

Abstract
When a genetic marker and a quantitative trait locus (QTL) are in linkage disequilibrium (LD) in one population, they may not be in LD in another population or their LD phase may be reversed. The objectives of this study were to compare the extent of LD and the persistence of LD phase across multiple cattle populations. LD measures r and r2 were calculated for syntenic marker pairs using genomewide single-nucleotide polymorphisms (SNP) that were genotyped in Dutch and Australian Holstein–Friesian (HF) bulls, Australian Angus cattle, and New Zealand Friesian and Jersey cows. Average r2 was ∼0.35, 0.25, 0.22, 0.14, and 0.06 at marker distances 10, 20, 40, 100, and 1000 kb, respectively, which indicates that genomic selection within cattle breeds with r2 ≥ 0.20 between adjacent markers would require ∼50,000 SNPs. The correlation of r values between populations for the same marker pairs was close to 1 for pairs of very close markers (<10 kb) and decreased with increasing marker distance and the extent of divergence between the populations. To find markers that are in LD with QTL across diverged breeds, such as HF, Jersey, and Angus, would require ∼300,000 markers.
MARKER-ASSISTED selection in livestock breeding programs relies on linkage between quantitative trait loci (QTL) and genetic markers. Three types of genetic markers can be distinguished: (1) direct markers, loci that code for the functional mutation; (2) linkage disequilibrium (LD) markers, loci that are in populationwide LD with the functional mutation; and (3) linkage equilibrium markers, loci that are in populationwide linkage equilibrium with the functional mutation, but are linked to the functional mutation within some families (Dekkers 2004). Direct markers are very difficult to find and their functionality is hard to prove (Andersson 2001), whereas linkage-equilibrium markers have been found in many studies (Khatkar et al. 2004), but the application is complicated because they can be used only within families. LD markers are much easier to use in marker-assisted selection as their LD phase with the QTL is consistent throughout the population (Dekkers 2004). LD markers can be found after fine mapping genomic regions with dense markers (Meuwissen and Goddard 2000; Farnir et al. 2002) or from whole-genome QTL mapping experiments with dense markers (Macleod et al. 2006; Barendse et al. 2007). Instead of searching for LD markers and subsequently using them in marker-assisted selection, Meuwissen et al. (2001) proposed genomic selection, in which breeding values are predicted from all dense markers across the genome. Genomic selection also relies on sufficient LD between markers and QTL such that the marker allele–QTL allele phase persists across generations.
LD markers are always discovered in some reference population in which the initial experiment was conducted, for example, a genomewide association study. The value of the markers in populations other than the reference population will depend on the persistence of LD phase between the reference population and the second population (Dekkers and Hospital 2002). For example, a marker that has been identified as a LD marker in the Holstein-Friesian (HF) breed may not be in LD with the QTL in the Jersey breed. LD phase can be compared between two populations at many levels, for example, between breeds, between countries, or between populations of the same breed and within the same country but of different generations. If the marker and QTL are not in LD in the selection candidates, selecting for the marker will not lead to genetic improvement, and the genetic response may even be negative if the LD phase is reversed. The LD phase is more likely to be different between two populations when these populations have diverged for many generations, the effective population size is small, and distance between the marker and the QTL is large, as these factors will either break down the LD from the ancestral population or create new LD within the subpopulation (Hill and Robertson 1968).
For several purposes it is important to know the persistence of the LD phase across populations. For example, the persistence of LD phase between two populations may explain why LD markers that were discovered in one population could not be confirmed in a second population. Furthermore, if the persistence of LD phase is known for two subpopulations across a range of genomic distances, one can determine which marker-to-QTL distance will provide enough persistence of the LD phase across the two populations. This information is important to predict the required marker density for a fine-mapping experiment, for a genomewide association study or genomic selection (Meuwissen et al. 2001).
Goddard et al. (2006) studied the extent of LD and the persistence of LD phase between Australian HF and Angus cattle, whereas Gautier et al. (2007) compared 14 European and African cattle breeds. Both studies reported that the LD phase across these diverged breeds was consistent only when the marker distance was <10 kb. For populations that are more related to each other, for example, subpopulations of the same breed but in different countries, the persistence of LD phase is expected to extend across larger distances, but this information is not available in the literature.
The objective of this study was to compare linkage disequilibrium and persistence of LD phase of genetic markers in different subpopulations of cattle. This study is analogous to Goddard et al. (2006) but considers a wider range of cattle populations. The average LD at different genomic distances was used to infer the effective population size of cattle in the past, whereas the observed persistence of LD phase was used to infer time of divergence for the different populations.
MATERIALS AND METHODS
Genotypic data:
The data for this study were obtained from three independent experiments conducted in The Netherlands, Australia, and New Zealand. Within each experiment animals were genotyped for a set of single-nucleotide polymorphism (SNP) markers and LD measures r and r2 were calculated for all syntenic marker pairs. The underlying genotypic data were not shared for this study, which explains some differences in methodology.
The Dutch data were obtained from the Holland Genetics breeding program for HF dairy cattle, comprising 2430 animals that were genotyped for 3072 SNP markers across 30 chromosomes. The SNPs were selected from public databases on the basis of their genomic position and minor allele frequency, without preferences for SNPs from specific (QTL) regions. Genomic positions were based on the preliminary bovine sequence map obtained from the Baylor College of Medicine of the Human Genome Sequencing Center (Btau 3.1, http://www.hgsc.bcm.tmc.edu/). The SNPs were analyzed using the GoldenGate assay (Illumina, San Diego), in which two pools of 1536 oligos were used. The group of animals comprised 1485 progeny-tested bulls born between 1981 and 2002, 468 bull and heifer calves born in 2006, and 477 (grand)dams born between 1990 and 2004, which were either black and white (BW) or red and white (RW). These groups had 138, 44, and 158 different sires, respectively. The Dutch RW HF population can be characterized as a population that was originally a Dutch red dual-purpose breed but has been bred to North American Red HF for several generations. The SNPs used in the Dutch data, as well as the Australian and New Zealand data sets described below, were mapped to the National Center for Biotechnology Information bovine sequence map (Btau 3.1, http://www.ncbi.nlm.nih.gov/projects/genome/guide/cow/) by BLAST search (Altschul et al. 1997). Btau 4.0 was not yet available during this study, but this study focused mostly on pairs of close SNPs that in most cases did not change in distance to each other between Btau 3.1 and Btau 4.0. After exclusion of nonpolymorphic markers, markers with an unknown genomic position, and markers on the X and Y chromosomes, 2755 markers in the Dutch data were kept for further analysis. Genotypes that did not match the parents' genotypes were removed (<1%). In the Dutch data, haplotypes were constructed by comparing an animal's genotype at each marker locus to its parents' genotypes. If this was not informative, the animal's linkage phase with the nearest informative marker was assumed the same as in the majority of its progeny. After applying these rules, genotypes with unknown phase were removed from the data set.
The Australian data comprised 379 Angus animals and 383 HF progeny-tested bulls that were genotyped for 9323 and 9919 SNP markers, respectively, using Parallele (Affymetrix, Santa Clara, CA) (Goddard et al. 2006). The HF bulls were selected for either high or low estimated breeding values for the Australian selection index, which has primary emphasis on protein production. The Angus animals were selected from a research project based at Trangie Agricultural Research Centre, New South Wales, Australia and were born between 1993 and 2000. The Angus animals were selected for either high or low postweaning residual feed intake, which is a measure of feed efficiency (Arthur et al. 2001). All markers that were genotyped in the Angus animals were also genotyped in the HF bulls. Most SNPs were discovered in the bovine genome sequencing project by the Baylor College of Medicine of the Human Genome Sequencing Center (http://www.hgsc.bcm.tmc.edu/), and other SNPs were discovered as a result of the assembly of expressed sequence tags (Hawken et al. 2004). After exclusion of nonpolymorphic markers, markers with an unknown genomic position, markers that were genotyped only in the HF bulls, and markers on the X and Y chromosomes, the Angus and HF data set comprised 6927 markers. In the Australian data set, haplotypes were not inferred, but LD was measured directly from the genotypes, as explained later.
The New Zealand data were extracted from an F2 crossing experiment with Jersey and New Zealand HF cattle (Spelman and Coppieters 2006). From the HF × Jersey F1 animals, 430 Jersey maternal haplotypes and 365 HF maternal haplotypes were used for this analysis. The animals were genotyped for 9713 SNP markers, using Parallele. Markers with >50 inconsistencies of inheritance, with significant departure from Hardy–Weinberg equilibrium (P < 0.001), with minor allele frequency <5%, with an unknown genomic position, and on the X and Y chromosomes were removed, leaving 5928 markers for further analysis. The haplotypes were inferred using an expectation-maximization algorithm including information on the estimated sire phase, progeny genotype, and dam allele frequency.
After editing, the Australian and New Zealand data set had 5237 SNPs in common, whereas the Dutch data set had 1291 SNPs in common with the Australian data set and 1252 SNPs with the New Zealand data set. Comparisons between a Dutch population and either an Australian or a New Zealand population were therefore based on less markers. In all data sets, the distribution of SNPs was very uneven, which meant that many marker pairs were at close distances.
Subpopulations:
The data were categorized into Dutch BW HF bulls (HF_NLD), Dutch RW HF bulls (RW_NLD), Australian HF bulls (HF_AUS), Australian Angus animals (ANG_AUS), New Zealand Friesian maternal haplotypes (HF_NZL), and New Zealand Jersey maternal haplotypes (JER_NZL). Table 1 shows the number of animals, haplotypes, and markers for each category.
TABLE 1.
Number of animals, sires, haplotypes, and markers per country and breed
| Category | Abbreviation | No. animals | No. sires | Haplotypes usedb | No. haplotypes | No. SNPs before editing | No. SNPs after editing |
|---|---|---|---|---|---|---|---|
| Dutch BW HFa | HF_NLD | 1296 | 105 | Pat + mat | 2592 | 3072 | 2755 |
| Dutch RW HF | RW_NLD | 189 | 35 | Pat + mat | 378 | 3072 | 2755 |
| Australian HF | HF_AUS | 383 | 119 | Pat + mat | 766 | 9919 | 6927 |
| Australian Angus | ANG_AUS | 379 | 93 | Pat + mat | 758 | 9329 | 6927 |
| New Zealand HF | HF_NZL | 430 | 81 | Mat | 430 | 9713 | 5928 |
| New Zealand Jersey | JER_NZL | 365 | 67 | Mat | 365 | 9713 | 5928 |
BW, black and white; RW, red and white; HF, Holstein–Friesian.
Both paternal and maternal (pat + mat) or only maternal (mat) haplotypes were used.
To compare the persistence of LD phase across generations and between paternal and maternal haplotypes, the Dutch BW HF population was categorized into six groups: Dutch BW HF bulls, born before 1995, paternal haplotypes (Pre95_p, n = 348); Dutch BW HF bulls, born before 1995, maternal haplotypes (Pre95_m, n = 348); Dutch BW HF bulls, born after 1997, paternal haplotypes (Post97_p, n = 514); Dutch BW HF bulls, born after 1997, maternal haplotypes (Post97_m, n = 514); Dutch BW HF calves, born in 2006, paternal haplotypes (Calf_p, n = 369); and Dutch BW HF calves, born in 2006, maternal haplotypes (Calf_m, n = 369).
Comparison of linkage disequilibrium and phase:
Within each population or group, except for the Australian data, r was computed for each marker pair as 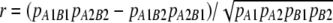 , where pA1B1 is the frequency of haplotypes with allele 1 at marker locus A and allele 1 at marker locus B and pA1 is the frequency of allele 1 at marker locus A (Hill and Robertson 1968). Marker alleles were numbered consistently across all data sets.
, where pA1B1 is the frequency of haplotypes with allele 1 at marker locus A and allele 1 at marker locus B and pA1 is the frequency of allele 1 at marker locus A (Hill and Robertson 1968). Marker alleles were numbered consistently across all data sets.
In the Australian data, r2 values were calculated for syntenic marker pairs using the LDMAX procedure of the GOLD program (Abecasis and Cookson 2000). The r values were calculated as the square root of r2 and were given the same sign as D, so the sign of r was consistent with the other data sets. D was calculated from the frequencies of all possible genotypes for markers A and B, as 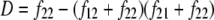 , where
, where
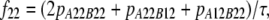 |
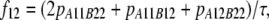 |
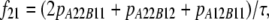 |
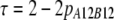 , and pA12B12 is the proportion of animals with genotype 12 at marker A and genotype 12 at marker B (Goddard et al. 2006).
, and pA12B12 is the proportion of animals with genotype 12 at marker A and genotype 12 at marker B (Goddard et al. 2006).
To determine the decay of LD with increasing distance between the markers, the average r2 within populations and the correlation of r across populations were expressed as a function of genomic distance. This was done by sorting the marker pairs on the basis of their distance and forming groups of n = 400 marker pairs with approximately equal genomic distance within group. Within each group of 400 marker pairs the average genomic distance, the average r2, and the correlation of r were calculated. The value of n = 400 was chosen to reduce stochastic variability across distance groups, but to still have enough distance groups to show the behavior of average r2 or the correlation of r as a function of distance. One exception was made, however, for calculation of average r2 for marker distance <100 kb; n = 200 was used. Standard errors of average r2 and correlation of r (corr) were calculated as 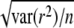 and
and  , respectively.
, respectively.
Past effective population size and time since divergence of breeds:
To interpret the observed average r2 at various distances, the effective population size at different stages in the past was estimated from the estimated average r2 at different marker distances, 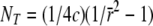 , where NT is the effective population size T generations ago, c is the marker distance in morgans, assuming 1 Mb = 1 cM, and
, where NT is the effective population size T generations ago, c is the marker distance in morgans, assuming 1 Mb = 1 cM, and 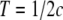 (Hayes et al. 2003; Goddard et al. 2006). These estimates are not extremely accurate because it is assumed that NT is constant, but they are approximately true if NT is changing linearly over time. Furthermore, various other factors influence the extent of LD as well (Ardlie et al. 2002), so the estimates should be regarded as approximations. Marker pairs with c < 10−6 (∼100 bp), i.e., T > 500,000, were not used because the approximation is valid only for c much larger than the mutation rate (∼10−8 per locus per generation).
(Hayes et al. 2003; Goddard et al. 2006). These estimates are not extremely accurate because it is assumed that NT is constant, but they are approximately true if NT is changing linearly over time. Furthermore, various other factors influence the extent of LD as well (Ardlie et al. 2002), so the estimates should be regarded as approximations. Marker pairs with c < 10−6 (∼100 bp), i.e., T > 500,000, were not used because the approximation is valid only for c much larger than the mutation rate (∼10−8 per locus per generation).
The decline in correlation of r between two breeds with increasing marker distance was used to estimate the number of generations since divergence of the breeds from a common ancestral population. We consider an ancestral population where two markers, A and B, are in LD with 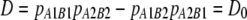 , where pA1B1 is the frequency of haplotypes with allele 1 at marker A and allele 1 at marker B, and
, where pA1B1 is the frequency of haplotypes with allele 1 at marker A and allele 1 at marker B, and 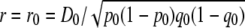 , where p0 and q0 are the initial allele frequencies at the markers. After T generations of divergence,
, where p0 and q0 are the initial allele frequencies at the markers. After T generations of divergence, 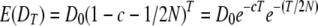 and
and 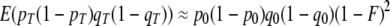 with
with 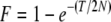 (Hill and Robertson 1968). This gives an expectation for r after T generations of divergence of
(Hill and Robertson 1968). This gives an expectation for r after T generations of divergence of 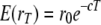 . New LD may be created in the two diverged lines, but this will arise independently, i.e., not contributing to the covariance of r between the diverged lines. Assuming that the variance of r remains constant in both breeds, the expected correlation of r is then
. New LD may be created in the two diverged lines, but this will arise independently, i.e., not contributing to the covariance of r between the diverged lines. Assuming that the variance of r remains constant in both breeds, the expected correlation of r is then 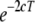 . The natural logarithm of the expected correlation of r then follows a linear decrease as a function of distance with slope −2T.
. The natural logarithm of the expected correlation of r then follows a linear decrease as a function of distance with slope −2T.
RESULTS
Linkage disequilibrium:
Average r2 decreased with increasing genomic distance for all defined populations (Figures 1 and 2). Each data point in Figures 1 and 2 represents the average r2 for 200 and 400 marker pairs, respectively. The lines for HF_NLD and RW_NLD have fewer data points because the number of markers in the Dutch data was much lower than those in the Australian and New Zealand data (Table 1). At marker distance <500 bp, average r2 was between 0.62 and 0.74 for HF_AUS, ANG_AUS, HF_NZL, and JER_NZ and between 0.40 and 0.61 for HF_NLD and RW_NLD. Around 5 kb, average r2 varied from 0.50 to 0.60 across all populations. For distances up to 100 kb, the populations showed a similar level and pattern of LD, with average r2 ∼ 0.35, 0.25, 0.22, 0.16, and 0.14 at marker distances 10, 20, 40, 80, and 100 kb, respectively (Figure 1). The largest difference in average r2 between populations was observed around 70–75 kb, where the average r2 was 0.10 for HF_NZL and 0.24 for ANG_AUS (Figure 1).
Figure 1.—

Average linkage disequilibrium (r2) as a function of average genomic distance for Dutch black-and-white Holstein–Friesian bulls (HF_NLD), Dutch red-and-white Holstein–Friesian bulls (RW_NLD), Australian Holstein–Friesian bulls (HF_AUS), Australian Angus animals (ANG_AUS), New Zealand Friesian cows (HF_NZL), and New Zealand Jersey cows (JER_NZL) for distances between 0 and 100 kb. Each data point was based on 200 marker pairs, resulting in standard errors ≤0.03.
Figure 2.—

Average linkage disequilibrium (r2) as a function of average genomic distance for Dutch black-and-white Holstein–Friesian bulls (HF_NLD), Dutch red-and-white Holstein–Friesian bulls (RW_NLD), Australian Holstein–Friesian bulls (HF_AUS), Australian Angus animals (ANG_AUS), New Zealand Friesian cows (HF_NZL), and New Zealand Jersey cows (JER_NZL) for distances between 100 and 1000 kb. Each data point was based on 400 marker pairs, resulting in standard errors ≤0.01.
For distances between 100 kb and 1 Mb (Figure 2), HF_NLD generally had the highest LD, followed by RW_NLD, then ANG_AUS and JER_NZL, and finally HF_AUS and HF_NZL. The average r2 for HF_NLD was generally twice that for HF_NZL. The average r2 was also calculated for Pre95_p, Pre95_m, Post97_p, Post97_m, Calf_p, and Calf_m, but these populations had almost identical average r2 to HF_NLD, and therefore these results are not shown.
The past effective population size, as estimated from the average r2 across genomic distances, showed that the effective population size of cattle was ≥10,000 at >10,000 generations ago (Figure 6). Around 1000 generations ago, the effective population size was already reduced to approximately a few thousand, whereas the most recent effective population size varies from 32 for RW_NLD to 135 for JER_NZL.
Figure 6.—

Effective population size along the population history, estimated from the average r2 at different marker distances, for Dutch black-and-white Holstein–Friesian bulls (HF_NLD), Dutch red-and-white Holstein–Friesian bulls (RW_NLD), Australian Holstein–Friesian bulls (HF_AUS), Australian Angus animals (ANG_AUS), New Zealand Friesian cows (HF_NZL), and New Zealand Jersey cows (JER_NZL). Data points were based on at least 400 marker pairs.
Persistence of LD phase:
As an example, Figure 3 shows the relationship between r obtained from HF_AUS and HF_NZL for 400 marker pairs with marker distance between 77 and 108 kb (average 93 kb). The correlation of r for HF_AUS vs. HF_NZL at this distance was 0.79. Across all populations, the correlation of r between populations decreased with increasing marker distance (Figure 4). For ease of reading, not all combinations of populations were included in Figure 4 but only the combinations of HF populations across countries, the combinations of HF and other breeds within country, and ANG_AUS vs. JER_NZL. For marker pairs that were <5 kb apart, the correlation of r was >0.97 for HF_NLD vs. RW_NLD and HF_NZL vs. JER_NZL and between 0.83 and 0.90 for HF_AUS vs. ANG_AUS, HF_AUS vs. HF_NZL, and ANG_AUS vs. JER_NZL. This means that if two markers at this distance were in LD in one population they showed very similar levels of LD in the other populations and that the LD phase of the marker alleles was the same. With increasing distance, the correlation of r decreased most rapidly for ANG_AUS vs. JER_NZL and decreased only slowly for HF_NLD vs. HF_AUS (Figure 4), which agrees with the greater divergence between the Angus and Jersey populations and the close genetic relationship between the HF populations. The correlation of r for HF_NLD vs. HF_NZL and HF_AUS vs. HF_NZL was lower than for HF_NLD vs. HF_AUS and HF_NLD vs. RW_NLD across all distances. This means that marker pairs that were in LD in the New Zealand HF population were more often not in LD in the other HF populations, or the LD phase was different.
Figure 3.—

Relationship between r in Australian Holstein–Friesian bulls (HF_AUS) and New Zealand Friesian cows (HF_NZL) for marker pairs with distance between 77 and 108 kb, averaging 93 kb (n = 400).
Figure 4.—

Correlation of r between populations as a function of genomic distance, for Dutch black-and-white Holstein–Friesian bulls (HF_NLD), Dutch red-and-white Holstein–Friesian bulls (RW_NLD), Australian Holstein–Friesian bulls (HF_AUS), Australian Angus animals (ANG_AUS), New Zealand Friesian cows (HF_NZL), and New Zealand Jersey cows (JER_NZL). Each data point was based on 400 marker pairs, resulting in standard errors of 0.02, 0.03, and 0.05 for correlations around 0.9, 0.8, and 0.2, respectively.
The correlation of r among different groups of Dutch black-and-white HF animals (Figure 5) showed that correlations of r among maternal haplotypes from different age groups, e.g., Pre95_m vs. Post97_m, were higher than the correlation of r among paternal haplotypes for the same age groups (Pre95_p vs. Post97_p). This means that LD that is observed in dams of bulls born before 1995 is very consistent with the LD in the dams of bulls born after 1997 and dams of calves born in 2006, whereas the LD that is observed in the sires of these groups is less consistent. Furthermore, Figure 5 shows that the correlation of r, for both maternal and paternal haplotypes, was highest for Post97 vs. Calf, followed by Pre95 vs. Post97, and was lowest for Pre95 vs. Calf. This corresponds to a decrease in correlation of r over time.
Figure 5.—

Correlation of r between groups of Dutch black-and-white Holstein–Friesian animals as a function of genomic distance, for progeny-tested bulls born before 1995 (Pre95), progeny-tested bulls born after 1997 (Post97), and calves born in 2006 (Calf), using maternal (extension “_m”) or paternal (extension “_p”) haplotypes. Each data point was based on 400 marker pairs, resulting in standard errors of 0.02, 0.03, and 0.05 for correlations around 0.9, 0.8, and 0.2, respectively.
DISCUSSION
Linkage disequilibrium:
Average r2's for JER_NZL were consistent with the values reported by Spelman and Coppieters (2006), which were based on a subset of the data used in this study. McKay et al. (2007) presented the extent of LD for eight cattle breeds (Dutch and American HF, Angus, Charolais, Limousin, Japanese Black, and Bos indicus breeds Brahman and Nelore) and observed quite similar average r2's across all B. taurus breeds that were also consistent with our results for HF_NLD and ANG_AUS. The average r2's for HF_AUS and ANG_AUS, however, were lower than those presented by Goddard et al. (2006), based on the same data. Further analysis revealed that this difference was completely caused by a technical error in their calculation of average r2. Consistent with Goddard et al. (2006), the average r2 in ANG_AUS was higher than that in HF_AUS.
Farnir et al. (2000) and Khatkar et al. (2006) used the absolute value of D′ (Lewontin 1964) to present LD in Dutch and Australian dairy bulls, respectively. For comparison, D′ was also calculated for HF_NLD (data not shown). The average absolute value of D′ in HF_NLD was very consistent with that in Farnir et al. (2000). Both Farnir et al. (2000) and Khatkar et al. (2006) concluded from their observations that useful LD extended for several centimorgans in cattle. However, the extent of useful LD is overestimated when using D′, where useful LD is defined as the proportion of QTL variance explained by a marker (Zhao et al. 2005; McKay et al. 2007).
The estimated average r2 for HF_AUS was substantially lower than that for HF_NLD, although both the Dutch and the Australian HF have been largely derived from the North American HF (Zenger et al. 2007). The reasons behind this result may be that the bulls used in HF_AUS came from a broader timeframe than the bulls used in HF_NLD and were selected for extremely high or low genetic merit. The estimated effective population size in the most recent generation was 64 for HF_NLD and 90 for HF_AUS (Figure 6). This is higher than estimates reported by Weigel (2001) and Sørensen et al. (2005), who calculated effective population sizes of 39 and 49 for U.S. and Danish HF, respectively, on the basis of rates of inbreeding, but lower than an effective population size of ∼100 using the rate of inbreeding for U.S. HF presented by Young and Seykora (1996). HF_NZL showed lower average r2 (Figures 1 and 2) and greater estimated effective population size (Figure 6) than HF_AUS and HF_NLD. The reason for this may be that in New Zealand the importation of North American HF bulls was not as extensive as in Australia and The Netherlands. The New Zealand Friesian population may therefore represent an admixture of a few breeds, including also other Friesian breeds (e.g., Dutch or British) that were imported into New Zealand at the end of the 19th and beginning of the 20th century (Jasiorowski et al. 1988), leading to a broader genetic base. The effect of these factors may be more pronounced in the HF_NZL data that were used in this study than in the whole New Zealand HF population because the proportion of imported HF genes in the HF_NZL data was only 20%, which is considerably smaller than the national average of 50%.
Past effective population size:
Figure 6 indicates that the effective size of the ancient cattle population was between 10,000 and 100,000 after the divergence of B. taurus and B. indicus, >100,000 generations ago (Machugh et al. 1997). Other support for this theory is the high average expected nucleotide diversity in the cattle genome, ∼0.0005 (M. E. Goddard, unpublished data), which corresponds to an effective population size of ∼10,000, averaged over time and assuming a mutation rate per locus per generation of 10−8. Given that the effective population size is ≪10,000 in the last 1000 generations, it must have been much larger before. Furthermore, some polymorphisms in cattle were also found in yak and bison, from which cattle diverged ∼2 million years ago (Maceachern 2007). MacEachern (2007) concluded from these observations that the effective population size of ancient cattle was ≫50,000. If the ancient cattle population had a population size of just a few thousand, drift would have moved many of these polymorphisms to fixation, although also hybridization of (domesticated) cattle with their ancestors may explain some of these polymorphisms (Beja-Pereira et al. 2006). After domestication, ∼10,000 generations ago, the effective population size decreased to a few thousand, whereas breed formation and artificial breeding techniques have decreased the effective population sizes to ∼100 over the last 50 generations (Figure 6), which is consistent with estimates of past effective population size by Gautier et al. (2007) for 14 European and African cattle breeds. Thévenon et al. (2007) used the same approach to estimate effective population size in a B. indicus × B. taurus cattle population in western Africa and obtained values ∼2000 for 50 generations in the past and ∼500 for recent generations. These higher estimates of effective population size were probably caused by the absence of intensive selection and inbreeding.
Persistence of LD phase:
The correlations of r for HF_AUS vs. ANG_AUS corresponded well with those reported by Goddard et al. (2006), which were based on the same data. Gautier et al. (2007) also observed that the correlation of r between European cattle breeds was on average 0.77 for markers <10 kb apart, but much lower for more distant markers. The correlations of r between populations are a result of the genetic relationship between the populations. Given that HF_NLD and HF_AUS are both largely derived from the same North American HF population (Zenger et al. 2007), it is not surprising that these populations have very high correlations of r, even for marker distances of >3 Mb. Because the genotypes were not shared for this study, it was not possible to calculate FST values between the populations. The RW_NLD population also had high correlations of r with HF_NLD, but less than with HF_AUS. The genetic relationships among other populations were much lower, with the lowest correlations of r for ANG_AUS vs. JER_NZL (Figure 4). The genetic relationship of HF_NZL with the other HF populations was surprisingly low, maybe because of the lower proportion of North American genes in the New Zealand HF population and especially in the animals used in this study. Correlations of r for HF_AUS vs. HF_NZL were only slightly higher than for HF_NZL vs. JER_NZL, which indicates that the HF_NZL animals in this study had almost the same genetic relationship to the North American HF population as to the New Zealand Jersey population. This may be because the New Zealand HF population was to some extent bred from Jerseys that were crossed to other dairy breeds, such as British Friesians and HF. This theory is supported by the correlations of r for HF_AUS vs. JER_NZL, which were much lower than for HF_NZL vs. JER_NZL.
The time since divergence between breeds (T) was approximated from the linear regression of the natural logarithm of the expected correlation of r on genomic distance. The slope of the regression is an approximation of −2T; i.e., T can be estimated from the slope divided by −2. For HF_AUS vs. ANG_AUS a value of T = 364 was estimated from the correlation of r (using data points with c < 400 kb), indicating that the HF and the Angus population have diverged ∼364 generations ago (Figure 7). Given that the effective size of both populations has decreased over this period, the variance of r has probably increased; i.e., there is more new LD, which will result in lower correlations of r and therefore a slight overestimation of T. For most other pairs of populations the decline in correlation of r did not follow this exponential function, for any T. A possible reason for this is that the populations have not really diverged from each other, but there has been some migration between the populations. In that case the new LD appears in both populations and causes a higher than expected correlation of r. In the same way that LD over long distances is representative of effective population size in the recent history (Hayes et al. 2003), the correlation of r over long distances may reflect migration in recent history. For example, for HF_NZL vs. JER_NZL the observed correlation of r followed the expected correlation of r for T = 191 for distances <400 kb. However, for distances between 1 and 10 Mb the expected correlation of r with T = 191 would be zero, whereas the observed correlation of r was 0.20 and 0.10, respectively (Figure 8). This may indicate that there are Jersey haplotypes that remained in the HF_NZL population, originating from somewhere between 5 and 50 generations ago.
Figure 7.—

Expected and realized correlation of r as a function of genomic distance (c) between Australian Holstein–Friesian bulls and Australian Angus animals, with expected correlation following exp(−2Tc) with T = 364.
Figure 8.—

Expected and realized correlation of r as a function of genomic distance (c) between New Zealand Friesian cows and New Zealand Jersey cows, with expected correlation following exp(−2Tc) with T = 191.
The persistence of LD phase among groups of paternal haplotypes was lower than among groups of maternal haplotypes (Figure 5). The reason for this may be that the sires within a generation represent a very small effective population, because only few elite bulls are used as sires of sons, whereas the dams represent a larger effective population. This may cause extensive LD within a group of sires from the same generation, but less correspondence of the LD phase over generations. The decay of LD phase over generations was relatively small for close markers, which means that the effects of LD markers can be used in marker-assisted selection for a number of generations. This slow decay in LD phase across generations was also observed in chicken populations (Heifetz et al. 2005) and is consistent with the expected decay of LD over generations (Hill and Robertson 1968).
Implications for QTL mapping and genomic selection:
The extent of “useful” LD in a population is often used to determine the appropriate marker density for QTL fine mapping or genomic selection, but the criterion for what level of LD is useful varies (Pritchard and Przeworski 2001; Ardlie et al. 2002; Zhao et al. 2005). Farnir et al. (2000) found the average absolute value of D′ > 0.50 in cattle for markers that were <5 cM apart and suggested that ∼1500 microsatellite markers could be sufficient for an initial LD screening. Meuwissen et al. (2001) predicted breeding values from dense markers across the whole genome and obtained accuracies up to 0.85 in simulation. Their simulation resulted in an average r2 between adjacent markers of 0.20. To obtain similar LD between adjacent markers requires a marker interval of ∼70 kb for HF_NLD and RW_NLD, ∼60 kb for ANG_AUS, ∼50 for HF_AUS, and ∼40 kb for HF_NZL and JER_NZL (Figure 1), which corresponds to between 43,000 (HF_NLD and RW_NLD) and 75,000 (HF_NZL and JER_NZL) evenly distributed markers across the genome. An alternative method using 10-marker haplotypes with identical-by-descent probabilities based on LD and linkage to model their covariance (Meuwissen and Goddard 2004) gave similar accuracies, using a sparser marker map (average r2 > 0.15), whereas single-marker regression needed a slightly denser map (average r2 > 0.21; Calus et al. 2008). A threshold of r2 > 0.15 reduces the required number of markers by a factor of 1.5–2, compared to r2 > 0.20 (Figure 1).
Zhao et al. (2007) compared the power and precision of several methods for LD mapping based on simulated data and concluded that single-marker regression was equal or superior to other regression methods and comparable to LD mapping using haplotypes and identical-by-descent probabilities. Their results, however, were based on simulated data with very high LD; i.e., average r2 was 0.41 for markers within 0.5 cM and 0.15 for markers within 1.5–2 cM, which is much higher than in the cattle populations analyzed in this study. Ardlie et al. (2002) and Du et al. (2007) propose thresholds of r2 > 0.33 and r2 > 0.3 for usable LD, respectively, because lower values of r2 would increase the required sample size of an association study to unfeasible numbers. To obtain these levels of LD between adjacent markers, the marker intervals should be reduced to 10–15 kb (Figure 1), or 200,000–300,000 markers genomewide, which is not much less than what has been proposed for genomewide association studies in humans (Kruglyak 1999; Pritchard and Przeworski 2001; Ardlie et al. 2002).
LD markers that are found in HF_NLD, RW_NLD, or HF_AUS will have very similar effects across these populations because correlations of r remained above 0.90 for hundreds of kilobases (Figure 4). For other pairs of populations, however, the persistence of LD phase extended for much shorter distances, i.e., tens of kilobases for HF_NLD vs. HF_NZL, HF_AUS vs. HF_NZL, and HF_NZL vs. JER_NZL and <10 kb for HF_AUS vs. ANG_AUS and ANG_AUS vs. JER_NZL. If the aim is to find markers that work consistently (i.e., correlation of r > 0.80) in HF and New Zealand Friesians, the marker-to-QTL interval should not be > ∼30 kb, which corresponds to at least ∼50,000 markers, equally distributed across the genome. This value is consistent with the proposed marker density when aiming for an average r2 > 0.20 between adjacent markers (43,000–75,000, depending on population). To obtain also similar LD phase in JER_NZL and ANG_AUS, the marker-to-QTL interval should be < ∼5 kb, or ∼300,000 genomewide markers.
Many studies have identified significant LD over long distances in cattle (Farnir et al. 2000; Tenesa et al. 2003; Khatkar et al. 2006) and other livestock species (McRae et al. 2002; Heifetz et al. 2005; Du et al. 2007). This study showed that LD in cattle decays rapidly over short distances (Figure 1), but remains above zero for great distances (Figure 2), which is consistent with the decreasing effective population size in cattle (Figure 6; Hayes et al. 2003). This implies that one or more markers may explain the variation in a QTL, even if they are quite distant from the QTL. As a result, Meuwissen et al. (2001) obtained high accuracies of genomic breeding values with an average r2 between adjacent markers of only 0.20. The extent of some LD over long distances, however, negatively affects precision in QTL mapping (Pritchard and Przeworski 2001). A potential solution to this problem is to map QTL in multiple populations simultaneously (Barendse et al. 2007), as the LD phase between marker and QTL will persist across multiple populations only if the distance between the QTL and a marker is small (Figure 4). For populations with higher effective size, such as humans, there is much less LD over long distances, which means that only markers very close to QTL may explain the variation of the QTL. For these populations, whole-genome association studies may require the average r2 between adjacent markers to be >0.20, as used by Meuwissen et al. (2001).
Conclusions:
In the cattle populations studied, LD decayed rapidly with increasing genomic distance, but remained present for great distances. The decay in LD indicated that the effective population size in cattle decreased from ∼10,000 ∼10,000 generations ago, then decreased to a few thousand after domestication, and further decreased to ∼100 for modern cattle populations. Within populations the marker distance at which r2 dropped below 0.20 varied from 30–60 kb in New Zealand Friesian and Jersey to 50–90 kb in Dutch HF. The persistence of LD phase extended for hundreds of kilobases between Dutch and Australian HF, but only for tens of kilobases between Dutch or Australian HF and New Zealand Friesians, and for <10 kb between Angus and Jersey. The persistence of LD phase for Holstein and Angus indicated that these breeds diverged ∼300–400 generations ago. The results imply that for genomic selection within HF, Jersey, or Angus ∼50,000 markers may be required, but ∼300,000 markers are needed to obtain consistent marker effects across these breeds.
References
- Abecasis, G. R., and W. O. C. Cookson, 2000. GOLD—graphical overview of linkage disequilibrium. Bioinformatics 16 182–183. [DOI] [PubMed] [Google Scholar]
- Altschul, S. F., T. L. Madden, A. A. Schäffer, J. Zhang, Z. Zhang et al., 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson, L., 2001. Genetic dissection of phenotypic diversity in farm animals. Nat. Rev. Genet. 2 130–138. [DOI] [PubMed] [Google Scholar]
- Ardlie, G. A., L. Kruglyak and M. Seielstad, 2002. Patterns of linkage disequilibrium in the human genome. Nat. Rev. Genet. 3 299–309. [DOI] [PubMed] [Google Scholar]
- Arthur, P. F., J. A. Archer, D. J. Johnston, R. M. Herd, E. C. Richardson et al., 2001. Genetic and phenotypic variance and covariance components for feed intake, feed efficiency, and other postweaning traits in Angus cattle. J. Anim. Sci. 79 2805–2811. [DOI] [PubMed] [Google Scholar]
- Barendse, W., A. Reverter, R. J. Bunch, B. E. Harrison, W. Barris et al., 2007. A validated whole genome association study of efficient food conversion in cattle. Genetics 176 1893–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beja-Pereira, A., D. Caramelli, C. Lalueza-Fox, C. Vernesi, N. Ferrand et al., 2006. The origin of European cattle: evidence from modern and ancient DNA. Proc. Natl. Acad. Sci. USA 103 8113–8118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calus, M. P. L., T. H. E. Meuwissen, A. P. W. De Roos and R. F. Veerkamp, 2008. Accuracy of genomic selection using different methods to define haplotypes. Genetics 178 553–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dekkers, J. C. M., 2004. Commercial application of marker- and gene-assisted selection in livestock: strategies and lessons. J. Anim. Sci. 82(E. Suppl.): E313–E328. [DOI] [PubMed] [Google Scholar]
- Dekkers, J. C. M., and F. Hospital, 2002. The use of molecular genetics in the improvement of agricultural populations. Nat. Rev. Genet. 3 22–32. [DOI] [PubMed] [Google Scholar]
- Du, F.-X., A. C. Clutter and M. M. Lohuis, 2007. Characterizing linkage disequilibrium in pig populations. Int. J. Biol. Sci. 3 166–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farnir, F., W. Coppieters, J.-J. Arranz, P. Berzi, N. Cambisano et al., 2000. Extensive genomie-wide linkage disequilibrium in cattle. Genome Res. 10 220–227. [DOI] [PubMed] [Google Scholar]
- Farnir, F., B. Grisart, W. Coppieters, J. Riquet, P. Berzi et al., 2002. Simultaneous mining of linkage and linkage disequilibrium to fine map quantitative trait loci in outbred half-sib pedigrees: revisiting the location of a quantitative trait locus with major effect on milk production on bovine chromosome 14. Genetics 161 275–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautier, M., T. Faraut, K. Moazami-Goudarzi, V. Navratil, M. Foglio et al., 2007. Genetic and haplotypic structure in 14 European and African cattle breeds. Genetics 177 1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goddard, M. E., B. J. Hayes, H. McPartlan and A. J. Chamberlain, 2006. Can the same genetic markers be used in multiple breeds? Proceedings of the 8th World Congress on Genetics Applied to Livestock Production, Belo Horizonte, Brazil, August 13–18, 2006. CD-ROM communication no. 22-16.
- Hawken, R. J., W. C. Barris, S. M. McWilliam and B. P. Dalrymple, 2004. An interactive bovine in silico SNP database (IBISS). Mamm. Genome 15 819–827. [DOI] [PubMed] [Google Scholar]
- Hayes, B. J., P. M. Visscher, H. C. McPartlan and M. E. Goddard, 2003. Novel multilocus measure of linkage disequilibrium to estimate past effective population size. Genome Res. 13 635–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heifetz, E. M., J. E. Fulton, N. O'Sullivan, H. Zhao, J. C. M. Dekkers et al., 2005. Extent and consistency across generations of linkage disequilibrium in commercial layer chicken breeding populations. Genetics 171 1173–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, W. G., and A. Robertson, 1968. Linkage disequilibrium in finite populations. Theor. Appl. Genet. 38 226–231. [DOI] [PubMed] [Google Scholar]
- Jasiorowski, H. A., M. Stolzman and Z. Reklewski, 1988. The international Friesian strain comparison trial: a world perspective. Food Agriculture Organization of the United Nations, Rome.
- Khatkar, M. S., P. C. Thomson, I. Tammen and H. W. Raadsma, 2004. Quantitative trait loci mapping in dairy cattle: review and meta-analysis. Genet. Sel. Evol. 36 136–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatkar, M. S., A. Collins, J. A. L. Cavanagh, R. J. Hawken, M. Hobbs et al., 2006. A first-generation metric linkage disequilibrium map of bovine chromosome 6. Genetics 174 79–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruglyak, L., 1999. Prospects for whole-genome linkage disequilibrium mapping of common disease genes. Nat. Genet. 22 139–144. [DOI] [PubMed] [Google Scholar]
- Lewontin, R. C., 1964. The interaction of selection and linkage. I. General considerations; heterotic models. Genetics 49 49–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maceachern, S. A., 2007. Molecular evolution of the domesticated cow (Bos taurus). Ph.D. Thesis, La Trobe University, Bundoora, Australia.
- Machugh, D. E., M. D. Shriver, R. T. Loftus, P. Cunningham and D. G. Bradley, 1997. Microsatellite DNA variation and the evolution, domestication and phylogeography of taurine and zebu cattle (Bos taurus and Bos indicus). Genetics 146 1071–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macleod, I. M., B. J. Hayes and M. E. Goddard, 2006. Efficiency of dense bovine single-nucleotide polymorphisms to detect and position quantitative trait loci. Proceedings of the 8th World Congress on Genetics Applied to Livestock Production, Belo Horizonte, Brazil, August 13–18, 2006. CD-ROM communication no. 20-04.
- McKay, S. D., R. D. Schnabel, B. M. Murdoch, L. K. Matukumalli, J. Aerts et al., 2007. Whole genome linkage disequilibrium maps in cattle. BMC Genet. 8 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRae, A. F., J. C. McEwan, K. G. Dodds, T. Wilson, A. M. Crawford et al., 2002. Linkage disequilibrium in domesticated sheep. Genetics 160 1113–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., and M. E. Goddard, 2000. Fine-mapping of quantitative trait loci using linkage disequilibria with closely linked markers. Genetics 155 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., and M. E. Goddard, 2004. Mapping multiple QTL using linkage disequilibrium and linkage analysis information and multitrait data. Genet. Sel. Evol. 36 261–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuwissen, T. H. E., B. J. Hayes and M. E. Goddard, 2001. Prediction of total genetic value using genome-wide dense marker maps. Genetics 157 1819–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K., and M. Przeworski, 2001. Linkage disequilibrium in humans: models and data. Am. J. Hum. Genet. 69 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen, A. C., M. K. Sørensen and P. Berg, 2005. Inbreeding in Danish dairy cattle breeds. J. Dairy Sci. 88 1865–1872. [DOI] [PubMed] [Google Scholar]
- Spelman, R. J., and W. Coppieters, 2006. Linkage disequilibrium in the New Zealand Jersey population. Proceedings of the 8th World Congress on Genetics Applied to Livestock Production, Belo Horizonte, Brazil, August 13–18, 2006. CD-ROM communication no. 22-21.
- Tenesa, A., S. A. Knott, D. Ward, D. Smith, J. L. Williams et al., 2003. Estimation of linkage disequilibrium in a sample of the United Kingdom dairy cattle population using unphased genotypes. J. Anim. Sci. 81 617–623. [DOI] [PubMed] [Google Scholar]
- Thévenon, S., G. K. Dayo, S. Sylla, I. Sidibe, D. Berthier et al., 2007. The extent of linkage disequilibrium in a large cattle population of western Africa and its consequences for association studies. Anim. Genet. 38 277–286. [DOI] [PubMed] [Google Scholar]
- Weigel, K. A., 2001. Controlling inbreeding in modern breeding programs. J. Dairy Sci. 84(E. Suppl.): E177–E184. [Google Scholar]
- Young, C. W., and A. J. Seykora, 1996. Estimates of inbreeding and relationship among registered Holstein females in the United States. J. Dairy Sci. 79 502–505. [DOI] [PubMed] [Google Scholar]
- Zenger, K. R., M. S. Khatkar, J. A. L. Cavenagh, R. J. Hawken and H. W. Raadsma, 2007. Genome-wide genetic diversity of Holstein Friesian cattle reveals new insights into Australian and global population variability, including impact of selection. Anim. Genet. 38 7–14. [DOI] [PubMed] [Google Scholar]
- Zhao, H., D. Nettleton, M. Soller and J. C. M. Dekkers, 2005. Evaluation of linkage disequilibrium measures between multi-allelic markers as predictors of linkage disequilibrium between markers and QTL. Genet. Res. 86 77–87. [DOI] [PubMed] [Google Scholar]
- Zhao, H. H., R. L. Fernando and J. C. M. Dekkers, 2007. Power and precision of alternate methods for linkage disequilibrium mapping of quantitative trait loci. Genetics 175 1975–1986. [DOI] [PMC free article] [PubMed] [Google Scholar]