

Abstract
Alzheimer’s disease (AD) is characterized by the accumulation of extracellular insoluble amyloid, primarily derived from polymerized amyloid-β (Aβ) peptides. We characterized the chemical composition of the Aβ peptides deposited in the brain parenchyma and cerebrovascular walls of triple transgenic Tg-SwDI mice that produce a rapid and profuse Aβ accumulation. The processing of the N- and C-terminal regions of mutant AβPP differs substantially from humans because the brain parenchyma accumulates numerous, diffuse, nonfibrillar plaques, whereas the thalamic microvessels harbor overwhelming amounts of compact, fibrillar, thioflavine-S- and apolipoprotein E-positive amyloid deposits. The abundant accretion of vascular amyloid, despite low AβPP transgene expression levels, suggests that inefficient Aβ proteolysis because of conformational changes and dimerization may be key pathogenic factors in this animal model. The disruption of amyloid plaque cores by immunotherapy is accompanied by increased perivascular deposition in both humans and transgenic mice. This analogous susceptibility and response to the disruption of amyloid deposits suggests that Tg-SwDI mice provide an excellent model in which to study the functional aftermath of immunotherapeutic interventions. These mice might also reveal new avenues to promote amyloidogenic AβPP processing and fundamental insights into the faulty degradation and clearance of Aβ in AD, pivotal issues in understanding AD pathophysiology and the assessment of new therapeutic agents.
Alzheimer’s disease (AD) dementia is affecting an escalating proportion of the elderly population because of a dramatic increase in life expectancy. This neurodegenerative disorder is characterized by the profuse accumulation of extracellular insoluble amyloid in cerebral vessels and senile plaques, which are mainly composed of amyloid fibrils derived from polymerized amyloid-β (Aβ) peptides. Amyloid-β40/42 peptides are generated from the proteolytic degradation of the amyloid-β precursor protein (AβPP) by the action of the β- and γ-secretases. Amyloid-β peptides are very insoluble and resistant to subsequent proteolytic degradation because they contain a segment of the hydrophobic transmembrane domain of AβPP. The prominent amyloid buildup associated with AD has resulted in the amyloid cascade hypothesis in which amyloid plays a fundamental mechanistic role in the pathogenesis and the emergence of dementia. A second relevant lesion in the AD brain is the accumulation of neurofibrillary tangles and neuropil threads that are mainly composed of hyperphosphorylated tau, a microtubule-associated protein.
To determine the mechanisms leading to amyloid deposition and its clearance as well as its pathophysiological effects on brain tissue, several transgenic (Tg) mice have been engineered that carry familial AD AβPP mutations with suitable promoters to accelerate the deposition of Aβ peptides. The Tg-SwDI triple Tg mice1 express an AβPP with Aβ flanking, double Swedish mutations (Lys670→Asn/ Met671→Leu), the Dutch (Glu693→Gln), and the Iowa (Asp694→Asn) mutations (sequence numbering in the AβPP770 isoform notation), under the control of the mouse Thy-1 promoter. In humans, the AβPP Swedish mutations are responsible for an increased production and deposition of the Aβ40/42 peptides,2 whereas the Dutch and Iowa AβPP variants, which occur at positions 22 and 23 of the Aβ peptide, respectively, independently produce a phenotype in which the Aβ40 species accumulation prevails.3,4 The massive accumulation of Aβ in the cerebrovasculature of patients carrying the Dutch and Iowa mutations results in vascular fragility with cerebral hemorrhages and dementia.5,6 An important feature of the Tg-SwDI mice is that detectable amyloid begins to accumulate at ∼2 to 3 months of age and deposits are extensive by 12 months of age. A peculiar trait of the Tg-SwDI, in contrast with other AβPP Tg mice, is that the rapid and abundant Aβ accumulation occurs despite a very low expression of the human AβPP construct relative to the endogenous mouse wild-type form.1 This situation suggests the Tg-SwDI paradigm may provide a means to investigate Aβ degradation and clearance, pivotal to understanding AD Aβ pathophysiology and the design and assessment of potential therapeutic interventions.7 The prolific deposition of vascular Aβ in the microvascular walls of the Tg-SwDI mice elicit a severe inflammatory reaction that is accompanied by reactive microgliosis and astrocytosis, vascular degeneration, and apoptosis.8 Another unique characteristic of the Tg-SwDI model is that the brain parenchymal tissue accumulates numerous diffuse nonfibrillar plaques, whereas the microvessels harbor an overwhelming amount of compact fibrillar and thioflavine-S-positive amyloid deposits. The vasculotropic preponderance of the Aβ fibrillar deposits emphasizes the importance of the Tg-SwDI model for the study of cerebrovascular amyloidosis at the capillary-arteriolar level and its effects on the integrity of the microvasculature as well as the blood-brain barrier alterations associated with AD. We investigated the characteristics of the AβPP and Aβ peptides produced and deposited in the brains of the Tg-SwDI mice and established the differences between this model and human amyloid pathology observed in AD.
Materials and Methods
Mice and Human Tissues
All work with mice followed the National Institutes of Health guidelines and was approved by the Stony Brook University Institutional Animal Care and Use Committee. Generation of Tg-SwDI mice on a pure C57BL/6 background was previously described.1 These mice express low levels of human Swedish/Dutch/Iowa mutant AβPP in neurons under the control of the mouse Thy1.2 promoter. Homozygous Tg-SwDI and age-matched non-Tg C57BL/6 mice were used in the present study (Table 1). Human brain specimens were obtained from our Brain and Body Donation Bank at Sun Health Research Institute in Sun City, AZ. From an ethical point of view, all donations are voluntary. The donors are clinically followed by periodic examinations by a neurologist during their lifetime and signed a legal consent form bestowing their brains for research (Table 1).
Table 1.
Human and Mouse Specimens
| Gender | Age | Aβ40 (μg/g wet weight) | Aβ42 (μg/g wet weight) | ||
|---|---|---|---|---|---|
| Mice used in experiments and Aβ ELISA values | |||||
| I. ELISAs and Westerns | |||||
| Tg-SwDI (n = 4) | M | 18 (months) | 0.109 | 1.209 | |
| M | 18 | 0.129 | 1.368 | ||
| M | 18 | 0.097 | 1.209 | ||
| M | 18 | 0.072 | 1.076 | ||
| C57BL6 (n = 4) | M | 18 | 0.018 | BLD | |
| M | 18 | 0.023 | BLD | ||
| M | 18 | 0.010 | BLD | ||
| M | 18 | 0.024 | BLD | ||
| II. Formic acid extraction of Aβ peptides | |||||
| Tg-SwDI (n = 10) | M | 25 | |||
| M | 25 | ||||
| M | 24 | ||||
| M | 24 | ||||
| M | 20 | ||||
| M | 20 | ||||
| F | 21 | ||||
| F | 17 | ||||
| F | 22 | ||||
| F | 22 | ||||
| III. Purification of parenchymal and vascular Aβ | |||||
| Tg-SwDI (n = 14) | M | 21 | |||
| M | 23 | ||||
| M | 23 | ||||
| M | 22 | ||||
| M | 22 | ||||
| M | 22 | ||||
| M | 23 | ||||
| F | 24 | ||||
| F | 20 | ||||
| F | 17 | ||||
| F | 22 | ||||
| F | 22 | ||||
| F | 22 | ||||
| F | 22 | ||||
| IV. Characterization AβPP C-terminal peptides | |||||
| Tg-SwDI (n = 5) | M | 25 | |||
| M | 23 | ||||
| F | 17 | ||||
| F | 22 | ||||
| V. Purification of vascular Aβ for thioflavine-S stain | |||||
| Tg-SwDI (n = 2) | M | 24 | |||
| M | 20 | ||||
| VI. Purification of cerebral blood vessels for immunohistochemistry | |||||
| Tg-SwDI (n = 2) | F | 24 | |||
| M | 23 | ||||
| Human cases used in Western blots
|
Age (years)
|
Gender
|
Apo E genotype
|
Braak stage
|
Total plaque score
|
| 9-AD | 80 | M | 3/3 | V | 14 |
| 10-AD | 81 | M | 3/4 | VI | 13 |
| 11-AD | 77 | M | 3/4 | VI | 14 |
| 12-NDC | 79 | M | 3/3 | I | 3.5 |
| 13-NDC | 81 | M | 3/3 | I | 0 |
| 14-NDC | 78 | M | 3/3 | II | 0 |
BLD, below the limit of detection; M, male. F, female; AD, Alzheimer’s disease; NDC, nondemental control; ELISA, enzyme-linked immunosorbent assay.
Quantification of Aβ Peptides by Immunoassay
Aβ40 and Aβ42 peptides were immunoassayed in four Tg-SwDI mice and four C57BL/6 wild-type mice. All mice were 18-month-old males (Table 1, section I). In each case, the whole cerebrum was finely ground and one-third of this tissue was homogenized in 8 vol of 5 mol/L guanidine hydrochloride prepared in 50 mmol/L Tris-HCl, pH 8.0. The homogenates were shaken for 3 hours at 4°C and centrifuged at 60,000 × g for 30 minutes and the supernatants were collected for analysis. The enzyme-linked immunosorbent assays for the Aβ40 and Aβ42 were obtained from Immunobiological Laboratories, Minneapolis, MN, and from Innogenetics, Gent, Belgium, respectively. The enzyme-linked immunosorbent assays were performed following the manufacturers’ instructions.
Western Blot Analysis
The entire cerebrum from four Tg-SwDI mice and four C57BL/6 wild-type mice (Table 1, section I), was finely ground and one-half of this tissue was homogenized in 10 vol of the denaturing RIPA buffer (Sigma, St. Louis, MO) containing a protease inhibitor cocktail (Roche Diagnostics, Mannheim, Germany) at 4°C. In addition, the frontal cortex of three AD cases and three human nondemented controls (Table 1) was homogenized in the same manner. The homogenates were centrifuged at 18,000 × g for 10 minutes at 4°C. The supernatants were collected and total protein quantified using the BCA protein assay (Pierce, Rockford, IL). The samples were placed in NuPage 2× LDS buffer (Invitrogen, Carlsbad, CA) containing 50 mmol/L dithiothreitol and heated for 10 minutes at 80°C. The specimens were separated on 4 to 12% Bis-Tris gels developed with NuPage 2-morpholinoethanesulfonic acid running buffer (Invitrogen). The separated proteins were transferred onto nitrocellulose membranes and blocked with 5% nonfat milk in phosphate-buffered saline (PBS) containing 0.5% Tween 20 (Fluka, St. Louis, MO). Proteins were probed with 22C11 (Millipore, Billerica, MA) to detect AβPP amino acid residues 66 to 81 and, CT9APP antibody (Millipore) to detect the last nine C-terminal amino acids of AβPP. In addition, BC2, a polyclonal antibody against amino acid residues 97 to 273 of the rat insulin-degrading enzyme (IDE), was created as previously described.9 Goat anti-mouse IgG-conjugated horseradish peroxidase was used as the secondary antibody for 22C11 and goat anti-rabbit IgG-conjugated horseradish peroxidase was used as the secondary antibody for CT9APP and anti-IDE. The blots were developed with SuperSignal WestPico chemiluminescent substrate and CL-Xpose film (Pierce) and Kodak GBX developer (Sigma). The films were scanned with a GS-800 calibrated densitometer (Bio-Rad, Hercules, CA) and analyzed with the Quantity One software (Bio-Rad).
Formic Acid Extraction and Analysis of Total Aβ Peptides
Ten Tg-SwDI mice cerebrums (Table 1, section II) were minced and homogenized in 10 ml of 90% glass-distilled formic acid (GDFA) using a Tenbroek frosted tissue grinder. The homogenate was allowed to stand for 20 minutes at room temperature before centrifugation at 250,000 × g in a SW 41Ti Beckman rotor (Fullerton, CA) for 1.5 hours. The fatty dense plug collected at the top of the tube and the insoluble pellet were discarded and the clear supernatant was divided into aliquots of 500 μl, which were submitted to size exclusion fast protein liquid chromatography (FPLC) on a Superose 12 column (1 × 30 cm; GE Healthcare, Piscataway, NJ). The chromatography was developed at room temperature with an 80% GDFA mobile phase at a flow rate of 15 ml/hour. The Superose 12 columns were precalibrated using the Aβ40-1 reverse sequence peptide (California Peptide Inc., Napa, CA). The fractions encompassing the 10-2-kDa range were pooled and after the addition of 10 μl of 10% betaine in water, the specimens were reduced to 125 μl by vacuum centrifugation (Savant/Thermo, Waltham, MA). To further separate the 10-2-kDa fraction from flanking contaminating peptides, four chromatographic runs were pooled and separated by an additional round of FPLC under the same conditions and the 10-2-kDa-containing fraction volumes reduced to 250 μl. After the addition of 250 μl of fresh 80% GDFA, the sample was submitted to reverse-phase high performance liquid chromatography (HPLC) system (Thermo Fisher Scientific, West Palm Beach, FL) on a C8 column (9.4 mm × 250 mm, Zorbax 300SB-C8; Agilent Technologies, Santa Clara, CA). The chromatography was performed with the column maintained at 80°C using a linear gradient from 20 to 40% solvent B [acetonitrile, 0.1% trifluoroacetic acid (TFA)] concentration formed by solvent A (water, 0.1% TFA) developed for 90 minutes at a flow rate of 1.5 ml/minute. All collected fractions were submitted to Western blot analysis for identification of the Aβ peptides as described above. Formic acid or acetonitrile solutions were removed by vacuum centrifugation (Savant) and replaced with 200 μl of distilled water and dried. This operation was repeated three times. The peptides of interest were detected with the primary anti-Aβ40 and Aβ42 antibodies (Invitrogen). Goat anti-rabbit IgG-conjugated horseradish peroxidase was used as the secondary antibody (Pierce). Those peaks that were positive for Aβ were subsequently investigated by surface-enhanced laser desorption ionization-time of flight (SELDI-TOF).
Preparation and Purification of Parenchymal Amyloid
Fourteen Tg-SwDI mice brains (Table 1, section III) were minced and suspended in 10 ml of an 8 mol/L urea, 50 mmol/L Tris-HCl, pH 8.0, solution and homogenized (five strokes, homogenizer clearance 150 μm). The homogenate was stirred overnight at 4°C. To remove the urea-insoluble blood vessels, the homogenate was filtered through 25-μm nylon mesh. Further removal of capillaries was achieved by submitting the filtered urea homogenate to centrifugation (3000 × g for 20 minutes). The blood vessels were set aside for further analysis (see below). The supernatant was dialyzed against four changes, 4 L each, of 0.1 mol/L ammonium bicarbonate. The precipitated insoluble material was recovered by centrifugation at 100,000 × g. The pellet and the supernatant were labeled as urea/ammonium bicarbonate - insoluble fraction and urea/ammonium bicarbonate-soluble fraction, respectively. The latter fraction was directly lyophilized yielding 137 mg. The pelleted portion was suspended in 50 ml of ammonium bicarbonate and lyophilized yielding 114 mg. The urea/ammonium bicarbonate-soluble fraction was dissolved in 8 ml of 80% GDFA, and the urea/ammonium bicarbonate-insoluble fraction was dissolved in 6 ml of 80% GDFA. Both fractions were aliquoted into 500-μl fractions and submitted to size exclusion FPLC under the conditions described above and the fractions were further purified by HPLC.
Characterization of Tg-SwDI AβPP C-Terminal Peptides
Five cerebral hemispheres of Tg-SwDI mice (Table 1, section IV) were homogenized in 10 ml of 98% GDFA, centrifuged at 250,000 × g for 1 hour at 4°C. Fractions of 500 μl were submitted to FPLC, as described above, and the 10-2-kDa fractions collected. The specimens were further separated by HPLC on a C8 reverse-phase column using an acetonitrile gradient from 0 to 100% acetonitrile in 0.1% TFA for 60 minutes. The chromatography was developed at 80°C. All separated fractions were submitted to Western blot analysis using the CT9APP antibody (Millipore) to localize AβPP C-terminal peptides.
Preparation and Purification of Vascular Amyloid
Two Tg-SwDI mice brains (Table 1, section V) were cut coronally and medially to yield four equal volume portions. They were stirred in a solution of 5% sodium dodecyl sulfate (SDS) and Tris (50 mmol/L, pH 7.5) for 72 hours. The insoluble white tufts of blood vessels remaining were rinsed with distilled water and a small portion placed on a glass slide, dried at 55°C, fixed with absolute ethanol for 20 minutes, and stained for 10 minutes with 1% aqueous thioflavine-S. Excessive stain was removed by rinsing with 70% ethanol. The remaining tufts of blood vessels from the parenchymal amyloid preparation (see above) were washed with distilled water and centrifuged at 3000 × g (Table 1, section III). The pellet was dissolved in 2.5 ml of 90% GDFA, centrifuged at 100,000 × g in a TLX Beckman ultracentrifuge for 20 minutes and the acid supernatant containing the Aβ peptides submitted in aliquots of 500 μl to FPLC on a size exclusion Superose 12 column, as described above. The 10-2 kDa fractions from the five chromatography runs were collected, pooled, and the volume reduced to 250 μl by vacuum centrifugation (Savant). These specimens were submitted to HPLC on a C8 reverse-phase column (4.6 mm × 250 mm, Zorbax 300SB-C8; Agilent Technologies). The chromatography was performed with a linear gradient of acetonitrile, 0.1% TFA mobile phase ranging from 20 to 60% concentration developed in 90 minutes at a flow rate of 0.8 ml/minute. The column was maintained at a constant temperature of 80°C. The collected fractions were concentrated by vacuum centrifugation and submitted to matrix-assisted laser desorption/ionization-time of flight (MALDI-TOF) mass spectrometry (MS).
Immunohistochemistry of Cerebral Blood Vessels
Tufts of cerebral blood vessels from two Tg-SwDI mice brains (Table 1, section VI) were air-dried on glass slides, fixed in absolute ethanol for 30 minutes, stained with 1% aqueous thioflavine-S for 10 minutes, rinsed with 70% ethanol to remove excessive nonbound fluorochrome, and mounted with Difco FA mounting fluid. Air-dried tufts of purified blood vessels were also fixed for 30 minutes in absolute ethanol and stained for apolipoprotein E (polyclonal antibody, 1:2000 dilution; Calbiochem, San Diego, CA), followed by reaction with Alexa-568 anti-goat IgG (1:1000 dilution, Invitrogen). Sections were incubated in 0.1% thioflavine-S prepared in 70% ethanol, rinsed with distilled water, and counterstained with 1% Sudan black to block autofluorescence. Sections were coverslipped with Vector Shield mounting media (Vector Laboratories, Burlingame, CA) and examined by dual-color argon/krypton lasers using an IX70 confocal microscope (Olympus, Center Valley, PA).
In Vitro Degradation Assays of Aβ
Recombinant rat IDE 42-1019 (rIDE) was subcloned from pECE-IDE (kindly provided by Dr. Richard Roth, Stanford University, CA) into pET-30a (+) (Novagen, Darmstadt, Germany), expressed in Escherichia coli BL21 and purified using a Hi Trap Ni2+ chelating column as described.9 Further purification was obtained by size exclusion chromatography on a Superdex 200 column (Amersham Biosciences, Arlington Heights, IL). Protein concentration was determined by absorbance at 280 nm (extinction coefficient 115,810 cm−1 mol/L−1) and by a BCA protein assay (Pierce) and expressed as monomeric rIDE. Lyophilized Aβ1-40 wild-type and Aβ1-40 Dutch/Iowa were dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) at 5 mg/ml and sonicated to disaggregate oligomers as described.10 Three μg of each synthetic peptide from the HFIP solution were brought to dryness under vaccum centrifugation (Savant) and incubated with 800 ng of purified rIDE in 10 μl of 100 mmol/L Na phosphate buffer, pH 7, at 37°C for 30 to 60 minutes. The reaction was stopped by adding 20 μl of 8% SDS sample buffer containing 0.1 mol/L dithiothreitol followed by boiling for 3 minutes. One sample was immediately boiled after mixing the peptides with rIDE and taken as the dead time, or time 0, of the experiment. After incubation, samples were analyzed by 15% Tris-tricine SDS-polyacrylamide gel electrophoresis and Western blot with monoclonal antibody 6E10 (Signet, Dedham, MA) as described above. Densitometric analysis was done after scanning with a STORM 840 phosphorimager and Image Quant software (GE Healthcare, Piscataway, NJ). The densities of both monomeric and dimeric Aβ were used to estimate the extent of degradation. The results represent three independent experiments.
MALDI-TOF MS
The HPLC fractions in which Western blot analysis revealed the presence of Aβ peptides were reduced to ∼10 μl vol and 5-μl samples were added to 5 μl of a saturated solution of α-cyano-4-hydroxycinnamic acid (CHCA) dissolved in 50% acetonitrile and 0.1% TFA. Aliquots of 0.5 μl were spotted on a sample plate and mass spectra obtained. A MALDI-TOF Voyager DE STR mass spectrometer (Applied Biosystems, Foster City, CA) fitted with a nitrogen laser that generated 337 nm pulses of 3-ns duration at a repetition rate of 20 Hz was used to acquire data. The system setting was in the positive mode using delayed extraction in the linear mode and averaging 100 laser shots. Calibration was made using the CalMix 2 mixture (Applied Biosystems). The detailed composition of the calibration markers is presented elsewhere.11
SELDI-TOF MS
Selected HPLC peaks in which Western blot analysis revealed the presence of Aβ peptides were reduced to 1 ml and dialyzed in 1000-MW cutoff bags for 1 hour against 50 mmol/L Tris-HCl, pH 8.0, and 1 hour against 10 mmol/L Tris-HCl, pH 8.0. Dialyzed sample volumes were then reduced to ∼10 μl before application on ProteinChip arrays and analyzed using SELDI-TOF MS (Ciphergen/Bio-Rad, Hercules, CA). All protein chip manipulations were performed in a humidity chamber to prevent solution evaporation. The PS20 ProteinChip Arrays were preconditioned by loading 4 μl of PBS onto the chip spots and incubating for 15 minutes at room temperature. A volume of 4 μl of the polyclonal anti-Aβ40, and anti-Aβ42 antibodies (Invitrogen) was loaded onto the chip at 0.38 mg/ml and 0.57 mg/ml (manufacturer provided concentrations), respectively. The antibody-treated protein chips were incubated for 1 hour at room temperature. The chip surfaces were blocked with 4 μl of ethanolamine (1 mol/L, pH 8.0) for 30 minutes. The sample spots were individually washed once with 4 μl of PBS plus 0.5% Triton X-100 and twice with 4 μl of PBS. The protein chips were loaded with either 4 μl of sample or 4 μl of Aβ 1-40 or 1-42 peptide standard (California Peptide Research Inc.) in dimethyl sulfoxide as positive controls and incubated overnight at 4°C. After the sample binding solutions were removed, the spots were washed individually, twice with 4 μl of PBS plus 0.5% Triton X-100, once with 4 μl of PBS, and once with 4 μl of distilled water. The chips were allowed to air-dry and a 20% saturated solution of CHCA in acetonitrile with 0.1% TFA was applied twice (0.5 μl per application) allowing the chip spots to dry between applications. The molecular mass assignments were made by 100 averaged shots in a Ciphergen Seldi Protein Biology System II with external calibration attained using the Ciphergen All-In-One peptide standard.
Results
Quantification of Aβ peptides present in the brains of the Tg-SwDI mice (n = 4) by immunoassay demonstrated that Aβ42 was substantially more abundant than Aβ40 (Table 1, section I). The average quantity of the former peptide was 1.214 μg per g of wet tissue (range, 1.076 to 1.368) and that of the latter, 0.102 μg per g of wet tissue (range, 0.072 to 0.129). In comparison, the C57BL/6 mice (n = 4) contained an average of 0.019 μg per g of wet tissue (range, 0.010 to 0.024) Aβ40, whereas the Aβ42 levels were below the limit of detection.
Western blots of the whole Tg-SwDI cerebral tissue that were developed with the 22C11 antibody raised against residue 66 to 81 of the AβPP molecule revealed a peptide of ∼55 kDa that was not present in the human brain of AD and control patients and is very faint in the C57BL/6 wild-type mouse brains. Furthermore, two bands between ∼25 and 30 kDa, representing AβPP N-terminal (NT) peptides, were more abundant in the Tg-SwDI than in the human brain and in the wild-type mouse brain (Figure 1A). Probing the Western blots with the CT9APP antibody that identifies the C-terminal (CT), nine amino acid residues of the AβPP molecule also demonstrated differences between humans and Tg or wild-type mice (Figure 1B). In the Tg-SwDI and wild-type mice, there were bands at ∼20 kDa, ∼22 kDa, and ∼26 kDa that were very faint in the human brains. Although the CT99/CT83 fragments were present in less abundant quantities in the Tg mice, two novel bands of ∼8 to 10 kDa were present in the Tg mice that were absent in the human brain and the wild-type mouse tissue. These bands probably correspond to the CT human AβPP sequence encompassing peptides from the γ-secretase cleavage at residue 40 or 42 (CT57/59) or correspond to the CT50 AβPP intracellular domain (AICD). In the Western blots of IDE, we observed that in both Tg and wild-type mice, the amount of full length IDE (115 kDa) did not differ significantly. However, both the human AD and ND control brains demonstrated reduced amounts of IDE when compared to the rodents, with the AD brains considerably lower overall (Figure 1C).
Figure 1.

Western blots of human and Tg-SwDI AβPP peptides and IDE. A: Western blot of the N-terminal peptides detected by the 22C11 monoclonal antibody raised against residues 66 to 81 of the human AβPP molecule. B: Western blot of the C-terminal peptides detected by the polyclonal CT9APP raised against the last nine amino acids of the AβPP. C: Western blot of BC2, an antibody raised against IDE. In both A and B, 25 μg of total protein were loaded and in C 40 μg of total protein were loaded. Lanes 1 to 4, Tg-SwDI mice; lanes 5 to 8, C57BL/6, wild-type mice; lane 9, male AD, 80 years old, with Apo E ε3/ε3 genotype; lane 10, male AD, 81 years old, with Apo E ε3/ε4 genotype; lane 11, male AD, 77 years old, with Apo E ε3/ε4 genotype; lane 12, male ND control, 79 years old, with Apo E ε3/ε3 genotype; lane 13, male ND control, 81 years old, with Apo E ε3/ε3 genotype; lane 14, male ND control, 78 years old, with Apo E ε3/ε3 genotype.
Complete lysis of whole Tg-SwDI brains by GDFA followed by centrifugation and FPLC separation of the acid-soluble fraction permitted the enrichment of the Aβ-containing 10-2-kDa fraction (Figure 2A). This fraction was further refined using reverse-phase HPLC that yielded six fractions reactive with Aβ40 and Aβ42 antibodies (Figure 2B). These specimens were submitted to SELDI-TOF MS. The resulting spectra are shown in Figure 2, C and D, and their equivalent masses and peptide assignments are given in Table 2, sections II and III. The Aβ-related peptides represented species with N-termini at residues 1, 2, 3, 4, 5, and 10 and C-termini at residues 36, 39, 40, and 42. SELDI-TOF also identified Aβ peptides ending at residues 48, 52, and 54 (Aβ sequence notation). In addition, the chemical separation of the CTAPP peptides was performed by reverse-phase chromatography on a C8 column at 80°C. The CT peptides were identified by Western blot using the CT9APP antibody that localized the CT peptides at peaks 5, 6, and 7 (Figure 2E). These peptides were submitted to SELDI-TOF and MALDI-TOF and their mass spectra are presented in Table 2, sections IV and V, respectively.
Figure 2.

Chromatographic profiles and mass spectra of Aβ-related peptides directly solubilized by GDFA from the Tg-SwDI mice. A: FPLC elution patterns of the acid-solubilized whole brains. The solid line corresponds to the initial FPLC and the horizontal bar indicates the collected material containing the Aβ peptides. To enrich the Aβ fraction and eliminate flanking contaminations, four of the initial FPLC chromatographies were pooled and resubmitted to FPLC under same conditions (hyphened line). B: Reverse-phase HPLC of the FPLC Aβ-enriched material showing the elution pattern and the fractions that contained the Aβ peptides. The Aβ40/Aβ42 and related peptides were characterized by SELDI-TOF and their mass spectra are shown in C and D. The spectrum of each of the individual HPLC peaks (1 to 6) is shown in C and D. The number at the top right corner on each spectral strip represents the HPLC peak analyzed. The standards for Aβ40 and Aβ42 are shown at the bottom of C and D. The observed and calculated mass spectral values are given in Table 2, sections II and III. E: Elution profile of the reverse-phase acid-soluble fraction of the Tg-SwDI. After GDFA-FPLC the Aβ-containing fractions were submitted the HPLC C8 column (0 to 100% acetonitrile gradient). The resolved peaks were investigated by Western blot (see inset) and SELDI-TOF (fractions 5 to 7) using the CT9APP as capture antibody. Their spectral values are shown in Table 2, section IV.
Table 2.
Mass Spectrometry of Tg-SwDI AβPP and Aβ Peptides
| Observed | Calculated | Peptide sequence |
|---|---|---|
| I. MALDI-TOF mass spectra of vascular amyloid peptides | ||
| 4031.3 | 4031.5 | 2-38 (ox) |
| 4089.8 | 4089.6 | 1-37 (ox) |
| 4148.5 | 4146.6 | 1-38 (ox) |
| 4248.6 | 4245.8 | 2-40 (2ox) |
| 284.0 | 4285.7 | 1-39 (f) |
| 4347.7 | 4344.9 | 1-40 (ox) |
| 4383.7 | 4384.9 | 1-40 (2f) |
| II. SELDI-TOF mass spectra of acid soluble fraction (Aβ40 captured) | ||
| 4344.4 | 4346.8 | 1-40 (ox) |
| 4028.8 | 4031.6 | 4-40 (ox) |
| 4330.4 | 4330.8 | 1-40 |
| 3881.9 | 9884.4 | 5-40 (ox) |
| 4140.4 | 4144.7 | 3-40 |
| 5023.3 | 5026.8 | 2-48 |
| III. SELDI-TOF mass spectra of acid soluble fraction (Aβ42 captured) | ||
| 4344.4 | 4346.8 | 1-40 (ox) |
| 3343.8 | 3345.9 | 10-42 (2ox) |
| 4245.2 | 4242.7 | 1-39 (ox) |
| 4060.8 | 4060.5 | 1-36 (ox/f) |
| 4214.9 | 4213.8 | 2-40 |
| 3882.3 | 3882.4 | 5-40 (ox) |
| 3229.5 | 3230.7 | 10-39 (ox) |
| 5030.3 | 5026.8 | 2-48 |
| 5740.7 | 5739.8 | 2-54 |
| 5477.6 | 5476.4 | 3-52 (4ox) |
| IV. SELDI-TOF mass spectra of APP CT peptides (Aβ notation) | ||
| 7355.9 | 7355.4 | 36-99 (5ox/f) |
| 7314.0 | 7316.3 | 37-99 (6f) |
| 7355.9 | 7355.2 | 38-99 (6ox/3f) |
| 7355.9 | 7358.7 | 42-99 (3ox/6f) |
| 6697.5 | 6695.7 | 43-99 (ox/f) |
| 6600.7 | 6606.5 | 44-99 (2f) |
| 6600.7 | 6599.4 | 45-99 (4ox/2f) |
| 6586.9 | 6586.3 | 46-99 (5ox/3f) |
| 6615.7 | 6615.1 | 47-99 (6ox/4f) |
| 6093.5 | 6091.7 | 50-99 (6ox/2f) |
| 5751.4 | 5752.2 | 53-99 (ox/3f) |
| 5767.2 | 5763.9 | 55-99 (2ox/5f) |
| V. MALDI-TOF mass spectra of APP CT peptides (Aβ notation) | ||
| 6737.6 | 6737.8 | 41-98 (ox) |
| 7320.4 | 7318.9 | 41-99 (4ox/5f) |
| 6722.7 | 6722.7 | 42-99 |
| 6292.9 | 6294.2 | 43-96 (ox) |
| 6043.4 | 6040.9 | 49-99 (ox) |
| 6043.4 | 6043.7 | 50-99 (3ox/2f) |
ox, oxygen; f, formyl.
Western blots of Aβ peptides present in the diffuse parenchymal plaques that were purified by FPLC and HPLC revealed the presence of abundant Aβ42 with trace amounts of Aβ40 (Figure 3, A–C). Interestingly, most of these Aβ peptides were in the dimeric conformation. Although all of the pathogenic Aβ1-40 variants have been shown to be degraded by rIDE, the Dutch (AβE22Q) variant is more resistant to proteolysis than Aβ1-40 wild-type in vitro.9 To extend these observations to Aβ1-40 Dutch/Iowa, synthetic peptides were incubated with rIDE in a time course experiment and the remaining dimeric and monomeric peptides, after SDS-polyacrylamide gel electrophoresis and Western blot, were quantified (Figure 4). Although 75% of Aβ1-40 wild-type monomer and 30% of dimer were degraded after 1 hour, Aβ1-40 Dutch/Iowa was extremely resistant to degradation (less than 20% of monomer and virtually no degradation of dimer).
Figure 3.
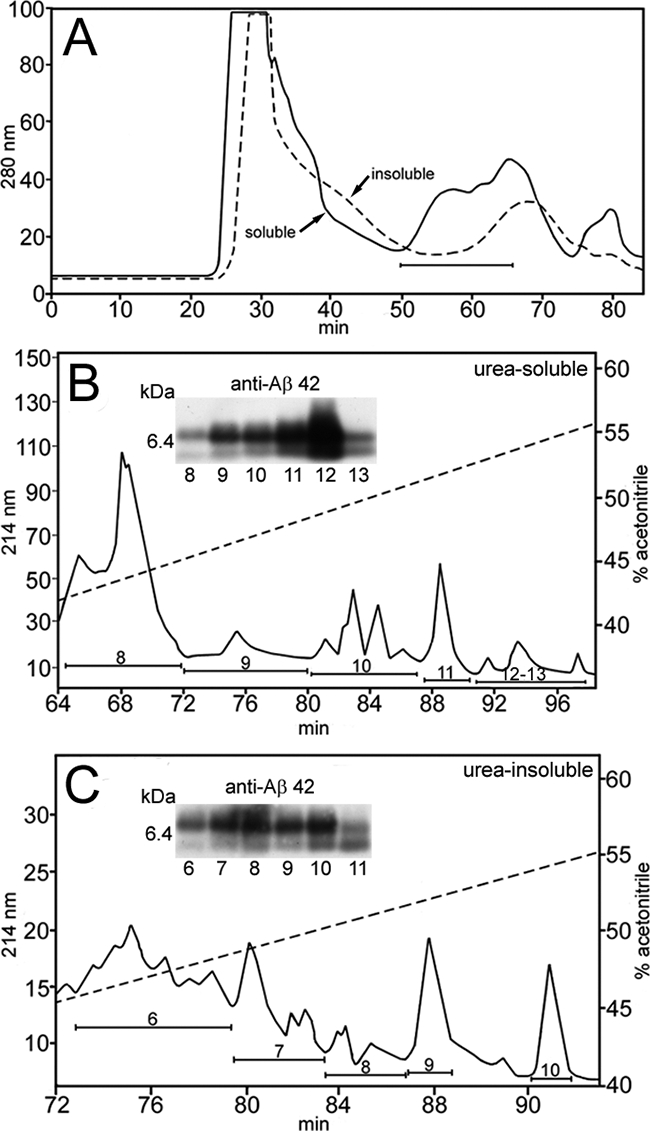
Purification and analysis of Aβ peptides isolated by urea and GDFA from the Tg-SwDI mice brain parenchymal tissue. A: FPLC elution profiles of the urea-soluble (solid line) and urea-insoluble fractions (hyphened line). The horizontal bar indicates those fractions that were subsequently purified by reverse-phase HPLC. B and C: HPLC profile of the urea-soluble and urea-insoluble fractions, respectively, recovered from the FPLC separations. The hyphened diagonal lines indicate the concentration of elution gradient. The insets show the Western blots probed with Aβ42 antibody as well as their corresponding fractions in the chromatograms. Under the experimental conditions used, the Western blots demonstrate a preponderance of dimers over monomers (∼9:1). Western blots were also reacted with Aβ40 antibody. Longer exposure times demonstrated the presence of very small amounts of Aβ40 (data not shown).
Figure 4.

Western blot with monoclonal anti-Aβ 6E10 after incubation of Aβ1-40 wild-type and Aβ1-40 Dutch/Iowa with rIDE for the indicated times (t0 refers to the dead time of the experiment). Monomers (m) and dimers (d) are indicated. On top, Coomassie blue staining of the upper part of the same gel showing the amount of rIDE in each sample.
Complete purification of the cerebral vessel tufts was achieved by the total lysis of the brain parenchyma by SDS. In an amyloid-free area from the Tg mice, as can be seen in Figure 5A, the sole remaining microscopically visible structures are the insoluble vascular connective tissue. However, in the vessels laden with amyloid cores at different stages of development, these structures were intimately attached to the microvasculature appearing similar to that of pussy willows (Figure 5B). The vascular amyloid cores exhibit a compact array of radiating fibrils with borders affecting a brush-like surface. A characteristic of the fibrillar amyloid cores attached to the microvasculature is their intense staining with thioflavine-S and apolipoprotein E (Figure 6).
Figure 5.

Tufts of purified Tg-SwDI microvessels. A: Control area devoid of amyloid deposits. B: The cores of amyloid, at different stages of development, appear to be closely attached to the microvessel basal laminae. Both micrographs were stained with thioflavine-S. Scale bars = 100 μm. Original magnifications, ×200.
Figure 6.

Confocal microscopy of amyloid fibrils and Apo E. A: Thioflavine-S-stained amyloid cores attached to the microvasculature (green fluorescent stain). B: Apolipoprotein E immunocytochemically-stained tufts of vessels loaded with amyloid cores (red fluorescent stain). C: An overlap of A and B to yield the yellow color that shows co-localization of both proteins. Scale bar = 20 μm. Original magnifications, ×200.
The chemical nature of the vascular-associated cores of amyloid was investigated after isolation of parenchyma-free pussy willows that were prepared by SDS lysis and filtration of the Tg-SwDI mice brains. Isolated vascular amyloid cores were solubilized with GDFA, submitted to FPLC and reverse-phase HPLC separations (Figure 7, A and B) with the resulting Aβ-containing peaks submitted to MALDI-TOF MS. The peaks and masses of these peptides are shown in Figure 7C and the observed and calculated masses as well as their assignments are given in Table 2, section 1. The Aβ-related peptides represented species having N-termini at Aβ residues 1 and 2 and C-termini at residues 37, 38, 39, and 40.
Figure 7.

Chromatographic separations and MALDI-TOF MS from Tg-SwDI mice isolated microvessels. A: The FPLC chromatographic profile of microvessels that were solubilized in GDFA. The bar represents the 10–2-kDa proteins that were collected. B: HPLC chromatographic elution pattern of the proteins collected in A. The fractions between the 37- and 48-minute elution times were submitted to MALDI-TOF. The hyphened diagonal line indicates the concentration of the elution gradient. Bars 1, 2, and 3 represent the Aβ-containing peptide fractions. C: The MALDI-TOF spectra, indicated by bars 1, 2, and 3, identify the Aβ peptides shown in B, respectively. The amino acid sequence numbering is indicated at the top of each peak. The calculated and observed masses of these peptides are shown in Table 2, section I.
Discussion
Tg-SwDI triple Tg mice produce rapid and disparate amyloid deposits. Thioflavine-S-positive amyloid core deposits are associated with the microvasculature whereas diffuse Aβ plaques accumulate in the brain parenchyma.8 These pathological accumulations occur rapidly despite the low level of SwDI AβPP transcription. Enzyme-linked immunosorbent assay analysis of Aβ peptides revealed that Aβ42 was substantially more abundant than Aβ40. However, this difference is probably an artifact of the 5 mol/L guanidine hydrochloride extraction method reflecting the fact that the Aβ40 present in amyloid cores attached to the cerebral microvessels is only soluble in harsher denaturants such as formic acid. As expected, the C57BL/6 (wild-type) mice contained lower amounts of Aβ40 and Aβ42 peptides than the Tg mice.
AβPP processing in the Tg-SwDI mice differs from that observed in humans. The Tg-SwDI mice generated an ∼55-kDa NT peptide that appears to be complementary to CT peptides with Mr ∼30 kDa and suggests the initial points of hydrolysis of the mutant AβPP lie upstream of the normal cleavage sites. In addition, the NT 27-kDa fragment is more abundant in the Tg mice than in the human counterpart. Differences were also evident in the CT degradation of AβPP in which the Tg mice generated ∼20, ∼22, and ∼26 kDa peptides that are present at minimal levels in the humans. The CT99/83 fragment (∼13-kDa band in the Western blot) is less abundant in the Tg-SwDI mice than in humans. Western blotting also revealed novel CT fragments (∼8 to 10 kDa) that could account for a heterogeneous mixture of CT remnant peptides probably produced by concerted secretase cleavages at the γ- and ε-sites or the combined activities of alternative endopeptidase(s). MS suggested that these shorter CT peptides had their N-termini between residues 36 through 55 and ending at residue 99, the CT residue of AβPP (all in the Aβ sequence notation). In humans, the predominant CT peptides on Western blots are CT99 and CT83, the products of the β- and α-secretases, that are degraded to yield Aβ and P3. A major difference between humans and rodents is that the latter has an increased number of proteases with a different substrate specificity, which might partially explain the different patterns of hydrolysis between the species.12 Together, these findings indicate that mice process human AβPP differently than humans.
The specific vascular affinity of the Aβ species in humans carrying the Dutch and Iowa mutations at positions Aβ22 and Aβ23, in which wild-type Glu and Asp have been mutated to Gln and Asn, must reflect critical changes in secondary and tertiary structure of the mutant Aβ peptide. These conformational alterations probably induce the rapid generation of Aβ dimers (the most common polymer we observed in the Tg-SwDI mice), possibly attributable to the lack of Aβ22 and Aβ23 carboxylic groups that impart intermolecular ionic repulsion in the middle domain of Aβ. Abundant accretion of Aβ in the cerebral vessels of Tg-SwDI despite a low APP transgene expression strongly suggests that defective clearance of the peptide could be a key pathogenic factor in this animal model. Mice in which neprilysin (NEP), IDE, and endothelin-converting enzyme genes have been altered suggest these proteases are involved in the removal of Aβ.13,14,15,16 From a purely quantitative point of view, our experiments demonstrated that IDE levels did not significantly differ between the Tg-SwDI and wild-type mice. However, there were substantial differences between the levels of IDE in rodents and humans. Interestingly, the AD patients contain lesser amounts of IDE than in the ND controls as has been previously reported.17 Several biochemical features of both mutant Aβ peptides and this group of Zn2+ metallo-endo peptidases may explain apparent proteolytic failure in Tg-SwDI brains. The tendency of Aβ containing the 22Glu→Gln Dutch mutation to form denaturant-resistant dimers may impede degradation, as has been shown for IDE in vitro.9,18 Although NEP is reportedly capable of degrading wild-type Aβ oligomers, peptides carrying the Dutch mutation are resistant to NEP hydrolysis.19,20 Therefore, despite the widespread expression of IDE and NEP in cerebral microvessels, mutation-induced conformational changes may impair Aβ proteolysis. In addition, the strong and stable interaction between Aβ-including Aβ22Glu→Gln and apolipoprotein E (Apo E)21 in the cerebral microvasculature of Tg-SwDI mice, might produce steric hindrance at the protease active center. Because it has been demonstrated that only monomeric forms of Aβ are rIDE substrates,9 the apparent degradation of dimeric Aβ1-40 wild-type in these experiments likely reflects a subpopulation of Aβ in a monomer-dimer equilibrium whereas the double-mutant species aggregated rapidly into stable, resistant dimers. Whether the specific activity of IDE and NEP is compromised in Tg-SwDI mice, as shown in humans, remains to be established.22,23,24
The Dutch/Iowa mutant Aβ peptides exhibit marked vascular tropism and low efficiency for LRP-1-mediated transport across the blood-brain barrier.1,25 These alterations result in early and robust brain accumulation of Aβ, particularly in the cerebral vasculature, and absence of Dutch/Iowa mutant Aβ in the blood.8 The vasculotropic predilection may also be related to the extracellular matrix composition and in particular to the microvascular basal membrane, which is rich in glycosaminoglycans and gangliosides. GM2 and GM3 gangliosides have been shown to markedly enhance the fibrillogenesis of Dutch and Iowa mutant Aβ peptides.26 Fibrillar vascular Aβ deposits generate compact amyloid cores resistant to solubilization by chaotropic agents and detergents. In agreement with previous chemical studies, the vasculotropic Dutch and Iowa amyloids are composed chiefly of Aβ ending at residue 40,1,18 whereas the Swedish double mutations increase the production of Aβ ending at residues 40 and 42.27 Although immunohistochemical studies in tissue sections reproduced the distribution characteristic of the specific mutations, they also revealed that the Tg-SwDI mice deposits represent a hybrid condition in which Aβ40 and Aβ42 are also found in vascular and parenchymal loci, respectively.
The Tg-SwDI mice also demonstrated a distinctive Aβ distribution because the cerebral cortex contains a strikingly low number of amyloid cores attached to the microvasculature and such structures were abundant in the thalamic area.8 In contrast, the cortex in the Tg-SwDI mice was loaded with diffuse amyloid deposits.1 In humans with rich vascular amyloid deposits, cores are far more abundant in the cerebral cortex and poorly represented in the subcortical nuclei where most of them are diffuse plaques.28,29,30,31 In the mouse brain, the number of astrocytes in the thalamus increases with age32,33 and astrocytes are producers of Aβ. In AβPP and PS1 Tg mice, the thalamic deposition of vascular amyloid is mediated by glial Apo E expression.34 Our observations indicate that in the Tg-SwDI mice there is an intimate association between vascular amyloid deposits and Apo E. Furthermore, the lack of endogenous mouse Apo E, as in the Tg-SwDI with Apo E knockout mice, severely reduces thalamic microvascular amyloid deposition.35 Interestingly, the C57BL/6 mouse, the background strain used in this study, appears to be more susceptible to age-dependent atherosclerosis and arteriosclerosis than other mice.36 It is noteworthy that in the Tg-SwDI mice the presence of vascular amyloid evokes vascular degeneration and inflammation with induction of complement proteins that is accentuated in the thalamic region.8,37 In vivo measurements of cerebral blood volumes in the AβPP TgCRND8 Tg mice showed that the thalamus had a significant reduction in cerebral blood volume, suggesting specific changes in the microvasculature of this region.38
In humans, the AβPP Swedish, Dutch, and Iowa mutations have been characterized separately. The Tg-SwDI mice incorporate all three mutations suggesting that the observations in humans might not completely reflect the complexity of these triple Tg animals. However, the Tg-SwDI mice clearly recreate many of the neuropathological and biochemical alterations observed in human patients carrying these mutations in substantially shorter time periods. The rapid deposition of vasculotropic and parenchymal Aβ, whether fibrillar or diffuse, and the neuroinflammatory reaction that it provokes, makes the Tg-SwDI mice an attractive choice to study the pathophysiological mechanisms that underlie Aβ accumulation. In particular, the recalcitrance of the Tg-SwDI mice microvascular deposits to denaturation and dispersion makes this model very attractive to study experimental immunotherapeutic interventions.39 Initial experiments demonstrated that amyloid plaque cores were removed by active Aβ immunization in humans. However, the disappearance of the parenchymal Aβ deposits was also accompanied by an increase of perivascular amyloid deposits in humans and experimental animals.40,41,42 Vaccination before Aβ deposition in the Tg-SwDI mice had no effect on subsequent amyloid accumulation. The failure to prevent amyloid accumulation in the Tg-SwDI mice may be ascribed to inadequate peripheral antibody titers, which consequently limits the levels of Aβ antibodies reaching the brain. Direct injection of Aβ antibodies resulted in the removal of diffuse Aβ deposits, but failed to disrupt amyloid in Tg-SwDI blood vessels.39 These observations suggest that Tg-SwDI mice offer a good analog to humans with substantial amyloid burdens and are an important tool for the investigation of new immunological approaches to control or eliminate amyloid deposits. Despite the very low expression of the mutant human AβPP gene in the mice, Aβ deposits accumulate rapidly in the vasculature and parenchyma. These observations suggest that Tg-SwDI mice might reveal both new avenues that promote amyloidogenic AβPP processing and fundamental insights into the faulty degradation and clearance of Aβ in Alzheimer’s disease.
Footnotes
Address reprint requests to Alex E. Roher, M.D., Ph.D., Sun Health Research Institute, Sun City AZ 85351. E-mail: alex.roher@sunhealth.org.
Supported by the National Institute on Aging (grant RO1-AG 19795), the National Institute of Neurological Disorders and Stroke (grant RO1-NS 55118), the Arizona Alzheimer’s Disease Core Center (grant NIA P30 AG 19610), the State of Arizona Alzheimer’s Disease Research Consortium, and the Argentina Agencia Nacional de Promoción Científica y Tecnológica (grant PICT 2354).
References
- Davis J, Xu F, Deane R, Romanov G, Previti ML, Zeigler K, Zlokovic BV, Van Nostrand WE. Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J Biol Chem. 2004;279:20296–20306. doi: 10.1074/jbc.M312946200. [DOI] [PubMed] [Google Scholar]
- Kalaria RN, Cohen DL, Greenberg BD, Savage MJ, Bogdanovic NE, Winblad B, Lannfelt L, Adem A. Abundance of the longer A beta 42 in neocortical and cerebrovascular amyloid beta deposits in Swedish familial Alzheimer’s disease and Down’s syndrome. Neuroreport. 1996;7:1377–1381. doi: 10.1097/00001756-199605310-00009. [DOI] [PubMed] [Google Scholar]
- Levy E, Prelli F, Frangione B. Studies on the first described Alzheimer’s disease amyloid beta mutant, the Dutch variant. J Alzheimers Dis. 2006;9:329–339. doi: 10.3233/jad-2006-9s337. [DOI] [PubMed] [Google Scholar]
- Shin Y, Cho HS, Fukumoto H, Shimizu T, Shirasawa T, Greenberg SM, Rebeck GW. Abeta species, including IsoAsp23 Abeta, in Iowa-type familial cerebral amyloid angiopathy. Acta Neuropathol. 2003;105:252–258. doi: 10.1007/s00401-002-0639-0. [DOI] [PubMed] [Google Scholar]
- Levy E, Carman MD, Fernandez-Madrid IJ, Power MD, Lieberburg I, van Duinen SG, Bots GT, Luyendijk W, Frangione B. Mutation of the Alzheimer’s disease amyloid gene in hereditary cerebral hemorrhage Dutch type. Science. 1990;248:1124–1126. doi: 10.1126/science.2111584. [DOI] [PubMed] [Google Scholar]
- Grabowski TJ, Cho HS, Vonsattel JP, Rebeck GW, Greenberg SM. Novel amyloid precursor protein mutation in an Iowa family with dementia and severe cerebral amyloid angiopathy. Ann Neurol. 2001;49:697–705. doi: 10.1002/ana.1009. [DOI] [PubMed] [Google Scholar]
- Davis J, Xu F, Miao J, Previti ML, Romanov G, Ziegler K, Van Nostrand WE. Deficient cerebral clearance of vasculotropic mutant Dutch/Iowa double A beta in human A betaPP transgenic mice. Neurobiol Aging. 2006;27:946–954. doi: 10.1016/j.neurobiolaging.2005.05.031. [DOI] [PubMed] [Google Scholar]
- Miao J, Xu F, Davis J, Otte-Holler I, Verbeek MM, Van Nostrand WE. Cerebral microvascular amyloid beta protein deposition induces vascular degeneration and neuroinflammation in transgenic mice expressing human vasculotropic mutant amyloid beta precursor protein. Am J Pathol. 2005;167:505–515. doi: 10.1016/s0002-9440(10)62993-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morelli L, Llovera R, Gonzalez SA, Affranchino JL, Prelli F, Frangione B, Ghiso J, Castano EM. Differential degradation of amyloid beta genetic variants associated with hereditary dementia or stroke by insulin-degrading enzyme. J Biol Chem. 2003;278:23221–23226. doi: 10.1074/jbc.M300276200. [DOI] [PubMed] [Google Scholar]
- Wood SJ, Maleeff B, Hart T, Wetzel R. Physical, morphological and functional differences between pH 5.8 and 7.4 aggregates of the Alzheimer’s amyloid peptide. J Mol Biol. 1996;256:870–877. doi: 10.1006/jmbi.1996.0133. [DOI] [PubMed] [Google Scholar]
- Esh C, Patton L, Kalback W, Kokjohn TA, Lopez J, Brune D, Newell AJ, Beach T, Schenk D, Games D, Paul S, Bales K, Ghetti B, Castano EM, Roher AE. Altered APP processing in PDAPP (Val717 –> Phe) transgenic mice yields extended-length Abeta peptides. Biochemistry. 2005;44:13807–13819. doi: 10.1021/bi051213+. [DOI] [PubMed] [Google Scholar]
- Puente XS, Lopez-Otin C. A genomic analysis of rat proteases and protease inhibitors. Genome Res. 2004;14:609–622. doi: 10.1101/gr.1946304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, Gerard C, Hama E, Lee HJ, Saido TC. Metabolic regulation of brain Abeta by neprilysin. Science. 2001;292:1550–1552. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch MP, Eckman CB, Tanzi RE, Selkoe DJ, Guenette S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci USA. 2003;100:4162–4167. doi: 10.1073/pnas.0230450100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller BC, Eckman EA, Sambamurti K, Dobbs N, Chow KM, Eckman CB, Hersh LB, Thiele DL. Amyloid-beta peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc Natl Acad Sci USA. 2003;100:6221–6226. doi: 10.1073/pnas.1031520100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckman EA, Watson M, Marlow L, Sambamurti K, Eckman CB. Alzheimer’s disease beta-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J Biol Chem. 2003;278:2081–2084. doi: 10.1074/jbc.C200642200. [DOI] [PubMed] [Google Scholar]
- Pérez A, Morelli L, Cresto JC, Castano EM. Degradation of soluble amyloid beta-peptides 1-40, 1-42, and the Dutch variant 1-40Q by insulin degrading enzyme from Alzheimer disease and control brains. Neurochem Res. 2000;25:247–255. doi: 10.1023/a:1007527721160. [DOI] [PubMed] [Google Scholar]
- Castano EM, Prelli F, Soto C, Beavis R, Matsubara E, Shoji M, Frangione B. The length of amyloid-beta in hereditary cerebral hemorrhage with amyloidosis Dutch type. Implications for the role of amyloid-beta 1-42 in Alzheimer’s disease. J Biol Chem. 1996;271:32185–32191. doi: 10.1074/jbc.271.50.32185. [DOI] [PubMed] [Google Scholar]
- Kanemitsu H, Tomiyama T, Mori H. Human neprilysin is capable of degrading amyloid beta peptide not only in the monomeric form but also the pathological oligomeric form. Neurosci Lett. 2003;350:113–116. doi: 10.1016/s0304-3940(03)00898-x. [DOI] [PubMed] [Google Scholar]
- Tsubuki S, Takaki Y, Saido TC. Dutch, Flemish, Italian, and Arctic mutations of APP and resistance of Abeta to physiologically relevant proteolytic degradation. Lancet. 2003;361:1957–1958. doi: 10.1016/s0140-6736(03)13555-6. [DOI] [PubMed] [Google Scholar]
- Castano EM, Prelli F, Wisniewski T, Golabek A, Kumar RA, Soto C, Frangione B. Fibrillogenesis in Alzheimer’s disease of amyloid beta peptides and apolipoprotein E. Biochem J. 1995;306:599–604. doi: 10.1042/bj3060599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpentier M, Robitaille Y, DesGroseillers L, Boileau G, Marcinkiewicz M. Declining expression of neprilysin in Alzheimer disease vasculature: possible involvement in cerebral amyloid angiopathy. J Neuropathol Exp Neurol. 2002;61:849–856. doi: 10.1093/jnen/61.10.849. [DOI] [PubMed] [Google Scholar]
- Morelli L, Llovera RE, Mathov I, Lue LF, Frangione B, Ghiso J, Castano EM. Insulin-degrading enzyme in brain microvessels: proteolysis of amyloid {beta} vasculotropic variants and reduced activity in cerebral amyloid angiopathy. J Biol Chem. 2004;279:56004–56013. doi: 10.1074/jbc.M407283200. [DOI] [PubMed] [Google Scholar]
- Miners JS, Van Helmond Z, Chalmers K, Wilcock G, Love S, Kehoe PG. Decreased expression and activity of neprilysin in Alzheimer disease are associated with cerebral amyloid angiopathy. J Neuropathol Exp Neurol. 2006;65:1012–1021. doi: 10.1097/01.jnen.0000240463.87886.9a. [DOI] [PubMed] [Google Scholar]
- Deane R, Wu Z, Sagare A, Davis J, Du YS, Hamm K, Xu F, Parisi M, LaRue B, Hu HW, Spijkers P, Guo H, Song X, Lenting PJ, Van Nostrand WE, Zlokovic BV. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron. 2004;43:333–344. doi: 10.1016/j.neuron.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Yamamoto N, Hirabayashi Y, Amari M, Yamaguchi H, Romanov G, Van Nostrand WE, Yanagisawa K. Assembly of hereditary amyloid beta-protein variants in the presence of favorable gangliosides. FEBS Lett. 2005;579:2185–2190. doi: 10.1016/j.febslet.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Steinhilb ML, Turner RS, Gaut JR. ELISA analysis of beta-secretase cleavage of the Swedish amyloid precursor protein in the secretory and endocytic pathways. J Neurochem. 2002;80:1019–1028. doi: 10.1046/j.0022-3042.2002.00764.x. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Alzheimer’s disease: striatal amyloid deposits and neurofibrillary changes. J Neuropathol Exp Neurol. 1990;49:215–224. [PubMed] [Google Scholar]
- Braak H, Braak E. Alzheimer’s disease affects limbic nuclei of the thalamus. Acta Neuropathol. 1991;81:261–268. doi: 10.1007/BF00305867. [DOI] [PubMed] [Google Scholar]
- Cataldo AM, Barnett JL, Mann DM, Nixon RA. Colocalization of lysosomal hydrolase and beta-amyloid in diffuse plaques of the cerebellum and striatum in Alzheimer’s disease and Down’s syndrome. J Neuropathol Exp Neurol. 1996;55:704–715. doi: 10.1097/00005072-199606000-00004. [DOI] [PubMed] [Google Scholar]
- Brilliant MJ, Elble RJ, Ghobrial M, Struble RG. The distribution of amyloid beta protein deposition in the corpus striatum of patients with Alzheimer’s disease. Neuropathol Appl Neurobiol. 1997;23:322–325. [PubMed] [Google Scholar]
- Mandybur TI, Ormsby I, Zemlan FP. Cerebral aging: a quantitative study of gliosis in old nude mice. Acta Neuropathol. 1989;77:507–513. doi: 10.1007/BF00687252. [DOI] [PubMed] [Google Scholar]
- Sturrock RR. A quantitative histological study of the anterodorsal thalamic nucleus and the lateral mammillary nucleus of ageing mice. J Hirnforsch. 1989;30:191–195. [PubMed] [Google Scholar]
- Van Dooren T, Muyllaert D, Borghgraef P, Cresens A, Devijver H, Van der Auwera I, Wera S, Dewachter I, Van Leuven F. Neuronal or glial expression of human apolipoprotein e4 affects parenchymal and vascular amyloid pathology differentially in different brain regions of double- and triple-transgenic mice. Am J Pathol. 2006;168:245–260. doi: 10.2353/ajpath.2006.050752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao J, Vitek MP, Xu F, Previti ML, Davis J, Van Nostrand WE. Reducing cerebral microvascular amyloid-beta protein deposition diminishes regional neuroinflammation in vasculotropic mutant amyloid precursor protein transgenic mice. J Neurosci. 2005;25:6271–6277. doi: 10.1523/JNEUROSCI.1306-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paigen B, Morrow A, Brandon C, Mitchell D, Holmes P. Variation in susceptibility to atherosclerosis among inbred strains of mice. Atherosclerosis. 1985;57:65–73. doi: 10.1016/0021-9150(85)90138-8. [DOI] [PubMed] [Google Scholar]
- Fan R, DeFilippis K, Van Nostrand WE. Induction of complement proteins in a mouse model for cerebral microvascular A beta deposition. J Neuroinflammation. 2007;4:22–29. doi: 10.1186/1742-2094-4-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu EX, Tang H, Asai T, Yan SD. Regional cerebral blood volume reduction in transgenic mutant APP (V717F. K670N/M671L) mice. Neurosci Lett. 2004;365:223–227. doi: 10.1016/j.neulet.2004.05.004. [DOI] [PubMed] [Google Scholar]
- Vasilevko V, Xu F, Previti ML, Van Nostrand WE, Cribbs DH. Experimental investigation of antibody-mediated clearance mechanisms of amyloid-beta in CNS of Tg-SwDI transgenic mice. J Neurosci. 2007;27:13376–13383. doi: 10.1523/JNEUROSCI.2788-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll JA, Wilkinson D, Holmes C, Steart P, Markham H, Weller RO. Neuropathology of human Alzheimer disease after immunization with amyloid-beta peptide: a case report. Nat Med. 2003;9:448–452. doi: 10.1038/nm840. [DOI] [PubMed] [Google Scholar]
- Patton RL, Kalback WM, Esh CL, Kokjohn TA, Van Vickle GD, Luehrs DC, Kuo YM, Lopez J, Brune D, Ferrer I, Masliah E, Newel AJ, Beach TG, Castano EM, Roher AE. Amyloid-beta peptide remnants in AN-1792-immunized Alzheimer’s disease patients: a biochemical analysis. Am J Pathol. 2006;169:1048–1063. doi: 10.2353/ajpath.2006.060269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelinas DS, DaSilva K, Fenili D, St George-Hyslop P, McLaurin J. Immunotherapy for Alzheimer’s disease. Proc Natl Acad Sci USA. 2004;101(Suppl 2):14657–14662. doi: 10.1073/pnas.0404866101. [DOI] [PMC free article] [PubMed] [Google Scholar]