

Abstract
Divergent populations are intrinsically reproductively isolated when hybrids between them either fail to develop properly or do not produce viable offpring. Intrinsic isolation may result from Dobzhansky–Muller (DM) incompatibilities, in which deleterious interactions among genes or gene products lead to developmental problems or underdominant chromosome structure differences between the parents. These mechanisms can be tested by studying marker segregation patterns in a hybrid mapping population. Here we examine the genetic basis of abnormal development in hybrids between two geographically distant populations of the moss Ceratodon purpureus. Approximately half of the hybrid progeny exhibited a severely reduced growth rate in early gametophyte development. We identified four unlinked quantitative trait loci (QTL) that interacted asymmetrically to cause the abnormal development phenotype. This pattern is consistent with DM interactions. We also found an excess of recombination between three marker pairs in the abnormally developing progeny, relative to that estimated in the normal progeny. This suggests that structural differences in these regions contribute to hybrid breakdown. Two QTL coincided with inferred structural differences, consistent with recent theory suggesting that rearrangements may harbor population divergence alleles. These observations suggest that multiple complex genetic factors contribute to divergence among populations of C. purpureus.
DIVERGENT populations become intrinsically reproductively isolated when hybrids between them either fail to develop properly or do not produce viable offpring. Intrinsic barriers may result from either deleterious interactions among heterospecific genes or gene products or chromosome structure differences that are deleterious in heterozygotes. The genic model, independently proposed by Bateson (1909), Dobzhansky (1937), and Muller (1942) and typically referred to as the Dobzhansky–Muller (DM) model, proposes that complementary alleles at interacting loci have normal fitness in their native genetic background, but low fitness in a heterospecific background. The DM model makes several clear predictions regarding the nature of these deleterious interlocus interactions (reviewed in Coyne and Orr 2004). The simplest DM incompatibility involves interactions between alleles at two loci. Consider an ancestral population of haploids with a two-locus genotype AB. In one daughter population, a derived mutation is fixed at one interacting partner (aB), while in a second daughter population a mutation is fixed at the complementary locus (Ab). The DM model proposes that the ab genotype, which has not been tested by natural selection, has a lower fitness. In this example, the ancestral A and B alleles each may be introgressed between the two daughter populations, but the derived a and b alleles cannot. While more complex interlocus interactions may occur, this asymmetry is the hallmark of DM interactions (Orr 1995; Turelli and Moyle 2007). Importantly, the DM model does not imply that a low-fitness valley must be crossed in the process of population divergence.
Alternatively, hybrid F1's between individuals with different karyotypes may have reduced fertility because recombination in rearranged chromosomal segments leads to the production of aneuploid gametes (King 1993). Thus, gametes with parental karyotypes are functional, but recombinant meiotic products often lack chromosome arms or whole chromosomes and generally do not contribute to the next generation. Closely related species frequently differ in karyotype, suggesting that rearrangements may be an important cause of intrinsic isolation (Stebbins 1971; White 1978; King 1993). However, whether such rearrangements contribute to the speciation process is a matter of debate (Sites and Moritz 1987; Coyne and Orr 2004). For a particular rearrangement to fix in a population, it initially must pass through a heterozygous phase where it confers reduced fertility. Several authors have noted that a rearrangement with strong underdominant fitness effects is unlikely to increase in frequency, unless the population size is sufficiently small such that it can fix by drift, the inbreeding rate is high, or the rearrangement is selectively favored (Lande 1979; Hedrick 1981; Walsh 1982; King 1993). Smaller rearrangements with a limited effect on fertility are more likely to fix, but these are generally incapable of forming a strong intrinsic barrier to gene flow. Thus, finding that intrinsic isolation between populations or species is due to structural rearrangements suggests that the divergence was associated with a reduction in effective population size, due to either selective or demographic processes.
Numerous studies report that complementary DM interactions cause hybrid sterility or inviability in a wide variety of eukaryotes (Coyne and Orr 2004), but sterility due to chromosome structure differences appears to be more common in plants than animals (Stebbins 1971; Rieseberg 2001). For example, in angiosperm hybrids fertility is often restored by experimentally inducing tetraploidy, suggesting that meiotic recombination between different karyotypes led to F1 hybrid sterility (Stebbins 1958). More recent studies have used genetic mapping approaches to identify genes or chromosomal regions that contribute to hybrid sterility among angiosperm species pairs (Rieseberg et al. 1999; Fishman and Willis 2001; Lai 2005; Moyle and Graham 2005; Sweigart et al. 2006; Bomblies et al. 2007; reviewed in Rieseberg and Willis 2007). However, few have examined the phenomenon in other plant lineages, like bryophytes, where the dominant phase of the life cycle is haploid (but see Nakazato et al. 2007). Like all land plants, the moss life cycle consists of a haploid gametophyte generation that alternates with a morphologically distinct diploid sporophyte generation. Recombinant haploid spores are produced in a single cross between two moss gametophytes. Thus, in a bryophyte system we can observe hybrid development directly on the products of meiosis (i.e., haploid gametophytes derived from a single sporophyte).
In a cross between divergent bryophyte populations or species, hybrid incompatibility due to DM interactions should be detectable by mapping quantitative trait loci (QTL) associated with abnormal development. Markers linked to DM loci generally will exhibit asymmetries in allelic effects and epistasis with markers linked to other interacting loci. In contrast, chromosome structural differences between the parents, like chromosomal inversions, will cause the F1 sporophyte to produce either parental-type or dead spores, which will leave no genetic signature. Several authors have noted that the effective reduction in recombination in structural heterozygotes may favor the accumulation of population-divergence mutations in inversions, particularly where gene flow may introduce maladaptive migrant alleles (Noor et al. 2001; Rieseberg 2001; Ortiz-Barrientos et al. 2002; Navarro and Barton 2003; Kirkpatrick and Barton 2006). However, in a genetic mapping study in a haploid organism like a bryophyte, inversions containing DM incompatibility loci will behave like any other DM incompatibility (although several markers may cosegregate with the incompatibility allele if the inversion is large).
Alternatively, recombination in structural heterozygotes may cause abnormal development rather than spore lethality. In the context of hybrid incompatibility, we expect that abnormally developing hybrid gametophytes may be aneuploid, as a result of meiotic crossovers in structurally diverged chromosomal regions, while normal hybrid gametophytes will have parental haplotypes in such regions. In contrast to chromosomal speciation models where recombinants are absent, this will be manifest as increased recombinational distances among markers flanking rearranged regions in abnormally developing progeny, relative to the intermarker distances in normally developing progeny. Studying marker segregation patterns in a hybrid mapping cross, therefore, provides a first approximation of the kind of mutations, genic or structural, that intrinsically isolate populations. Since these mutations potentially evolve under different demographic or selective conditions, identifying the genetic basis of hybrid breakdown may provide insight into the evolutionary histories of the diverging populations.
In many bryophytes inbreeding is frequent (Eppley et al. 2007), suggesting that chromosomal variants may make a significant contribution to speciation in this group. Analyses of several natural interspecies F1 hybrids between reproductively isolated mosses suggest that both structural and genic isolation may occur (reviewed in Natcheva and Cronberg 2004). For example, the chromosomes failed to undergo proper meiotic pairing in F1 hybrids between Ditrichum pallidum and Pleuridium acuminatum, which Anderson and Snider (1982) interpreted as chromosomal isolation. On the other hand, genic factors must contribute to the F1 inviability in crosses between Polytrichum commune and P. uliginosum, because the phenotype is evident in premeiotic sprorophyte tissues (Van Der Velde and Bijlsma 2004). Apart from these few examples, the genetic basis of hybrid sterility or inviability in the majority of natural (Anderson and Lemmon 1972; Natcheva and Cronberg 2007) or experimentally produced (Wettstein 1924, 1928, 1932) hybrids in mosses is equivocal.
Here we examine the genetic basis of abnormal hybrid development in the progeny of a cross between two isolates of the moss Ceratodon purpureus (Hedw.) Brid. Approximately half of the progeny from this cross grew abnormally slowly, a phenotype absent from natural populations. Loci in four chromosomal regions were significantly associated with the abnormal development phenotype. The effects of introgressing these loci were strongly asymmetric, consistent with the DM model. Additionally, the number of recombination events was significantly elevated in three mapped intervals in the abnormally developing progeny, relative to the normal progeny, consistent with structural differences contributing to hybrid incompatibility. Two loci associated with variation in early developmental growth rate coincided with putative structurally differentiated genomic regions. Collectively, these results suggest that genic and structural mechanisms may operate simultaneously, and potentially synergistically, in the process of divergence among conspecific populations of C. purpureus.
MATERIALS AND METHODS
Study system:
In the field, gametophytes of C. purpureus are variable in appearance and ecology (Crum and Anderson 1981). In the most recent taxonomic revision of the genus, Burley and Pritchard (1990) recognized six species or subspecies, largely on the basis of latitudinal variation in continuously varying traits (gametophyte leaf shape, sporophyte shape, and ornamentation). Subsequent studies, however, have failed to find genealogical support for the independently evolving lineages suggested by the morphological data, but rather have found evidence of generally high within-population nucleotide variation and limited among-population differentiation (McDaniel and Shaw 2005). Common garden experiments indicate that populations of C. purpureus contain genetic variation for gametophytic morphological and life history traits, and trait means and patterns of covariation may be significantly different between nearby populations (Shaw and Beer 1999; McDaniel 2005).
Germination of haploid moss spores, from field collected or experimentally generated sporophytes, gives rise to protonemal filaments. Protonema are comprised of two morphologically distinct cell types, chloronema and caulonema. The mature leafy gametophore develops from a bud on caulonemal cells. At sexual maturity, the gametophore of C. purpureus produces either sperm-producing antheridia or egg-producing archegonia within specialized perigonial or perichaetial buds, respectively. The sperm swim through water to fertilize the egg, forming a zygote that remains attached to the female parent gametophyte. Meiosis takes place in the sporangium (capsule) of the mature sporophyte, which ultimately ruptures, releasing the haploid spores.
Diploid sporophytes from populations of C. purpureus near Otavalo, Ecuador (Ec), and Ithaca, New York (NY), were chosen to represent extremes of the morphological distribution. The New York population was found on asphalt that was periodically inundated with rainwater (42°26′26″N, 76°29′49″W; elevation 250 m), while the Ecuador population was sampled from a grassy field evidently maintained by frequent burning (0°13′60″N, 78°17′60″W; elevation 2800 m). Spores from 48 sporophytes from each population were cultivated on agar using the method described in McDaniel et al. (2007). A single spore isolate (SSI) from each of the 48 sporophytes was isolated, and these individuals are maintained in the permanent culture collection.
A single male SSI from the New York population and a single female SSI from the Ecuador population were chosen to create a mapping population, as described in McDaniel et al. (2007). C. purpureus has sexually dimorphic chromosomally determined sexes (Shaw and Gaughan 1993; Shaw et al. 1997; Shaw and Beer 1999; McDaniel 2005). The diploid sporophyte is always heterogametic (by convention labeled XY), and the X and Y chromosomes segregate to haploid female and male gametophytes, respectively, following meiosis. The two parents were crossed to create a hybrid diploid F1 sporophyte, from which we isolated 288 recombinant (F2-like) haploid spores. The 288 recombinant progeny were grown for several weeks to generate tissue for a common garden experiment. The recombinant F1 gametophytes were initiated on 12-well agar plates containing standard media with ammonium tartrate (Knight et al. 2002). Each 12-well plate was replicated three times. Approximately 50–100 cells of protonemal tissue were used to start each replicate. The 48 genotypes from the two parental populations were similarly clonally replicated. The plates were randomly distributed on four light benches and grown for 20 days under 40 μmol/m2/sec of continuous light at ∼25°. At the end of the experiment each 12-well plate was photographed, and the digital image was imported into the analysis software ImageJ (available from http://www.rsb.info.nih.gov/ij/). The color of the protonema was generally homogeneous and sufficiently different from that of the agar so that the size of the protonema could be reliably estimated by summing the number of pixels having a color darker than 95 (on a scale of 0–255 with 0 being the darkest).
Statistical analysis of protonemal size variation:
To estimate the amount of variation associated with genetic differences within and between the parental NY and Ec populations, we performed a random-effects ANOVA with protonemal size as the dependent variable, with populations, and individuals nested within populations as the explanatory variables using the software JMP 6.0 (SAS Institute, Carey, NC). Because not all individuals were sexed, we did not include sex in this analysis. Nor did we include effects resulting from our experimental design (plate, light bench). Individuals in the mapping population were classed as either parental phenotype (>17,000 pixels) or small (<17,000 pixels), on the basis of a natural break in the size-class distribution (Figure 2). This phenotypic distribution is a hallmark of hybrid breakdown (see discussion); therefore, from here on we refer to the progeny with small protonema as the abnormally developing class. We performed another random-effects ANOVA on the segregating hybrid progeny, with sex and individual nested within sex as the explanatory variables. Sex was determined using an MseI or XbaI restriction-site difference between the male and female copies of the sex-linked des6 gene (McDaniel et al. 2007).
Figure 2.—

Size-class histogram of progeny from a cross between Ecuador (female) and New York (male). Individuals measuring >18,000 pixels were considered normal, while those less than this threshold were classed as abnormally developing. Least squared means for males and females are indicated by vertical arrows (male = 34,139; female mean = 39,375; F = 136.1; d.f. = 1, 150; P < 0.0001).
Surveying recombinational differences in developmentally abnormal hybrids:
If aneuploidy causes abnormal development, then the slow growing progeny will be recombinants. To determine whether recombination was associated with the slow growing phenotype, we counted the number of recombinants between all pairs of adjacent loci in abnormally developing progeny and assessed deviations from the proportion of recombinants in the parental-type progeny using a χ2-test with a Bonferroni correction. Previously, we constructed a linkage map by genotyping the 288 recombinant haploid progeny from a cross between a male from the NY population and a female from the Ec population at 124 molecular markers and sex (for complete details on map construction and marker segregation patterns, see McDaniel et al. 2007). In a map of 730 cM, we resolved 15 linkage groups (C. purpureus n = 13), each containing an average of more than four markers. Here we refer to these as LG1 to -14, and X and Y for the sex chromosomes. We estimated that the map covers ∼74% of the genome and that 94% of the genome was within 20 cM of a mapped marker. Although there was significant segregation distortion at many loci, >70 recombinant progeny had the rarer allele at the most distorted markers, giving us a sufficient sample size for testing recombinational differences between the two phenotypic classes.
Mapping loci causing Dobzhanky–Muller interactions:
To identify genic factors associated with abnormal development, we mapped QTL underlying variation in protonemal growth using composite interval mapping (CIM) (Zeng 1994) in the Windows version of QTL Cartographer (Wang et al. 2007) with all program options set to default values. This method generates a probability that variation at a particular chromosomal region is associated with trait variation. The CIM output includes LOD scores across the entire linkage map, which facilitates comparisons between different partitions of the hybrid progeny. Permutation tests with 1000 replicates were used to identify a threshold value of the test statistic to give an experimentwise type I error rate of P < 0.05 (Churchill and Doerge 1994).
To determine whether loci causing quantitative variation among individuals with a parental phenotype (i.e., size >17,000 pixels) also were responsible for the difference between normal and abnormally developing hybrids, we separately mapped QTL associated with variation in size in the parental-type individuals alone and those associated with the binary difference between parental-type and abnormally developing progeny. The mean size from the three replicates of each SSI was used for all analyses. In both the parental-type and hybrid-breakdown analyses, CIM analyses were conducted on each sex separately, in addition to the total data set (all parental-type individuals or all segregating progeny), since males and females are sexually dimorphic. Several significant QTL that were found in one of the single-sex analyses were absent or not significant in the other sex or the total analyses. To test whether such QTL had significant sex-specific effects on protonemal size variation, we used an ANOVA, implemented in the software JMP 6.0 (SAS Institute) with the marker most closely linked to each QTL, sex, and the interaction between these two effects as explanatory effects in the model.
To specifically examine the genetic basis of hybrid breakdown, we coded individuals as either parental type or abnormally developing and used CIM to identify loci contributing to the difference between individuals in these two classes. To test for higher order multilocus interactions (epistasis) among loci contributing to hybrid breakdown, we conducted a full-factorial ANOVA, with marker genotype at a locus linked to each QTL, sex, and all possible interaction terms included in the model, using JMP 6.0.
To test whether QTL colocalized with regions of the genome showing elevated numbers of recombination events in the abnormally developing progeny, we used the hypergeometric probability distribution function, following Paterson (2002). This approach generates a probability (P) that associations between mapped features arise by chance, using the formula:
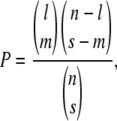 |
where l is the number of intervals containing the more frequent feature, s is the number of intervals containing the less frequent feature, m is the number of intervals containing both features, and n is the number of intervals that can be compared. We conservatively used n = 28, approximately half the number of intervals in the linkage map, because several intervals were very short.
Another expectation of the DM model is segregation distortion at loci causing hybrid lethality. To test whether loci that were associated with hybrid breakdown also caused spore lethality, we used a linear regression to test for a relationship between marker LOD score for hybrid breakdown, derived from the CIM analysis of the total data set, and the significance of markers' segregation distortion, estimated by a χ2-value (see McDaniel et al. 2007).
RESULTS
Patterns of phenotypic variation:
The NY and Ec parental populations exhibited considerable variation for protonemal growth rates (Figure 1). The individual mean protonemal sizes for the genotypes ranged from 20,000 pixels to 70,000 pixels for both populations. Approximately 75% of the variation evident in the progeny was due to differences among genotypes rather than environmental variance (R2 = 0.80; F = 8.50; d.f. = 93, 188; P < 0.0001). Although the distributions of trait values in the two populations were broadly overlapping, the least squared mean protonemal size in the NY population was slightly but significantly larger at the end of the experiment (Ec = 39,630; NY = 45,538; F = 5.58; d.f. = 1, 92; P = 0.01).
Figure 1.—

Size-class histogram of field-collected individuals sampled from the Ecuador (Ec) (open bars) and New York (NY) (solid bars) populations, with population least squared means indicated by vertical arrows (Ec = 39,630; NY = 45,538; F = 5.58; d.f. = 1, 92; P = 0.01).
A total of 288 recombinant F1 gametophytes were scored from the cross between a female from the Ecuador population and a male from the New York population. The sizes of the progeny were bimodally distributed, with 129 between ∼3000 and 16,500 pixels, and 159 between ∼18,500 and 70,000 pixels (Figure 2). The larger class of hybrid progeny were indistinguishable from individuals sampled from the two parental populations, although the progeny size distribution was more similar to the Ecuador (maternal) parental population (Figures 1 and 2).
Protonemal growth rate was significantly sexually dimorphic (Figure 2), with males in the parental type of hybrids reaching on average 85% of female size (male mean = 34,139; female mean = 39,375; F = 136.1; d.f. = 1, 150; P < 0.0001). Because we made the cross in only one direction, sex and population are confounded in our analysis. However, we believe that the sex effect (i.e., smaller males) is significant because it was opposed to the population effect, as the male parent (NY) came from the population with larger mean size. We found significant male-biased sex ratio (60%) in the total mapping population, although males were more overrepresented in the small size class (65%) than in the normal size class (56%). Additional details on segregation patterns are reported in McDaniel et al. (2007).
Recombination in abnormally developing progeny:
The chromosomal rearrangement model predicts that recombination in structural heterozygotes leads to aneuploidy and abnormal development. We found three regions of the genome where crossovers were significantly more frequent in the abnormally developing progeny than in the total mapping population (P > 0.0001; Figure 3; supplemental Table 1). We identify these intervals (and QTL localizations, see below) using the linkage group designations from McDaniel et al. (2007). On LG3 (interval 7) and LG8 (interval 2), the recombination was doubled (from 0.10 to 0.23 and 0.12 to 0.24, respectively) and on the sex chromosome (interval 3) recombination was elevated by >25% (from 0.34 to 0.43). In all cases the sample sizes were >120 individuals.
Figure 3.—

Comparison of centimorgan distances between all pairs of adjacent markers in the abnormally developing progeny to distances in the parental-type progeny is shown on the y-axis, with the χ2-values for each interval shown with a shaded diamond. Negative χ2-values indicate increased recombination in the parental-type progeny. The thick horizontal lines represent linkage groups (numbered below) with the hashes showing marker locations. Significance thresholds are shown with thin horizontal lines (solid, P < 0.001; dashed, P < 0.01). Three intervals showed significant deviation following a Bonferroni correction from 1:1 map distances between the two samples (P < 0.0001).
QTL analyses of protonemal growth:
We detected QTL that contributed to variation in protonemal growth in the progeny of the NY–Ec cross at eight unique chromosomal locations (Table 1, Figure 4). With the exception of QTL 5 (LG12), no QTL were found in the analyses of both parental-type variation and abnormal hybrid development (Table 1).
TABLE 1.
Protonemal-size QTL positions and effects
| Parental type (n = 159)
|
Hybrid breakdown (n = 288)
|
||||
|---|---|---|---|---|---|
| QTL | Chromosome, marker, position | LODa | Effect | LODa | Effect |
| 1b | LG3, 8, 1.51 | 7.42 | −1649 | — | — |
| 2 (spa2) | LG7, 1, 0.1–0.12 | — | — | 33.1 | −0.195 |
| 3 (spa1) | LG8, 3, 0.43 | — | — | 39.1 | −0.193 |
| 4b | LG9, 3, 0.41 | — | — | 11.0 | −0.089 |
| 5 (spa3) | LG12, 4, 0.27–0.39 | 15.7 | −2702 | 33.2 | +0.176 |
| 6 (spa4) | LG13, 1, 0.09 | — | — | 14.7 | +0.105 |
| 7b | LGXY, 2, 0.04 | — | — | 12.0 | −0.090 |
| 8 | LGXY, 9, 1.40–1.47 | 20.7 | +2642 | — | — |
LOD scores were generated by permutation.
QTL detected only in females.
Figure 4.—

Position of significant QTL detected in the CIM analyses of normal variation (a, total data set, n = 159; b, males, n = 89; c, females, n = 70) and hybrid breakdown (d, total data set, n = 288; e, males, n = 161; f, females, n = 127). Chromosomal position, with markers indicated by triangles, is shown on the x-axis, and LOD score is shown on y-axis. The horizontal lines indicate significance threshold values of P = 0.05 generated by permutation (see materials and methods).
Three QTL were found in the CIM analysis of parental-type variation. At QTL 1 on LG3, the Ec allele had a negative effect. This QTL was significant in females, but not in the analyses of males or the combined normal individuals (Figure 4, a–c). QTL 1 colocalized with an inferred chromosome structure difference, on the basis of patterns of recombination in the low fitness progeny. At QTL 5 on LG12 the Ec allele had the expected negative effect on protonemal size variation. This QTL was significant in the analyses of all normal individuals and males alone, but not females (Figure 3, b and c). Neither of these QTL exhibited significant genotype × sex interactions, on the basis of an ANOVA using the genotype of the markers most closely linked to either of these. QTL on LG12 and the sex-determining region of the XY were significant in the analysis of all parental-type progeny (threshold = 12.1). The X chromosome (i.e., Ec allele) had a positive effect on protonemal growth, consistent with females having significantly greater protonemal growth than males.
Six QTL contributed to the genetic basis of abnormal development (Table 2). Four hybrid-breakdown QTL were statistically significant in the total CIM analysis (threshold = 10.6; Figure 4, d–f), which we denote small-protonema 1–4 (spa1–4), numbered according to the strength of their association with the abnormal development phenotype. The marker intervals surrounding spa1, -2, and -4 (on LG8, LG7, and LG13, respectively) had no effect on protonemal variation in the normal individuals, but spa3 (on LG12) colocalized with a major QTL underlying variation in normal individuals. Two additional QTL were found in the analysis of females alone (Figure 4f; LG9 and XY marker 3). Of these, only the QTL in the pseudoautosomal region (PAR) of the sex chromosome had a significant genotype × sex interaction (P = 0.03).
TABLE 2.
Nominal logistic fit for four-locus DM model of hybrid breakdown
| Term | Estimate | SE | χ2 | P |
|---|---|---|---|---|
| Intercept | −0.29 | 0.17 | 2.83 | 0.092 |
| spa1 | −0.82 | 0.18 | 21.5 | <0.0001 |
| spa2 | 0.51 | 0.18 | 8.49 | 0.004 |
| spa3 | 0.42 | 0.16 | 6.64 | 0.010 |
| spa4 | −0.38 | 0.17 | 4.97 | 0.026 |
| spa2*spa1 | −0.25 | 0.17 | 2.21 | 0.14 |
| spa3*spa1 | −0.64 | 0.17 | 13.8 | 0.0002 |
| spa4*spa1 | 0.22 | 0.16 | 1.80 | 0.18 |
| spa3*spa2 | −0.06 | 0.17 | 0.13 | 0.72 |
| spa4*spa2 | −0.23 | 0.16 | 2.12 | 0.15 |
| spa3*spa4 | −0.32 | 0.16 | 3.88 | 0.049 |
| Whole model r2 = 0.23 | <0.0001 | |||
The DM model predicts that alleles at abnormal-development QTL should have asymmetric effects, so comparisons of complementary genotypes at such QTL can be used to test this model. The contributions of spa1–4 to the hybrid-breakdown phenotype are shown in Figure 5. Contrasting the frequency of low fitness hybrids in genotypes C and D in Figure 5, it is clear that the effect of introducing a NY allele spa1 into an Ec background at the remaining QTL has a significantly more severe effect than the converse introgression. Likewise, at spa2 and spa3 the allelic effects are significantly asymmetric (compare genotypes E and F and G and H), although at these loci introgressing an Ec allele into a NY background has a stronger effect. While spa4 alone has a limited effect on hybrid breakdown (genotypes I and J), the state at this locus alters the expression of the hybrid-breakdown phenotype; for example, compare genotypes N and G or D and P, where the severity of the complementary introgression of spa4 is asymmetric.
Figure 5.—

Proportion of progeny with a parental phenotype at all 16 possible genotypes at spa1–4, using the marker most closely linked to the QTL. Chromosomal regions from NY are solid and those from Ec are white, with parental genotypes (A and B), single introgressions (C–J), and double introgressions (K–P); reciprocal introgressions are paired to illustrate the asymmetry of allelic effects. Whiskers indicate standard errors, and number of progeny with each genotype is shaded to the right of the bars.
The DM model additionally predicts that significant epistasis among loci should contribute to the genetic architecture of hybrid breakdown. In testing interactions contributing to the difference between parental-type and abnormally developing progeny (Table 2), we found significant interactions between spa1 and spa3 (P < 0.001) and spa3 and spa4 (P < 0.05). Weaker interactions were found between spa1 and spa2 (P = 0.14), spa1 and spa4 (P = 0.18), and spa2 and spa4 (P = 0.15). In total, this model explained ∼25% of the variation between the two progeny classes. No epistasis was detected between QTL for normal growth.
QTL on LG3 and LG8 (spa1) coincided with intervals with elevated numbers of recombination events in the abnormally developing progeny, an arrangement that was unlikely to have arisen by chance (P < 10−10), on the basis of the hypergeometric probability function. To understand whether the QTL associated with the abnormal-development phenotype also caused spore lethality, we plotted the relationship between the strength of association of a marker to the slow-growth phenotype (measured by LOD score) and the significance of segregation distortion at that locus (measured by χ2-value; McDaniel et al. 2007). Figure 6 illustrates there was no relationship between these quantities.
Figure 6.—

A plot of segregation-distortion χ2-values and LOD scores (for association with the hybrid-breakdown phenotype) for all mapped markers (r2 = 0.0, P = 0.81). The solid vertical line indicates the threshold for significant segregation distortion (α = 0.05, χ2 = 3.84), and the dashed vertical line shows the significance threshold for association with the hybrid-breakdown phenotype generated by permutation (LOD = 10.6).
DISCUSSION
The genetic basis of abnormal development in interpopulation hybrids:
The most striking result from our analysis of the hybrid phenotypes in the NY–Ec cross was the clear discontinuity between the parental-type and the abnormally developing progeny (Figures 1 and 2). The slow-growth phenotype was evident almost immediately after spore germination, and preliminary growth experiments indicate that individuals with slow growing protonema were less likely than normal individuals to reach sexual maturity (data not shown). This phenotype was rare in natural populations (1–4%; Shaw and Beer 1999; McDaniel 2005; Figure 1) and is likely to be disadvantageous in the habitats where C. purpureus is typically found. Preliminary analyses of other crosses between the Ec and NY populations indicate that they also show partial hybrid breakdown, while within population crosses, like natural crosses, they do not. The manifestation of hybrid breakdown at this stage is unique to organisms with haploid gene expression, like land plants, where the products of meiosis undergo mitotic cell divisions before producing gametes themselves. Thus, while a failure to produce viable meiotic products is typically termed sterility, we prefer abnormal development for the slow-growth-gametophyte phenotype and reserve sterility for gametophytes that fail to produce functional gametes.
The QTL analyses clearly indicate that DM interactions contribute to hybrid breakdown in this cross. Three of the four chromosomal regions that were significantly associated with the difference between the parental-type and the abnormally developing progeny displayed strong asymmetries in allelic effects (spa1–3; Figure 5). This asymmetry is characteristic of DM interactions (Muller 1942; Orr 1995; Turelli and Moyle 2007). Additionally, we detected strong statistical interactions between the genotypes at spa1 and spa3, and between spa3 and spa4 (Figure 4). Several authors have noted that the probability of evolving a DM genetic incompatibility increases as more loci participate in the interaction (Cabot et al. 1994; Orr 1995; Porter and Johnson 2002; Gavrilets 2004). Indeed, the effect of single introgressions was either moderated or elevated by the allelic states at the other incompatibility QTL; for example, an NY allele at spa4 is particularly deleterious only with an NY allele at spa1 (genotype P in Figure 4). The absence of statistically significant higher-order interactions, however, suggests we may lack the power to detect more complex epistasis.
Finding DM interactions that cause abnormal hybrid development between conspecific populations of C. purpureus is not surprising, particularly given that even nearby populations are strongly differentiated in morphological and life-history traits (Shaw and Beer 1999; McDaniel 2005). Here we showed that the NY and Ec populations exhibited a slight but significant difference in mean protonemal growth rate under laboratory conditions, although the range of phenotypes was broadly overlapping (Figure 1). We would like to know, however, what processes are involved in the evolution of the genetic factors underlying the abnormal-development phenotype. To test whether hybrid breakdown was a pleiotropic effect of evolution at loci controlling protonemal growth differences between the parents, we mapped QTL for growth in the parental-type progeny alone. We detected significant QTL on LG3, LG12, and the sex chromosome (Figure 4, a–c). Consistent with patterns in natural populations, hybrid males grew ∼85% as fast as their female siblings (Shaw and Beer 1999; McDaniel 2005). While sex and population of origin are confounded in this analysis, the female X chromosome came from the slower-growing Ec population (Figure 1), suggesting that the population effect is smaller than the sex effect and that variation in sexual dimorphism may not contribute to population divergence in early development. However, only one of the six abnormal-development QTL (spa3) had pleiotropic effects on parental-type variation. Thus, parental-size variation and abnormal development are largely independent traits. We are continuing to analyze segregating phenotypic variation in the mapping population to identify other traits that share a common genetic basis with the abnormal-development phenotype.
While the evidence for DM interactions is strong, the four-locus DM model does not fully explain the abnormal-development phenotype. First, although certain genotypic combinations of spa1–4 were very deleterious (e.g., genotype P, Figure 5), a model including these four loci (and the interactions among them) explained ∼23% of the phenotypic variance between the parental-type and abnormal-developing hybrids (Table 2). Second, only 80% of the progeny with the parental genotypes (using the marker most closely linked to spa1–4 to infer the genotype) had parental phenotypes, suggesting some other factor caused 20% to show abnormal development. Clearly this unexplained variance could be a result of other undetected QTL that fell outside of mapped intervals or incomplete linkage between the markers and the underlying QTL. We expect the latter source of error to be small because the markers we used were within 5 cM of the QTL. It is also worth pointing out that while 96% of the individuals with genotype P exhibited abnormal development, this genotype accounts for ∼20% of the abnormally developing progeny. Although the various single-QTL introgressions may have variable degrees of penetrance, the modest contribution of genotype P to the total hybrid breakdown suggests that the abnormal-development phenotype may have multiple causes.
To test whether chromosome structure differences between the two parents also were associated with hybrid breakdown, we compared the number of recombination events between all pairs of adjacent markers in the abnormally developing and parental-type progeny. If aneuploidy causes the slow-growth phenotype, we expect to find an association between recombination in particular intervals and abnormal development. Consistent with this model, we found an excess of crossovers in three genomic regions in the abnormally developing progeny (Figure 3; supplemental Table 1). This is not due to an overall increase in recombination rate in these individuals, but rather is a reflection that recombinants in these three regions are more likely to show abnormal development. The individual contributions of recombination within each of these intervals to the abnormal-development phenotype are likely to be small; the recombination-fraction differences in these regions between the parental-type and abnormal progeny were 0.13, 0.12, and 0.09 (on LG3, LG8, and the recombining portion of the sex chromosome, respectively). However, combined they could cause approximately one-third of the hybrid progeny to exhibit slow growth. Another region of the genome on LG1 also contained an elevated number of crossovers in the abnormally developing progeny, but this increase was not significant because the number of events in either class was small. Thus, we suspect that a complete explanation of the genetic basis of the abnormal-development phenotype requires both DM interactions and structural differences between the parental genomes.
The evolution of underdominant mutations, structural or otherwise, generally requires small population sizes so that the mutations may increase in frequency due to genetic drift (Lande 1979; Hedrick 1981; Walsh 1982; King 1993). However, given the levels of polymorphism within populations and the lack of population structure across much of the distribution of C. purpureus (McDaniel and Shaw 2005), we suspect that drift alone is unlikely to govern the frequency of any underdominant mutations within the species. In addition, the nature of these mutations is uncertain. Interestingly, two QTL that we detected mapped to regions of putative structural differences between the NY and Ec populations (QTL 1 and spa1). The association between the inferred structural differences and population-difference QTL is unlikely to have arisen by chance, on the basis of the hypergeometric probability distribution function (P = 0.0). In some species pairs, particularly in regions of sympatry where gene flow is common, inverted chromosomal regions harbor population-difference QTL (reviewed in Hoffmann et al. 2004). However, recombination within inverted regions should lead to the loss of all or significant portions of the inversion-containing chromosomes, which we suspect would have a more severe phenotype than what we observed in the haploid progeny of the NY–Ec cross. We should stress that our current data regarding these putative structural differences are indirect. More detailed physical and genetic maps of the C. purpureus genome and a more complete understanding of the nature of divergence among these populations are necessary to assess the significance of these findings.
Population divergence in C. purpureus:
Our principle aim was to study the genetic basis of population divergence in C. purpureus; the NY and Ec populations were chosen to represent phenotypic extremes within the species. Taxonomically, these populations belong to different subspecies, on the basis of the distribution of continuously varying morphological traits (Burley and Pritchard 1990). Indeed, the genetic architecture of hybrid incompatibility that we describe here is similar to that separating other recently diverged plant species pairs (Hollingshead 1930; Stephens 1946; Avers 1953; MacNair and Christie 1983; Christie and MacNair 1984; Kubo and Yoshimura 2002, 2005; Moyle and Graham 2005; Sweigart et al. 2006; reviewed in Bomblies and Weigel 2007). The patterns of marker segregation distortion indicate that additional factors unrelated to the abnormal-development phenotype also participate in the overall intrinsic isolation between these two individuals (McDaniel et al. 2007). We had expected that hybrid breakdown might represent incomplete penetrance of lethality; that is, that spore lethality and slow protonemal growth shared a common genetic basis. In contrast, the QTL underlying the abnormal-development phenotype represent only a relatively small proportion of the distorted loci in this cross (Figure 6) and therefore a small proportion of the combined intrinsic barriers between the NY and Ec populations. Moreover, multiple potentially conflicting genetic interactions operate on single genomic regions in this hybrid cross. For example, a NY spa2 allele had a negative effect on spore germination (McDaniel et al. 2007), but individuals with the alternate (Ec) allele at spa2 were more likely to exhibit abnormal development.
Collectively, these observations suggest that the genetic control of early development was sufficiently diverged such that, although the F1 sporophyte was not obviously aberrant in development or morphology, the NY and Ec populations may be partially reproductively isolated. However, without additional interpopulation crossing data from other populations, it is unclear whether C. purpureus is composed of a complex geographic matrix of partially isolated genotypes or allopatric cryptic reproductively isolated groups defined by a few differentiated, and potentially rearranged, portions of the genome.
This study represents the first step in the process of identifying the genetic factors that cause abnormal hybrid development in crosses between populations of C. purpureus. Determining whether mutations underlying the DM interactions represented major adaptive substitutions when they arose in different populations or became major only in the context of a hybrid genetic background will be critical for understanding the role of epistasis in population divergence (Malmberg et al. 2005); that is, did the order of origin of the alleles underlying the abnormal-development QTL matter, or did the QTL act additively in the process of divergence and could have assembled in any order? What is the role of karyotypic change in the evolution of such loci or of hybrid breakdown? Identifying any generalities in both the kind of incompatibilities that first isolate diverging lineages and the evolutionary processes that govern their frequency within populations will be important steps toward understanding the genetics of speciation and diversification.
Acknowledgments
Past and present members of the Willis lab, in particular A. Bouck, A. L. Case, L. Fishman, and M. C. Hall, provided helpful guidance at various stages of this research. D. J. Cove, P.-F. Perroud, and two anonymous reviewers made numerous comments that improved this manuscript. S.F.M. thanks his dissertation committee at Duke University, P. S. Manos, M. D. Rausher, A. J. Shaw, R. Vilgalys, and J. H. Willis, for advise and encouragement. A collecting trip to Ecuador was supported by a National Geographic Society grant to A.J.S. Support for this research was provided by a National Science Foundation Doctoral Dissertation Improvement grant DEB-0206508 to S.F.M.
References
- Anderson, L. E., and B. E. Lemmon, 1972. Cytological studies of natural intergeneric hybrids and their parental species in the moss genera, Astomum and Weissia. Ann. Mo. Bot. Gard. 59 382–416. [Google Scholar]
- Anderson, L. E., and L. A. Snider, 1982. Cytological and genetic barriers in mosses. J. Hattori Bot. Lab. 52 241–254. [Google Scholar]
- Avers, C. J., 1953. Biosystematic studies in Aster, II: Isolating mechanisms and some phylogenetic considerations. Evolution 7 317–327. [Google Scholar]
- Bateson, W., 1909. Darwin and Modern Science. Cambridge University Press, Cambridge, UK.
- Bomblies, K., and D. Weigel, 2007. Hybrid necrosis: autoimmunity as a potential gene-flow barrier in plant species. Nat. Rev. Genet. 8 382–393. [DOI] [PubMed] [Google Scholar]
- Bomblies, K., J. Lempe, P. Epple, N. Warthmann, C. Lanz et al., 2007. Autoimmune response as a mechanism for a Dobzhansky-Muller-type incompatibility syndrome in plants. PLoS Biol. 5 e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burley, J. S., and N. M. Pritchard, 1990. Revision of the genus Ceratodon (Bryophyta). Harv. Pap. Bot. 2 1–76. [Google Scholar]
- Cabot, E. L., A. W. Davis, N. A. Johnson and C. I. Wu, 1994. Genetics of reproductive isolation in the Drosophila simulans clade: complex epistasis underlying hybrid male sterility. Genetics 137 175–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie, P., and M. R. MacNair, 1984. Complementary lethal factors in two North American populations of the yellow monkey flower. J. Hered. 75 510–511. [Google Scholar]
- Churchill, G. A., and R. W. Doerge, 1994. Empirical threshold values for quantitative trait mapping. Genetics 138 963–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne, J. A., and H. A. Orr, 2004. Speciation. Sinauer Associates, Sunderland, MA.
- Crum, H., and L. Anderson, 1981. Mosses of Eastern North America. Columbia University Press, New York.
- Dobzhansky, T. H., 1937. Genetics and the Origin of Species. Columbia University Press, New York.
- Eppley, S. M., P. T. Taylor and L. K. Jesson, 2007. Self-fertilization in mosses: a comparison of heterozygote deficiency between species with combined versus separate sexes. Heredity 98 38–44. [DOI] [PubMed] [Google Scholar]
- Fishman, L., and J. H. Willis, 2001. Evidence for Dobzhansky-Muller incompatibilites contributing to the sterility of hybrids between Mimulus guttatus and M. nasutus. Evol. Int. J. Org. Evol. 55 1932–1942. [DOI] [PubMed] [Google Scholar]
- Gavrilets, S., 2004. Fitness Landscapes and the Origin of Species. Princeton University Press, Princeton, NJ.
- Hedrick, P. W., 1981. The establishment of chromosomal variants. Evolution 35 322–332. [DOI] [PubMed] [Google Scholar]
- Hoffmann, A. A., C. M. Sgro and A. R. Weeks, 2004. Chromosomal inversion polymorphisms and adaptation. Trends Ecol. Evol. 19 482–488. [DOI] [PubMed] [Google Scholar]
- Hollingshead, L., 1930. A lethal factor in Crepis effective only in interspecific hybrids. Genetics 15 114–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King, M., 1993. Species Evolution: The Role of Chromosomal Change. Cambridge University Press, Cambridge, UK.
- Kirkpatrick, M., and N. Barton, 2006. Chromosome inversions, local adaptation, and speciation. Genetics 173 419–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knight, C. D., D. J. Cove, A. C. Cuming and R. S. Quatrano, 2002. Moss gene technology, pp. 285–301 in Molecular Plant Biology, edited by P. M. Gilmartin and C. Bowler. Oxford University Press, Oxford.
- Kubo, T., and A. Yoshimura, 2002. Genetic basis of hybrid breakdown in a Japonica/Indica cross of rice, Oryza sativa L. Theor. Appl. Genet. 105 906–911. [DOI] [PubMed] [Google Scholar]
- Kubo, T., and A. Yoshimura, 2005. Epistasis underlying female sterility detected in hybrid breakdown in a Japonica-Indica cross of rice (Oryza sativa L.). Theor. Appl. Genet. 110 346–355. [DOI] [PubMed] [Google Scholar]
- Lai, Z., T. Nakazato, M. Salmaso, J. M. Burke, S. Tang et al., 2005. Extensive chromosomal repatterning and the evolution of sterility barriers in hybrid sunflower species. Genetics 171 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lande, R., 1979. Effective deme sizes during long-term evolution estimated from rates of chromosomal rearrangement. Evolution 33 234–251. [DOI] [PubMed] [Google Scholar]
- MacNair, M. R., and P. Christie, 1983. Reproductive isolation as a pleiotropic effect of copper tolerance in Mimulus guttatus. Heredity 50 295–302. [Google Scholar]
- Malmberg, R. L., S. Held, A. Waits and R. Mauricio, 2005. Epistasis for fitness-related quantitative traits in Arabidopsis thaliana grown in the field and in the greenhouse. Genetics 171 2013–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDaniel, S. F., 2005. Genetic correlations do not constrain the evolution of sexual dimorphism in the moss Ceratodon purpureus. Evol. Int. J. Org. Evol. 59 2353–2361. [PubMed] [Google Scholar]
- McDaniel, S. F., and A. J. Shaw, 2005. Selective sweeps and intercontinental migration in the cosmopolitan moss Ceratodon purpureus (Hedw.) Brid. Mol. Ecol. 14 1121–1132. [DOI] [PubMed] [Google Scholar]
- McDaniel, S. F., J. H. Willis and A. J. Shaw, 2007. A linkage map reveals a complex basis for segregation distortion in an interpopulation cross in the moss Ceratodon purpureus. Genetics 176 2489–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyle, L. C., and E. B. Graham, 2005. Genetics of hybrid incompatibility between Lycopersicon esculentum and L. hirsutum. Genetics 169 355–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller, H. J., 1942. Isolating mechanisms, evolution, and temperature. Biol. Symp. 6 71–125. [Google Scholar]
- Nakazato, T., M.-K. Jung, E. A. Housworth, L. H. Rieseberg and G. J. Gastony, 2007. A genomewide study of reproductive barriers between allopatric populations of a homosporous fern, Ceratopteris richardii. Genetics 177 1141–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natcheva, R., and N. Cronberg, 2004. What do we know about hybridization among bryophytes in nature. Can. J. Bot. 82 1687–1704. [Google Scholar]
- Natcheva, R., and N. Cronberg, 2007. Recombination and introgression of nuclear and chloroplast genomes between the peat mosses, Sphagnum capillifolium and Sphagnum quinquefarium. Mol. Ecol. 16 811–818. [DOI] [PubMed] [Google Scholar]
- Navarro, A., and N. Barton, 2003. Accumulating postzygotic isolation genes in parapatry: a new twist on chromosomal speciation. Evolution 57 447–459. [DOI] [PubMed] [Google Scholar]
- Noor, M. A., K. L. Grams, L. A. Bertucci and J. Reiland, 2001. Chromosomal inversions and the reproductive isolation of species. Proc. Natl. Acad. Sci. USA 98 12084–12088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr, H. A., 1995. The population-genetics of speciation: the evolution of hybrid incompatibilities. Genetics 139 1805–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Barrientos, D., J. Reiland, J. Hey and M. A. F. Noor, 2002. Recombination and the divergence of hybridizing species. Genetica 116 167–178. [PubMed] [Google Scholar]
- Paterson, A. H., 2002. What has QTL mapping taught us about plant domestication? New Phytol. 154 591–608. [DOI] [PubMed] [Google Scholar]
- Porter, A. H., and N. A. Johnson, 2002. Speciation despite gene flow when developmental pathways evolve. Evolution 56 2103–2111. [DOI] [PubMed] [Google Scholar]
- Rieseberg, L. H., 2001. Chromosomal rearrangements and speciation. Trends Ecol. Evol. 16 351–358. [DOI] [PubMed] [Google Scholar]
- Rieseberg, L. H., and J. H. Willis, 2007. Plant Speciation. Science 317 910–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieseberg, L. H., J. Whitton and K. Gardner, 1999. Hybrid zones and the genetic architecture of a barrier to gene flow between two sunflower species. Genetics 152 713–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw, J., and S. C. Beer, 1999. Life history variation in gametophyte populations of the moss Ceratodon purpureus (Ditrichaceae). Am. J. Bot. 86 512. [PubMed] [Google Scholar]
- Shaw, A. J., and J. F. Gaughan, 1993. Control of sex-ratios in haploid populations of the moss, Ceratodon Purpureus. Am. J. Bot. 80 584–591. [DOI] [PubMed] [Google Scholar]
- Shaw, A. J., B. S. Weir and F. H. Shaw, 1997. The occurrence and significance of epistatic variance for quantitative characters and its measurement in haploids. Evolution 51 348–353. [DOI] [PubMed] [Google Scholar]
- Sites, J. W., and C. Moritz, 1987. Chromosomal evolution and speciation revisited. Syst. Zool. 36 153–174. [Google Scholar]
- Stebbins, G. L., 1958. The inviability, weakness, and sterility of interspecific hybrids. Adv. Genet. 9 147–215. [DOI] [PubMed] [Google Scholar]
- Stebbins, G. L., 1971. Chromosomal Evolution in Higher Plants. Edward Arnold, London.
- Stephens, S. G., 1946. The genetics of “corky.” I. The new world alleles and their possible role as an interspecific isolating mechanism. J. Genet. 47 150–161. [DOI] [PubMed] [Google Scholar]
- Sweigart, A. L., L. Fishman and J. H. Willis, 2006. A simple genetic incompatibility causes hybrid male sterility in mimulus. Genetics 172 2465–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turelli, M., and L. C. Moyle, 2007. Asymmetric postmating isolation: Darwin's corollary to Haldane's rule. Genetics 176 1059–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Velde, M., and R. Bijlsma, 2004. Hybridization and asymmetric reproductive isolation between the closely related bryophyte taxa Polytrichum commune and P. uliginosum. Mol. Ecol. 13 1447–1454. [DOI] [PubMed] [Google Scholar]
- Walsh, J. B., 1982. Rate of accumulation of reproductive isolation by chromosome rearrangements. Am. Nat. 120 510–532. [Google Scholar]
- Wang, S., C. J. Basten and Z.-B. Zeng, 2007. Windows QTL Cartographer. North Carolina State University, Raleigh, NC. http://statgen.ncsu.edu/qtlcart/WQTLCart.htm.
- Wettstein, F., 1924. Morphologie und physiologie des formwechsels der moose auf genetischer grundlage I. Zeitschr. fur induct. Abstammungs vererbungeslehr 33 1–236. [Google Scholar]
- Wettstein, F., 1928. Morphologie und physiologie des formwechsels der moose II. Biblio. Genetica 10 1–216. [Google Scholar]
- Wettstein, F., 1932. Genetik, pp. 233–272 in Manual of Bryology, edited by F. Verdoorn. Martinus Niehoff, The Hague, The Netherlands.
- White, M. J. D., 1978. Modes of Speciation. W. H. Freeman, San Francisco.
- Zeng, Z. B., 1994. Precision mapping of quantitative trait loci. Genetics 136 1457–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]