

Abstract
Mitochondrial complex I (NADH:ubiquinone oxidoreductase) undergoes reversible deactivation upon incubation at 30–37 °C. The active/deactive transition could play an important role in the regulation of complex I activity. It has been suggested recently that complex I may become modified by S-nitrosation under pathological conditions during hypoxia or when the nitric oxide:oxygen ratio increases. Apparently, a specific cysteine becomes accessible to chemical modification only in the deactive form of the enzyme. By selective fluorescence labeling and proteomic analysis, we have identified this residue as cysteine-39 of the mitochondrially encoded ND3 subunit of bovine heart mitochondria. Cysteine-39 is located in a loop connecting the first and second transmembrane helix of this highly hydrophobic subunit. We propose that this loop connects the ND3 subunit of the membrane arm with the PSST subunit of the peripheral arm of complex I, placing it in a region that is known to be critical for the catalytic mechanism of complex I. In fact, mutations in three positions of the loop were previously reported to cause Leigh syndrome with and without dystonia or progressive mitochondrial disease.
Proton-translocating NADH:ubiquinone oxidoreductases (EC 1.6.5.3, complex I or type I NADH dehydrogenase, NDH-1) are the largest components of mitochondrial and bacterial respiratory chains. Structural information about complex I is still very limited, although recently, the atomic structure of the peripheral arm of the enzyme from Thermus thermophilus comprising eight subunits and the location of the iron-sulfur centers and flavine mononucleotide were determined at 3.3 Å resolution (1).
The mammalian enzyme is extraordinarily complex; it is composed of 45 different subunits (2) and contains one non-covalently bound flavine mononucleotide and eight iron-sulfur clusters as prosthetic groups (3, 4). Two ubisemiquinone species were detected during turnover (5, 6). Complex I catalyzes transfer of two electrons from NADH to ubiquinone coupled to the translocation of four protons across the inner membrane of mitochondria (7, 8) or the membrane of submitchondrial particles (SMP5 (9)). Over the last 40 years, numerous hypothetical schemes for the coupling mechanism of complex I have been published (see Refs. 10 and 11 for a review). Those proposed most recently involve long range conformational changes as the coupling principle (11) rather than variations of classical redox loops as originally proposed by Peter Mitchell (12).
A unique functional property of complex I later operationally called
active/deactive (A/D) transition was originally observed many years ago
(13,
14). Exposure of
membrane-bound (15) or
detergent-solubilized and purified
(16) enzyme to elevated
temperature (30–37 °C) results in the so-called deactive or dormant
form of mitochondrial complex I that is characterized by a considerable lag
phase in the onset of NADH:ubiquinone oxidoreductase or reverse
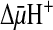 -dependent
ubiquinol:NAD+ oxidoreductase activity (see Ref.
17 for a review). This lag
phase is abolished after brief preincubation of the enzyme under conditions
where NAD(P)H:ubiquinone oxidoreductase turnover is permitted, thus providing
reactivation of the enzyme
(15,
16). This slow A/D transition
has been demonstrated with purified complex I
(16), coupled SMP
(15), intact mitochondria
(18), and in reperfused rat
hearts (19). Remarkably, the
A/D transition of complex I was found to be a prominent feature of several
other vertebrates and strictly aerobic yeasts with variable kinetic
characteristics (20) but was
not observed in membranes of the prokaryote Paracoccus denitrificans.
Only the rotenone-sensitive NADH:ubiquinone oxidoreductase activities in
different species undergo the A/D transition, whereas the non-physiological
NADH-dehydrogenase activities using ferricyanide or hexaammineruthenium (III)
as electron acceptors are the same for active and deactive enzyme.
-dependent
ubiquinol:NAD+ oxidoreductase activity (see Ref.
17 for a review). This lag
phase is abolished after brief preincubation of the enzyme under conditions
where NAD(P)H:ubiquinone oxidoreductase turnover is permitted, thus providing
reactivation of the enzyme
(15,
16). This slow A/D transition
has been demonstrated with purified complex I
(16), coupled SMP
(15), intact mitochondria
(18), and in reperfused rat
hearts (19). Remarkably, the
A/D transition of complex I was found to be a prominent feature of several
other vertebrates and strictly aerobic yeasts with variable kinetic
characteristics (20) but was
not observed in membranes of the prokaryote Paracoccus denitrificans.
Only the rotenone-sensitive NADH:ubiquinone oxidoreductase activities in
different species undergo the A/D transition, whereas the non-physiological
NADH-dehydrogenase activities using ferricyanide or hexaammineruthenium (III)
as electron acceptors are the same for active and deactive enzyme.
It was initially observed by Estabrook's group (21) that the NADH-oxidase reaction of complex I becomes sensitive to N-ethyl maleimide (NEM) upon incubation of SMP at 37 °C. Later, it was established that treatment of the deactive form with sulfhydryl reagent completely blocks the turnover-dependent activation of complex I (22, 23). In contrast, the steady-state NADH:ubiquinone oxidoreductase activity of the active form is entirely insensitive to thiol reagents, suggesting that the A/D transition involves conformational changes of complex I. In the deactive form, specific labeling by thiol reagents of only one subunit with an apparent molecular mass of 15 kDa has been observed with bovine SMP (24). This subunit is not labeled in the active form and was reported to be susceptible to modification from the matrix side only in the D-form of the enzyme (23).
Here we report the identification of the cysteine residue selectively accessible only in the D-form of complex I in the loop connecting two transmembrane helices of the mitochondrially encoded ND3 subunit. Implications for the mechanism and the role of the active/deactive transition are discussed.
EXPERIMENTAL PROCEDURES
Bovine heart SMP were prepared as described previously (15) and stored in liquid nitrogen. Labeling of the A/D-transition-specific sulfhydryl group was performed as reported previously (24), with minor modifications. All precipitation and wash steps were performed by ultracentrifugation at 4 °C 60 min 120,000 × g. SMP (1 mg/ml) were suspended at pH 8.0 in SET buffer (0.25 m sucrose, 0.2 mm EDTA, 50 mm Tris-Cl). 0.4 mm NADPH was added, and the suspension was incubated aerobically with vigorous stirring for 30 min at 23 °C for complex I activation (25). Activated SMP were washed at pH 8.0 with SET buffer once, resuspended to 10 mg/ml at pH 9.0 in SET buffer, and treated with 30 mm NEM at 15 °C for 30 min. The reaction was terminated by adding 10 volumes of SET buffer (pH 7.5) containing 35 mm cysteine. After NEM treatment that blocked all thiol groups accessible in the active form of complex I, SMP were precipitated, washed with the same buffer without cysteine, and resuspended in SET buffer (pH 8.0). The SMP suspension was divided into two equal parts that were incubated synchronously for 90 min at 30 °C (deactive) or 4 °C (active). Afterward, both samples were treated with 0.5 mm N-fluoresceinyl maleimide (NFM) for 20 min at 15 °C in the dark. The reaction was terminated by 10-fold dilution with SET buffer (pH 7.5) containing 35 mm cysteine, and the SMP samples were washed twice with SET buffer (pH 8.0). Each sample was resuspended and diluted to a protein concentration of about 20 mg/ml and stored in liquid nitrogen. Although the sample retaining the active form by incubation at 4 °C kept their NADH oxidase or NADH:ubiquinone-1 oxidoreductase activities, the activities of samples that had been deactivated by incubation at elevated temperature before NFM labeling were irreversibly inhibited by 95%. Both preparations catalyzed the non-physiological oxidation of NADH by hexaammineruthenium with the same rates (not shown).
The purification of complex I from each sample was carried out by blue native-PAGE (BN-PAGE) of SMP solubilized by 3 mg/mg of n-dodecyl-β-d-maltoside as described previously (26). The band containing complex I was cut out, and the protein was extracted from the gel by electroelution. This procedure allowed us to obtain around 30–50 μg of the enzyme from 1 mg of SMP protein.
Doubled SDS-PAGE (dSDS-PAGE) using 10% acrylamide gel containing 6 m urea for first dimension and 16% acrylamide gel for second dimension Tricine-SDS-PAGE of complex I (in the absence of mercaptoethanol) was performed as described before (27). The gels were scanned for fluorescence using 488 nm excitation and 520 nm emission in a Typhoon scanner (GE Healthcare) after the end of the first run, and the final two-dimensional gels were scanned before and after silver staining. The fluorescent spot and a few neighboring spots were excised, and proteins were digested “in-gel” by trypsin, chymotrypsin, and elastase essentially as described (28, 29), except that shrinking by acetonitrile before enzyme digestion was omitted.
For mass spectrometry (MS) and MS/MS, dried samples were dissolved in 5 μl of 50% (v/v) acetonitrile, 0.5% (v/v) trifluoroacetic acid. 1 μl of sample was mixed with 1 μl of matrix (1 mg of α-cyano-4-hydroxycinnamic acid in 1 ml of 50% (v/v) acetonitrile, 0.5% (v/v) trifluoroacetic acid) and dried on a target plate (Bruker Daltonics). The crystals were washed briefly with ice-cold 5% (v/v) formic acid. MS experiments were performed on an Ultraflex tandem time-of-flight instrument (Bruker Daltonics, Bremen, Germany). The low mass gate was adjusted to 650 Da. The acquisition range was set to 700–5,000 Da. Approximately 3,000 scans were accumulated for each mass spectrum. The obtained resolution was around 15,000 at m/z = 2,000. Using external calibration, mass accuracy was at least 100 ppm. A standard peptide calibration mix (Applied Biosystems, MDS SCIEX) that contained six peptides covering the acquired mass range was used for calibration. Selected peaks were fragmented by the LIFT method to verify Biotools v2.2 calculations with a mass accuracy of 100 ppm for the precursor and 0.6 Da for the fragments. The isolation width for the precursor was adjusted manually (0.5–1% of the precursor mass), and the following fragmentation was laser-induced using ∼15,000 scans.
All MS spectra were labeled according to monoisotopic peptide masses using Flex analysis v2.2 software (Bruker Daltonics; Sophisticated Numerical Annotation Procedure (SNAP) algorithm). Signal-to-noise ratios larger than three were used as cut-off values. MS spectra of the chymotrypsin and elastase digests were cleared from background signals, i.e. signals also found in MS spectra of blank gel pieces (listed in supplemental Tables S5 and S7). Using Biotools v2.2 software (Bruker Daltonics), theoretically possible fragments of the presumed protein sequence in the mass range 700–5,000 Da were calculated and automatically compared with the actual mass list. Mass tolerance was set to 100 ppm. Specific modifications of the protein sequence by NEM (molecular mass = 125.048 Da), NEM + H2O (molecular mass = 143.058 Da), NFM (molecular mass = 427.069 Da), N-fluoresceinyl-maleimide + H2O (NFMhyd; molecular mass = 445.08 Da), Mox (oxidized methionine; molecular mass = 15.999 Da), and methanesulfenic acid (molecular mass = 63.998 Da (30)) were considered. Trypsin specificity was defined by C-terminal cleavage of lysine and arginine except if proline was the next amino acid, and the maximum number of missed cleavages was two. Two definitions for C-terminal cleavage by chymotrypsin were used: cleavage following Phe, Trp, and Tyr or following Phe, Trp, Tyr, Leu, Met, Ala, Asp, and Glu (no cleavage if proline was the next amino acid; maximum number of missed cleavages was 20). Limited proteolysis by elastase varied too widely to be useful for calculating MS spectra by Biotools v2.2 software. Therefore, these MS spectra were analyzed manually, i.e. searched for characteristic signals of maleimide-modified fragments. For the analysis of the subsequent MS/MS experiments, a survey of all fragments potentially present in the MS spectra was required. Fragment masses of candidate protein sequences were therefore calculated without defining enzyme specificity and compared with the actual masses in the MS spectra. This approach was also used to search the MS data from chymotrypsin and trypsin cleavage to potentially identify further maleimide-modified peptides following unspecific cleavage.
All MS/MS spectra were processed by Flex analysis v2.2 Software (Bruker Daltonics) similar to MS analysis but with further smoothing and baseline correction. Biotools v2.2 software was used to calculate the laser-induced fragmentation of peptide sequences. The predicted fragmentation pattern for a specific peptide corresponding to a signal in the MS spectrum was then compared with the actual MS/MS spectrum.
All fine chemicals were from Sigma, and the enzymes were obtained as follows: modified trypsin, sequence grade (Sigma); modified chymotrypsin, sequence grade (Princeton Separations, Inc.); elastase (Roche Applied Science). Matrix-assisted laser desorption/ionization (MALDI) matrix was α-cyano-4-hydroxycinnamic acid (Bruker Daltonics).
RESULTS
Specific labeling of the thiol group involved in the A/D transition was achieved by a two-step procedure (24). First, all thiol groups of membrane-bound complex I in the active form were blocked by incubating bovine SMP with large molar excess of NEM. Although the blocking reaction was performed at a moderate temperature of 15 °C, some spontaneous deactivation of the enzyme inevitably occurred during the 1-h procedure necessary to wash and centrifuge, resuspend, and finally incubate the activated SMPs with NEM. Thus, as found previously (15), some complex I had already converted back into the deactive form, and the thiol group only accessible in this state of the enzyme became already modified in a fraction of the sample during the blocking reaction. It is important to note, however, that after subsequent conversion of the enzyme into the D-form by incubation at 30 °C, exclusively sulfhydryl groups specifically exposed in this state became labeled with the fluorescent analogue of NEM (NFM). As a control, a portion of the NEM-blocked SMP was kept on ice to stabilize the active state and was also incubated with the fluorescent NEM derivative.
Complex I from labeled and control SMP was extracted from the corresponding bands following blue native-PAGE by electroelution (Fig. 1A). 10% Tricine/SDS-PAGE of the concentrated complex I samples revealed a prominent difference in fluorescence between the active and deactive forms in a single band around 15 kDa (Fig. 1B). This observation was in accordance with previously published results (24). However, in the earlier study, it had not been possible to identify the labeled subunit since complex I contains several candidate subunits with apparent masses of around 15 kDa, namely 18-kDa, B18, B17.2, B16.6, ESSS, B17, SGDH, 15-kDa, ND3, B15, B14.7, B14.5a, B14.5b, and B14 (2).
FIGURE 1.

BN-PAGE of labeled bovine SMP and identification of the labeled band after SDS-PAGE separation of complex I subunits. A, first-dimensional BN-PAGE of bovine SMP was used for separation of complex I. Numbers on the lanes correspond to the n-dodecyl-β-d-maltoside/protein ratio (w/w). B, fluorescence scan of 10% acrylamide, 6 m urea SDS-gel of complex I electroeluted from BN-PAGE. 30 and 15 μg of complex I from both deactive (D) and active (A) SMP samples was loaded on the same gel. The arrow indicates subunit(s) labeled differently in deactive and active complex I.
To identify the labeled subunit, we further resolved the subunits of complex I by dSDS-PAGE (27) using a second dimension 16% Tricine/SDS-polyacrylamide gel (Fig. 2). A single fluorescent spot was observed in the dSDS-gel of the labeled complex I sample from deactivated SMP (Fig. 2D) that was absent in the sample from active SMP (Fig. 2C). Comparison with the silver-stained gels (Fig. 2, B and A) revealed that the fluorescent protein spot was only present in the deactivated sample and was well separated from other proteins. The position of the fluorescent spot (Fig. 2, B and D, spot 1) in dSDS-PAGE above a diagonal of hydrophilic proteins suggested that a highly hydrophobic protein had been labeled (27). The corresponding hydrophobic protein without fluorescent label was expected to have a 320 Da lower mass. Indeed, an appropriate spot was found in the silver-stained gels of the active and deactivated samples (Fig. 2, A and B, spot 2). As discussed before, the presence of the second spot in the deactivated sample was expected due to partial modification of the subunit in question by NEM during the blocking procedure because a fraction of the enzyme spontaneously deactivated, making the sulfhydryl group only accessible in this state amenable to chemical modification.
FIGURE 2.

Separation of the labeled protein by dSDS-PAGE. Second dimension 16% SDS-PAGE was performed using gel strips excised from the lanes of the 10% acrylamide, 6 m urea SDS-gel shown in Fig. 1B. Representative silver-stained gel of labeled complex I from deactive (A) and active (B) SMP and fluorescence scans of the same gels after silver staining (C and D) are shown.
We then went on to identify the protein that had been labeled specifically in the deactive form by mass spectrometry. The MALDI-MS spectrum of a tryptic digest of spot 2 revealed a doublet of signals (supplemental Fig. S1A and supplemental Tables S1 and S2) that could be assigned to the NEM-labeled peptide TSPYECGFDPMGSAR of ND3 (methionine-oxidized and not modified). A similar but extended (due to one missed cleavage at arginine) NFM-labeled peptide TSPYECGFDPMGSARLPFSMK of ND3 was revealed by MS analysis of a tryptic digest of spot 1 (supplemental Fig. S1B). However, mass accuracy of the NFM-labeled peptide (120 ppm) just missed the mass tolerance of external calibration (100 ppm), and the available small amounts of peptide were not sufficient for verification of the result by tandem mass spectrometry (MS/MS). Moreover, further tryptic fragments were unlikely to be detected because only five potential fragments are in the m/z range 700–5,000 and small peptides are not detectable in MALDI, whereas large peptides are hardly extractable from the 16% acrylamide gels used. Therefore, we tried to verify the involvement of subunit ND3 by using other proteases for limited hydrolysis.
We next used chymotrypsin that produces a higher number of fragments with suitable size but cannot be used for common peptide mass fingerprint searches due to the lower specificity of the enzyme. It cleaves preferentially C-terminally at aromatic amino acids Phe, Trp, and Tyr and at a lower rate at amino acids Leu, Met, Ala, Asp, and Glu (32, 33). Analysis of chymotryptic digests of spots 1 and 2 using the Biotools v2.2 software and setting cleavage at Phe, Trp, and Tyr clearly identified ND3 in both cases (Fig. 3). The peaks with highest signal intensities (Fig. 3, boxed) were identified with a mass accuracy better than 20 ppm (see supplemental Tables S3 and S4) as the NEM- and NFMhyd-(N-fluorescein-5-yl maleimide + H2O) labeled peptides ECGFDPMGSARLPF (1667.8 and 1987.8 Da) and SEKTSPYECGFDPMGSARLPF (2460.1 and 2780.1 Da). The NEM- and NFMhyd-labeled peptides differed by the expected mass of 320.02 Da, and they always appeared as peak pairs with a 16-Da mass shift since methionine was detected unmodified and in the oxidized form. Minor peaks of non-labeled fragments ECGFDPMGSARLPF and SEKTSPYECGFDPMGSARLPF, potentially produced by in-source decay, could also be identified (see supplemental Tables S3 and S4). Further peaks and peak pairs could be assigned to NFMhyd-labeled peptides of ND3 that were either cleaved C-terminally to Leu, Met, Ala, Asp, and Glu (ECGFDPM and SEKTSPYECGFDPM) or cleaved non-specifically on one side of the peptide (Y)SEKTSPYECGFDPMG(S) as summarized in supplemental Table S3. MS/MS analyses of the NEM- and NFMhyd-labeled peptides ECGFDPMGSARLPF and SEKTSPYECGFDPMGSARLPF confirmed the MS results (supplemental Fig. S2).
FIGURE 3.

MALDI MS of chymotryptic digests identified NEM- and NFMhyd-labeled fragments of ND3. A, MS of protein spot 2 (Fig. 2) from NEM-labeled complex I. [a.u.], counts in absolute units. B, MS of fluorescent-labeled protein from spot 1 (Fig. 2). Signals that could be assigned to labeled fragments ECGFDPMGSARLPF and SEKTSPYECGFDPMGSARLPF of the ND3 subunit of complex I are boxed. Signals that could be assigned to chymotryptic ND3 fragments in general are marked with a strong dot on the tip. Non-assignable signals are marked with a smaller dot (see also supplemental Tables S3 and S4). Signals corresponding to background are not marked (see also “Experimental Procedures” and supplemental Table S5).
For further verification of subunit ND3 as the fluorescent-labeled protein, spot 1 was digested by elastase cleaving unspecifically at the C terminus of all small hydrophobic amino acids (Ala, Val, Leu, etc.). A signal pair with a 16-Da mass shift was identified in the MS spectrum as NFMhyd-labeled fragment SPYECGFDPMGS (Fig. 4A and supplemental Table S6). Mass accuracy was 15 ppm. The corresponding MS/MS experiment of this fragment revealed a short b-ion series (b5-b8) that could be assigned to the sequence tag GFD (Fig. 4B, inset) and two internal fragments still carrying the NFMhyd label (marked by arrows). The preferential cleavage of the D-P bond is characterized by the high intensity of the b8-ion signal. We conclude that MALDI-MS and MS/MS data unambiguously identified spots 1 and 2 as ND3 subunit labeled specifically at cysteine in position 39 by NFM and NEM, respectively.
FIGURE 4.

MS and MS/MS analysis of the elastase digest of the fluorescent-labeled ND3 subunit (Fig. 2, spot 1). A, MS spectrum. The signal pair (with unmodified and oxidized methionine) for the NFMhyd-labeled fragment SPYECGFDPMGS is boxed. [a.u.], counts in absolute units. B, MS/MS spectrum verifying the assignment of signals in Fig. 5A. For details, see supplemental Fig. S2.(y ions marked bold; nomenclature for fragment ions according to Roepstorff and Fohlman (Ref. (31)).
DISCUSSION
We have identified the mitochondrially encoded ND3 subunit as the protein that was specifically labeled in the deactive form of complex I by a fluorescent NEM derivative. The modified cysteine was found in position 39 of the ND3 subunit of bovine complex I. In fact, a previous study had suggested that the modified residue should reside close to the lipid bilayer (23). The sequence of the ND3 (NuoA/NQO7) subunit is highly conserved in all organisms, but the only highly conserved cysteine is replaced by serine in Escherichia coli and T. thermophilus. Remarkably, the ND3 subunits of complex I from earthworm (Lumbricus terrestris), crickets (Acheta domesticus), and lobster (Hommerus hommerus) do contain this cysteine but are known to show no A/D transitions. It follows that the presence of this residue is not indicative of the capability of complex I to form a deactive form and is not required for the active/deactive transition. However, the fact that it is only labeled in the deactive form clearly indicates that the transition between the two functional states of complex I is associated with conformational changes of or around the ND3 subunit.
The specifically labeled cysteine-39 resides in a hydrophilic loop connecting the first two of three predicted transmembrane helices of the ND3 subunit (Fig. 5). Notably, two acidic residues (Glu-38 and Asp-42, Fig. 5) are found nearby, consistent with the observation that the specifically labeled cysteine is unusually alkaline with its apparent pKA shifted to a value of 10.2 (24).
FIGURE 5.

Proposed topology of the ND3 subunit of complex I. The boundaries of the transmembrane helices were predicted using the TMPred software package. Residues discussed under “Discussion” are in bold and labeled with their sequence number. The arrow points to Cys-39 (black square), the residue selectively labeled in the D-form of complex I. Positions where pathogenic mutations have been found are highlighted by black circles. Acidic residues in the vicinity of Cys-39 are printed in boldface.
According to a recent study, the NEM-modified cysteine residue of deactive complex I was found to be accessible from the matrix side of the membrane (23). In agreement with this finding, the topology of subunit ND3 as predicted by TMPred software based on the “positive inside rule” also places the N terminus of the protein into the intermembrane space so that cysteine-39 in the loop between the first and the second transmembrane helices would in fact reside on the matrix side (Fig. 5). This topology of the ND3 subunit was recently confirmed experimentally for the homologue NuoA from E. coli.6 It should be noted, however, that the opposite orientation was proposed for the homologous Nqo7 subunit from P. denitrificans based on heterologously expressed glutathione S-transferase fusions (34). Although this has to be kept in mind, it seems likely that the cysteine-containing loop resides on the matrix side of the membrane. This would place it at the interface to the peripheral arm of complex I as the ND3 subunit could be cross-linked to the PSST subunit (35). Subunit PSST is the central subunit binding the terminal iron-sulfur cluster N2 that acts as the immediate electron donor to ubiquinone (11). Recent structural studies on the orientation of the peripheral arm relative to the membrane arm (36) suggest that the cysteine-39 loop of the ND3 subunit is part of the structure connecting the peripheral and the membrane arm of complex I.
Chemical modification of cysteine-39 of the ND3 subunit prevents transition into the active form and thereby effectively inhibits complex I activity. This suggests that the region around the matrix loop of the ND3 subunit is critically involved in functionally relevant conformational changes occurring during the catalytic turnover of complex I. This notion gains further support from the fact that pathogenic mutations were reported in three positions of the loop upstream and downstream of Cys-39 (Fig. 5). Mutations S34P (Thr-34 in Bos taurus), S45P (Gly-45 in B. taurus), and A47T cause Leigh syndrome with or without dystonia or progressive mitochondrial disease (37–39). Also, basal ganglia lesions associated with complex I deficiency were recently found in patients with heteroplasmic A47T mutations (40). Irreversible inhibitory S-nitrosation of the D-form has been demonstrated recently in vitro. This mechanism may be relevant in vivo under pathological conditions when the turnover of the enzyme is limited during hypoxia or when the nitric oxide:oxygen ratio increases (41) and may suggest a regulatory role of the ND3 subunit.
As a central subunit, the ND3 protein is present in complex I from all sources, but so far the A/D transition has only been observed with eukaryotic complex I that contains a large number of accessory subunits not present in the bacterial enzyme (20). Thus, although it seems clear that conformational changes around the cysteine-39 loop of the ND3 subunit play a critical role in the A/D transition, one or several of these accessory subunits may well be involved in exerting or controlling these conformational changes of complex I.
Supplementary Material
Acknowledgments
We thank Volker Zickermann, Stefan Kerscher, Stefan Dröse, and Annie Higgs for carefully reading the manuscript and helpful discussion.
This work was supported, in whole or in part, by National Institutes of Health Grant R03 TW07825 funded by the Fogarty International Center. This work was also supported by grants from Sonderforschungsbereich SFB628 of the Deutsche Forschungsgemeinschaft and the Cluster of Excellence EXC115 to U. B., M. K., and H. S. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains supplemental figures and supplemental tables.
Footnotes
The abbreviations used are: SMP, submitochondrial particles; A/D, active/deactive transitions; BN-PAGE, blue native-PAGE; MALDI, matrix-assisted laser desorption/ionization; MS, single stage mass spectrometry; MS/MS, tandem mass spectrometry; NEM, N-ethylmaleimide; NFM, N-fluorescein-5-yl maleimide; NFMhyd, N-fluorescein-5-yl maleimide + H2O; Tricine, N-[2-hydroxy-1,1-bis(hydroxymethyl)ethyl] glycine; dSDS-PAGE, doubled SDS-PAGE.
Cecilia Hägerhall, personal communication.
References
- 1.Sazanov, L. A., and Hinchliffe, P. (2006) Science 311 1430-1436 [DOI] [PubMed] [Google Scholar]
- 2.Carroll, J., Fearnley, I. M., Skehel, J. M., Shannon, R. J., Hirst, J., and Walker, J. E. (2006) J. Biol. Chem. 281 32724-32727 [DOI] [PubMed] [Google Scholar]
- 3.Ohnishi, T. (1998) Biochim. Biophys. Acta 1364 186-206 [DOI] [PubMed] [Google Scholar]
- 4.Yano, T., Magnitsky, S., Sled, V. D., Ohnishi, T., and Yagi, T. (1999) J. Biol. Chem. 274 28598-28605 [DOI] [PubMed] [Google Scholar]
- 5.Magnitsky, S., Toulokhonova, L., Yano, T., Sled, V. D., Hägerhall, C., Grivennikova, V. G., Burbaev, D. S., Vinogradov, A. D., and Ohnishi, T. (2002) J. Bioenerg. Biomembr. 34 193-208 [DOI] [PubMed] [Google Scholar]
- 6.Ohnishi, T., Johnson, J. E., Jr., Yano, T., LoBrutto, R., and Widger, W. R. (2005) FEBS Lett. 579 500-506 [DOI] [PubMed] [Google Scholar]
- 7.Di Virgilio, F., and Azzone, G. F. (1982) J. Biol. Chem. 257 4106-4113 [PubMed] [Google Scholar]
- 8.Wikström, M. K. F. (1984) FEBS Lett. 169 300-304 [DOI] [PubMed] [Google Scholar]
- 9.Galkin, A. S., Grivennikova, V. G., and Vinogradov, A. D. (1999) FEBS Lett. 451 157-161 [DOI] [PubMed] [Google Scholar]
- 10.Brandt, U. (1997) Biochim. Biophys. Acta 1318 79-91 [DOI] [PubMed] [Google Scholar]
- 11.Brandt, U. (2006) Annu. Rev. Biochem. 75 69-92 [DOI] [PubMed] [Google Scholar]
- 12.Mitchell, P. (1961) Nature 191 144-148 [DOI] [PubMed] [Google Scholar]
- 13.Minakami, S., Schindler, F. J., and Estabrook, R. W. (1964) J. Biol. Chem. 239 2042-2048 [PubMed] [Google Scholar]
- 14.Minakami, S., Schindler, F. J., and Estabrook, R. W. (1964) J. Biol. Chem. 239 2049-2054 [PubMed] [Google Scholar]
- 15.Kotlyar, A. B., and Vinogradov, A. D. (1990) Biochim. Biophys. Acta 1019 151-158 [DOI] [PubMed] [Google Scholar]
- 16.Maklashina, E. O., Sled, V. D., and Vinogradov, A. D. (1994) Biokhimiya 59 946-957 [PubMed] [Google Scholar]
- 17.Vinogradov, A. D. (1998) Biochim. Biophys. Acta 1364 169-185 [DOI] [PubMed] [Google Scholar]
- 18.Grivennikova, V. G., Kapustin, A. N., and Vinogradov, A. D. (2001) J. Biol. Chem. 276 9038-9044 [DOI] [PubMed] [Google Scholar]
- 19.Maklashina, E., Sher, Y., Zhou, H. Z., Gray, M. O., Karliner, J. S., and Cecchini, G. (2002) Biochim. Biophys. Acta 1556 6-12 [DOI] [PubMed] [Google Scholar]
- 20.Maklashina, E., Kotlyar, A. B., and Cecchini, G. (2003) Biochim. Biophys. Acta 1606 95-103 [DOI] [PubMed] [Google Scholar]
- 21.Estabrook, R. W., Tyler, D. D., Gonze, J., and Peterson, J. A (1968) in Flavines and Flavoproteins, pp. 268-279, University of Tokyo Press, Tokyo, Japan
- 22.Kotlyar, A. B., Sled, V. D., and Vinogradov, A. D. (1992) Biochim. Biophys. Acta 1098 144-150 [DOI] [PubMed] [Google Scholar]
- 23.Gostimskaya, I. S., Cecchini, G., and Vinogradov, A. D. (2006) Biochim. Biophys. Acta 1757 1155-1161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gavrikova, E. V., and Vinogradov, A. D. (1999) FEBS Lett. 455 36-40 [DOI] [PubMed] [Google Scholar]
- 25.Burbaev, D. S., Moroz, I. A., Kotlyar, A. B., Sled, V. D., and Vinogradov, A. D. (1989) FEBS Lett. 254 47-51 [DOI] [PubMed] [Google Scholar]
- 26.Wittig, I., Braun, H. P., and Schägger, H. (2006) Nat. Protoc. 1 418-428 [DOI] [PubMed] [Google Scholar]
- 27.Rais, I., Karas, M., and Schägger, H. (2004) Proteomics 4 2567-2571 [DOI] [PubMed] [Google Scholar]
- 28.Meyer, B., Wittig, I., Trifilieff, E., Karas, M., and Schägger, H. (2007) Mol. Cell. Proteomics 6 1690-1699 [DOI] [PubMed] [Google Scholar]
- 29.Shevchenko, A., Wilm, M., Vorm, O., and Mann, M. (1996) Anal. Chem. 68 850-858 [DOI] [PubMed] [Google Scholar]
- 30.Schmidt, F., Krah, A., Schmid, M., Jungblut, P. R., and Thiede, B. (2006) Rapid Commun. Mass Spectrom. 20 933-936 [DOI] [PubMed] [Google Scholar]
- 31.Roepstorff, P., and Fohlman, J. (1984) Biol. Mass Spectrom. 11 601. [DOI] [PubMed] [Google Scholar]
- 32.Blow, D. M. (1971) in The Enzymes (Boyer, P. D., ed), Vol. 3, pp. 185-212, Academic Press, New York [Google Scholar]
- 33.Kamp, R. M. (1986) in Advanced Methods in Protein Microsequence Analysis (Wittmann-Liebold, B., Salnikow, J., and Erdmann, V. A., eds), pp. 8-20, Springer-Verlag, Berlin
- 34.Di Bernardo, S., Yano, T., and Yagi, T. (2000) Biochem. 39 9411-9418 [DOI] [PubMed] [Google Scholar]
- 35.Kao, M. C., Matsuno-Yagi, A., and Yagi, T. (2004) Biochemistry 43 3750-3755 [DOI] [PubMed] [Google Scholar]
- 36.Clason, T., Zickermann, V., Ruiz, T., Brandt, U., and Radermacher, M. (2007) J. Struct. Biol. 159 433-442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sarzi, E., Brown, M. D., Lebon, S., Chretien, D., Munnich, A., Rotig, A., and Procaccio, V. (2007) Am. J. Med. Genet. A 143 33-41 [DOI] [PubMed] [Google Scholar]
- 38.Taylor, R. W., Singh-Kler, R., Hayes, C. M., Smith, P. E., and Turnbull, D. M. (2001) Ann. Neurol. 50 104-107 [DOI] [PubMed] [Google Scholar]
- 39.McFarland, R., Kirby, D. M., Fowler, K. J., Ohtake, A., Ryan, M. T., Amor, D. J., Fletcher, J. M., Dixon, J. W., Collins, F. A., Turnbull, D. M., Taylor, R. W., and Thorburn, D. R. (2004) Ann. Neurol. 55 58-64 [DOI] [PubMed] [Google Scholar]
- 40.Chae, J. H., Lee, J. S., Kim, K. J., Hwang, Y. S., Bonilla, E., Tanji, K., and Hirano, M. (2007) Pediatr. Res. 61 622-624 [DOI] [PubMed] [Google Scholar]
- 41.Galkin, A., and Moncada, S. (2007) J. Biol. Chem. 282 37748-37753 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.