

Abstract
Nucleotide-dependent conformational changes of the constitutively dimeric molecular chaperone Hsp90 are integral to its molecular mechanism. Recent full-length crystal structures (Protein Data Bank codes 2IOQ, 2CG9, AND 2IOP) of Hsp90 homologs reveal large scale quaternary domain rearrangements upon the addition of nucleotides. Although previous work has shown the importance of C-terminal domain dimerization for efficient ATP hydrolysis, which should imply cooperativity, other studies suggest that the two ATPases function independently. Using the crystal structures as a guide, we examined the role of intra- and intermonomer interactions in stabilizing the ATPase activity of a single active site within an intact dimer. This was accomplished by creating heterodimers that allow us to differentially mutate each monomer, probing the context in which particular residues are important for ATP hydrolysis. Although the ATPase activity of each monomer can function independently, we found that the activity of one monomer could be inhibited by the mutation of hydrophobic residues on the trans N-terminal domain (opposite monomer). Furthermore, these trans interactions are synergistically mediated by a loop on the cis middle domain. This loop contains hydrophobic residues as well as a critical arginine that provides a direct linkage to the γ-phosphate of bound ATP. Small angle x-ray scattering demonstrates that deleterious mutations block domain closure in the presence of AMPPNP (5′-adenylyl-β,γ-imidodiphosphate), providing a direct linkage between structural changes and functional consequences. Together, these data indicate that both the cis monomer and the trans monomer and the intradomain and interdomain interactions cooperatively stabilize the active conformation of each active site and help explain the importance of dimer formation.
Hsp90 plays a central role in the activation and folding of a wide variety of critical proteins including kinases, steroid hormone receptors, nitric oxide synthase, and telomerase as well as being involved in important processes such as mitochondrial import (1-6). Hsp90 is a constitutive dimer where each ∼90-kDa monomer consists of three domains: the N-terminal domain (NTD),4 which confers nucleotide binding, the middle domain (MD), which is necessary for ATP hydrolysis, and the C-terminal domain (CTD), which is required for dimerization (7). Although the exact mechanism of how Hsp90 interacts with its substrate proteins (client proteins) remains unclear, the structure of the full-length apo protein from Escherichia coli reveals that each domain contributes hydrophobic elements into a large central cleft, and these elements are likely to be involved in client recognition (8). Dramatic domain rearrangements occur in response to ATP (9) and ADP (8) binding and are thought to drive client protein remodeling and release (Fig. 1). Inhibition of the ATPase of Hsp90 by either mutation or small molecule inhibitors results in the degradation of client proteins in vivo, demonstrating the central importance of the ATPase to the function of the chaperone (10-12).
FIGURE 1.

Nucleotide-dependent conformational changes of Hsp90. Previous crystallographic studies show that the conformational equilibrium of Hsp90 is biased by nucleotide binding and results in large changes in quarternary structure. However, the structural requirements for activating the ATPase activity remain elusive. The apo structure was taken from the full-length structure of the bacterial homolog, htpG (PDB: 2IOQ). The ATP-bound Hsp90 was taken from the Hsp82/AMPPNP/Sba1 full-length structure (PDB: 2CG9); Sba1 was removed to emphasize the conformational changes in Hsp90 alone. The ADP-bound structure is modeled from the ADP-bound htpG full-length structure (PDB: 2IOP) (8). The domain architecture highlighting the NTD, MD, and CTD is outlined in black on one monomer to emphasize the domain rearrangements.
Hsp90 is a relatively slow ATPase with activities ranging from 0.1 to 1.2 min-1 across prokaryotic and eukaryotic homologs (13, 14). The NTD alone provides all of the structural requirements for nucleotide binding, including a catalytic glutamate, which activates the attacking water in the hydrolysis reaction (15). However, when isolated, the NTD has very low activity (14), suggesting that other parts of the molecule are required for hydrolysis, similar to Hsp90 superfamily members MutL and GyrB (16). Some of these determinants come from the middle domain (17), and others arise only in the context of the full-length structure. Removal of the constitutive C-terminal dimerization domain reduces the ATPase activity 6-10-fold depending on the homolog (14, 18). Replacing this domain with a disulfide bridge between the two monomers rescues activity, clearly demonstrating the requirement of dimerization for efficient ATP hydrolysis (19). Additionally, structural and biochemical data indicate that ATP binding leads to the transient association of the NTDs (13). Given the multiple domain interactions and dimer requirements, it has been quite surprising that kinetic data provide no evidence for cooperativity in either nucleotide binding or hydrolysis (18).
The structural determinants beyond the NTD that are required for ATPase activation are largely unknown. Mutation of several MD residues (Arg-376, Gln-380 in yeast Hsc82) has been shown to affect ATPase activity (17). The recently solved full-length structure of the yeast Hsp90 (Hsp82) in complex with Sba1 and AMPPNP (9) most likely represents a prehydrolysis state (as Sba1 binding inhibits ATP hydrolysis (20)); however, it has proven a useful guide for understanding these mutations. For example, it shows that the Arg-380 (Arg-376 in Hsc82) side chain is within 3 Å of the γ-phosphate of the AMPPNP bound to the same monomer, suggesting its role in the completion of the active site.
Our work is focused on discovering the key intra- and interdomain as well as the intra- and intermonomer interactions required to create a complete and functional ATPase active site (Fig. 1). Toward this end, we have developed a heterodimeric ATPase assay that allows us both to assess the importance of a particular residue to the Hsp90 ATP cycle and, for the first time, to determine whether its role is via an intra- or intermonomer interaction. We show that the MD loop containing Arg-376 interacts synergistically with a region of the NTD on the opposite monomer as well as the active site on the same NTD. Additionally, small angle x-ray scattering (SAXS) of the mutants is used to provide a direct linkage between functional and structural consequences of mutation. Together, these data indicate the necessity of cooperative intra- and interdomain and intra- and intermonomer interactions and provides a clear structural rationale for the functional role of dimerization. It further suggests that there is a distinct NTD-MD conformation required for catalysis that must be somewhat different from the crystal structure.
EXPERIMENTAL PROCEDURES
Materials—Geldanamycin was purchased from Sigma.
Hsc82 Constructs—Yeast hsc82 was cloned from a yeast cosmid containing hsc82 (ATCC). PCR was used to isolate the gene that was subsequently cloned into the Invitrogen vector pET151/D-TOPO, which includes a tobacco etch virus-cleavable N-terminal His tag. Site-directed mutagenesis was performed by PCR using 20-nucleotide primers encoding for the mutated amino acid. Parental DNA was then digested using Dpn1 after the completed reaction, and the resulting plasmid was transformed into DH5α (Stratagene) cells. All mutations were confirmed by sequencing of the appropriate regions.
Hsc82 Purification—Protein was purified from induced E. coli cultures using nickel-nitrilotriacetic acid affinity resin (Qiagen) followed by anion exchange and size exclusion chromatography on a Superdex S200 column (GE Healthcare). Protein was concentrated in 10 mm Tris, pH 7.5, 100 mm NaCl using Ultrafree Biomax concentrators (Millipore) to a final concentration of 1-5 mg/ml based on UV280 absorption. Protein was flash-frozen in liquid nitrogen and stored at -80 °C until use. If needed, His tags were removed by cleavage with tobacco etch virus protease, and uncleaved protein was removed via a nickel-nitrilotriacetic acid column.
Mass Spectrometry—Hsc82 with and without its N-terminal His tag were diluted to 1 mg/ml from their stock concentrations. Based on the tagged:untagged ratios desired, each protein was mixed and equilibrated at 30 °C for 30 min after which the proteins were set on ice. Sinapinic acid at 10 mg/ml in 60% acetonitrile, 0.3% trifluoroacetic acid was used as the matrix. Protein and matrix were mixed in a 1:1, 1:2, or 1:3 ratio and spotted onto a MALDI plate. A Voyager-DE STR (Applied Biosystems) mass spectrometer was used to analyze the mass ratios and to determine the relative amount of heterodimer formed.
ATPase Activity—This assay was adapted from Felts et al. (21). 2 μm protein was used in each assay with 1 mm ATP and 0.8 pm [γ-32P]ATP (6000 Ci/mmol) in solution. For the assays using geldanamycin as a control, a final concentration of 200 μm was used. 20-min time points were taken over the course of an hour with the samples shaking and incubating at 37 °C. Separation of Pi from ATP was performed using the thin layer chromatography method as described (21). Visualization of the radiolabeled spots was performed on a Typhoon Imager (GE Healthcare), and quantification was performed using the program ImageQuant (GE Healthcare). The amount of ATP hydrolyzed at each time point was calculated by taking the ratio of Pi to ATP in solution. This ratio was then multiplied by the total amount of ATP added to the reaction and normalized by the total protein in solution. A linear fit of the time points gave the rate for each reaction.
Heterodimer Assay—The total protein concentration in each reaction was kept at 2 μm. Depending on the ratio desired, homodimers of the appropriate constructs were mixed together and incubated at 30 °C for 30 min and put on ice. The ATPase activity was then measured using the assay mentioned above.
Predicted inter- and intramonomer rates were calculated by accounting for the activity contributions of each population in solution. To calculate the activity contributions of the two homodimers present in solution, we used a least squares fitting of the heterodimer activities measured and extrapolated the individual contributions (MATLAB), taking into account the fraction of each population in solution. Predicted activities were then calculated using the following equations given either an intermonomer or an intramonomer interaction.
Intermonomer (trans) activity =
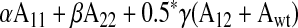 |
(Eq.1) |
Intramonomer (cis) activity =
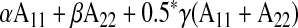 |
(Eq.2) |
where α, β, and γ represent the fraction homodimer 1, homodimer 2, and heterodimer in solution, and A11, A22, A12, and Awt represent the intrinsic ATPase activities of the homodimer 1, homodimer 2, the heterodimer, and wild-type proteins, respectively. α, β, and γ were calculated using the binomial theorem to simulate an equilibrium of mixing between two homodimers that have the same dimerization affinity. In the case of the intermonomer interaction, there are two contributions to the heterodimer activity: the activity of the double mutant (A12) and the WT ATPase activity (Awt). With respect to an intramonomer interaction, the two activity contributions arise from the activities of each of the original single mutations, A11 and A22. The heterodimer activity in each equation is multiplied by 0.5 since A11 and A22 are dimer activities.
The predicted activity curves were calculated using MATLAB and graphed in Kaleidagraph. We determined whether our residue of interest was involved in an inter- or intramonomer interaction by comparing the profiles of our experimentally measured heterodimers over a variety of concentrations with our predicted activities.
SAXS Data Collection and Data Analysis—Data reported here were collected at the Advanced Light Source (ALS) beamline 12.3.1 and the Stanford Synchrotron Radiation Laboratory (SSRL) beamline 4.2. To minimize aggregation, samples were spun in a tabletop microcentrifuge for 5 min before data collection. SAXS data were collected at 25 °C at 2-5 mg/ml. At the ALS, the samples were exposed for 6 and 60 s at a detector distance of 1.6 m. At the SSRL, samples were exposed for ten 30-s exposures at a detector distance of 2.5 m. Scattering data were recorded on a Mar165 CCD detector. The detector channels were converted to Q = 4πsinθ/λ, where 2O is the scattering angle and λ is the wavelength, using a silver behenate sample as a calibration standard. The data were circularly averaged over the detector and normalized by the incident beam intensity. The raw scattering data were scaled, and the buffers were subtracted. Individual scattering curves were then merged to provide the final averaged scattering curve. The interatomic distance distribution functions (P(r)) were then calculated using the program GNOM (22). Dmax was determined by constraining rmin to equal zero and then varying rmax between 150 and 250 Å. Rmax was then chosen so that the P(r) curve smoothly approached zero at the upper limit. Small changes in rmax (±10 Å) did not affect the overall shape of the P(r) curve. Radii of gyration were calculated from the P(r). Comparable results were obtained from the scattering curves using the Guinier approximation as implemented in the program PRIMUS (23); however, the calculated uncertainties were larger due to limitations in the low angle data.
RESULTS
Characterization of Heterodimers in Solution—To dissect the intra-versus intermonomer roles of specific residues with respect to the overall ATPase activity of Hsc82, it was necessary to create asymmetric dimers harboring different mutations on each monomer (Fig. 2). Because the KD for subunit dimerization is in the nm range (7), heterodimers can be formed simply by mixing homodimers containing different point mutations (14). By placing a mutation in one monomer that abolishes ATP hydrolysis without blocking nucleotide binding (E33A) (15), the role of a test mutation in the other monomer can be assayed by the effect of increasing proportions of the test mutation on the ATPase rate. First, test mutations are chosen that will alter ATPase rates within homodimeric Hsc82. If the test mutation functions by altering interactions with the active site on its own subunit (an intramonomer or “cis” interaction), then the activity of its own active site will be compromised by the mutation. If, however, the test mutation alters interactions with the active site on the opposite subunit (an intermonomer or “trans” interaction), which is already compromised by the E33A mutation, then it will not have a deleterious effect, and its own active site should be fully functional (Fig. 2). We can calculate the predicted hydrolysis rate of any mixture of the two homodimer concentrations using standard equilibrium measurements and homodimer activities (see “Experimental Procedures”). The hydrolysis rate of differentially mixed dimers can then be measured and compared with our predicted rates, allowing us to determine the role of the test residue independent of the level of its residual activity.
FIGURE 2.

Schematic of the heterodimer assay. Differentially mutated inactive homodimers are mixed together to form heterodimers. If the two residues mutated interact in an intermonomer interaction, a WT active site would form, and ATP hydrolysis would occur. If the residues interact in an intramonomer interaction, no activity would be measured.
Although it has previously been shown that Hsc82 heterodimers could form (14), in our hands, proof of heterodimer formation upon mixing in vitro was shown both physically and enzymatically. Using differentially tagged constructs of Hsc82 as indicators (supplemental Fig. 1), tagged and untagged Hsc82 were mixed in a 3:1, 1:1, or 1:3 ratio (untagged:tagged) and incubated at 30 °C for an hour. MALDI-time-of-flight was then used to probe the relative abundance of each dimer species in each of the reactions. As expected, three peaks are seen in the spectrum of the 1:1 ratio reaction corresponding to each of the pure homodimers and a peak between them representing the formation of heterodimer. When the untagged homodimer is in 3-fold excess, only two peaks are seen, corresponding to the excess untagged homodimer and the heterodimer. The converse result was obtained when the tagged homodimer was in excess. Although equilibrium calculations predict a small amount of the minor homodimer to be present in solution (12.5%), this species could not be observed in the experiment due to limited sensitivity when working with intact proteins of this size in a conventional MALDI instrument.
To confirm that the heterodimers still possessed the ability to hydrolyze ATP, a fixed concentration of wild-type Hsc82 was incubated with increasing amounts of the catalytically dead E33A mutant (Glu-33 is required to coordinate the hydrolytic water). Activity levels of the two homodimers are shown in Fig. 3B. ATP hydrolysis rates were unaffected by the amount of added inactive mutant (supplemental Fig. 2). When combined with the mass spectrometric data, these results show that under our conditions, heterodimers can be formed in solution and are fully able to hydrolyze ATP, although one of the monomers is catalytically dead. This confirms the independent activation of the individual NTD ATPases. In contrast, the removal of one of the NTDs reduces, but does not abolish, ATP activity (14) in the context of heterodimers, demonstrating the structural requirement of having both NTDs present in the dimer to fully activate each ATPase.
FIGURE 3.

Hydrolysis rates for hydrophobic mutants and the contribution of the middle domain arginine. A, five hydrophobic residues from both monomers come together and interact only in the ATP-bound closed state. One monomer is illustrated in brown, whereas the other monomer is shown in green. Two of the five residues lie on the same loop as Arg-376, which has been previously implicated in stabilizing the γ-phosphate of ATP. B, hydrolysis rates of each mutant used in this study. All rates have been normalized to the WT hydrolysis rate. C, heterodimer assay with R376A and E33A monomers. Ratios between the two homodimers were varied, whereas the total protein concentration was kept constant. Predicted inter- (black) and intramonomer (gray) interaction activities are shown with solid lines, and the closed circles represent the experimental data.
Influence of the Middle Domain on the ATPase Activity of Hsc82—Investigation of the apo and AMPPNP structures shows a group of residues (Thr-22, Val-23, Tyr-24, Leu-372, Leu-374, and Arg-376) that come together from distant parts of the apo structure to form an interacting cluster in the ATP state (Fig. 3A). Arg-376 (Arg-380 in Hsp82) was previously shown to be important for hydrolysis, making this entire cluster an attractive starting point for dissecting inter- and intramonomer interactions. Unlike the E33A mutation, the homomeric R376A mutant significantly reduces, but does not abolish, ATPase activity when compared with the E33A mutation previously described (Fig. 3B).
As a first test of our assay, we wanted to biochemically confirm that Arg-376 affects the ATPase of its own NTD, as suggested by the Hsp82/ATP/Sba1 structure. To do this, the mutant, R376A, was tested in our heterodimer assay in conjunction with the catalytically dead E33A mutation. Separately expressed and purified R376A and E33A were mixed in defined ratios while keeping the total protein concentration constant; after a period of incubation, their ATP hydrolysis rates were measured (Fig. 3C). Using the behaviors of the individual mutants as homodimers (Fig. 3B), the expected activity as a function of the mixture percentages can be readily predicted (Fig. 3C, trans = black, cis = gray; see also “Experimental Procedures”). From this, it is clear that Arg-376 has a required interaction with the NTD active site on the same monomer. Interestingly, we see no trans-like characteristics in these data, demonstrating that there is no catalytic cooperativity between the two NTDs, analogous to what was shown with human Hsp90 (18).
Contribution of Hydrophobic Interactions to Complete Active Site Formation—As discussed above, the Hsp82/ATP/Sba1 structure revealed a cluster of interacting hydrophobic and polar amino acids derived from the NTD and MD domains that come together in the ATP state to form both intermonomer and intramonomer interactions (Fig. 3A). The residues involved in this interaction are Thr-22, Val-23, and Tyr-24 from the NTD of one monomer and Leu-372 and Leu-374 from the MD of the other. Since the two leucines are located on the same loop and pack against Arg-376 (above) and make cross-monomer interactions in the ATP but not apo states (data not shown), this entire cluster seems appropriate for investigation.
To test whether these residues are involved in the hydrolysis of ATP, we systematically mutated each residue to alanine and measured the ATPase activity (Fig. 3B). All constructs were tested in the presence of the Hsp90 family specific inhibitor, geldanamycin, to confirm that the activity measured in these experiments was from Hsc82 and not from any contaminating ATPase activity (data not shown). Interestingly, as a homodimer, T22A showed no significant loss in activity when measured at 37 °C (data not shown); however, it has been previously shown that T22I is a temperature-sensitive mutation resulting in increased ATPase activity at elevated temperatures (24). To probe the importance of enhancing hydrophobicity at this site, we constructed the T22F mutation and saw a 3-fold increase in activity, proving that the hydrophobic nature of this interaction is important for its ATPase activity. Interestingly, when this mutation is modeled in the Hsp82/Sba1/AMPPNP crystal structure (PDB: 2CG9) (data not shown), steric clashes are observed, suggesting that the actual hydrolysis-competent structure must be somewhat different from the solved crystal structure.
As homodimers, V23A and Y24A both showed a significant loss of activity, whereas the L372A and L374A mutations had little effect (data not shown). Given the experience with Thr-22, we also explored the impact of polar mutations, changing Leu to either Asn or Asp. Both mutations had very pronounced effects, indicating the importance of hydrophobic residues in this network.
To determine the impact of these residues with respect to the two monomers and their ATP active sites, we again measured each residue in our heterodimer assay in conjunction with the catalytically dead E33A mutation. Both V23A and Y24A show a clear increase in ATP hydrolysis, strongly supporting a trans interaction (Fig. 4, A and B). We also note that the Y24A heterodimer data appear to reside between the cis and trans predictions, suggesting that it plays a role in the hydrolysis of both monomers. T22F showed a small but reproducible curvature in the mixture plots (Fig. 4C), demonstrating that it too participates in a trans interaction. Together, these results implicate this NTD region as providing interactions required for hydrolysis of the opposite monomer to proceed. As shown in Fig. 4, D and E, both leucine mutations interact in cis to the E33A mutation, suggesting that the role of these residues is to bridge the network between the N-terminal residues on the opposite monomer and the arginine that has been shown to interact with the γ-phosphate of ATP.
FIGURE 4.

Heterodimer assay on the hydrophobic residues. A-E, heterodimeric analysis of V23A (A), Y24A (B), T22F (C), L372D (D), and L374N (E). The T22F heterodimer assay has very small error bars that are occluded by the point size. Predicted inter- (black) and intramonomer (gray) interaction activities are shown with lines, and the experimental data are represented by closed circles. All of the residues that originate from the MD result in a cis-like interaction (L372D and L374N), whereas most of the residues originating from the NTD (V23A, Y24A, and T22F) result in a trans-like interaction with Tyr-24, showing evidence of being involved in both cis and trans.
The Hydrophobic Interaction Network Affects the Conformational Equilibrium of Hsc82—Given the evidence that the hydrophobic network affected the ATPase activity of Hsp90 and was shown to act in a cross-monomer interaction, we wanted to test whether or not these mutations had an effect on the conformational equilibrium of Hsc82 by preventing the stabilization of N-terminal dimerization when ATP is present. To directly assess the consequences of mutation on the solution structure of Hsc82, we used SAXS to probe the interatomic distance distribution in the presence and absence of the non-hydrolyzable ATP analog, AMPPNP.
Upon the addition of AMPPNP at saturating concentrations, we see a reproducible shift in the distance distribution to smaller distances when compared with the apo state, indicating that a more closed and compact conformation is being stabilized, analogous to what is seen in the AMPPNP-bound crystal structure (PDB: 2CG9) (Fig. 5A and Table 1). In the presence of AMPPNP, the observed solution structure is not quite as compact as the crystal structure (radii of gyration 47 versus 41 Å, respectively); however, whether this represents an equilibrium between open and closed states or an altered conformation will require further analysis. Representative mutations affecting the network in both cis (L374N) and trans (V23A) were then tested by SAXS. For both mutations, the radii of gyration indicated a slight expansion in the apo state. However, in sharp contrast to the wild-type enzyme, the addition of AMPPNP caused little or no change in the interatomic distances, P(r), and radii of gyration when compared with the apo state (Fig. 5, B and C, and Table 1). These data suggest that the network of interactions probed here is responsible for maintaining the conformational equilibrium of Hsc82 by stabilizing the closed state when nucleotide is present and perhaps altering the open-closed equilibrium in the absence of nucleotide. It also provides a mechanistic understanding for the loss of activity that is observed when these residues are mutated.
FIGURE 5.

SAXS analysis on cis and trans mutants. SAXS analysis was utilized to determine the effect of these mutations on the conformational equilibrium of Hsp90. A, WT Hsc82 in the absence (black line) and presence (red line) of saturating amounts (10 mm) of AMPPNP shows two distinct distance distribution functions with the nucleotide-bound state, showing a much narrower set of distances very similar to what is seen when the distance distribution function is calculated from the AMPPNP-bound crystal structure (blue line). B and C, V23A (B) and (C) L374N (apo = black line, AMPPNP = red line) show a loss of an effect when AMPPNP is in solution. D, however, T22F (apo = black line, AMPPNP = red line), with its increased ATP hydrolysis, shows a similar profile to that of the WT distribution in the presence of nucleotide (gray dashed line).
TABLE 1.
Radii of gyration of Hsc82 homomeric mutations as determined by SAXS
| apo | AMPPNP | |
|---|---|---|
| WT | 64.8 ± 0.1 Å | 46.5 ± 0.1 Å |
| L374N | 64.6 ± 0.1 Å | 66.0 ± 0.2 Å |
| V23A | 70.2 ± 0.2 Å | 70.2 ± 0.3 Å |
| T22F | 68.1 ± 0.3 Å | 51.1 ± 0.1 Å |
| Hsp82:AMPPNP | crystal structure | 41 Å |
Given its unique ability to increase ATP hydrolysis, we also examined the SAXS behavior of the T22F mutation. Although both states are slightly expanded when compared with the native state, T22F shows a response to AMPPNP binding comparable with the WT protein (Fig. 5D). This indicates that despite predictions from the crystal structure (PDB: 2CG9), the addition of a larger hydrophobic group still allows the closed state to be reached, in complete agreement with the observed ATPase activity of this mutant. Taken all together, the SAXS data confirm that the intra- and intersubunit network of hydrophobic residues is directly involved in the stabilization of the closed state.
The Middle and N-terminal Domains Act Synergistically—Although all of the residues tested show effects on the ATPase activity of Hsc82 via either intermonomer or intramonomer interactions, and simultaneously alter the open and closed equilibrium, whether these residues act cooperatively to stabilize the hydrolysis-competent conformation of Hsc82 is unknown. To determine this, we systematically made the homomeric double mutations (using the wild-type Glu-33) and measured the hydrolysis rates. If two residues interact in the same process, such as stabilizing the catalytic transition state, we expect to see an additive effect on the loss of activity when both residues are mutated (Fig. 6A). However, if the residues are involved in separate processes (e.g. one involved in hydrolysis and the other in stabilizing a conformational state), then only one step will be rate-limiting. As a consequence, the combined mutation will not have an additive effect (Fig. 6B). Instead, the observed rate will only reflect the rate of the mutation involved in the rate-limiting step of the reaction; the other mutation will be silent. To address this directly, we tested T22F, V23A, Y24A, and L374N each in the context of the R376A mutation and measured their ATP hydrolysis rates (Table 2). For each of the four double mutants tested, we observed a marked decrease in the ATPase rates corresponding to a nearly perfect additive effect. Thus despite their close proximity, these residues cannot be functioning primarily to position Arg-376 for better interaction with the ATP γ-phosphate. Instead, the reaction transition state for each active site is synergistically stabilized by residues from its own MD as well as those from the opposite NTD.
FIGURE 6.

Synergy schematized in energy diagram format. A, two residues involved in one process should have an additive effect on the reduction of activity. The WT energetic barrier will be raised by individual point mutants labeled A* and B*. If the residues are synergistic, the double mutant will show an additive increase in the activation barrier (dashed line). B, two residues not involved in the same process will exhibit the activity of the mutation in the rate-limiting process. The WT energetic landscape (black line) will be modulated differently by individual point mutants labeled A* and B*. The double mutant will show an energy landscape (dashed line) that reflects both modulations; however, the activity seen will only be dependent on the mutation involved in the rate-limiting step.
TABLE 2.
ATP hydrolysis rates of both single, double, and truncation mutants of Hsc82
| Protein | Activity | Fold reduction (with respect to WT) |
|---|---|---|
| pmol of ATP hydrolyzed/pmol 82/min | ||
| Hsc82 | 1.2 ± 0.04 | 1 |
| T22F | 3.14 ± 0.02 | 0.38 |
| V23A | 0.25 ± 0.08 | 4.8 |
| Y24A | 0.39 ± 0.05 | 4.8 |
| L372D | 0.04 ± 0.02 | 60 |
| L374N | 0.18 ± 0.02 | 3 |
| R376A | 0.18 ± 0.02 | 6.7 |
| T22F/R376A | 0.75 ± 0.07 | 1.6 |
| V23A/R376A | 0.06 ± 0.008 | 20 |
| Y24A/R376A | 0.05 ± 0.03 | 24 |
| L374N/R376A | 0.05 ± 0.004 | 24 |
| N599 | 0.18 ± 0.02 | 6.7 |
| N599 R376A | 0.05 ± 0.01 | 24 |
Given the synergy observed between the two monomers, we wanted to further dissect the role of a key NTD-MD interaction by investigating it in isolation from possible trans effects. Toward this end, we measured the activity of a constitutively monomeric NTD-MD truncation mutant (N599) in the presence and absence of the R376A mutant. We confirmed, as reported previously (14), that the WT N599 truncation mutant showed a substantial decrease in activity due to the loss of dimerization. However, when Arg-376 is mutated in this construct, we see a nearly complete loss of activity (Table 2). This further decrease in activity suggests that the arginine, and in turn, the entire role of the network, is to help stabilize a catalytically competent NTD-MD conformation.
DISCUSSION
ATP hydrolysis plays a critical role in Hsp90 function, ultimately providing the energy required to drive large scale conformational changes in Hsp90 and to remodel and/or release substrate client proteins. Thus developing a high resolution understanding of the linkage between conformational change and ATPase activity is a fundamental prerequisite for deciphering Hsp90 function. Although nucleotide hydrolysis is intrinsically an activity of the NTD and MD domains, it is greatly stimulated via CTD-mediated dimerization. In a mechanism analogous to other GHKL family members, it was argued that NTD dimerization was required. The recently solved structure of Hsp82 in the presence of AMPPNP and the Sba1 co-chaperone (8) confirmed the NTD dimerization and also gave the first structural understanding of why the MD is required for ATP hydrolysis by showing that Arg-380 (Arg-376 in Hsc82) is directed toward and interacts with the γ-phosphate of the bound AMPPNP. However, despite the obvious NTD dimerization seen in the crystal structure (PDB: 2CG9), kinetic data had suggested that the two ATPase active sites function independently (18).
To better understand the functional importance of dimerization, we focused on exploring the role of interdomain and intermonomer interactions in stabilizing the ATP hydrolysis-competent conformation of Hsc82. Because the residues investigated in this study are conserved among family members, the results obtained from Hsc82 should be directly applicable to the entire family. Using a novel heterodimeric assay, we identified Thr-22, Val-23, and Tyr-24 in the NTD as playing a role in the hydrolysis rate of the opposite monomer and MD residues Leu-372, Leu-374, and Arg-376 as being important for the ATPase activity of the same monomer. Through double mutations, these intra- and inter-subunit interactions appear to work not via Arg-376, but in parallel with it, to cooperatively stabilize the same hydrolysis-competent state (Fig. 7A). Significantly, our SAXS measurements indicate that mutations that alter the cis-trans interaction network also alter the hydrodynamic radius, suggesting that the two are causally linked. Destabilizing the network leads to a loss of AMPPNP-induced domain closure, which in turn leads to a loss of ATPase activity.
FIGURE 7.

Model for the ATPase activation of Hsp90. A, residues (shown with spheres) acting in trans are shaded blue, and those interacting in cis are shaded red. Tyr-24 is shown in purple to indicate its contribution both in cis and in trans as observed in the data. Arg-376 can be seen interacting with the bound ATP molecule (shown in cyan) on the same monomer. B, only upon nucleotide binding and formation of the hydrophobic cross-monomer network can the catalytically active NTD-MD conformation be stabilized and the Arg-376 correctly oriented at the γ-phosphate of ATP. The black arrow denotes the N-terminal dimerization region coming together. The blue and red arrows represent the trans and cis interactions required for stabilization of N-terminal dimerization, respectively.
The functional importance of this interaction network was also confirmed by the increase in activity observed when Thr-22 was substituted with Phe. Although the crystal structure (PDB: 2CG9) predicts that a Phe could not be accommodated at this site, both the functional and the SAXS data clearly indicate that T22F is compatible with a closed state. Although we initially imagined that it enhanced ATPase activity by shifting the open-closed equilibrium in favor of the active, closed state, the SAXS data suggest that both in the presence and in the absence of nucleotide, T22F slightly prefers a more open state. From this, it would be necessary to infer that the T22F closed state is even more catalytically active than initially appreciated.
We propose a model where stabilization of the ATP hydrolysis transition state of Hsp90 requires that a very specific NTD-MD conformation be obtained. Nucleotide binding and the cis interactions uncovered in this work act to directly stabilize this conformation. Although obviously important, from our data, it appears that NTD dimerization works largely in an indirect manner to reinforce the cis interactions that stabilize the active conformation of the opposite monomer. This model proposes that the functional role of the closed state and NTD dimerization is not to activate hydrolysis per se but to preferentially stabilize the catalytically correct NTD-MD conformation. In support of this, the N599 truncation mutant data indicate that cis stabilization interactions play an important role even in the isolated monomer.
This work confirms the lack of catalytic cooperativity previously observed (18); the ATP binding and hydrolysis of one monomer does not depend on the ability of the other monomer to bind or hydrolyze ATP. By contrast, our data show a clear structural cooperativity between the two monomers, where the interdomain and intersubunit interactions synergistically stabilize the active closed conformation. One possible explanation for this difference in cooperativity is that perhaps the catalytically active state is not symmetric as is generally thought but rather asymmetric where one ATP active site is correctly oriented, allowing hydrolysis to proceed and the other site unable to hydrolyze ATP. An asymmetric dimerization would still require structural cooperativity to orient one active site correctly but would not be cooperative with respect to ATP hydrolysis since only one site at a time would be functional. Notably, an asymmetric conformation has been observed by single particle electron microscopic studies on a yeast Hsp90/Cdc37/Cdk4 complex (25). Although the data presented here and all previously reported kinetic data fit with an asymmetric model, a more formal structural investigation is necessary to confirm such a hypothesis.
Co-chaperones play an important role both in client protein recruitment and in modulating the Hsp90 ATP cycle (26-30). By comparing the HtpG apo state and the Hsp82 AMPPNP-bound state, a movement of the MD loop by ∼7 Å is observed from a retracted position in the apo state to an extended conformation interacting with the opposite NTD. In contrast, no movement is seen in the NTD region containing residues 22-24 when the two states are compared. This supports a previous proposal suggesting that binding the co-chaperone Aha1 activates Hsp90 hydrolysis via alterations of the MD loop discussed here (31). Although based on crystallization of the Aha1 NTD with an isolated Hsp90 MD, it is clear from the present work that interactions of this loop with its own NTD and the trans NTD are vitally important and that even subtle changes could have significant effects on the hydrolysis rate. By contrast, whereas the crystal structure (PDB: 2CG9) of Sba1 bound to Hsp90 has been tremendously important, the mechanism of the inhibitory effects of Sba1 on the ATP hydrolysis rate (20, 32) has yet to be understood. Our data on the T22F mutant and its modeling provide the first direct evidence that an alternate conformation may be more relevant for ATP hydrolysis than the one seen in the crystal structure. Further experiments aimed at understanding the effects of the interactions between Hsp90 and its co-chaperones on its conformational stabilization and activation are required.
Supplementary Material
Acknowledgments
We thank L. Rice, D. Southworth, U. Boettcher, T. Street, and L. Lavery for many helpful discussions and comments.
Author's Choice—Final version full access.
This work was supported by grants from the Howard Hughes Medical Institute and a University of California Discovery Grant (bio03-10401/Agard). The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
The on-line version of this article (available at http://www.jbc.org) contains two supplemental figures.
This article was selected as a Paper of the Week.
Footnotes
The abbreviations used are: NTD, N-terminal domain; CTD, C-terminal domain; MD, middle domain; AMPPNP, 5′-adenylyl-β,γ-imidodiphosphate; SAXS, small angle x-ray scattering; MALDI, matrix-assisted laser desorption/ionization; WT, wild type; PDB, Protein Data Bank; N599, NTD-MD truncation mutant.
References
- 1.Picard, D. (2002) CMLS Cell Mol. Life Sci. 59 1640-1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Young, J. C., Hoogenraad, N. J., and Hartl, F. U. (2003) Cell 112 41-50 [DOI] [PubMed] [Google Scholar]
- 3.Pratt, W. B., and Toft, D. O. (1997) Endocr. Rev. 18 306-360 [DOI] [PubMed] [Google Scholar]
- 4.Freeman, B. C., and Yamamoto, K. R. (2002) Science 296 2232-2235 [DOI] [PubMed] [Google Scholar]
- 5.Pearl, L. H., and Prodromou, C. (2006) Annu. Rev. Biochem. 75 271-294 [DOI] [PubMed] [Google Scholar]
- 6.McClellan, A. J., Xia, Y., Deutschbauer, A. M., Davis, R. W., Gerstein, M., and Frydman, J. (2007) Cell 131 121-135 [DOI] [PubMed] [Google Scholar]
- 7.Harris, S. F., Shiau, A. K., and Agard, D. A. (2004) Structure (Camb.) 12 1087-1097 [DOI] [PubMed] [Google Scholar]
- 8.Shiau, A. K., Harris, S. F., Southworth, D. R., and Agard, D. A. (2006) Cell 127 329-340 [DOI] [PubMed] [Google Scholar]
- 9.Ali, M. M., Roe, S. M., Vaughan, C. K., Meyer, P., Panaretou, B., Piper, P. W., Prodromou, C., and Pearl, L. H. (2006) Nature 440 1013-1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Neckers, L., and Ivy, S. P. (2003) Curr. Opin. Oncol. 15 419-424 [DOI] [PubMed] [Google Scholar]
- 11.Maloney, A., and Workman, P. (2002) Expert Opin. Biol. Ther. 2 3-24 [DOI] [PubMed] [Google Scholar]
- 12.Neckers, L., and Neckers, K. (2002) Expert Opin. Emerg. Drugs. 7 277-288 [DOI] [PubMed] [Google Scholar]
- 13.Prodromou, C., Panaretou, B., Chohan, S., Siligardi, G., O'Brien, R., Ladbury, J. E., Roe, S. M., Piper, P. W., and Pearl, L. H. (2000) EMBO J. 19 4383-4392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Richter, K., Muschler, P., Hainzl, O., and Buchner, J. (2001) J. Biol. Chem. 276 33689-33696 [DOI] [PubMed] [Google Scholar]
- 15.Panaretou, B., Prodromou, C., Roe, S. M., O'Brien, R., Ladbury, J. E., Piper, P. W., and Pearl, L. H. (1998) EMBO J. 17 4829-4836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dutta, R., and Inouye, M. (2000) Trends Biochem. Sci. 25 24-28 [DOI] [PubMed] [Google Scholar]
- 17.Meyer, P., Prodromou, C., Hu, B., Vaughan, C., Roe, S. M., Panaretou, B., Piper, P. W., and Pearl, L. H. (2003) Mol. Cell 11 647-658 [DOI] [PubMed] [Google Scholar]
- 18.McLaughlin, S. H., Ventouras, L. A., Lobbezoo, B., and Jackson, S. E. (2004) J. Mol. Biol. 344 813-826 [DOI] [PubMed] [Google Scholar]
- 19.Wegele, H., Muschler, P., Bunck, M., Reinstein, J., and Buchner, J. (2003) J. Biol. Chem. 278 39303-39310 [DOI] [PubMed] [Google Scholar]
- 20.Richter, K., Walter, S., and Buchner, J. (2004) J. Mol. Biol. 342 1403-1413 [DOI] [PubMed] [Google Scholar]
- 21.Felts, S. J., Owen, B. A., Nguyen, P., Trepel, J., Donner, D. B., and Toft, D. O. (2000) J. Biol. Chem. 275 3305-3312 [DOI] [PubMed] [Google Scholar]
- 22.Svergun, D. I. (1992) J. Appl. Crystallogr. 25 495-503 [Google Scholar]
- 23.Konarev, P. V., Volkov, V. V., Sokolova, A. V., Koch, M. H. J., and Svergun, D. I. (2003) J. Appl. Crystallogr. 36 1277-1282 [Google Scholar]
- 24.Nathan, D. F., and Lindquist, S. (1995) Mol. Cell. Biol. 15 3917-3925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vaughan, C. K., Gohlke, U., Sobott, F., Good, V. M., Ali, M. M., Prodromou, C., Robinson, C. V., Saibil, H. R., and Pearl, L. H. (2006) Mol. Cell 23 697-707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roe, S. M., Ali, M. M., Meyer, P., Vaughan, C. K., Panaretou, B., Piper, P. W., Prodromou, C., and Pearl, L. H. (2004) Cell 116 87-98 [DOI] [PubMed] [Google Scholar]
- 27.McLaughlin, S. H., Sobott, F., Yao, Z. P., Zhang, W., Nielsen, P. R., Grossmann, J. G., Laue, E. D., Robinson, C. V., and Jackson, S. E. (2006) J. Mol. Biol. 356 746-758 [DOI] [PubMed] [Google Scholar]
- 28.Richter, K., Muschler, P., Hainzl, O., Reinstein, J., and Buchner, J. (2003) J. Biol. Chem. 278 10328-10333 [DOI] [PubMed] [Google Scholar]
- 29.Siligardi, G., Panaretou, B., Meyer, P., Singh, S., Woolfson, D. N., Piper, P. W., Pearl, L. H., and Prodromou, C. (2002) J. Biol. Chem. 277 20151-20159 [DOI] [PubMed] [Google Scholar]
- 30.Zhang, W., Hirshberg, M., McLaughlin, S. H., Lazar, G. A., Grossmann, J. G., Nielsen, P. R., Sobott, F., Robinson, C. V., Jackson, S. E., and Laue, E. D. (2004) J. Mol. Biol. 340 891-907 [DOI] [PubMed] [Google Scholar]
- 31.Meyer, P., Prodromou, C., Liao, C., Hu, B., Mark Roe, S., Vaughan, C. K., Vlasic, I., Panaretou, B., Piper, P. W., and Pearl, L. H. (2004) EMBO J. 23 511-519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Siligardi, G., Hu, B., Panaretou, B., Piper, P. W., Pearl, L. H., and Prodromou, C. (2004) J. Biol. Chem. 279 51989-51998 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.