

Abstract
Focal adhesion kinase (FAK) regulates cell migration, proliferation, and apoptosis. FAK protein is reduced at the edge of migrating gut epithelial sheets in vitro, but it has not been characterized in restitutive gut mucosa in vivo. Here we show that FAK and activated phospho-FAK (FAK397) immunoreactivity was lower in epithelial cells immediately adjacent to human gastric and colonic ulcers in vivo, but dramatically increased in epithelia near the ulcers, possibly reflecting stimulation by growth factors absent in vitro. Transforming growth factor (TGF)-β, but not fibroblast growth factor, platelet-derived growth factor, or vascular endothelial growth factor, increased FAK levels in Caco-2 and IEC-6 cells. Epithelial immunoreactivity to TGF-β and phospho-Smad3 was also higher near the ulcers, varying in parallel with FAK. The TGF-β receptor antagonist SB431542 completely blocked TGF-β-induced Smad2/3 and p38 activation in IEC-6 cells. SB431542, the p38 antagonist SB203580, and siRNA-mediated reduction of Smad2 and p38α prevented TGF-β stimulation of both FAK transcription and translation (as measured via a FAK promoter-luciferase construct). FAK397 levels were directly related to total FAK protein expression. Although gut epithelial motility is associated with direct inhibition of FAK protein adjacent to mucosal wounds, TGF-β may increase FAK protein near but not bordering mucosal ulcers via Smad2/3 and p38 signals. Our results show that regulation of FAK expression may be as important as FAK phosphorylation in critically influencing gut epithelial cell migration after mucosal injury.
Survival and function of epithelial cells depends critically on interactions between the extracellular matrix and cell surface integrins. These interactions are mediated through focal adhesions, multicomponent juxtamembrane structures that lie at the convergence of integrin adhesion, signaling, and the cytoskeleton.1 Focal adhesion kinase (FAK) is an important nonreceptor tyrosine kinase within the focal adhesion complex.2 Originally described as being rapidly tyrosine phosphorylated on integrin-mediated cell-matrix adhesion, FAK is now known to be activated during epithelial cell motility and phosphorylated at both serine and tyrosine residues by various factors.3,4,5 Once localized to sites of transmembrane integrin receptor clustering, tyrosine-phosphorylated FAK plays a central role in signal transduction triggered by diverse extracellular signals6 and represents a convergent point for synergistic interaction between signal pathways activated by growth factors and integrins.7 FAK binds to different signaling proteins via Src homology 2 (SH2) and SH3 recognition sites, acting as a scaffold and transmitting signals crucial to cell survival, motility, proliferation, and differentiation.8 FAK also regulates the cycle of focal contact formation and disassembly required for efficient cell movement and thus, mucosal restitution.9
Levels of FAK protein expression and/or activation have been correlated to phenotypic changes that affect cell differentiation and function, notably adhesion and migration, in a number of tissues.10,11,12,13 Targeted FAK deletion in vascular endothelial cells leads to apoptosis and aberrant cell movement whereas FAK overexpression results in increased angiogenesis.14,15 Total and activated FAK levels are also directly related to the state of differentiation in human colon cancer.16 Induction of differentiation in dedifferentiated, rounded, detached Colo201/205 human cancer cells increases both FAK protein and FAK activation and enhances cell adhesion.17 Furthermore, inhibition of FAK activation via transfection of FRNK, a nonphosphorylatable, truncated FAK, decreases adhesion and wound healing in 293 nonmalignant colon cancer cells exposed to gastrin-releasing peptide.18 Activated FAK397 levels vary with differentiation and cell migration in Caco-2 and HT-29 human colon cancer cells.19 In leukocytes, however, FAK overphosphorylation was associated with cessation of motility.20 In summary, modulating either FAK activity or abundance directly affects intestinal epithelial motility21,22 as well as tumor cell invasion.23,24 Finally, the level of expression of activated FAK has been linked to gastric wound healing in vivo,25,26 although these investigators did not evaluate levels of total FAK protein to determine whether the changes in phosphorylated FAK that they had observed might correlate with changes in total FAK protein.
Indeed, although most studies of FAK function have focused on rapid phosphorylation events,27 FAK protein levels may also be regulated, influencing the amount of FAK available to be phosphorylated, and such regulation may influence both normal and pathological states. FAK expression is altered in some cancers28,29 and increased during Caco-2 intestinal epithelial cell differentiation.30 We previously reported that immunoreactivity for FAK is decreased in chronically motile compared to static Caco-2 cells, so that although motility seems to activate the available FAK, there is actually far less activated FAK in motile cells because there is less FAK available to be activated.22 Indeed, intestinal epithelial cell motility modulates FAK protein abundance at the mRNA level in both human Caco-2 and rat nontransformed IEC-6 intestinal epithelial cells.31 Such changes in FAK protein levels dramatically alter the amount of phosphorylated (active) FAK available within motile cells independently of (and more substantially than) motility-driven changes in the proportion of activated FAK.
However, cell culture observations do not necessarily extrapolate to in vivo physiology. In this study, we therefore examined total and activated (FAK397) FAK immunoreactivity in archival specimens from human gastric and colonic ulcers. We found that FAK and FAK397 immunoreactivity were indeed lower in epithelial cells at the leading, migrating edge of the ulcer, consistent with our previously reported in vitro observations in Caco-2 and IEC-6 cells.5,22,31 However, the epithelial region immediately behind the ulcer margin exhibited dramatically enhanced FAK protein immunostaining that then tapered off distant from the ulcer. Phosphorylated FAK397 immunoreactivity followed the same pattern, suggesting that such changes in FAK protein critically regulate the amount of activated FAK present in vivo.
Efficient ulcer healing requires interplay between inflammatory cells, the extracellular matrix, and locally released cytokines and growth factors.26 In particular, tissue injury and wound healing spatially and temporally activate several growth factors, including epidermal growth factor (EGF), platelet-derived growth factor (PDGF), hepatocyte growth factor (HGF), transforming growth factor (TGF)-β, vascular endothelial growth factor (VEGF), trefoil peptides, and angiopoietins,32 as well as their receptors.33 We therefore further hypothesized that growth factors released at ulcer sites in vivo, but absent in vitro, might cause this increase in FAK protein levels near but not precisely at the ulcer border not identified in vitro in migrating intestinal epithelial cells. Of the growth factors we examined in vitro, only TGF-β increased FAK protein expression in both Caco-2 and IEC-6 cells. Indeed, TGF-β immunoreactivity also co-localized with FAK immunoreactivity in human gut mucosal ulcers as well as expanding intestinal epithelial IEC-6 rat cell monolayers, supporting a role for TGF-β in FAK protein induction. Additional studies further bolstered this model, demonstrating that TGF-β stimulates FAK expression via Smad and p38 MAP kinase pathways in IEC-6 cells.
Materials and Methods
Cells and Cell Culture
IEC-6 cells, originally derived from rat jejunum,34 were purchased from the American Type Culture Collection (Rockville, MD) and maintained as per their recommendations. Human colon cancer Caco-2BBE cells were originally obtained from Dr. Michelle Peterson.35,36 All cells were propagated in tissue culture flasks, but were studied in dishes precoated with type I collagen (Sigma, St. Louis, MO) as previously described.37
Growth Factor Treatment
TGF-β Signal Activation
For these experiments, IEC-6 cells grown to 70% confluence and serum-deprived for 6 hours were exposed to TGF-β (1 ng/ml; R&D Systems, Minneapolis, MN, and Sigma) in medium containing 0.5% fetal bovine serum for 1 hour before lysis. Preliminary dose response studies had demonstrated that this concentration of TGF-β was optimal for FAK induction (not shown). For pharmacological signal inhibition, cells were pretreated for 1 hour with either the TGF-β receptor I (ALK5) antagonist, SB431542 (0.5 μmol/L; Tocris, Ellisville, MO), or the p38 inhibitor, SB203580 (10 μmol/L; EMD Biosciences, Gibbstown, NJ), dissolved in dimethyl sulfoxide (DMSO) (0.1% final volume). Control cells were treated with equal volumes of DMSO. Signal activation was quantitated by Western immunoblot.
FAK Expression
To monitor FAK expression in response to growth factors, Caco-2 and IEC-6 cells were grown to 60 to 70% confluence before serum starvation (24 hours for Caco-2 cells and 6 hours for IEC-6 cells). The cells were exposed to basic FGF (5 ng/ml + 10 U heparin, EMD Biosciences), PDGF-BB (2.5 ng/ml; Oncogene Science, Cambridge, MA), TGF-β1 (1 ng/ml, Sigma) or VEGF (5 ng/ml, EMD Biosciences) in medium containing 0.5% fetal bovine serum for 24 hours before lysis. In some experiments, TGF-β signals were inhibited by pretreatment with SB431542 or SB203580. In other experiments, Smad2 and p38α protein levels were decreased with specific siRNA. Changes in FAK protein abundance and activated FAK397 levels were monitored by Western blot, FAK mRNA abundance by reverse transcriptase-polymerase chain reaction (RT-PCR), and FAK promoter activity by luciferase assay using a FAK promoter-luciferase construct created as described below.
Smad2 and p38α Protein Inhibition with siRNA
Rat Smad2 and p38α (MAPK14) SMARTpool and nontargeting (NT1) duplexes were purchased from Dharmacon (Dallas, TX). siRNA was introduced into IEC-6 (100 nmol/L) with a mix of Oligofectamine and Plus Reagent in OptiMEM by a slight modification of the manufacturer’s protocol (Invitrogen, Carlsbad, CA). After 6 hours, the OptiMEM was supplemented with an equal volume of 2× fetal bovine serum medium and the cells allowed to expand for 24 hours before serum starvation for 6 hours and treatment with 1 ng/ml of TGF-β in medium with 0.5% fetal bovine serum for a further 24 hours. At that time, cells were lysed for Smad2, p38α, total p38, and total FAK/FAK397 protein level analysis by Western immunoblot.
Western Immunoblot
For Western blotting, cells were collected in lysis buffer (20 mmol/L sodium phosphate, 50 mmol/L NaCl, 5 mmol/L ethylenediaminetetraacetic acid, 0.5% sodium dodecyl sulfate, pH 7.4, supplemented with 1 mmol/L phenylmethyl sulfonyl fluoride, 1 mmol/L Na3VO4, 10 μg/ml leupeptin, and 1 μg/ml aprotinin). After centrifugation, supernatant protein concentrations were measured by the BCA method (Pierce, Rockford, IL). Equal protein samples were resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and electrophoretically transferred onto nitrocellulose membranes. The following primary antibodies were used: 1) for FAK expression and activation, total FAK (BD Transduction, San Jose, CA), or FAK clone 4.47 (Upstate Biotechnology, Lake Placid, NY), FAKY397 (Biosource International, Camarillo, CA), and α-tubulin (EMD Biosciences) as control; 2) to ascertain Smad2 and p38α inhibition by siRNA, Smad2/3, and Smad2 antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, and Cell Signaling, Danvers, MA, respectively), p38α and total p38 antibody (Cell Signaling) and α-tubulin or GAPDH (Biodesign, Saco, ME) antibodies as control; and 3) for signal activation, a pSmad3 antibody that recognizes Smad2 and Smad3 phosphorylated at serines 423/425 and phospho-p38 antibody (both from Cell Signaling). To prevent skewing of the data because of problems with incomplete stripping, duplicate samples were analyzed for the total and activated forms of FAK, p38, and Smad and each normalized to the corresponding α-tubulin or GAPDH control. After incubation with the appropriate secondary antibody, the protein bands were visualized by enhanced chemiluminescence (GE Health Care, Little Chalfont, Buckinghamshire, UK). Imaging and analysis of band density were performed on a Kodak 440CF Image Station (Eastman Kodak, Rochester, NY). All exposures used for densitometric analysis were within the linear range of the system.
Luciferase Assay for FAK Promoter Activity
To assemble the FAK-promoter-luciferase construct we first obtained a human genomic DNA-BAC clone from BACPAC Resources (Children’s Hospital of the Oakland Research Institute, Oakland, CA) with an insert containing the human FAK promoter, exon 1, and intron 1 sequences as indicated in the GenBank data base. The BAC plasmid was cut with SacI and StuI (New England Biolabs, Ipswich, MA) releasing a fragment 1.65 kb in size that was subcloned into the pBluescript II SK (+) vector (Stratagene, La Jolla, CA) for amplification. The pBSK-FAK-promoter clone was subsequently cut with NarI, end-filled, and then cut with KpnI (New England Biolabs) to produce a 1.3-kb fragment that was subcloned into a pGL2 Basic vector (Promega, Madison, WI) to generate the FAK-promoter-luciferase construct used in the dual luciferase reporter assay System (Promega). The 1.3-kb fragment was sequenced using T3 and T7 primers (New England Biolabs) to verify that it matched the FAK genomic sequence in GenBank (not shown).
RT-PCR for FAK Expression
RT-PCR for FAK was performed as described previously.31 Briefly, RNA was isolated from control and TGF-β-treated Caco-2 and IEC-6 cells with the Qiagen Total RNA isolation kit (Qiagen, Valencia, CA) with digestion of DNA with DNase I. Complete digestion was tested for by standard PCR with primers for the control 18S gene (5′-CGGCTACCACATCCAAGGAA-3′ and 5′-GCTCGAATTACCGCGGCT-3′; Integrated DNA Technologies, Coralville, IA). The FAK primer pairs were designed with the help of the MIT Prime3 online primer designer protocol from the human FAK gene sequence.38 The primers: 5′-ATTGCTGCCTCGGAATGTRCT-3′ and 5′-GCTGAGGTAAAACGTCGAAAA-3′ (IDT) yielded a 167-base product. RT-PCR was performed on the cDNA template transcribed from total RNA extracted from Caco-2 and IEC-6 cells with the TaqDNA polymerase kit (New England Biolabs, Ipswich, MA) and 10 mmol/L DNTP Mix (Invitrogen). Identity of the FAK product was confirmed by sequencing. RT-PCR was performed with SYBR Green (Invitrogen) using the ABI 7700 sequence detection system (Applied Biosystems, Foster City, CA). Relative FAK mRNA levels were determined by the comparative CT method (Applied Biosystems User Manual) after ascertaining that the amplification efficiencies of the FAK and 18S control primers were similar (K = 0.147).
tFAK, FAK397, TGF-β, and Phospho-Smad3 (pSmad3) Immunohistochemistry in Human Gastric and Colonic Ulcers
A SNOMED search of archival stomach and colonic ulcers, followed by microscopic examination of the hematoxylin and eosin slides for each case was used to select appropriate paraffin blocks. The blocks were serially sectioned (4 μm) and collected on poly-l-lysine-coated slides. Sections were deparaffinized with xylene and treated with citrate antigen retrieval buffer (DAKO, Carpinteria, CA) in a steamer at 95°C for 20 minutes and 3% hydrogen peroxide for 15 minutes. Nonspecific sites were blocked with dilute horse serum (Vector Laboratories, Burlingame, CA). After washing with phosphate-buffered saline (PBS), the slides were incubated overnight at 4°C with anti-FAK antibody (3 μg/ml, clone 4.47; Upstate Biotechnology), activated FAK397 antibody (2 μg/ml, Biosource International), TGF-β antibody (12.5 μg/ml, R&D Systems), phospho-Smad3 antibody (Cell Signaling), or cytokeratin control antibodies (AE1, AE3, and broad spectrum; Zymed, South San Francisco, CA). Rabbit or mouse γ-globulin (2 μg/ml; Sigma and Invitrogen, respectively) was used as a negative control. After extensive washing in PBS, the slides were incubated with biotinylated secondary antibody, streptavidin-peroxidase and amino-ethyl carbazol chromogen (all from the VectaStain universal rapid kit, Vector). Intensity of staining was continuously monitored for maximal development before light counterstaining with Mayer’s hematoxylin (Sigma) and mounting with Geltol (ThermoShandon, Fisher Scientific, Hanover Park, IL). We obtained a waiver of the requirement for informed consent from our Human Studies Subcommittee for this research involving access to archival tissue.
A total of 224 slides were examined: 89 were immunostained for tFAK, 45 for FAK397, 58 for TGF-β, 85 for pSmad3, and 5 for cytokeratins as controls. In addition, numerous negative controls using mouse or rabbit isotype serum in place of the primary antibodies were performed simultaneously with immunostaining of specific marker proteins. Immunoreactivity was evaluated by examining each slide for areas of mucosal epithelium adjacent to ulcerated regions. A single reviewer assigned scores on a scale of 0 to 4 for each of the three defined ulceration zones. A score of zero meant that no immunostaining was observed; a score of 1 indicated mild but discernible immunostaining; a score of 2 represented moderate immunostaining, distinguishable from a score of 1 or 3; whereas a score of 3 meant more intense immunostaining, distinguishable from a score of 2 or 4; a score of 4 was assigned to maximum staining intensity, exemplified by cytokeratin controls. Some slides showed multiple areas acceptable for evaluation; all such areas were scored.
FAK, FAK397, and TGF-β Immunocytochemistry in Expanding IEC-6 Cell Monolayers
To assess FAK and TGF-β protein abundance in motile cells, confluent cell monolayers were scraped with a razor blade and the cells were allowed to migrate over the wound edge for 24 hours. The cell monolayers were fixed with PLP (periodate, lysine, formaldehyde), and then permeabilized with 0.2% Triton X on ice. Staining was accomplished using blocking serum, secondary antibody, streptavidin-peroxidase, and chromogen (3,3′-diaminobenzidine) from the Vectastain universal rapid kit (Vector Laboratories). Cells were incubated with specific primary antibodies for total FAK (Upstate), activated tyrosine-phosphorylated FAK397 (Biosource), and TGF-β (R&D Systems) for 2 hours at 37°C. After optimal color development, cells were counterstained with hematoxylin and mounted in aqueous medium (Geltol).
Statistical Analysis
Results are expressed as mean ± SEM; differences between groups were evaluated with the Student’s t-test. Western blot results are shown as percentage of DMSO/NT1 control but P values were calculated using the raw data, the densitometric ratio of the signal of interest to its specific control. Scored differences in immunostaining density of a given marker protein in the three ulcer zones of all slides were statistically assessed by χ2 analysis. Statistical significance was set a priori at P < 0.05.
Results
Total and Active FAK Immunoreactivity Are Lower at the Immediate Healing Edge of the Epithelium but Greatly Increased in Epithelium Adjacent to Benign Human Ulcers
We reported previously that immunoreactive total and activated FAK protein abundance is lower in motile Caco-2 and IEC-6 cells.5,22,31 We now sought to determine whether FAK protein at the edge of healing gut mucosa decreases similarly in vivo. We stained for FAK and activated FAK397 in 30 benign human gastric and ischemic and malignant colonic ulcers. Figures 1 and 2 illustrate representative examples.
Figure 1.

Benign human gastric ulcers exhibit three zones of FAK and phospho-FAK (FAK397) immunoreactivity in vivo. Top: In this representative ulcer, expression of total FAK (purple/brown stain) is extremely low immediately adjacent to the ulcer bed (zone 1), greatly elevated at the healing edge (zone 2), and again much lower in the distant mucosa (zone 3). Bottom left: Similarly, immunostaining for phosphorylated FAK397, lower at the tip of the migrating epithelial sheet (zone 1), is increased in the healing mucosa (zone 2). Staining is most prominent at the basal aspect of the cells where they contact the basement membrane matrix and where FAK would be expected to be functional. Bottom right: Intensity of staining in the cytokeratin control was similar throughout. Original magnifications: ×100 (top); ×200 (bottom).
Figure 2.

Colonic ulcers show a similar pattern of FAK immunoreactivity. A: Intensity of staining for total FAK shows an increasing gradient from low near the ulcer bed (zone 1) to high in the region adjacent to it (zone 2) (top). This transition (arrow) is demonstrated more clearly when the area is magnified (bottom left). Also magnified, zone 3, away from the ulcer bed, exhibits very low staining for total FAK (middle) while staining for the control cytokeratin remains even throughout (bottom right). B: FAK397 immunostaining is similarly reduced at the ulcer margin (zone 1) with a transition to higher staining in the adjacent zone (zone 2). Again, the transition zone is clearly seen under magnification (zone 2, bottom left), and FAK397 staining is reduced in cells distal to the ulcer (bottom right). In both the total and FAK397 micrographs, zone 3, although not contiguous to zones 1 and 2 was taken from the same section but removed slightly because mucosal sloughing obscured the transition between zones 2 and 3. Arrows point to enlarged transition areas in both the total and FAK397 immunostains. Hematoxylin was used as a counterstain in both the gastric and colonic ulcer micrographs, representative of 41 human ulcers. Original magnifications: ×100 (A, B; top), ×200 (A, B; bottom).
In gastric ulcers, immunohistochemical staining for total FAK (Figure 1, top) demonstrated a consistent pattern with three zones: zone 1: very low FAK immunoreactivity in the more flattened squamous-appearing epithelial sheet actually migrating over the ulcer bed; zone 2: considerably increased immunoreactivity in the epithelium adjacent to the ulcer; and zone 3: a zone of moderate FAK immunoreactivity within the epithelium distant from the ulcer (×100 magnification). Parallel stains for FAK397 displayed the same pattern (Figure 1, bottom left), with markedly lower levels in the squamous migrating epithelial sheet (zone 1) but higher levels of FAK397 near the ulcer zone (zone 2), suggesting that levels of activated FAK may reflect protein abundance under these conditions. Zone 3 lies beyond the margin of the micrograph in this higher magnification (×200) and is not shown. Cytokeratin staining intensity in migrating cells was not different from that behind the migrating front (Figure 1, bottom right).
Colonic epithelium also expresses FAK. Figure 2 depicts a representative nonmalignant colonic ulcer. Although somewhat lower in relative intensity from that observed in gastric ulcers, the distribution of FAK immunoreactivity was similar in colonic ulceration to that seen in gastric ulcers. We observed decreased total FAK immunoreactivity at the edge of the ulcer (zone 1) and much greater intensity in the epithelium adjacent to the ulcer (zone 2) (Figure 2A, top; ×100). Magnification (×200) clearly demonstrates the zone 1 to zone 2 transition in this micrograph (Figure 2A, bottom left). Although more distant from the colonic ulcer than in gastric tissue but similar in intensity, zone 3, the area behind the colonic ulceration, exhibits relatively low staining for total FAK (Figure 2A, middle). The cytokeratin control (Figure 2A, bottom right) shows equal intensity of staining throughout. Again, FAK397 immunostaining patterns (Figure 2B) paralleled those seen with total FAK with moderate staining at the ulcer margin (zone 1) and higher expression in the epithelium adjacent to the ulcer zone (zone 2) (Figure 2B, top; ×100) and a similar transition between zones (zone 2, bottom left; ×200). Staining was nearly absent in zone 3 distal to the ulcer (Figure 2B, bottom right; ×200).
Semiquantitative scoring of staining density in 67 tFAK and 43 FAK397 total gastric and colonic samples yielded the following median scores: 0 (no immunoreactivity) for zone 1, 2 (moderate immunoreactivity) for zone 2, and 1 (mild immunoreactivity) for zone 3. χ2 analysis, both overall and pair wise between zones 1 and 2 and 2 and 3, and zones 1 and 3 showed these differences to be highly significant (P < 0.0001). Score modes and ranges are shown in Table 1.
Table 1.
Immunohistochemistry Scores
| Antibody | Cases | Mode/range
|
||
|---|---|---|---|---|
| Zone 1 | Zone 2 | Zone 3 | ||
| Total FAK | 67 | 0/0–1 | 2/1–2 | 1/0–2 |
| FAK397 | 43 | 0/0–1 | 2/1–3 | 1/0–2 |
| TGF-β | 41 | 0/0–2 | 2/0–3 | 0/0–2 |
| pSmad3 | 81 | 0/0–1 | 1/0–2 | 0/0–2 |
TGF-β Increases FAK Protein Levels in IEC-6 and Caco-2 Cells
Several growth factors are released and activated at mucosal wound sites.26,39 Therefore, we next tested the possibility that one or more of these factors may be responsible for the elevated FAK immunoreactivity observed in epithelial cells immediately adjacent to the migrating front. Levels of immunoreactive FAK were quantitated by Western blot in lysates of Caco-2 and IEC-6 cells exposed to bFGF, PDGF-BB, TGF-β1, and VEGF for 24 hours. Of these growth factors, only TGF-β had any effect on FAK levels in IEC-6 (Figure 3A, bars on left) and Caco-2 (Figure 3A, bars on right) cells (P < 0.05 for each, n = 3 to 5). To determine whether the increased levels of tFAK resulted in greater FAK activation, membranes were also probed with an antibody specific for activated FAK397. As shown in Figure 3B, the extent of FAK activation 24 hours after exposure to TGF-β paralleled the increase in tFAK over control in both IEC-6 (145 ± 6% for tFAK and 138 ± 9% for FAK397, P < 0.02 for each, bars on left) and Caco-2 (207 ± 26% and 196 ± 16% of control for tFAK and FAK397, respectively, P < 0.01, bars on right) cells. The average ratio of the FAK397 to tFAK signal density was near 1 for each cell type (0.96 ± 0.08 for IEC-6 and 1.02 ± 0.23 for Caco-2 cells, n = 4), suggesting that the level of activated FAK is directly dependent on the amount of total FAK present under these in vitro conditions.
Figure 3.

A: TGF-β increases immunoreactive FAK protein levels in vitro. FAK protein abundance, expressed as the ratio of total FAK to α-tubulin and as a percentage of untreated control in cells treated with TGF-β (1 ng/ml), bFGF (5 ng/ml), PDGF-BB (2.5 ng/ml), or VEGF (5 ng/ml) for 24 hours, is significantly higher only in TGF-β-treated IEC-6 (left bars) and Caco-2 (right bars) cells (*P < 0.05, n = 3 to 5). B: Total and activated FAK are enhanced to a similar degree after 24 hours of exposure to TGF-β. Densitometric analyses of total FAK and FAK397:α-tubulin ratios as a percentage of their respective control are depicted for IEC-6 (left bars) and Caco-2 (right bars) cells (*P < 0.02, n = 4 for each). Representative blots are shown above the bars.
TGF-β and pSmad3 Immunoreactivity Parallel that of FAK in Human Colonic Ulcers
TGF-β expression has been reported to be increased in healing ulcers,40 but differential immunoreactive TGF-β distribution in the ulcer area has not been investigated to date. In another representative colonic ulcer, the pattern and distribution of TGF-β staining (Figure 4A) mirrored that of FAK seen previously: very light staining at the migrating front (zone 1), and increasing staining immediately behind the front (zone 2) that fades at a distance from the ulcer (zone 3). For comparison with Figure 4A, immediately adjacent sections of this ulcer were immunostained for total FAK (Figure 4B) and activated FAK397 (Figure 4C); staining distribution is similar in all three panels. To ascertain that the TGF-β visualized in the ulcer region was functional, both gastric and colonic ulcers were immunostained for pSmad3, a downstream effector of TGF-β signals. In the colonic ulcer shown, pSmad immunoreactivity followed the pattern seen with FAK, FAK397, and TGF-β: lightest staining at the ulcer margin (zone 1), darker staining in the cells near the ulcer (zone 2), and intermediate staining away from the ulcer (zone 3) (Figure 5, top). The transition area from zone 1 to zone 2 is shown at higher magnification in the lower left panel and a mouse isotype-negative control in the right lower panel of Figure 5. Similar localization patterns for TGF-β and pSmad 3 were observed in gastric ulcers (data not shown). Scored median values for 41 TGF-β immunostains were 0 for zone 1, 2 for zone 2, and 1 for zone 3 and 0, 1, and 0 for the corresponding zones in 85 specimens scored for intensity of pSmad3 immunoreactivity. Overall and pair-wise differences were highly significant (P < 0.0001 by χ2; Table 1). Although these results do not identify the source of the TGF-β, such co-localization would be consistent with the model that TGF-β induces epithelial cell FAK expression during ulcer healing.
Figure 4.

In vivo, the pattern of colonic ulcer epithelial cell TGF-β immunoreactivity mirrors that of FAK. A: In another colonic ulcer, TGF-β immunoreactivity exhibits a similar distribution: low intensity in cells at the migrating front (zone 1), higher intensity in zone 2, and lower intensity distal to the ulcer (zone 3). For comparison, total FAK immunostaining in an immediately adjacent section reveals the same low-high-lower pattern of staining intensity in epithelial cells bordering the ulcer (B), and FAK397 immunoreactivity is similarly low in the ulcer zone and elevated beyond it (C). Original magnifications: ×100 (A, C); ×200 (B).
Figure 5.

The pattern of colonic epithelial cell phospho-Smad3 (pSmad3) immunoreactivity mirrors that of FAK and TGF-β. Top: Immunoreactivity for pSmad3 is low immediately adjacent to the ulcer bed (zone 1), elevated at the healing edge (zone 2), and intermediate in intensity in the distant mucosa (zone 3). Bottom left: Higher magnification of the transition area from zone 1 at the tip of the migrating epithelial sheet to the healing mucosa (zone 2). Bottom right: A mouse isotype control showing lack of color development in epithelial cells. Original magnifications: ×100 (top, bottom right); ×200 (bottom left).
TGF-β Increases FAK Immunoreactivity in Motile Cells of Expanding IEC-6 Cell Monolayers
Intestinal epithelial motility itself decreases FAK expression in vitro.22,31 To determine whether exogenous TGF-β can alter FAK immunoreactivity in motile cells in vitro, we first determined whether TGF-β immunoreactivity was lower in motile cells and then examined FAK immunostaining intensity in TGF-β-treated IEC-6 cells migrating across a linear wound. As reported previously,31 FAK immunoreactivity was much lower in the cytoplasm of untreated cells that had migrated across the wound edge than in those behind it (Figure 6, top left). TGF-β immunoreactivity was also lower in the cells at the migrating edge of the monolayer (Figure 6, bottom left). However, FAK immunoreactivity was higher after TGF-β treatment both behind and at the migrating edge (Figure 6, top right). Motile cells exhibited little difference in control ERK immunoreactivity, corroborating previous results (Figure 6, bottom right).31 Thus, TGF-β, if present, can reverse the motility-induced decrease in FAK protein in this model of intestinal restitution.
Figure 6.

In vitro, TGF-β enhances FAK immunoreactivity in motile IEC-6 cells. Left: IEC-6 cells migrating across a linear wound were immunostained for total FAK (tFAK, top) or TGF-β (bottom). Lines delineate the location of the scrape; direction of migration is from left to right. Nuclei were counterstained with hematoxylin. tFAK and TGF-β immunoreactivity is decreased in motile cells: cytoplasmic immunostaining intensity is lower in cells migrating across the scrape than in those behind the front for either tFAK (top left) or TGF-β (bottom left). Right: TGF-β treatment increases tFAK immunoreactivity in motile cells, as evidenced by the level of cytoplasmic staining in the expanding monolayer (top). Intensity of staining for ERK as control (bottom) is essentially unchanged in motile cells (representative of three separate experiments). Original magnifications, ×10.
TGF-β Enhances FAK Promoter Activity and FAK mRNA Expression
We next sought to determine whether TGF-β increases FAK protein at the transcriptional level. The reported sequence of the FAK promoter and upstream region contains several known TGF-β-responsive transcriptional binding sites (Ap-1, Sp-1, p53, among others) and possible Smad binding elements.41 Both Caco-2 and IEC-6 cells transfected with a pGL2-FAK promoter luciferase construct exhibited an ∼1.5-fold increase in luciferase activity. Luciferase activity was normalized to the Renilla control; results are expressed as percentage of the respective nontreated control (n = 4, P = 0.007 for each; Figure 7A). To further confirm these findings, RNA was extracted for RT-PCR from both Caco-2 and IEC-6 cells 2 and 6 hours after TGF-β treatment. As shown in Figure 7B, FAK mRNA levels, as a ratio of the 18S control, were significantly higher after exposure to 1 ng/ml of TGF-β in IEC-6 cells. Similar results were obtained after 6 hours. In Caco-2 cells, mean CT was significantly lower in cells exposed to TGF-β (13.06 ± 0.8 versus 18.17 ± 1.9 in controls, P = 0.037, n = 5; data not shown), reflecting the greater mRNA abundance in the treated cells. Thus, FAK protein levels in intestinal epithelial cells appear to be regulated at the transcriptional level by either motility or TGF-β treatment.
Figure 7.

TGF-β enhances FAK protein expression at the level of transcription in Caco-2 and IEC-6 cells. A: Activity of the FAK promoter-luciferase construct, normalized to Renilla and expressed as percentage of the respective nontreated control, is significantly higher in IEC-6 and Caco-2 cells 24 hours after TGF-β treatment (*P = 0.007 versus nontreated respective control, n = 4 for each). B: FAK mRNA levels are higher in IEC-6 cells as early as 2 hours after treatment with TGF-β. RT-PCR results, normalized to the 18S control, are expressed as fold increase over nontreated control cells (*P < 0.05, n = 5).
TGF-β-Stimulated Smad2/3 Phosphorylation Is Inhibited by the TGF-β Receptor I (ALK5) Blocker SB431542 but Not by SB203580, a p38 Inhibitor
Several signaling pathways are activated after binding of TGF-β to its receptor.42,43,44,45 We initially examined the Smad pathway, which is most classically identified with TGF-β action. After 1 hour, TGF-β induced Smad 2/3 activation (∼15-fold, P = 0.0004), as assessed by Western blotting using an antibody that recognizes serine 423/425 phosphorylation on both Smad2 and Smad3 (Figure 8). A 1-hour pretreatment with 0.5 μmol/L SB431542 not only abolished this rise but also significantly lowered (to 30 ± 5%; P = 0.0001) basal Smad2/3 phosphorylation when compared to the DMSO control. The p38 inhibitor SB203580 had no effect on Smad2/3 phosphorylation in response to TGF-β, indicating that it did not have a nonspecific effect at the 10 μmol/L dose used46 and suggesting that p38 activation is not required for Smad2/3 phosphorylation in IEC-6 cells. In this, and all subsequent figures, densitometric analysis is shown in the bars and a typical Western blot is shown above the bars.
Figure 8.
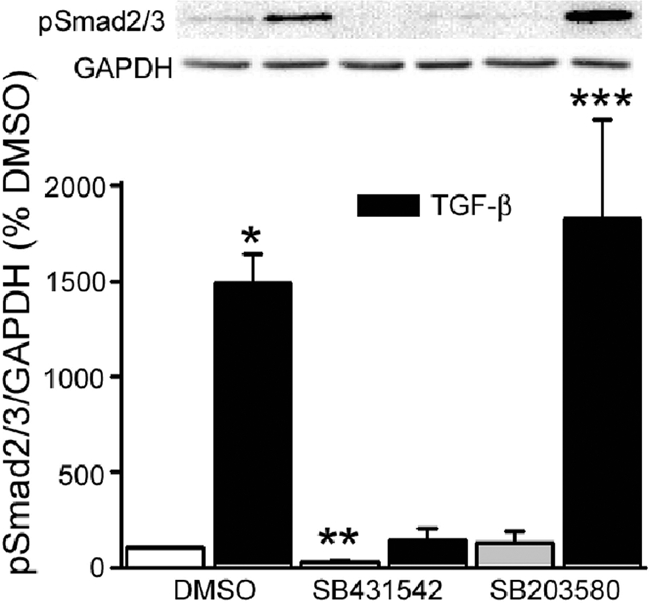
TGF-β-mediated Smad2/3 activation is blocked by a TGF-β receptor antagonist but not by p38MAPK inhibition. IEC-6 cells were pretreated with SB431542 (0.5 μmol/L) or SB203580 (10 μmol/L) for 1 hour before TGF-β exposure, also for 1 hour. The TGF-β-induced rise in Smad2/3 serine phosphorylation (∼15-fold; *P = 0.0004 versus DMSO vehicle-treated control) is totally abolished by SB431542. The TGF-β receptor blocker also significantly lowers basal Smad2/3 phosphorylation (**P = 0.0001). The p38 inhibitor, SB203580, has no effect on Smad2/3 activation (***P = 0.0001 versus SB203580 control). Bars represent densitometric analysis of five separate experiments; a representative Western blot is shown above. In this and subsequent figures, results are presented as percentage of control but statistics were calculated using the original densitometric ratios of the protein of interest and its respective GAPDH or α-tubulin control.
TGF-β-Stimulated p38 Activation Is Blocked by either SB431542 or SB203580
We then sought to determine whether activation of p38, a MAP kinase known to mediate the transcriptional effects of TGF-β in a number of cell types,47,48,49 may play a role in FAK induction in our cells. p38 phosphorylation was significantly enhanced (223 ± 51%, P = 0.05, n = 4) after 1 hour of exposure to TGF-β, an effect that was totally abolished by TGF-β receptor inhibition (Figure 9A). SB203580 inhibits p38 activity at a level subsequent to its phosphorylation but inhibition can be documented by assessing HSP27 accumulation as a surrogate.50,51,52 As shown in Figure 9B, the TGF-β-stimulated increase in HSP27 accumulation (189 ± 21% versus DMSO control, P = 0.006, n = 5) was completely negated by either the TGF-β receptor blocker or the specific p38 inhibitor.
Figure 9.

TGF-β receptor antagonism or p38MAPK inhibition blocks TGF-β-stimulated p38 activation. A: p38 phosphorylation, significantly increased by TGF-β (*P = 0.05 versus DMSO control, n = 4), is abolished by pretreatment with SB431542. SB203580, known to block p38 activity at a step beyond phosphorylation has no effect (**P = 0.001 versus SB203580). HSP27 accumulation was therefore used as a surrogate index of p38 activation/inhibition. B: TGF-β-stimulated accumulation of HSP27 (*P = 0.006 versus DMSO control, n = 5) is completely abrogated by either SB431542 or SB203580. Densitometric analysis is shown in the bars; representative Western blots are shown above.
Smad or p38 Signal Inhibition Abolishes the TGF-β-Mediated Increase in FAK Protein Expression/Activation and FAK Promoter Activity
We next examined the effects of chemical Smad and p38 signal inhibition on tFAK and FAK397 immunoreactivity in IEC-6 cells. As expected, tFAK protein levels were significantly increased (135 ± 9% as compared to the DMSO control, P = 0.001, n = 13) by TGF-β after 24 hours. FAK397 levels were enhanced by 150 ± 11% (P = 0.006, n = 10) over control. Exposure to either SB431542 or SB203580 completely blocked the increases in tFAK and FAK397 (Figure 10, A and B). Although changes in FAK397 levels were essentially parallel to those of tFAK, treatment with SB431542 led to consistent, nonsignificant elevations in FAK397 (Figure 10B). This activation was not seen with SB203580 or when either Smad2 or p38α protein expression was lowered with specific siRNA (data not shown), suggesting that this may be a response to TGF-β receptor blockade. To determine whether inhibition occurs at the level of the FAK promoter, cells transfected with the pGL2-FAK promoter luciferase construct were exposed to TGF-β, with and without SB431542 or SB203580 pretreatment. Cells transfected with the empty pGL2 vector exhibited little luciferase activity (Figure 10C). Luciferase activity, expressed as a ratio of the control Renilla activity, was higher in cells containing the FAK promoter construct. These levels were further increased by exposure to TGF-β (P = 0.008, n = 4). Luciferase activity was no different from non-TGF-β-treated control in cells treated with either inhibitor. Thus, TGF-β-mediated increases in FAK expression depend on TGF-β receptor activation and activation of at least one of its downstream effectors, p38 MAPK.
Figure 10.

Smad2/3 or p38 signal inhibition abolishes TGF-β-mediated FAK protein expression. A: A 24-hour exposure to TGF-β increases immunoreactive FAK protein levels in IEC-6 cells (135 ± 9% compared to DMSO-treated controls, *P = 0.001). Pretreatment with SB431542, a TGF-β receptor blocker, or SB203580 to block p38 activation, negates this effect. B: FAK397 levels are correspondingly elevated by TGF-β exposure and similarly blocked by either SB431542 or SB203580. Bars represent densitometric analysis of 6 to 12 individual experiments; typical Western blots for total FAK or FAK397, and α-tubulin, are shown above. C: IEC-6 cells transfected with the FAK-promoter-luciferase construct exhibit a fourfold greater luciferase activity over the empty pGL2 vector and a further increase (*P = 0.008) on exposure to TGF-β. Neither SB431542 nor SB203580 has an effect on basal FAK promoter activity; both inhibitors block the response to TGF-β. Bars represent luciferase activity, normalized to that of the Renilla control and expressed as fold increase over the pGL2 empty vector (n = 4).
Lowering Smad2 or p38α Protein Abundance with Specific siRNA also Inhibits the TGF-β Effect on FAK Protein Expression/Activation
Chemical inhibitors may have nonspecific effects. Also, although Smad2/3 phosphorylation is one of the primary events associated with TGF-β receptor activation, lack of FAK induction with TGF-β receptor I kinase inhibition only suggests that the Smad signaling pathway is involved because p38 activity is also decreased by such inhibition. Thus, to ascertain more specifically the contributions of Smad and p38 signaling to TGF-β induction of FAK, we blocked expression of each with the appropriate siRNA. Smad2 siRNA lowered Smad2 expression to 23 ± 5% (P = 0.0006, n = 19) of the nontargeting (NT1) control (data not shown). This decrease completely ablated the TGF-β-mediated increase in tFAK expression from154 ± 11% of the NT1 control (P < 0.001) to 89 ± 9% of that seen in cells treated with Smad2 siRNA alone (Figure 11A). FAK397 immunoreactivity was increased by 126 ± 8% by a 24-hour exposure to TGF-β, an effect that was also blocked by siRNA to Smad2 (to 109 ± 12% of its respective control, not shown). p38α expression was lowered to 15 ± 3%, and total p38 to 27 ± 5% of NT1 levels (P = 0.0008) by p38α siRNA (data not shown). p38 protein inhibition also blocked TGF-β-induced FAK expression to 90 ± 10% of the p38 siRNA-treated control (Figure 11B) and activation (to 87 ± 17% of its respective control, not shown). The combined results of chemical and siRNA inhibition of Smad and p38 signaling suggest that each of these signaling pathways plays an important role in FAK induction in response to TGF-β.
Figure 11.

Lowering Smad2 and p38α protein expression with specific siRNA also abolishes TGF-β-mediated FAK protein expression. A: Introduction of Smad2 siRNA into IEC-6 cells decreased Smad2 protein expression to 23 ± 5% of that in the nontargeting siRNA (NT1) treated control (P = 0.0003, not shown). This reduction in Smad2 protein completely inhibits the TGF-β-induced increase in FAK expression. B: p38α siRNA decreased p38α protein (to 15 ± 3%) and total p38 protein (to 27 ± 5%) levels compared to the NT1 siRNA-treated controls (P = 0.008 versus NT1, not shown). Lowering p38 expression also blocks the TGF-β effect. Densitometric analysis of total FAK, expressed as a ratio of α-tubulin and as a percentage of the NT1 control is shown in the bars. Representative Western blots for FAK, Smad2, and p38α and their respective α-tubulin or GAPDH controls are shown above the corresponding bars.
TGF-β Mediates FAK Protein Induction via Parallel Smad and p38 Pathways
Smad2/3 was maximally phosphorylated in response to short-term TGF-β stimulation in the presence of the p38 inhibitor, SB203580, indicating that p38 is not upstream of Smad. Lowering Smad2 expression with specific siRNA had no effect on total p38 protein levels (112 ± 15% of NT-treated control cells, n = 12). TGF-β-stimulated p38 phosphorylation was similar in NT1-treated (146 ± 15%, P = 0.002) and Smad2 siRNA-treated cells (138 ± 12%, P = 0.01) (Figure 12), suggesting that each pathway mediates the effect of TGF-β on FAK induction independently. Taken together, the data demonstrate that, although immunoreactive tFAK/activated FAK397 protein abundance is low in epithelial cells at the migrating edge of human gastric and colonic ulcers, it is higher in the area immediately adjacent to it and that this increase may be the result of TGF-β stimulation of FAK promoter activity via both Smad and p38 signaling pathways.
Figure 12.
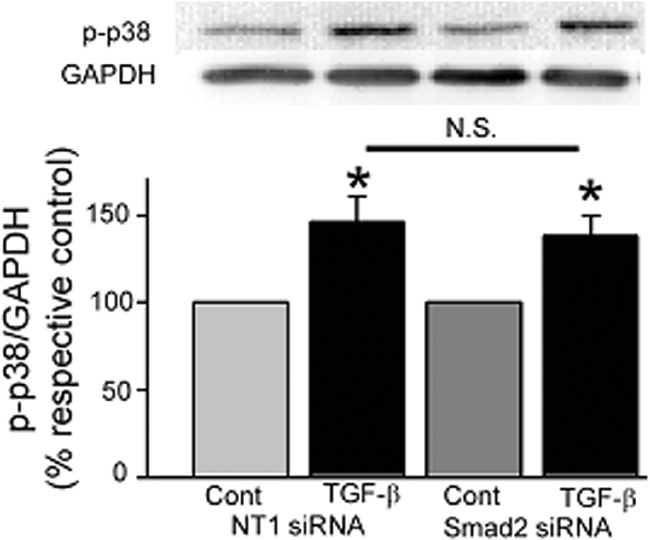
TGF-β-stimulated p38 phosphorylation is independent of Smad2/3 activation. p38 phosphorylation is enhanced equally in cells treated with nontargeting siRNA (NT1, P = 0.002) and specific siRNA directed at Smad2 (siSmad2, P = 0.01) when compared to their respective non-TGF-β-treated controls.
Discussion
Immunohistochemical examination of human gastric and colonic ulcers corroborated our in vitro findings that FAK and activated FAK397protein levels are decreased in motile gastrointestinal epithelial cells.5,22,31 Furthermore, these immunostains also delineated a zone of enhanced FAK/FAK397 protein expression behind the migrating front and a third zone of decreasing FAK/FAK397 immunoreactivity away from the ulcer. TGF-β and pSmad3, a downstream index of TGF-β function, exhibited identical immunostaining patterns. Indeed, TGF-β stimulated FAK expression in Caco-2 and IEC-6 intestinal epithelial cells and in migrating cells of IEC-6 wounded monolayers via Smad and p38 signaling. Although it would be interesting to repeat these studies in primary human intestinal epithelial cells, these are difficult to maintain in culture under sterile conditions, but both human Caco-2 cells and nontransformed rat IEC-6 cells are commonly used to model normal human intestinal epithelial biology.30,53,54,55,56,57
Cells modify focal adhesions in response to changes in composition, two- and three-dimensional structure, and physical forces of the extracellular matrix environment.58 During dynamic tissue conditions such as wound healing, cells may remain spread with clustered integrins but without mature focal adhesions to adapt to changing environmental conditions.59 FAK is a key component of focal adhesions and its activation is known to regulate focal adhesion maturation and disassembly and thus, cell adhesion and motility.2,3,21 Results of this study support data previously published by us and others31,60 that achievable levels of phosphorylated (active) FAK are dependent on the amount of total FAK available for activation. Further, the data presented above suggest a mechanism whereby epithelial cell responses to environmental challenges such as ulcer formation may be modulated by alterations in FAK protein expression that may significantly impact mucosal restitution and gastric wound healing.
Although it has been generally believed that FAK protein levels remain relatively constant and that FAK activation is most important to its function,61 several recent publications suggest that this may not always be the case. FAK expression in SW620 cells is enhanced by Eps8 (EGFR pathway substrate 8) affecting cell motility.62 In a neuroblastoma cell line, NMYC, a transcription factor important in cell survival, was shown to bind to the FAK promoter and increase FAK mRNA.63 On the other hand, eprhin-A1 down-regulates FAK In human glioma U251 cells.64 Our present and previous results demonstrate that FAK protein levels can be both down-regulated (by motility) and up-regulated (by exposure to TGF-β) in intestinal epithelial cells.
Growth factors are activated and active in both upper and lower gastrointestinal mucosal lesions.40 Some growth factors activate FAK. For example, EGF increased FAK397 levels and FAK at the lamellipodial edge but did not increase total FAK expression in MKN28 human gastric epithelial cells derived from gastric tubular adenocarcinoma.25 This is consistent with our observation that EGF failed to enhance FAK expression in either IEC-6 or Caco-2 cells. In myofibroblasts, TGF-β stimulated FAK397 in adherent cells. This was followed by induction of fibronectin and integrin receptor expression and activity.65
The precise localization of TGF-β in the gastrointestinal tract remains controversial. Some reports indicate that it is present mainly in goblet and endocrine cells in gastric tissue. Other authors suggest that TGF-β may be released from fibroblasts or macrophages in granulation tissue at the ulcer base.32 However, TGF-β1, -2, and -3 mRNA have been found in mouse jejunum and colon and localized by immunohistochemistry to the villus tip in the small intestine and the surface epithelium in the colon.66,67 TGF-β transcripts have been documented in IEC-6 cells.68 In the rat acetic acid gastric ulcer model, TGF-β1 levels reached a peak after 2 days by immunohistochemistry,69 but in the same model, TGF-β mRNA levels were reported to be elevated from days 3 to 18.70 Regardless of its source, TGF-β has been shown to mediate intestinal healing in vitro (IEC-6) and in vivo (TGF-βRII-DN transgenic mice).71 Indeed, both TGF-β and its receptors are strongly expressed in tissues from patients with well-healed ulcers via in situ hybridization and RT-PCR.40 Our immunohistochemistry results are consistent with the literature in that we identified TGF-β immunoreactivity in epithelial cells around gastrointestinal ulcers. However, to our knowledge, this is the first report of differential, co-localized TGF-β and FAK immunoreactivity in different zones along the ulcer margins.
Interestingly, TGF-β FAK interactions have been described in a number of cells, despite their apparent disparate actions, ie, growth inhibition and apoptosis with TGF-β versus cell differentiation and anti-apoptosis with FAK.50,72,73 All of these processes, however, involve interaction with the extracellular matrix and may thus share elements of the same signaling pathways. TGF-β/FAK interactions are often related to changes in cell phenotype and/or behavior. For example, TGF-β stimulation of FAK397 enhances migration in keratinocytes and hepatocarcinoma cells.74,75 TGF-β-induced fibroblast differentiation to myoblasts was shown to require adhesion- dependent FAK activation by some investigators,73 although others have suggested that in some cases FAK effects on differentiation may be mediated by interactions with another growth factor.76 Finally, TGF-β acts synergistically with integrins to activate FAK and induce phenotypic changes in rat podocytes.77 All of these studies examined FAK activation whereas the present investigation focuses on FAK protein expression. However, our results show that the amount of FAK protein available to be activated may be critical to the amount of phosphorylated/activated FAK generated by an activating stimulus. Thus, motility- and TGF-β-modulated changes in FAK protein per se may substantially regulate the amount of active FAK and thus in turn the eventual behavior and phenotype in the healing gut mucosa.
Our results also implicate both the Smad and the p38 MAPK pathways in the transduction of the TGF-β-induced increase in FAK protein expression. The best-described classical TGF-β signaling pathway involves TGF-β binding to its receptor, a type I/II heteromeric serine/threonine kinase, and induction of Smad 2/3 phosphorylation. Smads have intrinsic transcription-inducing activity.42 In the basal state, Smad2/3 are found in the cytoplasm but after activation, they form a complex with Smad4 and are shuttled to the cytoplasm where they bind DNA at a number of transcription sites such as AP-1 or Sp-1.78 A specific Smad3/4 binding sequence (SBE, a CAGA motif) has also been described.79 TGF-β has also been reported to regulate gene expression via activation of mitogen-activated protein kinases (MAPK), particularly p38.47,49,80
The published FAK promoter and upstream sequences contain several sites, AP-1, Sp-1, and putative SBEs among others, which may transduce TGF-β-mediated FAK expression.41 Indeed, when cloned into a luciferase construct, the promoter not only drove luciferase expression in response to TGF-β and in parallel to changes in FAK mRNA and protein levels but also proved sensitive to inhibition of this effect by specific TGF-β receptor I and p38 antagonists. Treatment of IEC-6 cells with TGF-β resulted in a significant increase in Smad2/3 and p38 phosphorylation that could be abolished with SB431542. This compound inhibits the activin receptor-like kinase (ALK) activity of the TGF-β receptor I and is closely related to the p38 inhibitor SB203580. Both chemicals act as competitive ATP-binding inhibitors but SB431542 is a selective inhibitor of Smad phosphorylation with an IC50 of 94 nmol/L whereas SB203580 only inhibits Smad phosphorylation at doses higher than required to inhibit p38.46,81 Thus, although SB203580, at the 10 μmol/L dose used here, inhibited TGF-β-stimulated p38 activation, it did not alter Smad phosphorylation. This is consistent with a previous analysis of ALK5 and p38 inhibition in vascular smooth muscle cells.82 Both compounds, however, abolished the TGF-β effect on FAK induction as assessed by Western blot and luciferase assays.
TGF-β- and p38-mediated gene expression may be either Smad-dependent or -independent, depending on the cell type studied.49,80 For instance, in renal epithelial carcinoma A498 cells Smad activation induced collagen 1α1 expression whereas p38 induced fibronectin.81 TGF-β enhanced fibronectin mRNA in transformed human proximal tubular epithelial HKC cells, an effect that was blocked by both the p38 inhibitor, SB202190, and the ALK5 inhibitor, SB431542 but not by Smad knockdown.48 On the other hand, in human pancreatic adenocarcinoma PANC-1 cells, TGF-β regulation of biglycan gene expression via p38 activation also required Smad activation.49 Results of the luciferase assays indicate that inhibition of either p38 or Smad signaling can abolish the TGF-β-induced stimulation of FAK expression, suggesting that each pathway plays an individual role at the level of FAK transcription. To further define the specific roles of Smad and p38 activation in the regulation of FAK protein expression by TGF-β, we lowered expression of each with specific siRNA. Reducing either Smad2 or p38α protein levels abrogated the TGF-β-induced increase in FAK protein. The fact that Smad2/3 was maximally phosphorylated in the presence of the p38 inhibitor SB203580 does argue against p38 being upstream of Smad. The possibility that Smad activation may be necessary for p38 activity was also excluded because TGF-β-stimulated p38 phosphorylation was not affected by Smad2 protein ablation. Thus, our results are consistent with reports that suggest a role for each of these signals, independently or in concert, in TGF-β stimulation of various proteins in other cells. They differ from those of Horowitz and colleagues83 who have reported that TGF-β activation of FAK was dependent on Smad but independent of p38 in myoblasts. Certainly, different cell types may exhibit different signal pathways. In addition, however, signals that activate a kinase such as FAK would not necessarily be expected to control its expression, as examined in our study, whereas signals that stimulate expression may not necessarily activate the expressed protein. Indeed, although FGF-2 was shown to up-regulate both FAK protein abundance and activation in rodent lenses, VEGF only stimulated FAK activation in cardiac myocytes.84,85
In summary, FAK seems critically regulated at the protein level in human gastric and colonic ulcers. Protein levels are low at the migrating edge, higher immediately adjacent to the leading edge, and lower further away from the ulcer. TGF-β immunoreactivity displays a similar distribution pattern, and TGF-β induces FAK expression in vitro via Smad and p38 signaling, suggesting that TGF-β may have an autocrine or paracrine effect on FAK protein abundance in vivo. The absence of TGF-β at the migrating epithelial edge may contribute to the decrease in FAK in vivo in these cells because our in vitro studies suggest that migrating intestinal epithelial cells may still be responsive to exogenous TGF-β. In zone 2, adjacent to the ulcer, FAK induction by TGF-β via Smad and p38 signaling may prevent anoikis or may induce differentiation to either a more migratory phenotype or one that is more sensitive to activation by other locally produced factors (Figure 13).86,87 Modulation of FAK, Smad, and p38 signaling might therefore be expected to modulate TGF-β effects, but this is beyond the scope of the current investigation and awaits future study.
Figure 13.
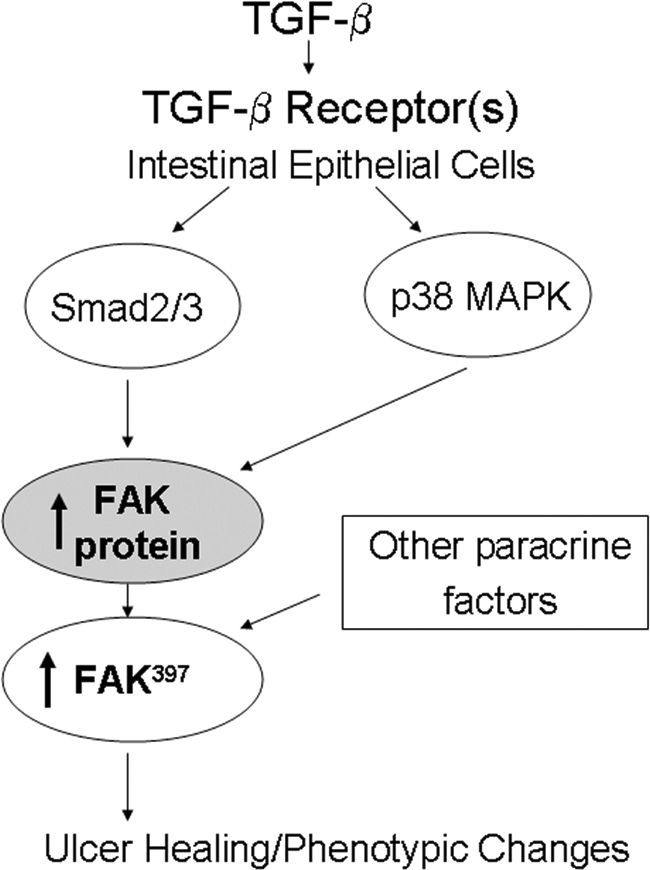
Schematic diagram illustrating TGF-β-induced FAK protein induction by parallel Smad2/3 and p38 signal pathways.
Re-epithelialization, a process initially dependent on cell migration or restitution is essential to gastrointestinal ulcer healing. Migrating epithelial cells in the vicinity of the ulcer maximize their response to the changes in their environment by modifying focal adhesion structure and function.43 Because FAK is an important component and regulator of focal adhesions, alterations in FAK protein levels, driven by motility and/or exposure to growth factors such as TGF-β, may contribute to this plasticity and, in concert with FAK activation, significantly impact the biology of mucosal restitution and wound healing.
Acknowledgments
We thank Matthew Sanders for performing the scrapes used for the IEC-6 immunostains.
Footnotes
Address reprint requests to Dr. Marc Basson, Chief, Surgical Service, John D. Dingell VA Medical Center, 4646 John R. St., Detroit, MI 48201-1932. E-mail: marc.basson@va.gov.
Supported in part by the National Institutes of Health (grant RO1DK067257 to M.D.B.) and the Veterans Administration (merit research support to M.D.B.).
References
- Burridge K, Nuckolls G, Otey C, Pavalko F, Simon K, Turner C. Actin-membrane interaction in focal adhesions. Cell Differ Dev. 1990;32:337–342. doi: 10.1016/0922-3371(90)90048-2. [DOI] [PubMed] [Google Scholar]
- Schaller MD, Borgman CA, Cobb BS, Vines RR, Reynolds AB, Parsons JT. pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc Natl Acad Sci USA. 1992;89:5192–5196. doi: 10.1073/pnas.89.11.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore AP, Romer LH. Inhibition of focal adhesion kinase (FAK) signaling in focal adhesions decreases cell motility and proliferation. Mol Biol Cell. 1996;7:1209–1224. doi: 10.1091/mbc.7.8.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar EP, Rozengurt E. Bombesin and platelet-derived growth factor induce association of endogenous focal adhesion kinase with Src in intact Swiss 3T3 cells. J Biol Chem. 1999;274:28371–28378. doi: 10.1074/jbc.274.40.28371. [DOI] [PubMed] [Google Scholar]
- Yu CF, Basson MD. Matrix-specific FAK and MAPK reorganization during Caco-2 cell motility. Microsc Res Tech. 2000;51:191–203. doi: 10.1002/1097-0029(20001015)51:2<191::AID-JEMT10>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Zachary I. Focal adhesion kinase. Int J Biochem Cell Biol. 1997;29:929–934. doi: 10.1016/s1357-2725(97)00008-3. [DOI] [PubMed] [Google Scholar]
- Reif S, Lang A, Lindquist JN, Yata Y, Gabele E, Scanga A, Brenner DA, Rippe RA. The role of focal adhesion kinase-phosphatidylinositol 3-kinase-akt signaling in hepatic stellate cell proliferation and type I collagen expression. J Biol Chem. 2003;278:8083–8090. doi: 10.1074/jbc.M212927200. [DOI] [PubMed] [Google Scholar]
- Sieg DJ, Hauck CR, Schlaepfer DD. Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J Cell Sci. 1999;112:2677–2691. doi: 10.1242/jcs.112.16.2677. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Mitra SK. Multiple connections link FAK to cell motility and invasion. Curr Opin Genet Dev. 2004;14:92–101. doi: 10.1016/j.gde.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Kovacic-Milivojević B, Roediger F, Almeida EA, Damsky CH, Gardner DG, Ilic D. Focal adhesion kinase and p130Cas mediate both sarcomeric organization and activation of genes associated with cardiac myocyte hypertrophy. Mol Biol Cell. 2001;12:2290–2307. doi: 10.1091/mbc.12.8.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korah R, Choi L, Barrios J, Wieder R. Expression of FGF-2 alters focal adhesion dynamics in migration-restricted MDA-MB-231 breast cancer cells. Breast Cancer Res Treat. 2004;88:17–28. doi: 10.1007/s10459-004-6006-2. [DOI] [PubMed] [Google Scholar]
- Hakim ZS, DiMichele LA, Doherty JT, Homeister JW, Beggs HE, Reichardt LF, Schwartz RJ, Brackhan J, Smithies O, Mack CP, Taylor JM. Conditional deletion of focal adhesion kinase leads to defects in ventricular septation and outflow tract alignment. Mol Cell Biol. 2007;27:5352–5364. doi: 10.1128/MCB.00068-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Govindarajan G, Eble DM, Lucchesi PA, Samarel AM. Focal adhesion kinase is involved in angiotensin II-mediated protein synthesis in cultured vascular smooth muscle cells. Circ Res. 2000;87:710–716. doi: 10.1161/01.res.87.8.710. [DOI] [PubMed] [Google Scholar]
- Braren R, Hu H, Kim YH, Beggs HE, Reichardt LF, Wang R. Endothelial FAK is essential for vascular network stability, cell survival, and lamellipodial formation. J Cell Biol. 2006;172:151–162. doi: 10.1083/jcb.200506184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Ueda H, Zhou H, Stokol T, Shen TL, Alcaraz A, Nagy T, Vassalli JD, Guan JL. Overexpression of focal adhesion kinase in vascular endothelial cells promotes angiogenesis in transgenic mice. Cardiovasc Res. 2004;64:421–430. doi: 10.1016/j.cardiores.2004.07.012. [DOI] [PubMed] [Google Scholar]
- Matkowskyj KA, Keller K, Glover S, Kornberg L, Tran-Son-Tay R, Benya RV. Expression of GRP and its receptor in well-differentiated colon cancer cells correlates with the presence of focal adhesion kinase phosphorylated at tyrosines 397 and 407. J Histochem Cytochem. 2003;51:1041–1048. doi: 10.1177/002215540305100807. [DOI] [PubMed] [Google Scholar]
- Nakagawa K, Sogo S, Hioki K, Tokunaga R, Taketani S. Acquisition of cell adhesion and induction of focal adhesion kinase of human colon cancer Colo 201 cells by retinoic acid-induced differentiation. Differentiation. 1998;62:249–257. doi: 10.1046/j.1432-0436.1998.6250249.x. [DOI] [PubMed] [Google Scholar]
- Glover S, Delaney M, Dematte C, Kornberg L, Frasco M, Tran-Son-Tay R, Benya RV. Phosphorylation of focal adhesion kinase tyrosine 397 critically mediates gastrin-releasing peptide’s morphogenic properties. J Cell Physiol. 2004;199:77–88. doi: 10.1002/jcp.10456. [DOI] [PubMed] [Google Scholar]
- Glover S, Nathaniel R, Shakir L, Perrault C, Anderson RK, Tran-Son-Tay R, Benya RV. Transient upregulation of GRP and its receptor critically regulate colon cancer cell motility during remodeling. Am J Physiol. 2005;288:G1274–G1282. doi: 10.1152/ajpgi.00108.2004. [DOI] [PubMed] [Google Scholar]
- Cohen-Hillel E, Yron I, Meshel T, Ben-Baruch A. Interleukin 8 and cell migration to inflammatory sites: the regulation of focal adhesion kinase under conditions of migratory desensitization. Isr Med Assoc J. 2007;9:579–583. [PubMed] [Google Scholar]
- Owen JD, Ruest PJ, Fry DW, Hanks SK. Induced focal adhesion kinase (FAK) expression in FAK-null cells enhances cell spreading and migration requiring both auto- and activation loop phosphorylation sites and inhibits adhesion-dependent tyrosine phosphorylation of Pyk2. Mol Cell Biol. 1999;19:4806–4818. doi: 10.1128/mcb.19.7.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu CF, Sanders MA, Basson MD. Human caco-2 motility redistributes FAK and paxillin and activates p38 MAPK in a matrix-dependent manner. Am J Physiol. 2000;278:G952–G966. doi: 10.1152/ajpgi.2000.278.6.G952. [DOI] [PubMed] [Google Scholar]
- Hauck CR, Sieg DJ, Hsia DA, Loftus JC, Gaarde WA, Monia BP, Schlaepfer DD. Inhibition of focal adhesion kinase expression or activity disrupts epidermal growth factor-stimulated signaling promoting the migration of invasive human carcinoma cells. Cancer Res. 2001;61:7079–7090. [PubMed] [Google Scholar]
- Westhoff MA, Serrels B, Fincham VJ, Frame MC, Carragher NO. SRC-mediated phosphorylation of focal adhesion kinase couples actin and adhesion dynamics to survival signaling. Mol Cell Biol. 2004;24:8113–8133. doi: 10.1128/MCB.24.18.8113-8133.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo IL, Pai R, Jones MK, Ehring GR, Kawanaka H, Tarnawski AS. Indomethacin delays gastric restitution: association with the inhibition of focal adhesion kinase and tensin phosphorylation and reduced actin stress fibers. Exp Biol Med (Maywood) 2002;227:412–424. doi: 10.1177/153537020222700607. [DOI] [PubMed] [Google Scholar]
- Tarnawski A, Szabo IL, Husain SS, Soreghan B. Regeneration of gastric mucosa during ulcer healing is triggered by growth factors and signal transduction pathways. J Physiol Paris. 2001;95:337–344. doi: 10.1016/s0928-4257(01)00046-8. [DOI] [PubMed] [Google Scholar]
- Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol. 1999;71:435–478. doi: 10.1016/s0079-6107(98)00052-2. [DOI] [PubMed] [Google Scholar]
- Agochiya M, Brunton VG, Owens DW, Parkinson EK, Paraskeva C, Keith WN, Frame MC. Increased dosage and amplification of the focal adhesion kinase gene in human cancer cells. Oncogene. 1999;18:5646–5653. doi: 10.1038/sj.onc.1202957. [DOI] [PubMed] [Google Scholar]
- Lark AL, Livasy CA, Calvo B, Caskey L, Moore DT, Yang X, Cance WG. Overexpression of focal adhesion kinase in primary colorectal carcinomas and colorectal liver metastases: immunohistochemistry and real-time PCR analyses. Clin Cancer Res. 2003;9:215–222. [PubMed] [Google Scholar]
- Lévy P, Robin H, Kornprobst M, Capeau J, Cherqui G. Enterocytic differentiation of the human Caco-2 cell line correlates with alterations in integrin signaling. J Cell Physiol. 1998;177:618–627. doi: 10.1002/(SICI)1097-4652(199812)177:4<618::AID-JCP12>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Basson MD, Sanders MA, Gomez R, Hatfield J, Vanderheide R, Thamilselvan V, Zhang J, Walsh MF. Focal adhesion kinase protein levels in gut epithelial motility. Am J Physiol. 2006;291:G491–G499. doi: 10.1152/ajpgi.00292.2005. [DOI] [PubMed] [Google Scholar]
- Tanigawa T, Pai R, Arakawa T, Higuchi K, Tarnawski AS. TGF-beta signaling pathway: its role in gastrointestinal pathophysiology and modulation of ulcer healing. J Physiol Pharmacol. 2005;56:3–13. [PubMed] [Google Scholar]
- Milani S, Calabro A. Role of growth factors and their receptors in gastric ulcer healing. Microsc Res Tech. 2001;53:360–371. doi: 10.1002/jemt.1104. [DOI] [PubMed] [Google Scholar]
- Quaroni A, Wands J, Trelstad RL, Isselbacher KJ. Epithelioid cell cultures from rat small intestine. Characterization by morphologic and immunologic criteria. J Cell Biol. 1979;80:248–265. doi: 10.1083/jcb.80.2.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson MD, Mooseker MS. An in vitro model for the analysis of intestinal brush border assembly. I. Ultrastructural analysis of cell contact-induced brush border assembly in Caco-2BBe cells. J Cell Sci. 1993;105:445–460. doi: 10.1242/jcs.105.2.445. [DOI] [PubMed] [Google Scholar]
- Peterson MD, Bement WM, Mooseker MS. An in vitro model for the analysis of intestinal brush border assembly. II. Changes in expression and localization of brush border proteins during cell contact-induced brush border assembly in Caco-2BBe cells. J Cell Sci. 1993;105:461–472. doi: 10.1242/jcs.105.2.461. [DOI] [PubMed] [Google Scholar]
- Basson CT, Kocher O, Basson MD, Asis A, Madri JA. Differential modulation of vascular cell integrin and extracellular matrix expression in vitro by TGF-beta 1 correlates with reciprocal effects on cell migration. J Cell Physiol. 1992;153:118–128. doi: 10.1002/jcp.1041530116. [DOI] [PubMed] [Google Scholar]
- Whitney GS, Chan PY, Blake J, Cosand WL, Neubauer MG, Aruffo A, Kanner SB. Human T and B lymphocytes express a structurally conserved focal adhesion kinase, pp125FAK. DNA Cell Biol. 1993;12:823–830. doi: 10.1089/dna.1993.12.823. [DOI] [PubMed] [Google Scholar]
- Tarnawski AS. Cellular and molecular mechanisms of gastrointestinal ulcer healing. Dig Dis Sci. 2005;50(Suppl 1):S24–S33. doi: 10.1007/s10620-005-2803-6. [DOI] [PubMed] [Google Scholar]
- Shih SC, Tseng KW, Lin SC, Kao CR, Chou SY, Wang HY, Chang WH, Chu CH, Wang TE, Chien CL. Expression patterns of transforming growth factor-beta and its receptors in gastric mucosa of patients with refractory gastric ulcer. World J Gastroenterol. 2005;11:136–141. doi: 10.3748/wjg.v11.i1.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golubovskaya V, Kaur A, Cance W. Cloning and characterization of the promoter region of human focal adhesion kinase gene: nuclear factor kappa B and p53 binding sites. Biochim Biophys Acta. 2004;1678:111–125. doi: 10.1016/j.bbaexp.2004.03.002. [DOI] [PubMed] [Google Scholar]
- Massagué J, Seoane J, Wotton D. Smad transcription factors. Genes Dev. 2005;19:2783–2810. doi: 10.1101/gad.1350705. [DOI] [PubMed] [Google Scholar]
- Massagué J. TGF-beta signal transduction. Annu Rev Biochem. 1998;67:753–791. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- Massagué J. The transforming growth factor-beta family. Annu Rev Cell Biol. 1990;6:597–641. doi: 10.1146/annurev.cb.06.110190.003121. [DOI] [PubMed] [Google Scholar]
- ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004;29:265–273. doi: 10.1016/j.tibs.2004.03.008. [DOI] [PubMed] [Google Scholar]
- Inman GJ, Nicolas FJ, Callahan JF, Harling JD, Gaster LM, Reith AD, Laping NJ, Hill CS. SB-431542 is a potent and specific inhibitor of transforming growth factor-beta superfamily type I activin receptor-like kinase (ALK) receptors ALK4, ALK5, and ALK7. Mol Pharmacol. 2002;62:65–74. doi: 10.1124/mol.62.1.65. [DOI] [PubMed] [Google Scholar]
- Brezniceanu ML, Wei CC, Zhang SL, Hsieh TJ, Guo DF, Hebert MJ, Ingelfinger JR, Filep JG, Chan JS. Transforming growth factor-beta 1 stimulates angiotensinogen gene expression in kidney proximal tubular cells. Kidney Int. 2006;69:1977–1985. doi: 10.1038/sj.ki.5000396. [DOI] [PubMed] [Google Scholar]
- Niculescu-Duvaz I, Phanish MK, Colville-Nash P, Dockrell ME. The TGFbeta1-induced fibronectin in human renal proximal tubular epithelial cells is p38 MAP kinase dependent and Smad independent. Nephron Exp Nephrol. 2007;105:e108–e116. doi: 10.1159/000100492. [DOI] [PubMed] [Google Scholar]
- Ungefroren H, Lenschow W, Chen WB, Faendrich F, Kalthoff H. Regulation of biglycan gene expression by transforming growth factor-beta requires MKK6-p38 mitogen-activated protein kinase signaling downstream of Smad signaling. J Biol Chem. 2003;278:11041–11049. doi: 10.1074/jbc.M300035200. [DOI] [PubMed] [Google Scholar]
- Walsh MF, Thamilselvan V, Grotelueschen R, Farhana L, Basson M. Absence of adhesion triggers differential FAK and SAPKp38 signals in SW620 human colon cancer cells that may inhibit adhesiveness and lead to cell death. Cell Physiol Biochem. 2003;13:135–146. doi: 10.1159/000071864. [DOI] [PubMed] [Google Scholar]
- Thamilselvan V, Basson MD. Pressure activates colon cancer cell adhesion by inside-out focal adhesion complex and actin cytoskeletal signaling. Gastroenterology. 2004;126:8–18. doi: 10.1053/j.gastro.2003.10.078. [DOI] [PubMed] [Google Scholar]
- Freshney NW, Rawlinson L, Guesdon F, Jones E, Cowley S, Hsuan J, Saklatvala J. Interleukin-1 activates a novel protein kinase cascade that results in the phosphorylation of Hsp27. Cell. 1994;78:1039–1049. doi: 10.1016/0092-8674(94)90278-x. [DOI] [PubMed] [Google Scholar]
- Turner DJ, Martin PC, Rao JN, Greenspon J, Zou T, Bass BL, Wang JY, Strauch ED. Substance P regulates migration in rat intestinal epithelial cells. Ann Surg. 2007;245:408–414. doi: 10.1097/01.sla.0000245549.57076.db. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenier E, Maupas FS, Beaulieu JF, Seidman E, Delvin E, Sane A, Tremblay E, Garofalo C, Levy E. Effect of retinoic acid on cell proliferation and differentiation as well as on lipid synthesis, lipoprotein secretion, and apolipoprotein biogenesis. Am J Physiol. 2007;293:G1178–G1189. doi: 10.1152/ajpgi.00295.2007. [DOI] [PubMed] [Google Scholar]
- Bulut K, Pennartz C, Felderbauer P, Ansorge N, Banasch M, Schmitz F, Schmidt WE, Hoffmann P. Vascular endothelial growth factor (VEGF164) ameliorates intestinal epithelial injury in vitro in IEC-18 and Caco-2 monolayers via induction of TGF-beta release from epithelial cells. Scand J Gastroenterol. 2006;41:687–692. doi: 10.1080/00365520500408634. [DOI] [PubMed] [Google Scholar]
- McKaig BC, Makh SS, Hawkey CJ, Podolsky DK, Mahida YR. Normal human colonic subepithelial myofibroblasts enhance epithelial migration (restitution) via TGF-beta3. Am J Physiol. 1999;276:G1087–G1093. doi: 10.1152/ajpgi.1999.276.5.G1087. [DOI] [PubMed] [Google Scholar]
- Rao JN, Liu L, Zou T, Marasa BS, Boneva D, Wang SR, Malone DL, Turner DJ, Wang JY. Polyamines are required for phospholipase C-gamma1 expression promoting intestinal epithelial restitution after wounding. Am J Physiol. 2007;292:G335–G343. doi: 10.1152/ajpgi.00282.2006. [DOI] [PubMed] [Google Scholar]
- Wozniak MA, Modzelewska K, Kwong L, Keely PJ. Focal adhesion regulation of cell behavior. Biochim Biophys Acta. 2004;1692:103–119. doi: 10.1016/j.bbamcr.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Orr AW, Pallero MA, Xiong WC, Murphy-Ullrich JE. Thrombospondin induces RhoA inactivation through FAK-dependent signaling to stimulate focal adhesion disassembly. J Biol Chem. 2004;279:48983–48992. doi: 10.1074/jbc.M404881200. [DOI] [PubMed] [Google Scholar]
- Salasznyk RM, Klees RF, Williams WA, Boskey A, Plopper GE. Focal adhesion kinase signaling pathways regulate the osteogenic differentiation of human mesenchymal stem cells. Exp Cell Res. 2007;313:22–37. doi: 10.1016/j.yexcr.2006.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg LJ, Galante LM, Bill HM, Mack CP, Taylor JM. An endogenous inhibitor of focal adhesion kinase blocks Rac1/JNK but not Ras/ERK-dependent signaling in vascular smooth muscle cells. J Biol Chem. 2003;278:29783–29791. doi: 10.1074/jbc.M303771200. [DOI] [PubMed] [Google Scholar]
- Maa MC, Lee JC, Chen YJ, Chen YJ, Lee YC, Wang ST, Huang CC, Chow NH, Leu TH. Eps8 facilitates cellular growth and motility of colon cancer cells by increasing the expression and activity of focal adhesion kinase. J Biol Chem. 2007;282:19399–19409. doi: 10.1074/jbc.M610280200. [DOI] [PubMed] [Google Scholar]
- Beierle EA, Trujillo A, Nagaram A, Kurenova EV, Finch R, Ma X, Vella J, Cance WG, Golubovskaya VM. N-MYC regulates focal adhesion kinase expression in human neuroblastoma. J Biol Chem. 2007;282:12503–12516. doi: 10.1074/jbc.M701450200. [DOI] [PubMed] [Google Scholar]
- Liu DP, Wang Y, Koeffler HP, Xie D. Ephrin-A1 is a negative regulator in glioma through down-regulation of EphA2 and FAK. Int J Oncol. 2007;30:865–871. [PubMed] [Google Scholar]
- Thannickal VJ, Lee DY, White ES, Cui Z, Larios JM, Chacon R, Horowitz JC, Day RM, Thomas PE. Myofibroblast differentiation by transforming growth factor beta1 is dependent on cell adhesion and integrin signaling via focal adhesion kinase. J Biol Chem. 2003;278:12384–12389. doi: 10.1074/jbc.M208544200. [DOI] [PubMed] [Google Scholar]
- Avery A, Paraskeva C, Hall P, Flanders KC, Sporn M, Moorghen M. TGF-beta expression in the human colon: differential immunostaining along crypt epithelium. Br J Cancer. 1993;68:137–139. doi: 10.1038/bjc.1993.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnard JA, Warwick GJ, Gold LI. Localization of transforming growth factor beta isoforms in the normal murine small intestine and colon. Gastroenterology. 1993;105:67–73. doi: 10.1016/0016-5085(93)90011-z. [DOI] [PubMed] [Google Scholar]
- Koyama SY, Podolsky DK. Differential expression of transforming growth factors alpha and beta in rat intestinal epithelial cells. J Clin Invest. 1989;83:1768–1773. doi: 10.1172/JCI114080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst H, Konturek PC, Brzozowski T, Konturek SJ, Hahn EG. Subserosal application of transforming growth factor-beta 1 in rats with chronic gastric ulcers: effect on gastric ulcer healing and blood flow. J Physiol Pharmacol. 1996;47:443–454. [PubMed] [Google Scholar]
- Tominaga K, Arakawa T, Kim S, Iwao H, Kobayashi K. Increased expression of transforming growth factor-beta1 during gastric ulcer healing in rats. Dig Dis Sci. 1997;42:616–625. doi: 10.1023/a:1018867630686. [DOI] [PubMed] [Google Scholar]
- Beck PL, Rosenberg IM, Xavier RJ, Koh T, Wong JF, Podolsky DK. Transforming growth factor-beta mediates intestinal healing and susceptibility to injury in vitro and in vivo through epithelial cells. Am J Pathol. 2003;162:597–608. doi: 10.1016/s0002-9440(10)63853-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier R, Harnois C, Drolet JF, Reed JC, Vezina A, Vachon PH. Human intestinal epithelial cell survival: differentiation state-specific control mechanisms. Am J Physiol. 2001;280:C1540–C1554. doi: 10.1152/ajpcell.2001.280.6.C1540. [DOI] [PubMed] [Google Scholar]
- Thomas PE, Peters-Golden M, White ES, Thannickal VJ, Moore BB. PGE(2) inhibition of TGF-beta1-induced myofibroblast differentiation is Smad-independent but involves cell shape and adhesion-dependent signaling. Am J Physiol. 2007;293:L417–L428. doi: 10.1152/ajplung.00489.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong HW, Kim IS. TGF-beta1 enhances betaig-h3-mediated keratinocyte cell migration through the alpha3beta1 integrin and PI3K. J Cell Biochem. 2004;92:770–780. doi: 10.1002/jcb.20110. [DOI] [PubMed] [Google Scholar]
- Cai T, Lei QY, Wang LY, Zha XL. TGF-beta 1 modulated the expression of alpha 5 beta 1 integrin and integrin-mediated signaling in human hepatocarcinoma cells. Biochem Biophys Res Commun. 2000;274:519–525. doi: 10.1006/bbrc.2000.3177. [DOI] [PubMed] [Google Scholar]
- Greenberg RS, Bernstein AM, Benezra M, Gelman IH, Taliana L, Masur SK. FAK-dependent regulation of myofibroblast differentiation. FASEB J. 2006;20:1006–1008. doi: 10.1096/fj.05-4838fje. [DOI] [PubMed] [Google Scholar]
- Chen CA, Hwang JC, Guh JY, Tsai JC, Chen HC. TGF-beta1 and integrin synergistically facilitate the differentiation of rat podocytes by increasing alpha-smooth muscle actin expression. Transpl Res. 2006;148:134–141. doi: 10.1016/j.trsl.2006.03.008. [DOI] [PubMed] [Google Scholar]
- Qing J, Zhang Y, Derynck R. Structural and functional characterization of the transforming growth factor-beta-induced Smad3/c-Jun transcriptional cooperativity. J Biol Chem. 2000;275:38802–38812. doi: 10.1074/jbc.M004731200. [DOI] [PubMed] [Google Scholar]
- Dennler S, Itoh S, Vivien D, ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. EMBO J. 1998;17:3091–3100. doi: 10.1093/emboj/17.11.3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Hebert MC, Zhang YE. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. EMBO J. 2002;21:3749–3759. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laping NJ, Grygielko E, Mathur A, Butter S, Bomberger J, Tweed C, Martin W, Fornwald J, Lehr R, Harling J, Gaster L, Callahan JF, Olson BA. Inhibition of transforming growth factor (TGF)-beta1-induced extracellular matrix with a novel inhibitor of the TGF-beta type I receptor kinase activity: SB-431542. Mol Pharmacol. 2002;62:58–64. doi: 10.1124/mol.62.1.58. [DOI] [PubMed] [Google Scholar]
- Seay U, Sedding D, Krick S, Hecker M, Seeger W, Eickelberg O. Transforming growth factor-beta-dependent growth inhibition in primary vascular smooth muscle cells is p38-dependent. J Pharmacol Exp Ther. 2005;315:1005–1012. doi: 10.1124/jpet.105.091249. [DOI] [PubMed] [Google Scholar]
- Horowitz JC, Rogers DS, Sharma V, Vittal R, White ES, Cui Z, Thannickal VJ. Combinatorial activation of FAK and AKT by transforming growth factor-beta1 confers an anoikis-resistant phenotype to myofibroblasts. Cell Signal. 2007;19:761–771. doi: 10.1016/j.cellsig.2006.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokkinos MI, Brown HJ, de Iongh RU. Focal adhesion kinase (FAK) expression and activation during lens development. Mol Vis. 2007;13:418–430. [PMC free article] [PubMed] [Google Scholar]
- Takahashi N, Seko Y, Noiri E, Tobe K, Kadowaki T, Sabe H, Yazaki Y. Vascular endothelial growth factor induces activation and subcellular translocation of focal adhesion kinase (p125FAK) in cultured rat cardiac myocytes. Circ Res. 1999;84:1194–1202. doi: 10.1161/01.res.84.10.1194. [DOI] [PubMed] [Google Scholar]
- Bouchard V, Demers MJ, Thibodeau S, Laquerre V, Fujita N, Tsuruo T, Beaulieu JF, Gauthier R, Vezina A, Villeneuve L, Vachon PH. Fak/Src signaling in human intestinal epithelial cell survival and anoikis: differentiation state-specific uncoupling with the PI3-K/Akt-1 and MEK/Erk pathways. J Cell Physiol. 2007;212:717–728. doi: 10.1002/jcp.21096. [DOI] [PubMed] [Google Scholar]
- Yao K, Tan J, Ye P, Wang K, Xu W, ShenTu X, Tang X. Integrin beta1-mediated signaling is involved in transforming growth factor-beta2-promoted migration in human lens epithelial cells. Mol Vis. 2007;13:1769–1776. [PubMed] [Google Scholar]