

Abstract
Formation of the mammalian secondary palate is a highly regulated and complex process whose impairment often results in cleft palate, a common birth defect in both humans and animals. Loss-of-function analysis has linked a growing number of genes to this process. Here we report that Lhx8, a recently identified LIM homeobox gene, is expressed in the mesenchyme of the mouse palatal structures throughout their development. To test the function of Lhx8 in vivo, we generated a mutant mouse with a targeted deletion of the Lhx8 gene. Our analysis of the mutant animals revealed a crucial role for Lhx8 in palatogenesis. In Lhx8 homozygous mutant embryos, the bilateral primordial palatal shelves formed and elevated normally, but they often failed to make contact and to fuse properly, resulting in a cleft secondary palate. Because development of other craniofacial structures appeared normal, the impaired palatal formation in Lhx8-mutant mice was most likely caused by an intrinsic primary defect in the mesenchyme of the palatal shelves. The cleft palate phenotype observed in Lhx8-mutant mice suggests that Lhx8 is a candidate gene for the isolated nonsyndromic form of cleft palate in humans.
The secondary palate in mammals forms through a complex process characterized by a series of events involving cell proliferation and migration, cell differentiation, production of extracellular matrix, and cell death (1). Recently, studies with targeted mutations in mice have revealed a growing number of genes that play crucial roles in that process. These genes encode a variety of molecules including transcription regulators (2–8), growth factors (9, 10), enzymes for signaling molecule synthesis (11, 12), and receptors (13–15). By association studies, some of these genes have also been identified as candidate genes (16, 17) whose mutations may cause cleft palate in humans, a common birth defect that results from impairments in palatal development.
LIM homeobox genes encode a family of transcription regulators that share common structural features. They all contain two tandemly repeated cysteine-rich double-zinc finger motifs called LIM domains, in addition to a homeodomain. Whereas the homeodomain is a DNA-binding domain, the LIM domains are essential for regulating the activity of these molecules by interacting with other proteins (18–22). Genetic studies in various organisms have shown that members of the LIM homeobox gene family are required for the patterning (23, 24) or the specification and differentiation of different cell types during embryonic development (25–38).
Lhx8, also called L3 (39) and Lhx7 (40), is a recently identified member of the LIM homeobox gene family. During mouse embryogenesis, Lhx8 has been shown to be expressed in several regions in the head, including the maxillary and mandibular processes and the ventral forebrain (39, 40). In situ chromosomal hybridization has revealed that the Lhx8 gene is localized on mouse chromosome 3, H3–4 (41). Homologs of genes in that region have been mapped to human chromosome 4, q25–31 (http://www.ncbi.nlm.nih.gov/Omim/Homology), a region that has been linked to craniofacial clefting (42). This raised the possibility that Lhx8 may be involved in development of the secondary palate.
To test whether Lhx8 has a role in palatal development, we further examined the expression of Lhx8 at different stages during palatogenesis and generated a mutant mouse with a targeted deletion of the gene. We show that Lhx8 is continuously expressed in the mesenchyme of the normal developing palatal structures. Disruption of Lhx8 gene function causes impairments in palatal shelf contact and fusion that lead to the formation of a cleft secondary palate.
Materials and Methods
Generation of Lhx8 Knockout Mice.
A plasmid containing the entire mouse Lhx8 gene was obtained by PCR screening of a mouse bacterial artificial chromosome library (GenomeSystems, St. Louis) with primers (sense, 5′-TGTTCCCGCTGTGGCAGGCACAT-3′; antisense, 5′-ACAGAGGACCTTCTCCTCCACCAA-3′) synthesized according to the Lhx8 cDNA sequence (39). To construct the targeting vector, exons 4–6 of the Lhx8 gene were replaced with a neomycin resistance gene flanked by 3.0-kb (5′) and 3.4-kb (3′) homologous sequences and by the thymidine kinase gene (see Fig. 2A). The vector was linearized and electroporated into the R1 line of embryonic stem (ES) cells (43). Targeted clones were double selected with G418 (350 μg/ml) and gancyclovir (2 mM), and screened by Southern blot analysis with both a 5′ probe and a 3′ probe outside the flanking homologous sequences (see Fig. 2A). The ES cells that were heterozygous for the targeted Lhx8 mutation were microinjected into C57BL/6 blastocysts to produce chimeric mice that carried the mutation into the germ line. These mice were mated with C57BL/6 wild-type mice to generate Lhx8 heterozygous animals that were subsequently crossed to produce F2 offspring for analysis.
Figure 2.

Targeted mutation of the Lhx8 gene by homologous recombination. (A) Schematic representation of the wild-type Lhx8 gene, the targeting vector, and the mutated Lhx8. (B) Southern (Left) and PCR (Right) analyses of genomic DNA derived from offspring of a cross between heterozygous parents. After SacI digestion, the 5′ probe detected an 11-kb band of the wild-type allele and a 9-kb band of the mutant allele (arrowheads). The 3′ probe detected a 6.6-kb band of the wild-type allele and a 4.6-kb band of the mutant allele (arrowheads) after PstI digestion. PCR amplified an 150-bp band from the wild-type allele and a 550-bp band from the mutant allele (arrowheads). B, BamHI; P, PstI; Sa, SacI; Sm, SmaI; neo, the neomycin resistance gene; tk, the thymidine kinase gene.
Genotyping of Lhx8 Knockout Mice.
Genotypes of the mice were determined by either Southern blot or PCR analysis of genomic DNA prepared from tail biopsies or yolk sacs of embryos. The probes used for Southern analysis were the same as those used for ES cell screening. For PCR analysis, a pair of primers from exon 5 of the Lhx8 gene (the same as those used for screening the mouse bacterial artificial chromosome library; see above) was used to amplify an 150-bp fragment from the wild-type allele, and a pair of primers from the neomycin resistance gene (sense, 5′-TCGTCCTGTACGGACCGCAGTTT-3′; antisense, GATCCCCTCAGAAGAACTCGT-3′) was used to amplify a 550-bp fragment from the mutant allele.
Histological Analysis and Skeletal Staining.
Lhx8 heterozygous mice were mated, and noon on the day when vaginal plugs were observed was considered as embryonic day 0.5 (E0.5). Embryos were isolated at different stages and fixed in 4% paraformaldehyde at 4°C overnight. The embryos were embedded in paraffin and sectioned at a thickness of 5 μm. The sections were mounted on silanated slides and stained with hematoxylin/eosin (H&E) or used for in situ hybridization. For skeletal staining, skinned and eviscerated newborn mice were fixed overnight in 95% ethanol and stained with alcian blue and alizarin red as previously described (5).
In Situ Hybridization.
In situ hybridization was performed on paraffin sections by previously published procedures (44). The 35S-labeled antisense RNA probe used was synthesized by in vitro transcription with a linearized pGEM-T Easy vector (Promega) that contained the full-length Lhx8 coding sequence.
Results
Expression of Lhx8 During Palatal Development.
We examined the expression of Lhx8 mRNA in the palatal region at different stages during development by in situ hybridization. At E11.5, Lhx8 mRNA was detected in the maxillary processes (Fig. 1A). From E13.5 to E16.5, while palatogenesis progressed through palatal shelf outgrowth, elevation, contact, and fusion, Lhx8 was continuously expressed in the mesenchyme of the palatal shelves (Fig. 1 B–D). This pattern of expression suggests that Lhx8 could play a role in palatal development. Consistent with previous studies (39, 40), we also detected Lhx8 mRNA in the ventral telencephalon and in other structures derived from the first branchial arch, such as the molar tooth buds and the tongue (Fig. 1 B–D).
Figure 1.
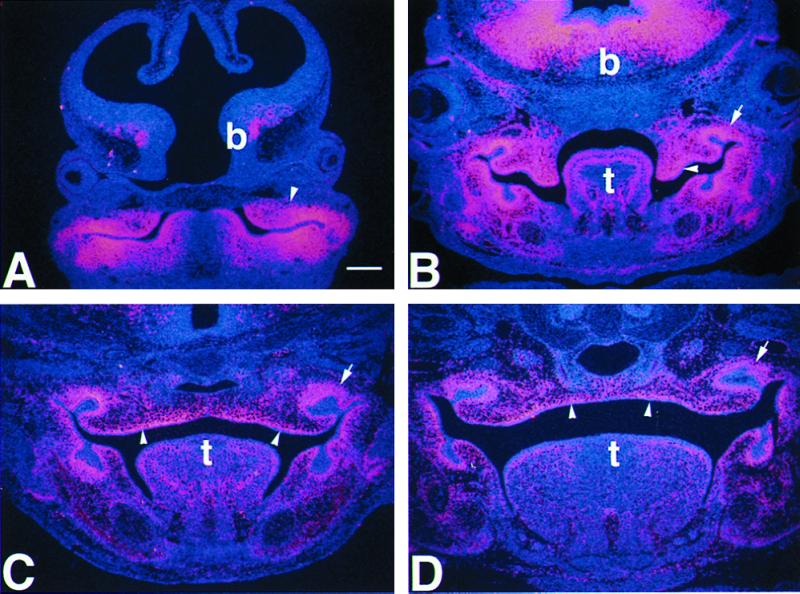
In situ hybridization, showing expression of Lhx8 mRNA at different stages of palatal development. (A) Expression of Lhx8 in the maxillary processes (arrowhead) of an E11.5 embryo. (B) Expression of Lhx8 in the palatal shelves (arrowhead) of an E13.5 embryo, before shelf elevation. (C) Expression of Lhx8 in the palate shelves (arrowheads) of an E14.5 embryo, after shelf elevation and contact. (D) Continuous expression of Lhx8 in the palate of an E16.5 embryo, after fusion of the palate shelves. Notice that Lhx8 was also expressed in the tooth bud (arrows), tongue (t), and the ventral forebrain (b). Scale bar in A represents 200 μm for all panels.
Cleft Palate in Lhx8 Knockout Mice.
To test the function of the Lhx8 gene in palatal development, we used homologous recombination in ES cells to generate mice with a targeted deletion of exons 4–6 of the Lhx8 gene (Fig. 2A). These exons encode part of the first LIM domain, the entire second LIM domain, and part of the homeodomain of the Lhx8 protein. Correctly targeted ES cells were selected by Southern blot analysis and were used to produce mutant mice. Mice heterozygous for the Lhx8 mutation appeared normal and fertile. These mice were subsequently crossed to generate offspring that were genotyped by Southern hybridization or PCR (Fig. 2B) and used for phenotypic analysis.
Lhx8 homozygous mutant mice were born alive and their gross morphology appeared normal (Fig. 3A). However, many of them were found dead (9 out of 19 or 47.4%) within 24 h. Genotyping of animals that survived after weaning showed that, of a total of 265 offspring from heterozygous crosses, only 23 (8.7%) were homozygous mutants. Gross examination of the oral cavity of the homozygous mice that died after birth revealed that they all had a complete cleft of the secondary palate (Fig. 3G), as compared with wild-type (Fig. 3F) or heterozygous controls. The secondary palate in homozygous animals that survived was normal. We also examined embryos at E18.5. Among 10 homozygous mutant embryos, 6 (60%) showed a cleft palate. Thus, disruption of the Lhx8 gene in mice resulted in a phenotype of cleft palate with incomplete penetrance.
Figure 3.

Cleft palate in Lhx8 knockout mice. (A) Two dead newborn Lhx8 homozygous mice (Left and Center) are compared with their wild-type littermate (Right). (B and C) Skeletal staining of the head of a newborn Lhx8 homozygous mutant with cleft palate (C) as compared with that of a wild-type control (B), viewed from the side. The craniofacial features of the mutant mouse with cleft palate appeared grossly normal. In the mutant, the mandible was lower because the mouth was more widely open when the animal was fixed. Arrows and arrowheads point at the tip and the middle part of the mandibles in both the wild-type (B) and the mutant (C) animals, respectively. (D and E) Top view of the mandible of a newborn Lhx8 mutant animal with cleft palate (E) as compared with that of a wild-type control (D). The mandible of the mutant appeared normal. (F and G) Ventral view of the upper jaw of a newborn Lhx8 homozygous mutant (G) as compared with that of a wild-type control (F). Notice a cleft of the secondary palate (indicated by arrowheads). (H and I) Ventral view of the base of skulls of newborn wild-type (H) and Lhx8 homozygous mutant (I) mice stained with alcian blue and alizarin red. The palatal bones in the mutant failed to grow toward the midline (indicated by arrowheads). The scale bar in C represents 6.6 mm for panel A and 1.3 mm for panels B–-I.
To analyze the defect of bone formation in Lhx8 mutant mice, skeletal staining of the skull was performed. Unlike those in wild-type controls (Fig. 3H), the palatal bones in the homozygous mutant animals with cleft palate failed to extend toward the midline at the base of the skull (Fig. 3I). This defect was restricted to the palatal region. Other bones of the skull appeared normal in mutants (Fig. 3 C and E) as compared with wild-type (Fig. 3 B and D) animals.
Impaired Palatal Development in Lhx8 Knockout Mice.
To better define the role of Lhx8 in palatogenesis, we monitored palatal development in Lhx8 mutant embryos at different stages. During normal development, palatal shelves were observed in E13.5 embryos as they grew out from the maxillary processes on either side of the tongue (Fig. 4A). At E14.5, the palatal shelves on both sides elevated to a horizontal position dorsal to the tongue and abutted each other (Fig. 4C). By E16.5, the medial-edge epithelia approximating the palatal shelves from the two sides completed fusion and then degenerated, resulting in an intact palate with continuous mesenchyme (Fig. 4E). In Lhx8-mutant embryos, the palatal shelves initially formed (Fig. 4B, eight embryos examined) and elevated (Fig. 4D, seven embryos examined), but they often failed to contact each other and fuse properly (Fig. 4D), resulting in a cleft of the palate (Fig. 4F). Despite the defect in palatal closure, development of other oral structures, including the mandible, the tongue, the molar teeth (Fig. 4 B, D, and F), the incisor teeth, and the Meckel's cartilage (Fig. 4H), all appeared normal in mutant embryos as compared with wild-type embryos (Fig. 4 A, C, E, and G).
Figure 4.
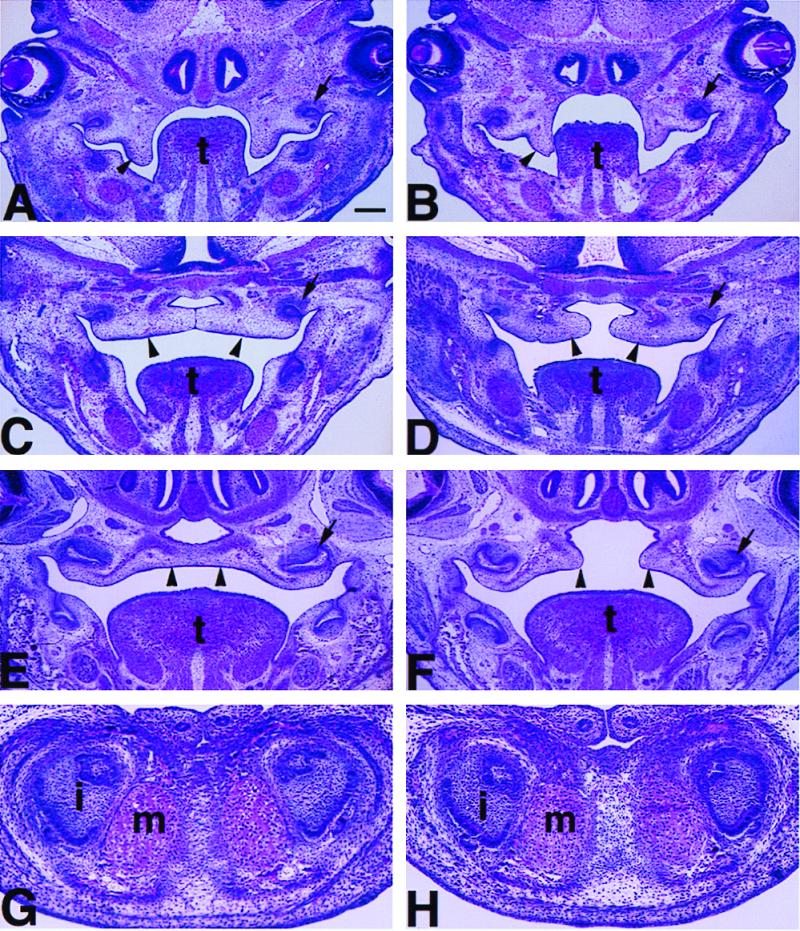
Palate shelves elevated but failed to make contact and fuse in Lhx8 homozygous mutant mouse without detectable defects in the mandible. H&E staining of coronal sections through the palatal region of the homozygous mutant embryos (B, D, and F) at different developmental stages as compared with that of wild-type controls (A, C, and E). (A and B) E13.5; (C and D) E14.5; (E and F) E16.5. Arrowheads point at the palatal shelves that were not in contact (A, B, D, and F), were in contact (C), or were completely fused (E). Arrows point at one of the upper molar tooth buds in all panels. (G and H) H&E-stained coronal sections through the distal region of the mandible of a wild-type (G) and an Lhx8 homozygous mutant embryo with cleft palate at E16.5, showing normal development of the mandible, the incisor teeth (i), and the Meckel's cartilage (m) in the mutant. Scale bar in A represents 200 μm for panels A–F and 80 μm for panels G–H. t, tongue.
Discussion
Expression of Lhx8 mRNA during mouse embryogenesis has been characterized previously by in situ hybridization. It has been reported that Lhx8 transcripts are expressed in the neural crest-derived ectomesenchyme of the first branchial arch from E9.5 to E12; the expression persists in the developing teeth until the postnatal period. In addition, Lhx8 is also expressed in the medial ganglionic eminence of the developing ventral telencephalon (39, 40). In this study, a similar pattern of Lhx8 expression was observed. However, our experiments further revealed that Lhx8 mRNA was also continuously expressed in the developing secondary palate, suggesting that Lhx8 may play a role in palatal development.
Our analysis of mice with a targeted deletion of the Lhx8 gene showed that Lhx8 was indeed involved in palatal development. However, other than the developing palate, histological staining and in situ hybridization analysis of expression of molecular markers such as Dlx1, Dlx2, and Msx1 revealed no clear defects in the rest of the Lhx8-expressing structures (data not shown). It is possible that the function of Lhx8 is partially redundant and can be substituted for by other genes. One likely candidate is Lhx6, another LIM homeobox gene that shares a similar expression pattern and a high sequence homology with Lhx8 (40).
Formation of the mammalian secondary palate is a dynamic process (1). It begins with migration of the cranial neural crest cells from the posterior midbrain and rhombomeres 1 and 2 of the hindbrain to form the first branchial arch (45, 46). The primordial palatal shelves emerge as a bilateral outgrowth from the maxillary processes of the first arch. The palatal shelves first grow vertically downward along the sides of the tongue. They later elevate to a horizontal position, make contact, and finally fuse at the midline to form a complete palate. The phenotype of cleft palate can result from impairments at any of the steps in palatal development. In Lhx8 homozygous mutant mice, formation and elevation of the palatal shelves appeared to proceed normally. However, the palatal shelves often failed to make contact and to fuse. This indicates that Lhx8 plays a specific role at this particular stage in palatal development.
Cleft palate has been observed in a number of mice carrying mutations in various genes. Although the phenotype may appear similar, the mechanisms that cause this phenotype could be different. Impairments in palatal development can derive from intrinsic defects of the palatal shelves themselves. For example, in mice with mutations of genes encoding transforming growth factor-β3 (9, 10), epidermal growth factor receptor (15), or transcription factor TTF2 (8), the palatal shelves are able to elevate and abut each other, but they fail to fuse because the medial-edge epithelia overlying the mesenchyme between the opposing palatal shelves do not adhere and disappear promptly. In addition, palate formation is also dependent on normal development of other oral or craniofacial structures. The cleft palate observed in many mice with mutations of homeobox genes, like Hoxa2 (2, 3), Msx1 (4), Mhox (5), Dlx1 (6), Dlx2 (6), and Pax9 (7), is accompanied by gross craniofacial and skeletal malformations. The palatal defect in those mice can be attributed, at least partially, to those malformations, which often cause mechanical hindrance in elevation, contact, or fusion of the palatal shelves. In Lhx8-mutant mice, despite the defect in the secondary palate, the gross morphology of the skull and other oral structures such as the mandible, tongue, molar and incisor teeth, and Meckel's cartilage all appeared normal. This indicates that the cleft palate observed in those mutant mice is most likely caused by an intrinsic primary defect in development of the palatal shelves.
The expression of Lhx8 mRNA in the mesenchymal cells of the developing palatal shelves suggests that its role is played out in those cells. Contact and fusion of the palatal shelves can be impaired by a reduction of their size, which in turn can result from a reduction in cell proliferation (1). Previously, the LIM homeobox gene Lhx2 has been shown to be involved in controlling the proliferation of neural precursor cells in the developing forebrain (32). However, we analyzed the palatal mesenchymal cell proliferation with bromodeoxyuridine pulse labeling and detected no clear differences between Lhx8 homozygous mutant and wild-type embryos (data not shown). Fusion of the palatal shelves also requires interactions between the medial-edge epithelia and the underlying mesenchyme (1, 47). Previous knockout studies have demonstrated that molecules expressed in the palatal epithelia, like transforming growth factor-β3 (9, 10), epidermal growth factor receptor (15), and TTF2 (8), are required for the fusion of the palatal shelves. The cleft palate phenotype observed in the Lhx8-mutant mouse is similar to those observed in transforming growth factor-β3 (9, 10), epidermal growth factor receptor (15), and TTF2 (8) knockout mice, which suggests that Lhx8 may play a role in the mesenchyme in mediating the epithelia-mesenchyme interactions that are required for the fusion of the palatal shelves. It has been shown, in fact, that the expression of Lhx8 in the ectomesenchyme of the developing tooth bud is induced by signals from the overlying oral epithelia (40). Further studies of the Lhx8 and other mutant mice that have been generated will help to elucidate the molecular and cellular mechanisms underlying the formation of the mammalian secondary palate.
The incomplete penetrance of the cleft palate phenotype observed in the Lhx8-mutant mouse suggests that the Lhx8 gene may interact with other cleft palate susceptibility genes or teratogens. Cleft palate phenotype with incomplete penetrance has also been observed in other mutant mice (15, 48). Consistent with these studies in mice, isolated nonsyndromic cleft palate in humans has been found to have a highly complex multifactorial etiology that involves both genetic and environmental factors (49). Previous studies have identified the association of cleft palate with TGF-A and MSX1 loci (16, 17). The cleft palate phenotype we observe here identifies Lhx8 as another potential candidate gene for human cleft palate.
Acknowledgments
We thank O. Birk, X. Jian, and K. Pfeifer for comments on the manuscript; N. Malik for discussion; and M. Yarolin for assistance.
Abbreviation
- ES
embryonic stem
References
- 1.Ferguson M W. Development (Cambridge, UK) 1988;103,Suppl.:41–60. doi: 10.1242/dev.103.Supplement.41. [DOI] [PubMed] [Google Scholar]
- 2.Gendron-Maguire M, Mallo M, Zhang M, Gridley T. Cell. 1993;75:1317–1331. doi: 10.1016/0092-8674(93)90619-2. [DOI] [PubMed] [Google Scholar]
- 3.Rijli F M, Mark M, Lakkaraju S, Dierich A, Dolle P, Chambon P. Cell. 1993;75:1333–1349. doi: 10.1016/0092-8674(93)90620-6. [DOI] [PubMed] [Google Scholar]
- 4.Satokata I, Maas R. Nat Genet. 1994;6:348–356. doi: 10.1038/ng0494-348. [DOI] [PubMed] [Google Scholar]
- 5.Martin J F, Bradley A, Olson E N. Genes Dev. 1995;9:1237–1249. doi: 10.1101/gad.9.10.1237. [DOI] [PubMed] [Google Scholar]
- 6.Qiu M, Bulfone A, Ghattas I, Meneses J J, Christensen L, Sharpe P T, Presley R, Pedersen R A, Rubenstein J L. Dev Biol. 1997;185:165–184. doi: 10.1006/dbio.1997.8556. [DOI] [PubMed] [Google Scholar]
- 7.Peters H, Neubuser A, Kratochwil K, Balling R. Genes Dev. 1998;12:2735–2747. doi: 10.1101/gad.12.17.2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.De Felice M, Ovitt C, Biffali E, Rodriguez-Mallon A, Arra C, Anastassiadis K, Macchia P E, Mattei M G, Mariano A, Scholer H, Macchia V, Di Lauro R. Nat Genet. 1998;19:395–398. doi: 10.1038/1289. [DOI] [PubMed] [Google Scholar]
- 9.Kaartinen V, Voncken J W, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J. Nat Genet. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- 10.Proetzel G, Pawlowski S A, Wiles M V, Yin M, Boivin G P, Howles P N, Ding J, Ferguson M W, Doetschman T. Nat Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Asada H, Kawamura Y, Maruyama K, Kume H, Ding R G, Kanbara N, Kuzume H, Sanbo M, Yagi T, Obata K. Proc Natl Acad Sci USA. 1997;94:6496–6499. doi: 10.1073/pnas.94.12.6496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Condie B G, Bain G, Gottlieb D I, Capecchi M R. Proc Natl Acad Sci USA. 1997;94:11451–11455. doi: 10.1073/pnas.94.21.11451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Culiat C T, Stubbs L J, Woychik R P, Russell L B, Johnson D K, Rinchik E M. Nat Genet. 1995;11:344–346. doi: 10.1038/ng1195-344. [DOI] [PubMed] [Google Scholar]
- 14.Homanics G E, DeLorey T M, Firestone L L, Quinlan J J, Handforth A, Harrison N L, Krasowski M D, Rick C E, Korpi E R, Makela R, et al. Proc Natl Acad Sci USA. 1997;94:4143–4148. doi: 10.1073/pnas.94.8.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miettinen P J, Chin J R, Shum L, Slavkin H C, Shuler C F, Derynck R, Werb Z. Nat Genet. 1999;22:69–73. doi: 10.1038/8773. [DOI] [PubMed] [Google Scholar]
- 16.Shiang R, Lidral A C, Ardinger H H, Buetow K H, Romitti P A, Munger R G, Murray J C. Am J Hum Genet. 1993;53:836–843. [PMC free article] [PubMed] [Google Scholar]
- 17.Lidral A C, Romitti P A, Basart A M, Doetschman T, Leysens N J, Daack-Hirsch S, Semina E V, Johnson L R, Machida J, Burds A, Parnell T J, Rubenstein J L, Murray J C. Am J Hum Genet. 1998;63:557–568. doi: 10.1086/301956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jurata L W, Kenny D A, Gill G N. Proc Natl Acad Sci USA. 1996;93:11693–11698. doi: 10.1073/pnas.93.21.11693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Agulnick A D, Taira M, Breen J J, Tanaka T, Dawid I B, Westphal H. Nature (London) 1996;384:270–272. doi: 10.1038/384270a0. [DOI] [PubMed] [Google Scholar]
- 20.Bach I, Carriere C, Ostendorff H P, Andersen B, Rosenfeld M G. Genes Dev. 1997;11:1370–1380. doi: 10.1101/gad.11.11.1370. [DOI] [PubMed] [Google Scholar]
- 21.Morcillo P, Rosen C, Baylies M K, Dorsett D. Genes Dev. 1997;11:2729–2740. doi: 10.1101/gad.11.20.2729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Breen J J, Agulnick A D, Westphal H, Dawid I B. J Biol Chem. 1998;273:4712–4717. doi: 10.1074/jbc.273.8.4712. [DOI] [PubMed] [Google Scholar]
- 23.Taira M, Otani H, Saint-Jeannet J P, Dawid I B. Nature (London) 1994;372:677–679. doi: 10.1038/372677a0. [DOI] [PubMed] [Google Scholar]
- 24.Shawlot W, Behringer R R. Nature (London) 1995;374:425–430. doi: 10.1038/374425a0. [DOI] [PubMed] [Google Scholar]
- 25.Way J C, Chalfie M. Cell. 1988;54:5–16. doi: 10.1016/0092-8674(88)90174-2. [DOI] [PubMed] [Google Scholar]
- 26.Freyd G, Kim S K, Horvitz H R. Nature (London) 1990;344:876–879. doi: 10.1038/344876a0. [DOI] [PubMed] [Google Scholar]
- 27.Cohen B, McGuffin M E, Pfeifle C, Segal D, Cohen S M. Genes Dev. 1992;6:715–729. doi: 10.1101/gad.6.5.715. [DOI] [PubMed] [Google Scholar]
- 28.Bourgouin C, Lundgren S E, Thomas J B. Neuron. 1992;9:549–561. doi: 10.1016/0896-6273(92)90192-g. [DOI] [PubMed] [Google Scholar]
- 29.Pfaff S L, Mendelsohn M, Stewart C L, Edlund T, Jessell T M. Cell. 1996;84:309–320. doi: 10.1016/s0092-8674(00)80985-x. [DOI] [PubMed] [Google Scholar]
- 30.Sheng H Z, Zhadanov A B, Mosinger B, Jr, Fujii T, Bertuzzi S, Grinberg A, Lee E J, Huang S P, Mahon K A, Westphal H. Science. 1996;272:1004–1007. doi: 10.1126/science.272.5264.1004. [DOI] [PubMed] [Google Scholar]
- 31.Sheng H Z, Moriyama K, Yamashita T, Li H, Potter S S, Mahon K A, Westphal H. Science. 1997;278:1809–1812. doi: 10.1126/science.278.5344.1809. [DOI] [PubMed] [Google Scholar]
- 32.Porter F D, Drago J, Xu Y, Cheema S S, Wassif C, Huang S P, Lee E, Grinberg A, Massalas J S, Bodine D, Alt F, Westphal H. Development (Cambridge, UK) 1997;124:2935–2944. doi: 10.1242/dev.124.15.2935. [DOI] [PubMed] [Google Scholar]
- 33.Thor S, Thomas J B. Neuron. 1997;18:397–409. doi: 10.1016/s0896-6273(00)81241-6. [DOI] [PubMed] [Google Scholar]
- 34.Hobert O, Mori I, Yamashita Y, Honda H, Ohshima Y, Liu Y, Ruvkun G. Neuron. 1997;19:345–357. doi: 10.1016/s0896-6273(00)80944-7. [DOI] [PubMed] [Google Scholar]
- 35.Hobert O, D'Alberti T, Liu Y, Ruvkun G. J Neurosci. 1998;18:2084–2096. doi: 10.1523/JNEUROSCI.18-06-02084.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Duggan A, Ma C, Chalfie M. Development (Cambridge, UK) 1998;125:4107–4119. doi: 10.1242/dev.125.20.4107. [DOI] [PubMed] [Google Scholar]
- 37.Sharma K, Sheng H Z, Lettieri K, Li H, Karavanov A, Potter S, Westphal H, Pfaff S L. Cell. 1998;95:817–828. doi: 10.1016/s0092-8674(00)81704-3. [DOI] [PubMed] [Google Scholar]
- 38.Zhao Y, Sheng H Z, Amini R, Grinberg A, Lee E, Huang S, Taira M, Westphal H. Science. 1999;284:1155–1158. doi: 10.1126/science.284.5417.1155. [DOI] [PubMed] [Google Scholar]
- 39.Matsumoto K, Tanaka T, Furuyama T, Kashihara Y, Mori T, Ishii N, Kitanaka J, Takemura M, Tohyama M, Wanaka A. Neurosci Lett. 1996;204:113–116. doi: 10.1016/0304-3940(96)12341-7. [DOI] [PubMed] [Google Scholar]
- 40.Grigoriou M, Tucker A S, Sharpe P T, Pachnis V. Development (Cambridge, UK) 1998;125:2063–2074. doi: 10.1242/dev.125.11.2063. [DOI] [PubMed] [Google Scholar]
- 41.Kitanaka J, Takemura M, Matsumoto K, Mori T, Wanaka A. Genomics. 1998;49:307–309. doi: 10.1006/geno.1998.5203. [DOI] [PubMed] [Google Scholar]
- 42.Mitchell L E, Healey S C, Chenevix-Trench G. Am J Hum Genet. 1995;57:1130–1136. [PMC free article] [PubMed] [Google Scholar]
- 43.Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder J C. Proc Natl Acad Sci USA. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Robinson G W, Wray S, Mahon K A. New Biol. 1991;3:1183–1194. [PubMed] [Google Scholar]
- 45.Osumi-Yamashita N, Ninomiya Y, Doi H, Eto K. Dev Biol. 1994;164:409–419. doi: 10.1006/dbio.1994.1211. [DOI] [PubMed] [Google Scholar]
- 46.Kontges G, Lumsden A. Development (Cambridge, UK) 1996;122:3229–3242. doi: 10.1242/dev.122.10.3229. [DOI] [PubMed] [Google Scholar]
- 47.Sharpe P M, Ferguson M W. J Cell Sci Suppl. 1988;10:195–230. doi: 10.1242/jcs.1988.supplement_10.15. [DOI] [PubMed] [Google Scholar]
- 48.Wilson J B, Ferguson M W, Jenkins N A, Lock L F, Copeland N G, Levine A J. Cell Growth Differ. 1993;4:67–76. [PubMed] [Google Scholar]
- 49.Houdayer C, Bahuau M. Ann Genet. 1998;41:89–117. [PubMed] [Google Scholar]