

Abstract
Studies of mouse models of human cancer have established the existence of multiple tumor modifiers that influence parameters of cancer susceptibility such as tumor multiplicity, tumor size, or the probability of malignant progression. We have carried out an analysis of skin tumor susceptibility in interspecific Mus musculus/Mus spretus hybrid mice and have identified another seven loci showing either significant (six loci) or suggestive (one locus) linkage to tumor susceptibility or resistance. A specific search was carried out for skin tumor modifier loci associated with time of survival after development of a malignant tumor. A combination of resistance alleles at three markers [D6Mit15 (Skts12), D7Mit12 (Skts2), and D17Mit7 (Skts10)], all of which are close to or the same as loci associated with carcinoma incidence and/or papilloma multiplicity, is significantly associated with increased survival of mice with carcinomas, whereas the reverse combination of susceptibility alleles is significantly linked to early mortality caused by rapid carcinoma growth (χ2 = 25.22; P = 5.1 × 10−8). These data indicate that host genetic factors may be used to predict carcinoma growth rate and/or survival of individual backcross mice exposed to the same carcinogenic stimulus and suggest that mouse models may provide an approach to the identification of genetic modifiers of cancer survival in humans.
Cancer risk in humans is determined by a combination of several environmental and genetic factors. Exposure to chemical or physical carcinogens, as modified by the capacity of the individual to metabolize or nullify the effects of such exogenous agents, will result in the accumulation of multiple mutations in genes required for the development of neoplasia. In certain rare familial cancer syndromes, the inheritance of mutant forms of oncogenes or tumor suppressor genes through the germ line can accelerate the process of carcinogenesis, leading to early onset of multiple primary tumors. In such high-penetrance familial cancer syndromes, such as familial adenomatous polyposis or familial retinoblastoma, the probability of tumor development in affected individuals approaches 100%. Fortunately, these familial cases account for only about 5% or less of the total human cancer burden. The remaining 95% of human cancers are in the “sporadic” category, but this does not mean that there is no hereditary genetic determinant of susceptibility. Multiple low penetrance genes or tumor modifiers segregate within the human population and may be major contributing factors to individual tumor susceptibility (1, 2). It has been proposed that the overall complement of alleles that confer either resistance or susceptibility to tumorigenesis by modulating the effects of exposure to exogenous carcinogens or by controlling intrinsic rate-limiting steps in neoplastic cell growth determines individual cancer risk in humans (3, 4).
The identification of tumor modifier genes solely by studying human cancer patients is likely to prove difficult. As in other complex genetic traits, the absence of clear Mendelian inheritance patterns necessitates the development of alternative statistical approaches for the localization of genes associated with the disease. Moreover, for the human population, the environmental component of cancer risk is obviously impossible to control, thus adding to the complexities of the genetic analysis. The use of animal models such as the mouse may overcome these problems by almost completely removing the problem of environmental variation and exploiting the availability of genetically uniform strains or species that have wide variation in susceptibility to spontaneous or carcinogen-induced tumorigenesis (4).
In a previous report, three significant skin tumor modifier loci on chromosomes 5 and 7 were identified by using a large (NIH/Ola × Mus spretus)F1 backcross (NSP; ref. 5). Skts3 on chromosome 5 was linked to both promotion and progression, whereas Skts1 and Skts2 were mainly associated with benign papilloma multiplicity. Herein, we present an additional seven quantitative trait loci (QTLs) identified by extended hierarchical analysis of NSP mice involved in susceptibility to skin tumor development and show that a subset of three QTLs influences survival time of tumor-bearing mice.
Methods
Animals and Tumor Induction.
Inbred NIH/Ola mice were purchased from Harlan Olac (Bichester, U.K.). Outbred Mus spretus mice were obtained from S. Brown (Medical Research Council Radiobiology Unit, Harwell, England). Tumor number and size were assessed at least every other week until 80 weeks after tumor initiation. The data showing papilloma and carcinoma incidence in 326 NSP animals were reported previously (5). Papilloma onset was scored as the number of weeks after initiation when the papilloma diameter reached more than 2 mm. Mice that did not develop papillomas larger than 2 mm were ignored in this analysis. Survival time was taken as the period from initial treatment to the date when the mice were killed because of the progressive growth of a malignant tumor. Mice with carcinomas that showed signs of adverse effects on health status, that had a tumor more than 2 cm in diameter, or that had a total tumor mass more than 10% of body weight were killed, and tumor histology was examined after standard hematoxylin/eosin staining. Mice (n = 83) that developed carcinomas were used for the analysis of association with survival. Squamous carcinomas were classified as grade 1, 2, 3, or 4 (spindle cell carcinomas) according to published criteria (6).
DNA Preparation and Linkage Analysis.
DNAs were prepared from tails and amplified by standard methods. Microsatellite markers (n = 146) with an average genomic spacing of 10.1 centimorgans (cM) were employed. Tumor multiplicity in chemical carcinogen-induced mouse experiments frequently follows a negative binomial distribution (7), especially when the tumor number is overdispersed, such as in the Mus spretus/Mus musculus backcross, where more than 30% of animals have no papilloma. For QTL analysis, tumor multiplicity data are generally transformed by root-square transformation to improve the fit of negative binomial data to a normal distribution (7) or alternatively are analyzed by using a nonparametric test (8). However, transformation does not improve the analysis substantially in a case with overdispersion. Normally a nonparametric test is less powerful than a parametric test when the data are fit to a standard distribution. Because our papilloma multiplicity data fitted very well to a negative binomial distribution by testing for goodness of fit (P = 0.1; ref. 9), we employed a negative binomial multiple regression analysis to screen for predisposition loci associated with papilloma multiplicity. The negative binomial regression model is
 |
1 |
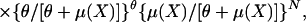 |
where Y is number of papillomas and θ indicates the heterogeneity of response,
 |
2 |
is the mean, and xi is the value of marker I. xi is 1 if marker I is homozygous. xi is 0 if marker I is heterozygous.
Of 146 markers on 12 chromosomes, 50 had a P value < 0.15 in the primary analysis of 96 NSP backcross animals. On chromosomes 1, 4–7, 9, 11–13, and 15–17, 73 markers were examined in the complete panel of 326 mice for association with papilloma multiplicity by using stepwise negative binomial regression analysis (stata, Stata, College Station, TX).
The association between carcinoma incidence and marker was analyzed by a logistic regression method as described (5). Cox multiple regression analysis, which is a multivariate analysis technique (10, 11), was used to detect the effect of multiple genotypes on papilloma onset and survival after carcinoma development. The combined effect of the loci was screened genome-wide by stepwise Cox regression analysis (stata, Stata, College Station, TX).
The statistical estimation of relationships between cancer grade and genotype was calculated by using a Trend test (stata, Stata, College Station, TX; ref. 12).
Results
Genome-Wide Scan of NSP Mice.
A genome scan carried out by using negative binomial regression analysis as described in Methods identified a number of markers that attained significant or suggestive P values for linkage to papilloma multiplicity (Table 1; Fig. 1; ref. 13). Significant linkages were detected at the single markers D1Mit47 on chromosome 1 (Skts8), D4Mit37 on chromosome 4 (Skts7), D9Mit76 on chromosome 9 (Skts6), D12Mit154 on chromosome 12 (Skts5), and D16Mit2 on chromosome 16 (Skts9) for Mus spretus papilloma-resistance loci. There was a suggestive Mus spretus papilloma-susceptibility locus (Skts10) at D17Mit7 on chromosome 17. Although loci that reach only suggestive significance would not normally be designated as skin tumor susceptibility loci (Skts), data presented below show that Skts10 does in fact reach significance in interaction with other genomic loci (see also refs. 14 and 15). In the subsequent discussion, any loci that fail to reach significance will be followed by an asterisk (*) to indicate that the designation is provisional until further data are obtained. The loci Skts1 and Skts3 were detected as significant papilloma-resistance loci in our previous analysis of this backcross by using the mapmaker qtl program (5). A second locus on chromosome 7 (Skts2) did not reach statistical significance in this negative binomial regression analysis of papilloma multiplicity, although markers close to Skts2 showed linkage with papilloma onset, survival, and with carcinoma susceptibility (see below). The percentage of the total variance in papilloma multiplicity accounted for by all detected loci is 27.4%. This value is increased further when genetic interactions involving these loci are taken into account (H.N., J.-H.M. and A.B., unpublished results).
Table 1.
Linkage between marker and papilloma multiplicity (negative binomial multiple regression method)
| Locus | Marker (cM) | Logarithm of odds score | Coefficient* | P value | Corrected P value |
|---|---|---|---|---|---|
| Skts8 | D1Mit47 (79.0) | 3.17 | 0.4856 ± 0.1270 | 1.3 × 10−4 | 0.018 |
| Skts7 | D4Mit37 (56.5) | 3.73 | 0.5176 ± 0.1248 | 3.4 × 10−5 | 5.3 × 10−3 |
| Skts3 | D5Mit7 (24.0) | 4.85 | 0.6096 ± 0.1286 | 2.1 × 10−6 | 4.1 × 10−4 |
| Skts1 | D7Mit87 (27.8) | 11.46 | 0.9524 ± 0.1310 | 3.7 × 10−13 | 1.6 × 10−10 |
| Skts6 | D9Mit76 (49.0) | 3.71 | 0.5256 ± 0.1270 | 3.5 × 10−5 | 5.5 × 10−3 |
| Skts5 | D12Mit154 (17.0) | 3.45 | 0.5249 ± 0.1316 | 6.7 × 10−5 | 9.8 × 10−3 |
| Skts9 | D16Mit2 (14.1) | 5.90 | 0.6590 ± 0.1263 | 1.8 × 10−7 | 4.3 × 10−5 |
| Skts10 | D17Mit7 (27.3) | 2.40 | −0.4256 ± 0.1280 | 8.8 × 10−4 | 0.095 |
*Data are shown as means ± SEM.
Figure 1.

Chromosomal positions of markers associated with cancer susceptibility identified in this and a previous report (5). There are 19 autosomal chromosomes shown, and their distances (in centimorgans) from the centromere are indicated. Markers within the gray regions show at least suggestive linkage to susceptibility or resistance. The asterisk (*) indicates that markers are provisional until further data are obtained. The locus Skts6 (chromosome 9) is very close to a previously reported promotion-susceptibility locus (19). Skts4 (20) is on chromosome 5 (42–69 cM) but may be distinct from Skts3, which is in a more proximal position (24 cM).
To look for linkage to carcinoma incidence, a logistic regression analysis was carried out as described (5) by using the carcinoma incidence as a dichotomous trait. This analysis showed strong linkage (logarithm of odds score = 4.3) to a marker only 7 cM distant from the previously detected resistance locus on chromosome 5 (Skts3), as well as suggestive linkage to a Mus spretus carcinoma-susceptibility locus at D6Mit9 on chromosome 6 (Skts11*) and a Mus spretus carcinoma-resistance locus at D7Mit14, only 3 cM telomeric from Skts2 (Fig. 1).
In summary, analysis of this backcross by using negative binomial and logistic regression methods has identified a total of seven significant and three suggestive linkages with either papilloma multiplicity or carcinoma incidence. Three of these loci are very close to, and probably identical to, the previously detected Skts1, 2, and 3 loci found by mapmaker qtl analysis. Although most of the loci correspond to Mus spretus resistance loci, as might be expected from the strong resistance phenotype, two of the Mus spretus loci (Skts10 and Skts11*, on chromosomes 17 and 6, respectively) nevertheless confer susceptibility to either papilloma or carcinoma development. Other studies have also shown that mice that have an overall resistance phenotype can harbor loci that confer tumor susceptibility (14–16).
Additive Effects of Loci That Determine Survival Time After Carcinoma Development.
Mice treated with initiators and promoters of carcinogenesis develop malignant carcinomas that become grossly detectable at any time between 4 and 18 months after initiation. Thereafter, the carcinomas can progress quite rapidly, leading to the death of the host after variable time periods depending on whether the tumor has spread to distant sites or grows locally to a size that necessitates the killing of the animal to avoid unnecessary suffering. We have taken the time between tumor initiation and death caused by carcinoma development as a measure of survival time, in an attempt to determine whether specific loci contribute to the risk of early death caused by malignancy. Animals (n = 83) that developed carcinomas and died or were killed during the course of the 18-month observation period were used for a study of linkage to loci that affect survival time. Malignant tumors were detected macroscopically between 16 weeks and 80 weeks, with a mean of 40 weeks after tumor initiation (Table 2).
Table 2.
Linkage between marker and survival time (Cox multivariate regression method)
| Marker | No. of subjects | Survival time, weeks
|
χ2‡ | P value | Corrected P value | ||
|---|---|---|---|---|---|---|---|
| 75%† | 50%† | 25%† | |||||
| Total | 83 | 31 | 38 | 45 | |||
| D6Mit15-D7Mit12-D17Mit7 | |||||||
| N-N : N-N : N-N | 11 | 34 | 39 | 44 | |||
| N-N : N-N : N-S | 13 | 29 | 39 | 46 | |||
| N-N : N-S : N-N | 9 | 44 | 56 | 68 | |||
| N-N : N-S : N-S | 6 | 27 | 32 | 45 | 25.22 | 5.1 × 10−8 | 3.7 × 10−5 |
| N-S : N-N : N-N | 10 | 29 | 32 | 45 | |||
| N-S : N-N : N-S | 14 | 24 | 31 | 32 | |||
| N-S : N-S : N-N | 5 | 40 | 42 | 44 | |||
| N-S : N-S : N-S | 15 | 36 | 39 | 47 | |||
| D6Mit15 (74.0 cM) | |||||||
| N-N | 39 | 32 | 41 | 55 | |||
| N-S | 44 | 30 | 33 | 40 | 8.70 | 0.0032 | 0.094 |
| D7Mit12 (66.0 cM) | |||||||
| N-N | 48 | 29 | 32 | 41 | |||
| N-S | 35 | 37 | 42 | 56 | 8.49 | 0.0036 | 0.11 |
| D17Mit7 (27.3 cM) | |||||||
| N-N | 35 | 34 | 41 | 55 | |||
| N-S | 48 | 29 | 33 | 44 | 3.56 | 0.0593 | 0.89 |
N-N, homozygous NIH/Ola Mus spretus; N-S, heterozygous NIH/Ola Mus spretus.
†75%, 50%, and 25% indicate the percentage of mice alive until the time (in weeks) after initiation.
‡χ2 values were obtained by the Cox regression method.
Previous genetic studies have shown strong positive cooperativity or negative interactions between loci that influence susceptibility to development of colon or lung tumors (14, 15, 17). We used a stepwise Cox multivariate regression method to look for linkage both to individual markers and for evidence of interactions between different loci associated with cancer survival in the mouse skin model. Mice carrying a combination of three alleles that confer longer survival (N-N:N-S:N-N, D6Mit15:D7Mit12:D17Mit7) survived significantly longer, and those with the corresponding susceptibility alleles at the same three loci (N-S:N-N:N-S haplotype) died or had to be killed substantially earlier than the mean survival time (P = 5.1 × 10−8; Table 2). The Kaplan–Meier survival curves in Fig. 2d confirm the strong association of three resistance alleles with long survival and of three susceptibility alleles with early mortality, whereas the other haplotypes show an intermediate pattern.
Figure 2.

Survival curves for mice of different genotypes at Skts12, Skts2, and Skts10. (a) The Kaplan–Meier survival curves for four groups with combinations of multiple resistance or susceptibility alleles: three resistance alleles (RRR), two resistance and one susceptibility (RRS), one resistance and two susceptibility (RSS), and three susceptibility alleles (SSS). Kaplan–Meier curves for survival time associated with homozygosity or heterozygosity at single markers of D6Mit 15 (b), at D7Mit12 (c), and at D17Mit7 (d) are also plotted by using the stata program. χ2 and P values estimated by Cox regression analysis are indicated in each graph. The dotted line curve shows the survival curve for the total of 83 mice. We also confirmed that the P values from the Cox model analysis were comparable to those obtained from nonparametric numerical tests (log-rank and Wilcoxon) and from the Kaplan–Meier analysis (data not shown).
Although the interaction between these loci was significant, only suggestive evidence was found of linkage between total survival time and genotype at single markers on chromosomes 6 and 7. Animals that were homozygous NIH/Ola (N-N) at D6Mit15 (Skts12) survived longer on average (P = 0.0032) than mice carrying a Mus spretus allele at this locus (N-S; Table 2; Fig. 2b). The inheritance of the Mus spretus allele at D7Mit12 (Skts2) conferred a longer survival time (P = 0.0036; Fig. 2c), whereas weak (below the suggestive level) association with decreased survival attributable to the Mus spretus allele was found with a marker on chromosome 17 [D17Mit7 (Skts10); P = 0.059; Fig. 2d; Table 2]. Two of the loci that affect survival (D7Mit12 and D17Mit7) were also found to be associated with carcinoma or papilloma incidence in the complete genome scan for linkage to these phenotypes. It is likely that the same loci are involved in determining both incidence and survival, because both the location and the direction of the effect on susceptibility are the same (i.e., Mus spretus alleles at D7Mit12 and D17Mit7 confer resistance and susceptibility, respectively, to tumor incidence and early mortality). The Mus spretus allele at the third survival locus (D6Mit15) also contributes positively to susceptibility, as does the locus Skts11* (D6Mit9) for carcinoma incidence. Although the same locus may be involved in both phenotypes, the relatively large distance between the markers that show the highest association with incidence or survival (D6Mit9 is at 36.5 cM and D6Mit15 is at 74 cM) suggests that more than one locus may be involved, and consequently, we have designated the survival locus as Skts12.
Linkage to Papilloma Latency.
Although we were unable to establish an exact timing for the appearance of carcinomas (carcinoma latency) because of the difficulty of estimating the stage of progression from papillomas, the same is not true for papilloma latency, because the time of first appearance could be accurately determined. We therefore carried out a survey for genomic loci associated with papilloma onset time by using the Cox proportional hazards model (Table 3). A minimum P value of 0.0024 was detected at D7Mit246 on chromosome 7. This linkage was the only suggestive one detected, either for a single marker or for interactions between loci linked to papilloma onset time, although weak linkage was detected to markers at D9Mit72 and D9Mit9 (48 cM; P = 0.0332 and 0.0365, respectively). The marker on chromosome 7 is within 13 cM of a locus with significant linkage to papilloma incidence, suggesting that the same loci are involved in both phenotypes. Interestingly, the combined haplotype of resistance or susceptibility alleles at all combinations of markers did not show any additive increase in association with papilloma onset, in contrast to the results observed for loci associated with survival.
Table 3.
Linkage between marker and papilloma onset (Cox multivariate regression method)
| Marker | No. of subjects | Survival time, weeks
|
χ2‡ | P value | Corrected P value | ||
|---|---|---|---|---|---|---|---|
| 75%† | 50%† | 25%† | |||||
| Total | 189 | 10 | 12 | 16 | |||
| D7Mit246 (15.0 cM) | |||||||
| N-N | 109 | 9 | 11 | 15 | |||
| N-S | 57 | 12 | 15 | 16 | 9.21 | 0.0024 | 0.071 |
†75%, 50%, and 25% indicate the percentage of mice alive until the time (in weeks) after initiation.
‡χ2 values were obtained by the Cox regression method.
Relationship Between Tumor Grade and Genotype.
The histological classification of tumors is sometimes used as a prognostic indicator of the probability of survival for individual cancer patients, although the usefulness of this parameter is by no means clear. The identification of different groups of animals with early or late death caused by carcinoma development enabled us to investigate the relationship between tumor grade and survival time. A total of 74 carcinomas were classified into the following groups: 24 grade I, 24 grade II, 9 grade III, and 17 grade IV carcinomas. When these were grouped according to genotype at the three loci linked to survival, it was seen that mice carrying a combination of three alleles that confer longer survival developed relatively high-grade cancers, whereas mice carrying the reverse combination of alleles that died or were killed earlier had relatively low-grade tumors (P = 0.0456; Fig. 3). The only individual marker that showed any linkage to tumor grade was D7Mit14 (P = 0.0128). We conclude that early mortality is not due to the early development of high-grade invasive carcinomas. The major factor that affects tumor grade seems to be the latency period during which carcinomas develop, because the animals that survive the longest have, at the time of death, on average higher-grade tumors.
Figure 3.

Histogram shows the percentage of cancers at each stage in mice of different genetic backgrounds as indicated. The white bar is grade 1; hatched bars are grade 2 and 3; and the black bar is grade 4. Genotypes are indicated at the bottom of the graph; abbreviations are defined for Fig. 2. The highest-grade tumors were found in the group of mice showing longest survival. The only marker that showed an association with tumor grade was D7Mit14. Animals carrying the Mus spretus allele at this locus had longer survival and more histologically advanced tumors.
Discussion
Cancer as a Combination of Multiple Phenotypes.
The development of cancer in mouse models can be quantified in a number of different ways. Cancer itself is a very complex phenotype comprising a number of distinct subphenotypes such as tumor multiplicity, tumor size or growth rate, degree of vascularization, histological grade, or invasive capacity. Theoretically, loci that control each of these subphenotypes (and many others) can be mapped in rodent cancer models, provided that functional polymorphisms exist in the parental strains chosen for the analysis. Most studies of cancer susceptibility in mice have compared tumor multiplicity and/or size in different strains of mice formed by the particular time point at which the animals are killed, but in many cases, the term “tumor” in fact refers to relatively benign lesions that develop over a fairly short time period after carcinogen exposure. Only in certain rare situations has linkage to loci that control tumor progression been investigated (3, 5), and no prior studies of linkage to survival time have been reported. In this study, we define the survival time as the length of time that genetically distinct animals survive after exposure to the same combination of chemical carcinogen and tumor-promoting agents. These mice were generally killed, because increasing tumor burden caused an adverse health status or because the tumor reached the maximum allowable size. The survival time after carcinogen exposure therefore comprises both tumor latency, i.e., the time until appearance of malignancy, and the time during which the carcinomas grow and begin to affect the overall health of the animals. Some of the animals had to be killed because they showed signs of weight loss or became moribund, possibly because of tumor spread. Metastasis, however, was not quantified in this particular study. Survival time of human cancer patients is generally assumed to be the length of time the patient remains alive after therapy, and our studies do not address the effect of host genetic background on this property. Nevertheless, survival time after carcinogen exposure is clearly a relevant parameter for studies of human cancer susceptibility, and identification of the loci that control this phenotype in the mouse may allow the development of approaches to retard or prevent the growth or progression of human malignancies.
In the present investigation, we have used a combination of negative binomial and logistic regression methods to investigate linkage to four distinct phenotypic traits: papilloma multiplicity and time of onset, as well as carcinoma incidence and overall survival time. This type of study has generally not been carried out with other model systems involving tumor development in internal organs such as the liver, lung, or colon, because of the overall effects of organ dysfunction on the viability of the host animal. We have detected a total of at least 10 loci that confer resistance or susceptibility to benign or malignant tumor multiplicity or incidence. In a separate genome scan for linkage to survival-controlling genes, a subset of three loci was identified, at least two of which also influence papilloma or carcinoma incidence. These loci on chromosomes 6, 7, and 17 specifically affect the survival time of mice with carcinomas. An additional marker on chromosome 7 was associated with the papilloma latency period in the same backcross. Because of the limited resolution of QTL mapping with these statistical approaches (about 10–20 cM for a population size of 200–500 mice and a QTL of intermediate strength; ref. 18), it is not possible to conclude definitively that the same loci are involved in the different phenotypes. However, their physical proximity and the similarity in the qualitative nature of their effects in promoting or repressing tumorigenicity support the conclusion that at least Skts2 (distal chromosome 7) and Skts10 (chromosome 17) affect multiple phenotypes. By the same reasoning, it seems likely that Skts1 affects both papilloma multiplicity and time of onset. The situation is less clear for chromosome 6, because a broad peak that covered several Mus spretus markers but centered at D6Mit9 (36.5 cM) showed suggestive linkage with carcinoma susceptibility, whereas a more discrete linkage at the distal marker D6Mit15 (74 cM) conferred higher risk of early mortality. It is therefore possible that more than one locus on chromosome 6 may be involved, and the distal survival locus has been designated Skts12. Further studies to identify these survival loci may provide us with tools for the prediction, prevention, or therapy of human cancers.
Comparison of Interspecific and Intraspecific Crosses.
Several other studies have been carried out on the susceptibility to skin tumor formation in intraspecific crosses between Mus musculus strains. In these cases, the phenotype of F1 hybrids generally is intermediate between the two parental strains, in contrast to the dominant resistance phenotype seen in the interspecific Mus spretus/Mus musculus crosses. These studies identified a “promotion-susceptibility” locus on chromosome 9 (D9Mit271; 48 cM; ref. 19) close to the Skts6 locus (D9Mit76; 49 cM) and a locus designated Skts4 on chromosome 5 (20). Although direct comparisons are complicated by differences in the methodologies used, these results involving intraspecific crosses contrast with the relatively large number of strong modifier loci detected in the Mus spretus/Mus musculus backcross described in this report. This is probably due to the substantial genetic divergence between these two species. It is therefore possible that interspecific backcrosses involving Mus spretus will provide a convenient route to the detection of multiple dominant resistance or modifier genes for the development of tumors of the skin and other tissues.
Of the candidate genes located in the vicinity of the skin tumor modifier loci identified in this report, of particular interest for the discussion here is the coincidence between the localizations of the subset of three loci that affect survival and three members of the cyclin-dependent kinase inhibitor family. Skts2 is close to p57Kip2 on distal chromosome 7 (21); Skts10 lies near p21Waf1 on chromosome 17 (22); and Skts12 is in a region of chromosome 6 that is syntenic with human chromosome 12p13, where p27Kip1 has been localized (23). The mouse pulmonary adenoma-susceptibility gene Pas1 has been localized in this region (24), and association studies have suggested that a gene or genes on human chromosome 12p can influence lung-cancer incidence (25). The expression level of human p27Kip1 has been shown to be a good prognostic indicator of survival in cases of human breast or lung cancer (26), and haploinsufficiency of p27Kip1 predisposes mice to the development of a number of carcinogen-induced tumors (27). Additional evidence exists for an association between a polymorphism in the p21/Waf1 gene and human cancer susceptibility (28). Further studies of the combinatorial effects of alleles of the cyclin-dependent kinase inhibitor genes on cancer survival are warranted.
Most studies on prediction of survival of human cancer patients have concentrated on the identification of somatic genetic alterations or gene expression patterns in tumors that may be correlated with prognosis. The results we have presented show that the host genetic background is a major determinant of carcinoma growth rate and survival. The characterization of similar host genetic factors in humans would be a prerequisite for the assessment of cancer risk for individual patients and eventually for the development of patient-based strategies for prevention or treatment.
Acknowledgments
We would like to thank Sheila Bryson, Frances Fee, and Kenneth Brown for useful discussions and technical support. H.N. is grateful to Michio Ogawa for his encouragement and support of this work. We are very grateful to Stephen Bell and the Cancer Research Campaign Beatson Labs Animal House staff for excellent assistance with animal husbandry. This work was funded by a grant from the Cancer Research Campaign (U.K.), and further support was provided by Onyx Pharmaceuticals and the University of California San Francisco Cancer Center. J.-H.M. was supported by a grant to A.B. from the European Community.
Abbreviations
- NSP
(NIH/Ola × Mus Spretus)F1 backcross
- QTL
quantitative trait locus
- cM
centimorgan
- N-N
homozygous NIH/Ola Mus spretus
- N-S
heterozygous NIH/Ola Mus spretus
References
- 1.Peto J. In: Predisposition to Cancer. Cairns J, Lyon J L, Skolnick M, editors. Plainview, NY: Cold Spring Harbor; 1980. pp. 203–213. [Google Scholar]
- 2.Ponder B A J. Trends Genet. 1990;6:213–218. doi: 10.1016/0168-9525(90)90181-5. [DOI] [PubMed] [Google Scholar]
- 3.Dragani T A, Canzian F, Pierotti M A. FASEB J. 1996;10:865–870. doi: 10.1096/fasebj.10.8.8666163. [DOI] [PubMed] [Google Scholar]
- 4.Balmain A, Nagase H. Trends Genet. 1998;14:139–144. doi: 10.1016/s0168-9525(98)01422-x. [DOI] [PubMed] [Google Scholar]
- 5.Nagase H, Bryson S, Cordell H, Kemp C J, Fee F, Balmain A. Nat Genet. 1995;10:424–429. doi: 10.1038/ng0895-424. [DOI] [PubMed] [Google Scholar]
- 6.Kruszewski F H, Conti C J, DiGiovanni J. Cancer Res. 1987;47:3783–3790. [PubMed] [Google Scholar]
- 7.Drinkwater N, Klotz J H. Cancer Res. 1981;41:113–119. [PubMed] [Google Scholar]
- 8.Kruglyak L, Lander E S. Genetics. 1995;139:1421–1428. doi: 10.1093/genetics/139.3.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kendall M G, Sutart A. Advanced Theory of Statstics. New York: Hafner; 1973. [Google Scholar]
- 10.Cox D R. J R Stat Soc. 1972;34:187–220. [Google Scholar]
- 11.Cox D R, Oakes D. Analysis of Survival Data. London: Chapman & Hall; 1984. [Google Scholar]
- 12.Fleiss J L. Statistical Methods for Rates and Proportions. 2nd Ed. New York: Wiley; 1981. [Google Scholar]
- 13.Lander E S, Schork N J. Science. 1994;265:2037–2047. doi: 10.1126/science.8091226. [DOI] [PubMed] [Google Scholar]
- 14.Fijneman R, de Vries S, Jansen R, Demant P. Nat Genet. 1996;14:465–467. doi: 10.1038/ng1296-465. [DOI] [PubMed] [Google Scholar]
- 15.van Wezel T, Stassen A, Moen C, Hart A, van der Valk M, Demant P. Nat Genet. 1996;14:468–470. doi: 10.1038/ng1296-468. [DOI] [PubMed] [Google Scholar]
- 16.Lee G, Bennett L, Carabeo R, Drinkwater N R. Genetics. 1995;139:387–395. doi: 10.1093/genetics/139.1.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fijneman R, Jansen R, van der Valk M, Demant P. Cancer Res. 1998;58:4794–4798. [PubMed] [Google Scholar]
- 18.Tanksley S D. Annu Rev Genet. 1993;27:205–233. doi: 10.1146/annurev.ge.27.120193.001225. [DOI] [PubMed] [Google Scholar]
- 19.Angel J M, Beltran L, Minda K, Rupp T, DiGiovanni J. Mol Carcinog. 1997;20:162–167. doi: 10.1002/(sici)1098-2744(199710)20:2<162::aid-mc2>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- 20.Mock B A, Lowry D T, Rehman I, Padlan C, Yuspa S H, Hennings H. Carcinogenesis. 1998;19:1109–1115. doi: 10.1093/carcin/19.6.1109. [DOI] [PubMed] [Google Scholar]
- 21.Hatada I, Mukai T. Nat Genet. 1995;11:204–206. doi: 10.1038/ng1095-204. [DOI] [PubMed] [Google Scholar]
- 22.Huppi K, Siwarski D, Dosik J, Michieli P, Chedid M, Reed S, Mock B, Givol D, Mushinski J F. Oncogene. 1994;9:3017–3020. [PubMed] [Google Scholar]
- 23.Polyak K, Lee M H, Erdjument-Bromage H, Koff A, Roberts J M, Tempst P, Massague J. Cell. 1994;78:59–66. doi: 10.1016/0092-8674(94)90572-x. [DOI] [PubMed] [Google Scholar]
- 24.Gariboldi M, Manenti G, Canzian F, Falvella F S, Radice M T, Pierotti M A, Porta G D, Binelli G, Dragani T A. Nat Genet. 1993;3:132–136. doi: 10.1038/ng0293-132. [DOI] [PubMed] [Google Scholar]
- 25.Manenti G, De Gregorio L, Pilotti S, Falvella F, Incarbone M, Ravagnani F, Pierotti M, Dragani T A. Carcinogenesis. 1997;18:1917–1920. doi: 10.1093/carcin/18.10.1917. [DOI] [PubMed] [Google Scholar]
- 26.Catzavelos C, Tsao M, DeBoer G, Bhattacharya N, Shepherd F, Slingerland J M. Cancer Res. 1999;59:684–688. [PubMed] [Google Scholar]
- 27.Fero M, Randel E, Gurley K, Roberts J, Kemp C J. Nature (London) 1998;396:177–180. doi: 10.1038/24179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Facher E, Becich M, Deka A, Law J C. Cancer. 1997;79:2424–2429. doi: 10.1002/(sici)1097-0142(19970615)79:12<2424::aid-cncr19>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]