomes in response to insulin. According to currently accepted views, insulin action mobilizes both endosomal and non-endosomal GLUT4-containing pools, including the specialized insulin-responsive pool that is the main reservoir of GLUT4 during the initial hormonal effect, the recycling endosomes that also contain GLUT1, and the GLUT4 inter-endosomal traffic away from early endosomes [2–5,33]. The fact that GLUT1 surface abundance remained unaltered by ArPIKfyve/PIKfyve depletion (Fig. 3F) supports the notion that as with the recycling pathway in other cell types [34,35], perturbations in PIKfyve function is not critical in the performance of insulin-regulated endosome recycling. Based on these data and considerations we suggest that the increased exit of GLUT4 from early endosomes, which is ultimately a prerequisite for replenishing the rapidly depleting insulin-sensitive GLUT4 pool during or after insulin challenge, is a step that requires a robust PIKfyve-catalyzed PtdIns 3,5-P2 production. Consistent with this idea, the insulin-dependent increase of PtdIns 3,5-P2, documented here upon labeling of isolated membranes with exogenous [32P]ATP was more pronounced on the microsomal fraction (Fig. 4), a fraction where PIKfyve was found recruited in response to insulin [21]. It should be emphasized, however, that whereas PIKfyve is responsible for the insulin-dependent increase in PtdIns 3,5-P2 biosynthesis, it is possible for additional level of regulation to occur at the PtdIns 3,5-P2 turnover. This point becomes increasingly interesting in the light of recent reports in yeast, where a Sac-domain-containing PtdIns 3,5-P2 phosphatase, named Fig 4, in concert with yeast ArPIKfyve (Vac14) and PIKfyve (Fab1) orchestrates an intricate regulation of the PtdIns 3,5-P2 pool [36,37]. A biochemically uncharacterized mammalian ortholog of Fig 4 is Sac3 [38]. Potential functional interactions of Sac3 with the ArPIKfyve/PIKfyve complex and a plausible insulin-dependent regulation are important objectives for future studies.
In mammalian cells, including 3T3-L1 adipocytes, PIKfyve can produce PtdIns 5-P, in addition to PtdIns 3,5-P2 [7]. Therefore, the RNAi-based ablation of endogenous PIKfyve or ArPIKfyve could presumably affect the intracellular PtdIns 5-P pool to some extent. This could potentially contribute to the decrease in the insulin-regulated glucose-transport stimulation observed here because insulin signaling to GLUT4 in 3T3-L1 adipocytes has been found to require PtdIns 5-P [8]. It should be emphasized that the PtdIns 5-P enzymology is rather complex, whereby, in addition to PIKfyve, PI phosphatases and Type II PIP kinases contribute to the steady-state or regulated PtdIns 5-P pool [7,9,38–40]. As each of these enzyme activities could potentially be regulated by insulin [21,41,42], their contribution to the increased PtdIns 5-P in response to insulin is unknown. Therefore, whether reduced PIKfyve-dependent biosynthesis of PtdIns 5-P enhances the effect of decreased PtdIns 3,5-P2 in the here observed loss of surface LUT4 and glucose-transport inhibition under ArPIKfyve/PIKfyve ablation is unclear.
The exact mode of ArPIKfyve/PIKfyve up-regulation and the downstream targets of PtdIns 3,5-P2 (or PtdIns 5-P) in the context of insulin signaling to GLUT4 translocation and glucose transport stimulation remains debatable and still unresolved. Given the fact that endogenous PIKfyve and GLUT4 or their overexpressed forms do not colocalize to a significant extent in biochemical and morphological studies [12–14,17,21], the functional interaction between them should be firmly regulated. Published data by our group are consistent with a role for an insulin-dependent phosphorylation step that causes PIKfyve recruitment and tethering to 3T3-L1 adipocyte low density microsomal fraction [21] but the kinase responsible remains unknown. Another study [17] links the insulin-dependent PIKfyve phosphorylation to PIKfyve activation rather than to enzyme localization. Reportedly, insulin-activated Akt renders PIKfyve phosphorylated on S318, thereby up-regulating PtdIns 3,5-P2 production [17]. It remains unclear why the PIKfyveS318A mutant that is Akt-phosphorylation deficient produces a gain of surface GLUT4 in response to insulin [17]. Plausible explanation of this paradox could be provided by our unpublished mass spectrometry analysis with in vitro phosphorylated recombinant GST-PIKfyve (15 mM ATP, 5 mM MnCl2, 50 mM Hepes, pH 7.4), where we determined phospho-S318 as an autophosphorylation site (Donald Hunt and Assia Shisheva, unpublished data). Because PIKfyve autophosphorylation and lipid kinase activity are inversely related [43], the reported gain-of-function of phosphorylation-deficient PIKfyveS318A [17] may reflect the release of this autoinhibition. Alternatively, Akt could be positioned downstream of PIKfyve. This possibility is consistent with our previous findings for increased basal or insulin-dependent Akt phosphorylation under high levels of recombinant PIKfyvewt activity [14]. This notion is further reinforced by the current studies, where we documented slight but significant decreases of insulin-activated Akt phosphorylation (Fig. 7) under PIKfyve/ArPIKfyve protein ablation and concomitant depletion of the PtdIns 3,5-P2 pool. Noteworthy, a recent report documents Akt activation by elevated PtdIns 5-P in several mammalian cell types [44]. Together, these data imply that in a cell context, the products of PIKfyve kinase are a cause, rather than a consequence, of Akt activation and that Akt functions as a plausible PtdIns 3,5-P2 downstream effector in GLUT4 signaling/translocation. Several candidate downstream protein effectors of PtdIns 3,5-P2 have been recently characterized [45–47]. It remains to be identified whether those or yet-to-be-identified targets of PtdIns 3,5-P2 fit into the insulin signaling network leading to GLUT4 translocation and glucose transport.
In summary, the present data demonstrate that ArPIKfyve and PIKfyve physically assemble to control PtdIns 3,5-P2 production in 3T3-L1 adipocytes and that the novel ArPIKfyve-PIKfyve-PtdIns 3,5-P2 pathway is among the mechanisms linking insulin signaling and its stimulation of glucose transport and GLUT4 translocation.
Supplementary Material
Suppl. Fig. 1. Undetectable32P-PtdIns 3,5-P2 on inner membranes under ArPIKfyve/PIKfyve knockdown. 3T3-L1 adipocytes were transfected with the siRNA duplexes as specified in the legend to Fig. 1. Serum-starved cells were stimulated or not with insulin and fractionated to PM and IM. Shown is an HPLC profile of extracted lipids following IM labeling with [γ-32P]ATP performed as indicated in the legend to Fig. 4.
Acknowledgments
We thank Drs. Alan Saltiel and Louise Chang, for valuable advice and reagents for the glucose transport assay, Drs. Mike Czech and Paul Pilch, for the antibodies, and Drs. Jeff Pessin and Tony Carruthers for GLUT4 and GLUT1 constructs. We are grateful to Linda McCraw for her excellent secretarial assistance.
GRANTS
This project was supported by Research Grants (to AS) from the National Institute of Health (DK58058) and American Diabetes Association (1-05-RA-120).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Czech MP. Molecular actions of insulin on glucose transport. Annu Rev Nutr. 1995;15:441–471. doi: 10.1146/annurev.nu.15.070195.002301. [DOI] [PubMed] [Google Scholar]
- 2.Bryant NJ, Govers R, James DE. Regulated transport of the glucose transporter GLUT4. Nat Rev Mol Cell Biol. 2002;3:267–277. doi: 10.1038/nrm782. [DOI] [PubMed] [Google Scholar]
- 3.Dugani CB, Klip A. Glucose transporter 4: cycling, compartments and controversies. EMBO Rep. 2005;6:1137–1142. doi: 10.1038/sj.embor.7400584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shisheva A. Regulating GLUT4 vesicle dynamics by phosphoinositide kinases and phosphoinositide phosphatases. Front Biosci. 2003;8:S 945–967. doi: 10.2741/1101. [DOI] [PubMed] [Google Scholar]
- 5.Watson RT, Kanzaki M, Pessin JE. Regulated membrane trafficking of the insulin-responsive glucose transporter 4 in adipocytes. Endo Rev. 2004;25:177–204. doi: 10.1210/er.2003-0011. [DOI] [PubMed] [Google Scholar]
- 6.Sbrissa D, Ikonomov OC, Shisheva A. PIKfyve, a mammalian ortholog of yeast Fab1p lipid kinase, synthesizes 5-phosphoinositides. J Biol Chem. 1999;274:21589–21597. doi: 10.1074/jbc.274.31.21589. [DOI] [PubMed] [Google Scholar]
- 7.Sbrissa D, Ikonomov OC, Deeb R, Shisheva A. Phosphatidylinositol 5-phosphate biosynthesis is linked to PIKfyve and is involved in osmotic response pathway in mammalian cells. J Biol Chem. 2002;277:47276–47284. doi: 10.1074/jbc.M207576200. [DOI] [PubMed] [Google Scholar]
- 8.Sbrissa D, Ikonomov OC, Strakova J, Shisheva A. Role for a novel signaling intermediate, phosphatidylinositol 5-phosphate, in insulin-regulated F-actin stress fiber breakdown and GLUT4 translocation. Endocrinology. 2004;145:4853–4865. doi: 10.1210/en.2004-0489. [DOI] [PubMed] [Google Scholar]
- 9.Tsujita K, Itoh T, Ijuin T, Yamamoto A, Shisheva A, Laporte J, Takenawa T. Myotubularin regulates the function of the late endosome through the GRAM domain-phosphatidylinositol 3,5-bisphosphate interaction. J Biol Chem. 2004;279:13817–13824. doi: 10.1074/jbc.M312294200. [DOI] [PubMed] [Google Scholar]
- 10.Shisheva A. PIKfyve: the road to PtdIns 5-P and PtdIns 3,5-P2. Cell Biol Internal. 2001;25:1201–1206. doi: 10.1006/cbir.2001.0803. [DOI] [PubMed] [Google Scholar]
- 11.Ikonomov OC, Sbrissa D, Mlak K, Deeb R, Fligger J, Soans A, Finley RL, Jr, Shisheva A. Active PIKfyve associates with and promotes the membrane attachment of the late endosome-to-TGN transport factor Rab9 p40. J Biol Chem. 2003;278:50863–50871. doi: 10.1074/jbc.M307260200. [DOI] [PubMed] [Google Scholar]
- 12.Shisheva A, DeMarco C, Ikonomov O, Sbrissa D. PIKfyve and acute insulin actions. Insulin Signaling: From Cultured Cells to Animal Models. 2002:89–205. [Google Scholar]
- 13.Shisheva A, Sbrissa D, Ikonomov O. Cloning, characterization, and expression of a novel Zn2+-binding FYVE finger-containing phosphoinositide kinase in insulin-sensitive cells. Mol Cell Biol. 1999;19:623–634. doi: 10.1128/mcb.19.1.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ikonomov OC, Sbrissa D, Mlak K, Shisheva A. Requirement for PIKfyve enzymatic activity in acute and long-term insulin cellular effects. Endocrinology. 2002;143:4742–4754. doi: 10.1210/en.2002-220615. [DOI] [PubMed] [Google Scholar]
- 15.Sbrissa D, Shisheva A. Acquisition of unprecedented phosphatidylinositol 3,5-bisphosphate rise in hyperosmotically stressed 3T3-L1 adipocytes, mediated by ArPIKfyve-PIKfyve pathway. J Biol Chem. 2005;280:7883–7889. doi: 10.1074/jbc.M412729200. [DOI] [PubMed] [Google Scholar]
- 16.Ikonomov OC, Sbrissa D, Mlak K, Kanzaki M, Pessin J, Shisheva A. Functional dissection of lipid and protein kinase signals of PIKfyve reveals the role of PtdIns 3,5-P2 production for endomembrane integrity. J Biol Chem. 2002;277:9208–9211. doi: 10.1074/jbc.M108750200. [DOI] [PubMed] [Google Scholar]
- 17.Berwick DC, Dell GD, Welsh GI, Heesom KJ, Hers I, Fletcher LM, Cooke KT, Tavaré JM. Protein kinase B phosphorylation of PIKfyve regulates the trafficking of GLUT4 vesicles. J Cell Sci. 2004;117:5985–5993. doi: 10.1242/jcs.01517. [DOI] [PubMed] [Google Scholar]
- 18.Sbrissa D, Ikonomov OC, Strakova J, Dondapati R, Mlak K, Deeb R, Silver R, Shisheva A. A mammalian ortholog of Saccharomyces cerevisiae Vac14 that associates with and up-regulates PIKfyve phosphoinositide 5-kinase activity. Mol Cell Biol. 2004;24:10437–10447. doi: 10.1128/MCB.24.23.10437-10447.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bonangelino CJ, Nau JJ, Duex JE, Brinkman M, Wurmser AE, Gary JD, Emr SD, Weisman LS. Osmotic stress-induced increase of phosphatidylinositol 3,5-bisphosphate requires Vac14, an activator of the lipid kinase Fab1p. J Cell Biol. 2002;156:1015–1028. doi: 10.1083/jcb.200201002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dove SK, McEwen RK, Mayes A, Hughes DC, Beggs JD, Michell RH. Vac14 controls PtdIns(3,5)P2 synthesis and Fab1-dependent protein trafficking to the multivesicular body. Curr Biol. 2002;12:885–893. doi: 10.1016/s0960-9822(02)00891-6. [DOI] [PubMed] [Google Scholar]
- 21.Shisheva A, Rusin B, Ikonomov OC, DeMarco C, Sbrissa D. Localization and insulin-regulated relocation of 5′-phosphonositide kinase PIKfyve in 3T3-L1 adipocytes. J Biol Chem. 2001;276:11859–11869. doi: 10.1074/jbc.M008437200. [DOI] [PubMed] [Google Scholar]
- 22.Garza LA, Birnbaum MJ. Insulin-responsive aminopeptidase trafficking in 3T3-L1 adipocyte. J Biol Chem. 2000;275:2560–2567. doi: 10.1074/jbc.275.4.2560. [DOI] [PubMed] [Google Scholar]
- 23.Kandror K, Pilch PF. Identification and isolation of glycoproteins that translocate to the cell surface from GLUT4-enriched vesicles in an insulin-dependent fashion. J Biol Chem. 1994;269:138–142. [PubMed] [Google Scholar]
- 24.Ross SA, Scott HM, Morris NJ, Leung WY, Mao F, Lienhard GE, Keller SR. Characterization of the insulin-regulated membrane aminopeptidase in 3T3-L1 adipocytes. J Biol Chem. 1996;271:3328–3332. doi: 10.1074/jbc.271.6.3328. [DOI] [PubMed] [Google Scholar]
- 25.Katz EB, Stenbit AE, Hatton K, DePinho R, Charron MJ. Cardiac and adipose tissue abnormalities but not diabetes in mice deficient in GLUT4. Nature. 1995;377:151–155. doi: 10.1038/377151a0. [DOI] [PubMed] [Google Scholar]
- 26.Rudich A, Konrad D, Törö D, Ben-Romano R, Huang C, Niu W, Garg RR, Wijesekara N, Germinario RJ, Bilan PJ, Klip A. Indinavir uncovers different contributions of GLUT4 and GLUT1 towards glucose uptake in muscle and fat cells and tissues. Diabetolgia. 2003;46:649–658. doi: 10.1007/s00125-003-1080-1. [DOI] [PubMed] [Google Scholar]
- 27.Kandror KV, Pilch PE. gp160, a tissue-specific marker for insulin-activated glucose transport. Proc Natl Acad Sci USA. 1994;91:8017–8021. doi: 10.1073/pnas.91.17.8017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keller SR. The insulin-regulated aminopeptidase: a companion and regulator of GLUT4. Front Biosci. 2003;8:s 410–420. doi: 10.2741/1078. [DOI] [PubMed] [Google Scholar]
- 29.Lampson MA, Racz A, Cushman SW, McGraw TE. Demonstration of insulin-responsive trafficking of GLUT4 and vpTR in fibroblasts. J Cell Sci. 2000;113:4065–4076. doi: 10.1242/jcs.113.22.4065. [DOI] [PubMed] [Google Scholar]
- 30.Liao W, Nguyen MTA, Imamura T, Singer O, Verma IM, Olefsky JM. Lentiviral short hairpin ribonucleic acid-mediated knockdown of GLUT4 in 3T3-L1 adipocytes. Endocrinology. 2006;147:2245–2252. doi: 10.1210/en.2005-1638. [DOI] [PubMed] [Google Scholar]
- 31.Zhou QL, Park JG, Jiang ZY, Holik JJ, Mitra P, Semiz S, Guilherme A, Powelka AM, Tang X, Virbasius J, Czech MP. Analysis of insulin signaling by RNAi-based gene silencing. Biochem Soc Trans. 2004;32:817–821. doi: 10.1042/BST0320817. [DOI] [PubMed] [Google Scholar]
- 32.Ikonomov OC, Sbrissa D, Shisheva A. Localized PtdIns 3,5-P2 synthesis to regulate early endosome dynamics and fusion. Am J Physiol Cell Physiol. 2006;291:C393–404. doi: 10.1152/ajpcell.00019.2006. [DOI] [PubMed] [Google Scholar]
- 33.Foster LJ, Li D, Randawa VK, Klip A. Insulin accelerates inter-endosomal GLUT4 traffic via phosphatidylinositol 3-kinase and protein kinase B. J Biol Chem. 2001;276:44212–44221. doi: 10.1074/jbc.M102964200. [DOI] [PubMed] [Google Scholar]
- 34.Ikonomov OC, Sbrissa D, Fligger J, Foti M, Carpentier J-L, Shisheva A. PIKfyve controls fluid-phase endocytosis but not recycling/degradation of endocytosed receptors or sorting of procathepsin D by regulating multivesicular body morphogenesis. Mol Biol Cell. 2003;14:4581–4591. doi: 10.1091/mbc.E03-04-0222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rutherford AC, Traer C, Wassmer T, Pattni K, Bujny MV, Carlton JG, Stenmark H, Cullen PJ. The mammalian phosphatidylinositol 3-phosphate 5-kinase (PIKfyve) regulates endosome-to-TGN retrograde transport. J Cell Sci. 2006;119:3944–3957. doi: 10.1242/jcs.03153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duex JE, Tang F, Weisman LS. The Vac14p-Fig4p complex acts independently of Vac7p and couples PI3,5P2 synthesis and turnover. J Cell Biol. 2006;172:693–704. doi: 10.1083/jcb.200512105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rudge SA, Anderson DM, Emr SD. Vacuole size control: regulation of PtdIns(3,5)P2 levels by the vacuole-associated Vac14-Fig 4 complex, a PtdIns(3,5)P2-specific phosphatase. Mol Biol Cell. 2004;15:24–36. doi: 10.1091/mbc.E03-05-0297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Minagawa T, Ijuin T, Mochizuki Y, Takenawa T. Identification and characterization of a Sac domain-containing phosphoinositide 5-phosphatase. J Biol Chem. 2001;276:22011–22015. doi: 10.1074/jbc.M101579200. [DOI] [PubMed] [Google Scholar]
- 39.Clague MJ, Lorenzo O. The myotubularin family of lipid phosphatases. Traffic. 2005;6:1063–1069. doi: 10.1111/j.1600-0854.2005.00338.x. [DOI] [PubMed] [Google Scholar]
- 40.Pagliarini DJ, Worby CA, Dixon JE. A PTEN-like phosphatase with a novel substrate specificity. J Biol Chem. 2004;279:38590–38596. doi: 10.1074/jbc.M404959200. [DOI] [PubMed] [Google Scholar]
- 41.Carricaburu V, Lamia KA, Lo E, Favereaux L, Payrastre B, Cantley LC, Rameh LE. The phosphatidylinositol (PI)-5-phosphate 4-kinase type II enzyme controls insulin signaling by regulating PI-3,4,5-trisphosphate degradation. Proc Natl Acad Sci USA. 2003;100:9867–9872. doi: 10.1073/pnas.1734038100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lamia KA, Peroni OD, Kim YB, Rameh LE, Kahn BB, Cantley LC. Increased insulin sensitivity and reduced adiposity in phosphatidylinositol 5-phosphate 4-kinase β −/− mice. Mol Cell Biol. 2004;24:5080–5087. doi: 10.1128/MCB.24.11.5080-5087.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sbrissa D, Ikonomov OC, Shisheva A. PIKfyve lipid kinase is a protein kinase: downregulation of 5′-phosphoinositide product formation by autophosphorylation. Biochemistry. 2000;39:15980–15989. doi: 10.1021/bi001897f. [DOI] [PubMed] [Google Scholar]
- 44.Pendaries C, Tronchère, Arbibe L, Mounier J, Gozani O, Cantley L, Fry MJ, Gaits-Iacovoni F, Sansonetti PJ, Payrastre B. PtdIns(5)P activates the host cell PI3-kinase/Akt pathway during Shigella flexneri infection. EMBO J. 2006;25:1024–1034. doi: 10.1038/sj.emboj.7601001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dove SK, Piper RC, McEwen, Yu JW, King MC, Hughes DC, Thuring J, Holmes AB, Cooke FT, Michell RH, Parker PJ, Lemmon MA. Svp1p defines a family of phosphatidylinositol 3,5-bisphosphate effectors. EMBO J. 2004;23:1922–1933. doi: 10.1038/sj.emboj.7600203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Whitley P, Reaves BJ, Hashimoto M, Riley AM, Potter BVL, Holman GD. Identification of mammalian Vps24p as an effector of phosphatidylinositol 3,5-bisphosphate-dependent endosome compartmentalization. J Biol Chem. 2003;278:38786–38795. doi: 10.1074/jbc.M306864200. [DOI] [PubMed] [Google Scholar]
- 47.Eugster A, Pécheur EI, Michel F, Winsor B, Letourneur F, Friant FS. Ent5p is required with Ent3p and Vps27p for ubiquitin-dependent protein sorting into the multivesicular body. Mol Biol Cell. 2004;15:3031–3041. doi: 10.1091/mbc.E03-11-0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl. Fig. 1. Undetectable32P-PtdIns 3,5-P2 on inner membranes under ArPIKfyve/PIKfyve knockdown. 3T3-L1 adipocytes were transfected with the siRNA duplexes as specified in the legend to Fig. 1. Serum-starved cells were stimulated or not with insulin and fractionated to PM and IM. Shown is an HPLC profile of extracted lipids following IM labeling with [γ-32P]ATP performed as indicated in the legend to Fig. 4.